

Abstract
Sporadic human mismatch repair-deficient colorectal cancers (MMR-deficient CRC) account for ∼12.5% of all cases of colorectal cancer. MMR-deficient CRCs are classically characterized by right sided location, multifocality, mucinous histology and lymphocytic infiltration. However, tumors in germline MMR deficient mouse models lack these histopathologic features. Mice lacking the heterotrimeric G-protein alpha subunit Giα2 develop chronic colitis and multifocal, right sided cancers with mucinous histopathology, similar to human MMR-deficient CRC. Young Giα2-/- colonic epithelium has normal MMR expression but selectively loses MLH1 and consequently PMS2 expression following inflammation. Giα2-/- cancers have microsatellite instability. MLH1 is epigenetically silenced, not by promoter hypermethylation, but by decreased histone acetylation. Chronically inflamed Giα2-/- colonic mucosa contains patchy hypoxia, with increased crypt expression of the hypoxia markers DEC-1 and BNIP3. Chromatin immunoprecipitation identified increased binding of the transcriptional repressor DEC-1 to the proximal MLH1 promoter in hypoxic YAMC cells and colitic Giα2-/- crypts. Treating Giα2-/- mice with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) significantly decreased colitis activity, and rescued MLH1 expression in crypt epithelial cells that was associated with increased acetyl H3 levels and decreased DEC-1 binding at the proximal Mlh1 promoter, consistent with an HDAC-dependent mechanism. These data link chronic hypoxic inflammation, epigenetic MMR protein downregulation, the development of MMR-CRC, and is the first mouse model of somatically acquired MMR-deficient CRC.
Keywords: Mismatch repair deficient colon cancer, Inflammatory Bowel Disease, Epigenetic silencing, Hypoxia, Histone deacetylase inhibitors
Introduction
DNA mismatch repair-deficient colorectal cancers (MMR-CRC) have distinct features that distinguish them from other CRC types (1). These include increased frameshift mutation rates (commonly called microsatellite instability (MSI)), multi-focality, increased likelihood of right-sided colon cancer location, non-polyposis pathology, mucinous histology and the presence of a Crohn's-like lymphocytic infiltrate in tumors (2, 3).
MMR-deficient CRCs arise through three distinct genetic mechanisms. First, monoallelic germline MMR mutation and somatic loss of the second MMR allele causes Hereditary Non Polyposis Colon Cancer, also known as Lynch Syndrome (LS). LS carriers body tissues are MMR-proficient because of the other allele. As predicted by the Knudson hypothesis, the 2nd MMR gene allele is somatically lost in some cells, which then become MMR-deficient and transform into CRC and other cancers. The second mechanism is biallelic germline constitutive MMR gene mutation, where all body tissues are MMR-deficient. These patients have a different clinical phenotype, with very early-onset hematological, brain, and small intestinal cancers (4-6). While CRCs can develop, penetrance is low and arise more commonly in the left colon (4, 5).
The third genetic mechanism, de novo spora dic MMR-deficient CRCs, account for ∼12.5% of CRC (6). Both MMR alleles are somatically mutated or epigenetically inactivated in some cells, which transform into cancer. Mlh1 promoter hypermethylation is the most common cause of sporadic MMR-deficient CRC (7-9). In vitro studies have shown that epigenetic Mlh1 inactivation can also occur via hypoxia-induced transcriptional repression of the proximal Mlh1 promoter by histone deacetylases (9). However, whether this mechanism is relevant to in vivo CRC tumorigenesis is unknown.
Multiple biallelic MMR germline knockout mouse models have been created (Table S1, supplemental data). These mutations primarily cause lymphomas and small intestine adenomas (10-16) Only rarely do these models develop CRC, and mucinous, non-polyposis or proximal colon CRCs essentially never arise. These models are therefore more similar to human biallelic germline constitutive MMR deficiency than somatically acquired MMR deficiency syndromes.
Giα2 is a heterotrimeric G-protein alpha subunit whose deletion in mice leads to spontaneous colitis and non-polyposis, right sided, multifocal CRCs with mucinous histology and a Crohn's-like inflammatory infiltrate (17-19). The similarities between Giα2-/- mouse CRC and somatically acquired human MMR-deficient CRC (Table S1) led us to investigate whether acquired MMR silencing might contribute as a mechanism of CRC tumorigenesis in Giα2-/- mice. We found that MLH1 and PMS2 protein levels in crypts are initially normal but are suppressed following the onset of hypoxic colitis. Hypoxia induces colonic expression of the bHLH transcriptional repressor DEC-1 and enhances its binding to the proximal Mlh1 promoter, thereby repressing MLH1 expression and leading to MSI. Treatment of Giα2-/- mice with histone deacetylase inhibitor (HDACi) decreases colitis and relieves epigenetic repression of MLH1 expression. In summary, we describe the first mouse model of somatically acquired MMR deficiency, and demonstrate that hypoxia-induced Mlh1 silencing may be another important epigenetic mechanism contributing to the development of MMR-CRC.
Materials and Methods
Mice
WT and Giα2-/- mice on a crossbred 129Sv/C57BL6 background were genotyped as previously described (19). All experiments were approved by the IACUC at UC Irvine.
In vivo assessment of apoptosis induction
Age-matched WT and Giα2-/- mice were injected i.p. with 20 mg/kg of 1,2 Dimethylhydrazine (1,2 DMH) for 24 hours. 5 μm sections of fixed control and DMH-treated colons were stained for apoptotic cells using an Apoptag staining kit (Chemicon, Temecula, CA). The data are expressed as the mean +/- SEM of the normalized ratio of TUNEL+/PI+ cells.
Colonic crypt isolation, SDS-PAGE and Western blotting
Rinsed small intestine and colon segments were everted on a Labconco vibratory stirrer and shaken in 30mM EDTA in HBSS. Crypts liberated between 20 and 40 minutes (>98% pure by cytospin analysis) were used for western blotting and qRT-PCR. Antibodies used include MLH-1 (BD Pharmingen, clone G168-728), PMS2 (BD Pharmingen, clone A16-4), MSH2 (Calbiochem, clone FE11), MSH-6 (Abcam, clone 44) DEC-1 (Santa Cruz, clone S-8), BNIP-3 (Santa Cruz, clone Ana40) and Beta actin (Sigma, clone 20-33).
Immunohistochemistry
WT (n=24) and Giα2-/- (n = 22) colon sections were microwaved (20 minutes) in citrate buffer. After peroxidase, biotin, and endogenous IgG blocking, anti-MMR antibodies (described above) were added overnight at 4°C then developed using peroxidase-based detection. 8 mucinous and 17 non-mucinous carcinomas located throughout the colon were stained.
Laser capture microdissection of mouse crypt epithelium and colon cancer cells
Formalin-fixed, deparaffinized WT and Giα2-/- colon sections on PENfoil slides (Leica), were microdissected using a Leica AS-LMD. Genomic DNA from regions of WT or inflamed Giα2-/- crypts and Giα2-/- cancers was purified using a PureGene DNA isolation kit (Qiagen) and used for MSI and promoter methylation studies
Microsatellite instability analysis
Individual microsatellite templates were analyzed for insertion/deletion mutations by single-molecule PCR using standard protocols (20, 21). Briefly, DNA isolated from laser capture-microdissected Giα2 -/- and WT colons was diluted to 0.5–1.5 single-genome equivalents followed by PCR amplification by mononucleotide microsatellite markers A33 and L24375.
DNA methylation analysis
Genomic DNA (650 ng) from WT and colitic Giα2-/- crypts (n=7 each) was modified using an Imprint® DNA Modification Kit (Sigma). PCR primers, complementary to the modified or to the unmodified sequence, were designed for the proximal Mlh1 promoter region. For the analysis of the proximal area of the Mlh1 promoter, a region expanding from -324bp to -288bp of the Mlh1 promoter was amplified by hemi-nested PCR using the following conditions:
Unmethylated Primers: 5′- ACTACAATCCTACGCCGTCT-3′(sense), 5′- TTCGACCGACTACAATCCTA -3′ (antisense for 1st PCR), 5′- TTTAGTTTGGTGTCGGGTC -3′ (antisense for 2nd PCR). Methylated Primers: 5′- AACTACAATCCTACACCATCTTC-3′ (sense), 5′- ACTTCAACCAACTACAATCCTA -3′ (antisense for 1st PCR), 5′- TTTTTAGTTTGGTGTTGGGTT- 3′ (antisense for 2nd PCR). Bisulfite-treated DNA was subjected to nested PCR and subsequent sequence verification. One microliter of the 1st PCR product was used as template for the second PCR reaction.
Semi-quantitative real-time PCR
Real-time PCR was performed using an Applied Biosystems Step One Plus instrument using SYBR Green detection reagents. Oligonucleotide sequences were as follows: murine MLH1 forward: 5′-GTTTTACTCCATTCGGAAGCAGT-3′; murine MLH1 reverse 5′-TGTGAGCGGAAGGCTTTATAGAT-3′; murine PMS2 forward 5′-ACCTCACGCAGGTTGACTT-3′; murine PMS2 reverse 5′- AGTCGAGTCCCAACGCTTG -3′; murine BNIP3 forward: 5′- CGAAGCGCACAGCTACTCTC-3′; murine BNIP3-reverse: 5′-GCCTTCCAATGTAGATCCCCAAG-3′; murine DEC-1 forward: 5′-TACAAGCTGGTGATTTGTCGG-3′; murine DEC-1 reverse: 5′-CTGGGAAGATTTCAGGTCCCG-3′. Normalization was performed using murine RPA2 (forward 5′ ATACCCAGACAACAGAGGG-3′; reverse: 5′- GCCAGTCCGCTCAATCACC-3′)
In vitro hypoxia treatment
Subconfluent cultures of YAMC cells (a kind gift of Dr. Robert Whitehead) were grown in RPMI with 5% FBS, 100 ng/μl IFNγ, 1× Insulin/Transferrin/Selenium and incubated in a Biospherix hypoxia chamber at 37°C in 1% O2 for 24 hours and total RNA analyzed by q-RT-PCR.
SAHA treatment of Giα2-/-mice
Groups of 32-36 week old Giα2-/- mice (n=4) were treated with daily gavage feedings with a 5 mg/ml solution of suberoylanilide hydroxamic acid (Cayman Chemical) solubilized in PBS containing a 5-molar excess of β-hydroxypropyl cyclodextrin (Sigma) for 7 days (final dose 50 mg/kg). Control animals received only cyclodextrin solution. Colons were harvested and crypts isolated as described above, and analyzed for MLH1 expression by western blot analysis.
Chromatin Immunoprecipitation
Cells and crypts were crosslinked in 1% formaldehyde for 10 minutes and then lysed in SDS lysis buffer and sonicated to a fragment length of 300 to 1000 bp. The sonicate was precleared and the supernatant incubated overnight at 4°C with the following antibodies: Anti-DEC-1 (Santa Cruz, clone S-8), Anti-acetyl Histone H3 (Millipore #06-599). Rabbit IgG was used as a negative control Ab. Following immunoprecipitation, washing, crosslink reversal, and DNA cleanup, the proximal Mlh1 promoter region was amplified for an optimized number of cycles using the following primers: Forward: 5′- GTTTAGAGCGGGACAGAGAT-3′; reverse 5′-CCTGCTACAAACGCCATATT-3′.
Histopathological assessment of Giα2-/- and SAHA-treated mice
The severity of colitis and assessment of cancer histology for Giα2-/- mice was performed as previously described (19).
Statistical analyses
For MSI analysis, statistically significant mutation frequency differences between genomic DNA from different genotypes were tested by Fisher's Exact Test. Statistical significance was assessed using Student's paired t-test for linear data and the Mann–Whitney U-test for nonlinear data.
Results
Previously, we detailed the natural history and histopathology of Giα2-/- CRC (19). We have now extended our CRC analysis in a cohort (N=47) of Giα2-/- mice (Table 1). Among Giα2-/- mice sacrificed after 20 weeks of age, 89.8% had at least one CRC present, with a mean tumor incidence of 1.89 CRCs/mouse. In contrast, germline Mlh1 and Msh2 MMR-deficient mice rarely develop colonic adenomas or CRC; fewer than 50% develop small intestine adenomas by 12 months, and only rarely develop CRC (22). Giα2-/- mice CRCs were predominantly right-sided (75%). Like human MMR-deficient CRCs, all Giα2-/- CRCs were non-polypoid and had a Crohn's like lymphocytic infiltrate. (See Supplemental Figure S3 for a histopathologic comparison of a LS CRC vs. Giα2-/- CRC) Mucinous CRC's are more common in MMR-deficient CRCs (15-25%) (23-25) than sporadic CRC patients (5-10%). Giα2-/- tumors develop a higher rate of mucinous CRCs (29%) than has previously been reported in any mouse model of CRC. In summary, Giα2-/- CRC are more similar to LS and sporadic MMR deficient CRC (26) than the existing MMR deficient mouse models (Table S1) or MMR-proficient CRC.
Table 1. Histopathology of CRC's in Giα2-/- mice.
Colons from 47 mice with cancer between 12 and 53 weeks of age were evaluated for the number, location and histologic differentiation of CRC's.
| Location/His tology | Non-mucinous | Mucinous | Total No. | % of total |
|---|---|---|---|---|
| Cecum | 39 | 7 | 46 | 51.7 |
| Proximal | 15 | 7 | 22 | 24.7 |
| Mid | 9 | 5 | 14 | 15.7 |
| Distal | 0 | 7 | 7 | 7.9 |
| Totals | 63 | 26 | 89 | 100 |
| % of Total | 70.7 | 29.3 | ||
Acquired suppression of MLH1 and PMS2 Protein in Giα2 Knockout Colon Epithelium
Epigenetic silencing of MLH1 causes the majority of sporadic MMR-defective CRC. (27, 28). Since Giα2-/- CRC phenocopy LS and sporadic MMR-deficient CRC, we stained colonic epithelium from young vs. old WT and Giα2-/- mice for MMR expression. Giα2-/- colons from young (<6 weeks) mice have no inflammation or CRC (19), and IHC performed on 8-week old Giα2-/- mice showed WT levels of MMR proteins MLH1, PMS2, MSH2, and MSH6 (Figure S1). By 40 weeks of age, IHC of Giα2-/- colitis and CRC showed a consistent, marked reduction in MLH1 and PMS2 protein levels in inflamed and cancerous Giα2-/- mucosa (Figure S2), both in the epithelium and fixed stromal cells (Panels F,J,G,K). MLH1 expression was preserved in infiltrating lymphocytes in the Giα2-/- tissues (white boxed region, panel F). PMS2 is an obligate heterodimeric partner of MLH1. With MLH1 silencing, un-dimerized PMS2 protein becomes unstable and is degraded (20). Expression of the MSH2 and MSH6 MMR proteins are not affected by MLH1 silencing.
To confirm the IHC results, we quantified MMR protein expression in purified epithelia from WT and inflamed Giα2-/- colons. In young mice, MMR protein expression was unchanged (Figure 1A, top). However, after 20 weeks, total crypt MLH1 and PMS2 were markedly decreased in Giα2-/- colonic epithelial cells. (Figure 1A, bottom) gain, consistent with the immunohistochemical result (Figure 1S-H), MSH2 expression was unchanged.
Figure 1. A.) Acquired suppression of MLH1 and PMS in Giα2-/- crypts.
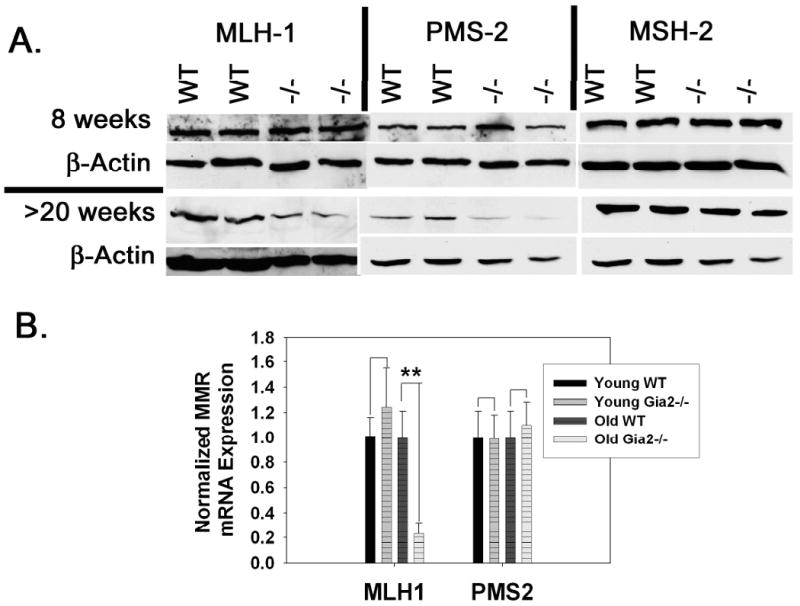
Purified 8 week old (top) and older than 20 week (bottom) WT and Giα2-/- crypt epithelial cells were immunoblotted for MLH1, PMS2, and MSH2. B.) qPCR for MLH1 and PMS2 in young and old crypts (n=3 each) reveals a significant downregulation of MLH1 in older Giα2-/- crypts. Results shown are representative of three separate experiments. **, p<0.01.
To corroborate the acquired loss of MLH1 expression between 8 and 20 weeks of age, qPCR analysis of MLH1 expression in 8 and 20-week old Giα2-/- crypt epithelium revealed normal MLH1 expression at the early timepoint, but a ∼75% drop in MLH1 expression in inflamed 20 week old Giα2-/- crypts (Figure 1B). Giα2-/- crypt PMS2 mRNA levels were not significantly different from WT at either timepoint.
In summary, both IHC and Western analysis show that MLH1 and PMS2 protein levels are markedly reduced in inflamed Giα2-/- colon. This decrease is somatically acquired, as it follows the onset of inflammation. MSH2 and MSH6 levels are not affected. Mlh1 is transcriptionally repressed, while PMS2 reduction is posttranslational, likely in response to decreased MLH1 protein levels.
MSI in inflamed Giα2-/- mucosa
MMR-deficient CRCs include increased MSI and impaired DNA damage response. Previously we had demonstrated hypomorphic MSI in Mlh3-/- and Pms2-/- mice (because of redundancy between Mlh3 and Pms2) and full-blown MSI in Mlh3-/-; Pms2-/- mice, indistinguishable from Mlh11-/- mice (29). Using mouse microsatellite markers previously studied in other MMR deficient mouse models (16, 30), we performed MSI testing on laser capture-microdissected Giα2-/- tumor DNA and WT colon epithelium DNA. Consistent with previous studies of both mouse and human, almost all MSI instability was caused by mononucleotide repeat deletions that shortened microsatellites (31). For each marker tested, Giα2-/- CRCs had significantly increased MSI vs. WT (both p<0.0001) (Table 2). At the same time, young precolitic Giα2-/- colon showed no MSI (data not shown).
Table 2. Microsatellite analysis.
Total mutation frequencies were determined using unique mouse mononucleotide microsatellite markers A33 and L24375 (A23). The total number of mutant size variant alleles divided by the total number of alleles is shown for each microsatellite marker. We compared the mutation frequency in colonic tumor DNA from Giα2-/-, Mlh3-/- Pms2-/-or normal wildtype tissue among pairs of genotypes. A Fisher's Exact Test determined statistically significant differences. * Considered significant when compared to Wildtype controls; P<0.0001.
| MSI Marker | Genotype | #N | # Variant Alleles | Total Mutation Freq. | p-value vs WT |
|---|---|---|---|---|---|
| A33 | WT | 235 | 8 | 0.03 | |
| Giα2-/- | 229 | 62 | 0.27 | < 0.0001 | |
| MLH3-/- | 235 | 107 | 0.46 | ||
| PMS2-/- | |||||
| L24375 (A23) | WT | 248 | 4 | 0.02 | |
| Giα2-/- | 275 | 91 | 0.33 | < 0.0001 | |
| MLH3-/- | 258 | 104 | 0.4 | ||
| PMS2-/- | |||||
MLH1 promoter methylation is not increased in Giα2-/- mice
Epigenetic silencing of the proximal Mlh1 promoter by CpG methylation is a common mechanism for sporadic MMR-deficient CRC tumorigenesis (32). We therefore studied the orthologous CpG island in the mouse Mlh1 promoter in Giα2-/- inflamed colon. Using bisulfate-treated WT and colitic Giα2-/- crypt gDNA, we observed no significant differences in the intensity of methylated vs. unmethylated PCR products between WT colon and Giα2-/- inflamed colon samples (Figure S4). Methylation of the proximal Mlh1 promoter does not appear to contribute to epigenetic silencing of Mlh1 in Giα2-/- inflamed colons and CRC.
Inflamed Giα2-/- colons and CRCs are hypoxic
The lack of promoter hypermethylation in the setting of suppressed MLH1 expression led us to test other potential mechanisms shown to silence Mlh1. In vitro studies have shown that hypoxia can repress MLH1 mRNA expression in a histone deacetylase-dependent manner. (33). Because other mouse models of colitis have been shown to have mucosal hypoxia (34), we evaluated whether the Giα2-/- inflamed colon might be hypoxic, and whether hypoxic repression of MLH1 might be occurring in vivo. DEC-1 and BNIP3 are well characterized markers of hypoxia (35, 36). First, we validated DEC-1 (37) and BNIP3 (38) as hypoxic markers in colonocytes by incubating YAMC cells in vitro under hypoxia (1% O2) or normoxia for 24 hours. Consistent with previous studies in human colon cancer cell lines and other cells types, exposure to hypoxic conditions resulted in a significant upregulation of BNIP3 (9.00 +/- 1.07 fold p = 0.009) and DEC-1 (2.11 +/- 0.27 fold, p = 0.033) mRNA expression. Western analysis confirmed proportional increases in DEC-1 and BNIP3 protein in hypoxic YAMC cells (Figure 2A), confirming that DEC-1 and BNIP3 are inducible markers of hypoxia in mouse colonocytes. Next, we determined that these hypoxia markers were upregulated in vivo in colonocytes in crypts from colitic Giα2-/- mice compared to WT mice.
Figure 2. Inflamed Giα2-/- crypts are hypoxic.
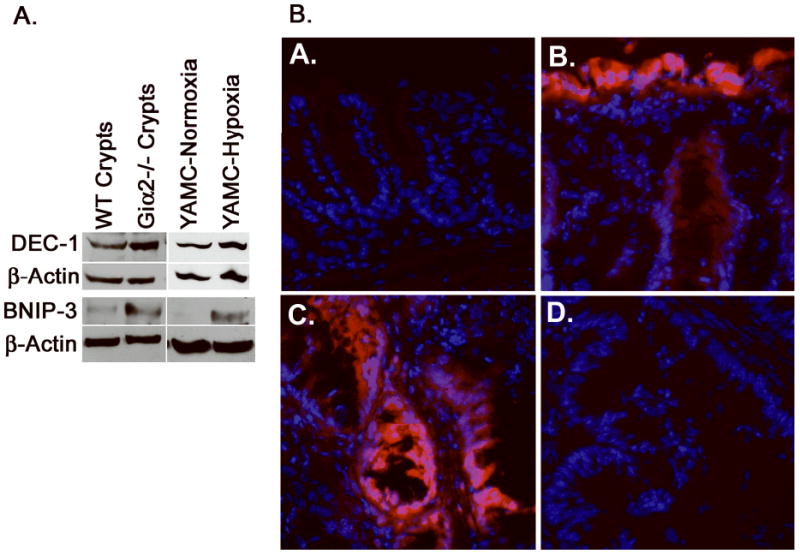
(A), YAMC cells grown in 1% O2 (right) upregulate the hypoxia markers DEC-1 and BNIP3, which are also upregulated in purified crypt lysates from colitic Giα2-/- mice (left). The data shown are representative of three separate YAMC and crypt isolation experiments. (B), Frozen sections from EF5-treated WT and Giα2-/- mice (age 20-27 weeks) were stained with Cy3-conjugated anti EF5. WT colon (A) had minimal EF5 staining, while colitic Giα2-/- mucosa (B) was profoundly hypoxic in surface epithelium and in some deeper crypt cells. EF5 staining of Giα2-/- cancers (C.) was patchy, and involved primarily malignant epithelial cells and the adjacent stroma. As a negative control, pre-adsorbed anti EF-5 (D.) revealed no staining.
To demonstrate directly that Giα2-/- inflamed colons are hypoxic, we used EF5 (kindly provided by Dr. Cameron Koch), a well-characterized direct probe of tissue hypoxia (39). We injected WT and colitic Giα2-/- mice with EF5. Four hours after IV injection, in WT colon tissues the surface epithelium contains a faint diffuse staining, difficult to capture in unenhanced immunofluorescence images (Figure 2B, panel A). In contrast, in Giα2-/- inflamed colon, hypoxia is greatly increased in the surface epithelium and in the deeper crypts in Giα2-/- mice. Notably, hypoxia is particularly prominent in areas with cancer and adjacent underlying stroma (panels B,C). Treatment of the same cancer sample with pre-competed anti-EF5 abrogated staining (D). The corresponding areas following hematoxylin counterstaining are shown in Figure S6. The pattern of EF5 staining seen in inflamed Giα2-/- colons is similar to that seen in the mouse model of TNBS colitis (34). In summary, DEC-1 and BNIP3 are biomarkers of hypoxia in mouse colonocytes and in Giα2-/- inflamed colon epithelium. Furthermore, EF5 dye studies confirm that, similar to TNBS-induced colitis, EF5 trapping in hypoxic tissue is increased in Giα2-/- inflamed colon.
Hypoxia modulates acetyl-histone H3 levels and DEC-1 association with the proximal MLH1 promoter in vitro and in vivo
Low levels of acetylated histone H3 are associated with transcriptional silencing of many genes. (40) Using chromatin immunoprecipitation (ChIP) we tested whether hypoxia decreases acetyl-H3 content at the proximal Mlh1 promoter in YAMC cells, and whether treatment with HDAC inhibitors (HDACi) can increase acetylated H3 levels. Figure 3A shows that hypoxia suppressed histone H3 acetylation at the proximal Mlh1 promoter, while pretreatment with trichostatin A enhanced Mlh1 promoter histone H3 acetylation in hypoxic (as well as normoxic) culture conditions. This indicates that hypoxia decreases acetyl-H3 content at the Mlh1 promoter in vitro which can be rescued by HDACi treatment. Consistent with other cell lines rendered hypoxic in vitro, hypoxia suppresses MLH1 expression at the mRNA and protein level by qPCR and western blot, respectively (data not shown).
Figure 3. Hypoxia modulates histone H3 acetylation, and DEC-1 and HDAC1 binding to the proximal MLH1 promoter.
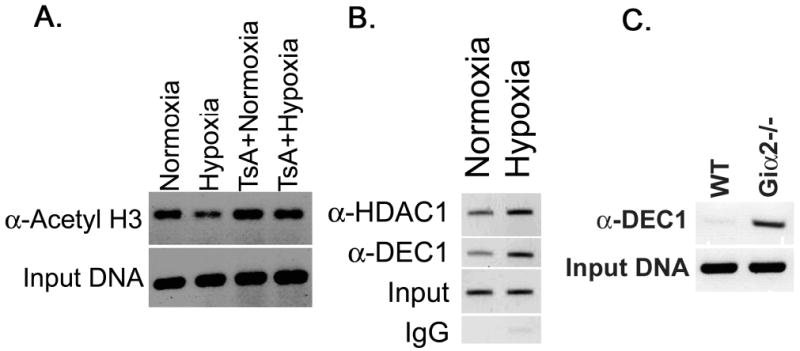
Hypoxia induces histone H3-deacetylation at the MLH1 promoter in a HDAC-dependent manner (A), enhances DEC-1 and HDAC1 binding to the proximal MLH1 promoter in YAMC cells (B) In colitic Giα2-/- crypts, DEC-1 binding to the MLH1 promoter is also enhanced.
Hypoxia is known to enhance DEC-1 binding to the proximal Mlh1 promoter in cell culture, suppressing Mlh1 expression (41). We performed chromatin immunoprecipitation on genomic DNA from hypoxic YAMC cells and from colonic crypt epithelium from 28 week-old WT and colitic Giα2-/- mice, and analyzed the proximal murine Mlh1 promoter using oligos spanning this region. We identified a marked increase in DEC-1 at the proximal Mlh1 promoter in hypoxic YAMC cells as well as Giα2-/- crypts mice in vivo (Figure 3B,C). Consistent with previous reports indicating DEC-1's ability to bind HDAC1 (42), we also found an increase in HDAC1 bound to the Mlh1 promoter in hypoxic YAMC cells. In hypoxic YAMC cells and Giα2-/- inflamed crypts, DEC-1 occupancy at the proximal Mlh1 promoter is increased, as is HDAC1 binding that may be responsible for decreased acetyl histone H3 levels.
HDAC inhibitors relieve hypoxia, reduce inflammation and normalize MLH1 expression in Giα2-/- CRCs
Recent studies have demonstrated both an anti-inflammatory and anti-neoplastic effect of HDAC inhibitors in mouse models of chronic colitis and colitis-associated CRC (43, 44). These results suggest that chronic inflammation alters histone acetylation in the inflamed colon, and this epigenetic change may contribute to tumorigenesis. Since HDAC inhibitors have been shown to block hypoxia-induced MLH1 silencing in vitro (33), we tested whether HDACi treatment could enhance MLH expression in vivo. We gavage-fed groups of 33-36 week old Giα2-/- mice with 50 mg/kg SAHA daily for 8 days, and evaluated MLH1 expression in crypt epithelium by western blot and qPCR. SAHA caused a significant reduction in colitis severity (Figure 5A), using a previously published histologic activity index (45). We found significant increases in MLH1 mRNA and protein expression (Figure 5B,C, and Figure S5). Overall acetyl histone H3 levels in SAHA-treated crypts were increased (data not shown). By ChIP analysis, SAHA treatment enhanced acetylated histone H3 levels and decreased DEC-1 occupancy at the Mlh1 promoter (Figure 4D). These experiments indicate that HDACis relieve Mlh1 transcriptional repression in chronically inflamed Giα2-/- crypts via epigenetic alterations of the Mlh1 promoter involving histone acetylation and DEC-1.
Figure 4. HDACi treatment improves colitis and rescues MLH1 expression by decreasing DEC-1 occupancy at the proximal MLH1 promoter.
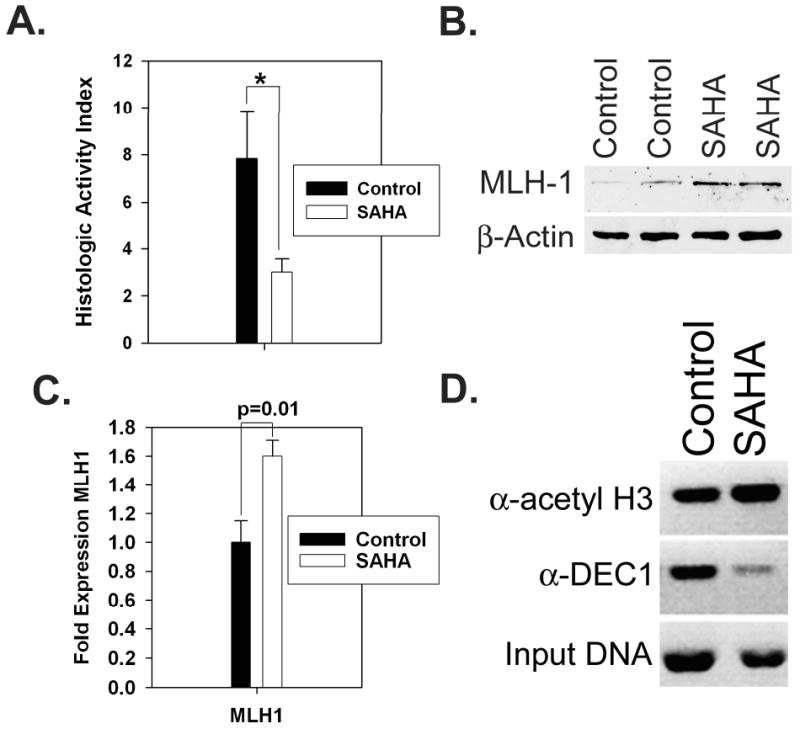
(A), Age matched 33-36 week old Giα2-/- mice were treated with vehicle or 50 mg/kg SAHA (n=4 each) for 8 days, then assessed for disease severity using a histologic activity scoring index. *, p < 0.05; data are representative of three separate experiments. Purified crypt lysates were evaluated for MLH1 protein (B) and mRNA (C) by western blotting and qPCR, respectively. (D), ChIP reveals an increase in acetyl H3 levels and decreased DEC-1 binding in SAHA-treated crypts.
Discussion
Human MMR-deficient CRCs have histopathologic features that distinguish them from MMR-proficient sporadic CRC (1), including right-sidedness, non-polyposis and mucinous histology, earlier age of onset, multi-focality, a Crohn's-like lymphocytic infiltrate, and MSI (2, 3). While multiple germline MMR-deficient mouse models have been generated, a persistent mystery is their very different histologic phenotype vs. that seen in human LS and MMR-deficient sporadic CRC.
The histopathology of Giα2-/- CRC is very similar to LS and MMR-deficient sporadic CRC. We speculate that the mechanism of inheritance of MMR deficiency may explain these observations. When all mouse/human body tissues are MMR-deficient, intestinal cells transform into cancers in a MMR-deficient microenvironment, in the absence of hypoxic inflammation. This may enhance tumor growth in the tissues seen in human and mouse biallelic germline MMR deficiency. When hypoxic inflammation-induced MMR-deficient cells transform into cancers surrounded by a partially MMR-proficient microniche (given the hypomorphic MSI that we observe), tumor growth is enhanced with the spectrum seen in LS, sporadic MMR deficient CRC and Giα2-/- mice. Determining the relative importance of hypoxia-induced signaling vs. inflammation-derived mediators in MMR silencing is under way. The MMR status of tumor microniche may also explain why Mlh3-/- mice (which are MMR-deficient hypomorphs) develop a higher proportion of CRCs vs. small intestinal adenomas than completely MMR-deficient mouse models (16), albeit with overall lower intestinal cancer penetrance.
Giα2-/- mice have somatically acquired MMR-deficiency that occurs both in inflamed and cancerous colon mucosa. This repression of MMR activity is specific to the MutL homologues Mlh1 and Pms2, as the MutS homologues Msh2 and Msh6 are not affected.
Consistent with this reduction in MLH1/PMS2 protein levels, inflamed Giα2-/- CRCs have MSI. However, some residual MLH1 protein remains. Therefore, Giα2-/- mice have a hypomorphic MSI phenotype. Hypomorphic MSI likely reflects two factors: (1) decreased but not absent MLH1/PMS2 protein, and (2) MSI arises only in older mice after colon inflammation, so mouse tissue is MMR-deficient for a shorter period of time. Together, these factors would be expected to cause lower MSI frequencies compared to germline MMR-deficient knockout mice. The same issue may also explain why the impaired DNA damage response in Giα2-/- inflamed colon (19) is also not as dramatic as that seen in germline MMR deficient models (46). Studies using inducible Mlh1 knockouts showed that low level MLH1 expression has ∼20% of the MSI mutation rate compared to complete loss of MLH1 expression and near-proficiency in cell extract-based single-base substitution repair assays. (47) Another example of hypomorphic MMR and MSI/frameshift capacity was seen in studies with Pms2-/- and Mlh3-/- mice. There is redundancy between Pms2 and Mlh3 in MSI/frameshift repair. Loss of each of these genes individually causes hypomorphic MSI, but together, Mlh3-/-;Pms2-/- mice have “full” MSI that is indistinguishable from Mlh1-/- mice. Since both Mlh3-/- and Pms2-/- mice develop tumors, these data are consistent with a mechanism whereby partial MMR loss causes cancer susceptibility. The precise relationship between the degree of MMR capacity and MSI rates is likely to depend on the cell lineage, residual MMR capacity and the specific repeat sequences analyzed to quantify MSI mutation rates. Future studies that use genome-wide assays of MSI mutation rates in MMR-proficient, hypomorphic and deficient colon will be helpful to resolve the relationship between MMR capacity, hypomorphic MMR and MSI mutations in CRC “driver” CAN genes and non-coding sequences important for CRC tumorgenesis.
Hypermethylation of the proximal Mlh1 promoter is a well established mechanism for MLH1 silencing in sporadic MMR-deficient CRC (reviewed in (48). In inflamed Giα2-/- crypt gDNA, we did not find increased methylation of the orthologous sequence in the mouse proximal Mlh1 promoter (Figure S4). We cannot, however, rule out that the Mhl1 promoter is methylated in Giα2-/- cancers. Future studies treating Giα2-/- mice with hypomethylating agents and assessing the impact on tumorigenesis and Mlh1 silencing may be helpful to address this issue.
Recently, tissue hypoxia has emerged as a mechanism of Mlh1 silencing and MMR deficiency in vitro, but has not yet been shown to occur in vivo. Using hypoxia biomarkers BNIP3, DEC-1 and EF5 (49), we show that Giα2-/- inflamed colon has regional hypoxia, likely resulting from the dense infiltrate of chronic inflammatory cells, and increased metabolic activity in damaged/regenerating epithelium. Hypoxia-induced epigenetic MLH1 silencing involves upregulation of the bHLH transcription factor DEC-1/Sharp2/Stra13 via a HIF-1α response element in its promoter (42). DEC-1 contains an N-terminal DNA binding domain that recognizes a CANNTG- E-box consensus sequence in the proximal Mlh1 promoter (41), and a C-terminal transactivation domain that binds the class 1 histone deacetylase HDAC1 (42). In vitro, DEC-1 expression inhibits MLH1 expression in an HDAC-dependent manner (41) (33, 50), as HDAC inhibitors prevent hypoxic DEC-1-mediated MLH1 repression. Consistent with predictions based on in vitro studies, we demonstrated increased DEC-1 expression in inflamed Giα2-/- crypts (Figure 2A), and increased DEC-1 occupancy at the proximal Mlh1 promoter in vivo (Figure 3C). Since colitis (and hypoxia) is patchy, the DEC-1 western and ChIP data likely underestimate the degree of hypoxia-induced DEC-1 expression and its binding to the Mlh1 promoter, as both techniques assay all crypts isolated from a segment of colon, not all of which is necessarily hypoxic. A model of epigenetic Mlh1 silencing by hypoxia is illustrated in Figure S7.
Recent reports indicate that treatment with HDACi have anti-inflammatory and anti-neoplastic effects in animal models of IBD and IBD-CRC. In DSS and TNBS colitis models, the HDACis VPA and SAHA dose-dependently increase acetyl- H3 in lamina propria mononuclear cells (LPMC), inhibit proinflammatory cytokines and reduce colitis severity (43). Another HDACi, ITF2357, can suppress IBD-CRC development in IL-10-/- and AOM-DSS-treated mice apparently by lowering NF-kB activity and enhanced apoptotic activity in LPMC (44). Interestingly, the anti-neoplastic effect was only seen in the right colon, suggesting differential sensitivity of right-sided and left-sided colitis-associated-CRC's to HDAC inhibitors. We found that daily oral gavage with 50 mg/kg SAHA caused a significant decrease in colitis and upregulated MLH1 expression, suggesting that decreased hypoxic inflammation results in increased a reversal of Mlh1 silencing via increased tissue oxygenation.
Chronic inflammation is a risk factor for CRC. While the suppressive effects of HDAC inhibitors on NF-kB activity and inflammation undoubtedly contribute to their chemopreventative effect in AOM-DSS and IL-10KO mice, our studies indicate that the hypoxic tissue microenvironment may also contribute to the development of microsatellite instability via Mlh1 suppression, and that HDACi treatment can reverse Mlh1 silencing. Most importantly, in concert with previous work, our studies provide additional pre-clinical mechanistic support that CRC prevention trials in IBD patients with HDAC inhibitors should be explored in the future.
Supplementary Material
Acknowledgments
This work was funded by NIH grants # K08 DK59816 and R21 DK071591, to R.A.E. and grants # CA98626 to S.M.L. This work was also supported in part by the Intramural Research Program of the NIH (Project Z01-ES101643). Neither author has any potential conflicts of interest.
References
- 1.Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–57. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenson JK, Bonner JD, Ben-Yzhak O, et al. Phenotype of microsatellite unstable colorectal carcinomas: Well-differentiated and focally mucinous tumors and the absence of dirty necrosis correlate with microsatellite instability. Am J Surg Pathol. 2003;27:563–70. doi: 10.1097/00000478-200305000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Jenkins MA, Hayashi S, O'Shea AM, et al. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: a population-based study. Gastroenterology. 2007;133:48–56. doi: 10.1053/j.gastro.2007.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Felton KE, Gilchrist DM, Andrew SE. Constitutive deficiency in DNA mismatch repair: is it time for Lynch III? Clin Genet. 2007;71:499–500. doi: 10.1111/j.1399-0004.2007.00801.x. [DOI] [PubMed] [Google Scholar]
- 5.Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. Jama. 2005;293:1979–85. doi: 10.1001/jama.293.16.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnetson RA, Tenesa A, Farrington SM, et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006;354:2751–63. doi: 10.1056/NEJMoa053493. [DOI] [PubMed] [Google Scholar]
- 7.Samowitz WS, Albertsen H, Sweeney C, et al. Association of smoking, CpG island methylator phenotype, and V600E BRAF mutations in colon cancer. J Natl Cancer Inst. 2006;98:1731–8. doi: 10.1093/jnci/djj468. [DOI] [PubMed] [Google Scholar]
- 8.Shen L, Issa JP. Epigenetics in colorectal cancer. Curr Opin Gastroenterol. 2002;18:68–73. doi: 10.1097/00001574-200201000-00012. [DOI] [PubMed] [Google Scholar]
- 9.Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 10.Baker SM, Bronner CE, Zhang L, et al. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–19. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- 11.de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–30. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 12.Edelmann W, Umar A, Yang K, et al. The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Research. 2000;60:803–7. [PubMed] [Google Scholar]
- 13.Edelmann W, Yang K, Kuraguchi M, et al. Tumorigenesis in Mlh1 and Mlh1/Apc1638N mutant mice. Cancer Res. 1999;59:1301–7. [PubMed] [Google Scholar]
- 14.Edelmann W, Yang K, Umar A, et al. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell. 1997;91:467–77. doi: 10.1016/s0092-8674(00)80433-x. [DOI] [PubMed] [Google Scholar]
- 15.Lipkin SM, Moens PB, Wang V, et al. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet. 2002;31:385–90. doi: 10.1038/ng931. [DOI] [PubMed] [Google Scholar]
- 16.Chen PC, Dudley S, Hagen W, et al. Contributions by MutL homologues Mlh3 and Pms2 to DNA mismatch repair and tumor suppression in the mouse. Cancer Res. 2005;65:8662–70. doi: 10.1158/0008-5472.CAN-05-0742. [DOI] [PubMed] [Google Scholar]
- 17.Rudolph U, Finegold MJ, Rich SS, et al. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat Genet. 1995;10:143–50. doi: 10.1038/ng0695-143. [DOI] [PubMed] [Google Scholar]
- 18.Hornquist CE, Lu X, Rogers-Fani PM, et al. G(alpha)i2-deficient mice with colitis exhibit a local increase in memory CD4+ T cells and proinflammatory Th1-type cytokines. J Immunol. 1997;158:1068–77. [PubMed] [Google Scholar]
- 19.Edwards RA, Wang K, Davis JS, Birnbaumer L. Role for epithelial dysregulation in early-onset colitis-associated colon cancer in Gi2-alpha-/- mice. Inflamm Bowel Dis. 2008 doi: 10.1002/ibd.20414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao X, Buermeyer AB, Narayanan L, et al. Different mutator phenotypes in Mlh1- versus Pms2-deficient mice. Proc Natl Acad Sci U S A. 1999;96:6850–5. doi: 10.1073/pnas.96.12.6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang G, Scherer SJ, Shell SS, et al. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004;6:139–50. doi: 10.1016/j.ccr.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 22.Taketo MM, Edelmann W. Mouse models of colon cancer. Gastroenterology. 2009;136:780–98. doi: 10.1053/j.gastro.2008.12.049. [DOI] [PubMed] [Google Scholar]
- 23.Alexander J, Watanabe T, Wu TT, Rashid A, Li S, Hamilton SR. Histopathological identification of colon cancer with microsatellite instability. Am J Pathol. 2001;158:527–35. doi: 10.1016/S0002-9440(10)63994-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jass JR. HNPCC and sporadic MSI-H colorectal cancer: a review of the morphological similarities and differences. Fam Cancer. 2004;3:93–100. doi: 10.1023/B:FAME.0000039849.86008.b7. [DOI] [PubMed] [Google Scholar]
- 25.Yearsley M, Hampel H, Lehman A, Nakagawa H, de la Chapelle A, Frankel WL. Histologic features distinguish microsatellite-high from microsatellite-low and microsatellite-stable colorectal carcinomas, but do not differentiate germline mutations from methylation of the MLH1 promoter. Hum Pathol. 2006;37:831–8. doi: 10.1016/j.humpath.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 26.Raut CP, Pawlik TM, Rodriguez-Bigas MA. Clinicopathologic features in colorectal cancer patients with microsatellite instability. Mutat Res. 2004;568:275–82. doi: 10.1016/j.mrfmmm.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 27.Whitelaw NC, Whitelaw E. Transgenerational epigenetic inheritance in health and disease. Curr Opin Genet Dev. 2008;18:273–9. doi: 10.1016/j.gde.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Ligtenberg MJ, Kuiper RP, Chan TL, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet. 2009;41:112–7. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 29.Chen PC, Kuraguchi M, Velasquez J, et al. Novel roles for MLH3 deficiency and TLE6-like amplification in DNA mismatch repair-deficient gastrointestinal tumorigenesis and progression. PLoS Genet. 2008;4:e1000092. doi: 10.1371/journal.pgen.1000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabbarah O, Mallon MA, Pfeifer JD, Edelmann W, Kucherlapati R, Goodfellow PJ. A panel of repeat markers for detection of microsatellite instability in murine tumors. Mol Carcinog. 2003;38:155–9. doi: 10.1002/mc.10157. [DOI] [PubMed] [Google Scholar]
- 31.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–8. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng G, Chen A, Hong J, Chae HS, Kim YS. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res. 1999;59:2029–33. [PubMed] [Google Scholar]
- 33.Mihaylova VT, Bindra RS, Yuan J, et al. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol Cell Biol. 2003;23:3265–73. doi: 10.1128/MCB.23.9.3265-3273.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koukourakis MI, Giatromanolaki A, Polychronidis A, et al. Endogenous markers of hypoxia/anaerobic metabolism and anemia in primary colorectal cancer. Cancer Sci. 2006;97:582–8. doi: 10.1111/j.1349-7006.2006.00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turley H, Wykoff CC, Troup S, Watson PH, Gatter KC, Harris AL. The hypoxia-regulated transcription factor DEC1 (Stra13, SHARP-2) and its expression in human tissues and tumours. J Pathol. 2004;203:808–13. doi: 10.1002/path.1585. [DOI] [PubMed] [Google Scholar]
- 37.Wykoff CC, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Identification of novel hypoxia dependent and independent target genes of the von Hippel-Lindau (VHL) tumour suppressor by mRNA differential expression profiling. Oncogene. 2000;19:6297–305. doi: 10.1038/sj.onc.1204012. [DOI] [PubMed] [Google Scholar]
- 38.Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. 2000;97:9082–7. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koch CJ. Measurement of absolute oxygen levels in cells and tissues using oxygen sensors and 2-nitroimidazole EF5. Methods Enzymol. 2002;352:3–31. doi: 10.1016/s0076-6879(02)52003-6. [DOI] [PubMed] [Google Scholar]
- 40.Vaissiere T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res. 2008;659:40–8. doi: 10.1016/j.mrrev.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura H, Tanimoto K, Hiyama K, et al. Human mismatch repair gene, MLH1, is transcriptionally repressed by the hypoxia-inducible transcription factors, DEC1 and DEC2. Oncogene. 2008;27:4200–9. doi: 10.1038/onc.2008.58. [DOI] [PubMed] [Google Scholar]
- 42.Ivanov SV, Salnikow K, Ivanova AV, Bai L, Lerman MI. Hypoxic repression of STAT1 and its downstream genes by a pVHL/HIF-1 target DEC1/STRA13. Oncogene. 2007;26:802–12. doi: 10.1038/sj.onc.1209842. [DOI] [PubMed] [Google Scholar]
- 43.Glauben R, Batra A, Fedke I, et al. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol. 2006;176:5015–22. doi: 10.4049/jimmunol.176.8.5015. [DOI] [PubMed] [Google Scholar]
- 44.Glauben R, Batra A, Stroh T, et al. Histone deacetylases: novel targets for prevention of colitis-associated cancer in mice. Gut. 2008;57:613–22. doi: 10.1136/gut.2007.134650. [DOI] [PubMed] [Google Scholar]
- 45.Edwards RA, Smock AZ. Defective arachidonate release and PGE2 production in Gi alpha2-deficient intestinal and colonic subepithelial myofibroblasts. Inflamm Bowel Dis. 2006;12:153–65. doi: 10.1097/01.MIB.0000201100.72191.19. [DOI] [PubMed] [Google Scholar]
- 46.Edelmann W, Cohen PE, Kane M, et al. Meiotic pachytene arrest in MLH1-deficient mice. Cell. 1996;85:1125–34. doi: 10.1016/s0092-8674(00)81312-4. [DOI] [PubMed] [Google Scholar]
- 47.Cejka P, Stojic L, Mojas N, et al. Methylation-induced G(2)/M arrest requires a full complement of the mismatch repair protein hMLH1. EMBO J. 2003;22:2245–54. doi: 10.1093/emboj/cdg216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grady WM. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004;23:11–27. doi: 10.1023/a:1025861527711. [DOI] [PubMed] [Google Scholar]
- 49.Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med. 2007;85:1295–300. doi: 10.1007/s00109-007-0277-z. [DOI] [PubMed] [Google Scholar]
- 50.Rodriguez-Jimenez FJ, Moreno-Manzano V, Lucas-Dominguez R, Sanchez-Puelles JM. Hypoxia causes downregulation of mismatch repair system and genomic instability in stem cells. Stem Cells. 2008;26:2052–62. doi: 10.1634/stemcells.2007-1016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.