

Summary
Brain circuits oscillate during sleep. The same circuits appear to generate pathological oscillations. In this review we discuss recent advances in our understanding of how epilepsy co-opts normal, sleep-related circuits to generate seizures.
Introduction
The synchronized firing among large populations of neurons is thought to underlie several fundamental processes in the brain ranging from stimulus encoding and memory consolidation to navigation and path integration (Buzsaki, 2006). Such coordinated firing, which often comes in the form of neural network oscillations, underscores the premise that the brain is designed to rapidly integrate and process signals from many sources. Under pathological conditions, however, these brain circuits can be hijacked to generate aberrant oscillatory network activity. Indeed, abnormal neural network oscillations are associated with several disease states including schizophrenia, Alzheimer's Disease and epilepsy (Herrmann and Demiralp, 2005).
Regarding epilepsy, the primary focus of this review, it has long been appreciated that seizure activity can be driven by hyperexcitable oscillatory networks, observations that have been summarized in several recent review articles (Huguenard and McCormick, 2007; McCormick and Contreras, 2001; Steriade, 2005). In this review we develop the thesis that normal brain circuits provide a template that epileptic circuits use to generate seizures. The assumption, then, is that normal and epileptic brain circuits share common features. Of course, it would be foolhardy to assume that these circuits are identical – something must distinguish an epileptic circuit from a normal circuit. However, approaching epilepsy without knowledge of the normal underlying physiology and circuitry in the brain appears likewise imprudent. To illustrate this point we highlight two well-described oscillatory sleep circuits – one in the thalamus and one in the hippocampus – that are linked to epilepsy. In each case we first describe the normal circuitry mediating the sleep-related oscillations. Then we highlight clinical features of the particular epilepsy associated with the brain structures. Finally, we develop the hypothesis that seizures are generated by mechanisms comparable to those subserving sleep-related oscillations.
The Thalamus and Sleep Spindles: Stitching Together Memories?
The thalamus is a subcortical structure that is critical for the generation of both normal and pathological synchronized oscillatory activity in the cortex (Steriade et al., 1993). A well-described thalamocortical oscillation is the sleep spindle, an intermittent 10-15 Hz oscillation persisting for 1-3 seconds that is so-named because it's waxing and waning appearance resembles a spindle of wound string or thread. Spindles are regularly observed during non-REM sleep in normal individuals using electroencephalogram (EEG) recording techniques. EEG electrodes are placed on the scalp and, therefore, primarily record the activity of cortical neurons. As the skull/scalp functions as a low-pass filter that filters out high-frequency events such as action potentials, the voltage deflections in EEG recordings primarily reflect the slower synaptic events driven by action potentials. Thus, the synchronized occurrence of synaptic events in cortical neurons - presumably resulting from the coordinated presynaptic firing of large populations of neurons - underlies the periodic voltage deflections observed during spindle oscillations (Figure 1A). It is noteworthy that the action potentials that drive the synaptic events recorded by cortical EEG electrodes can originate from subcortical structures, a point that will be expanded upon below.
Figure 1.

EEG recordings of sleep spindles (A) and spike-wave discharges (B) from the same patient. A1. A 10 Hz low-amplitude spindle oscillation lasting two seconds was recorded on all 4 channels during stage 2 sleep (demarked by the horizontal bar). B1. EEG activity during an absence seizure as evidenced by the hallmark 3 Hz spike-wave discharge on all 4 channels. Traces correspond to same 4 channels recorded in A. A2 & B2. The section of recordings demarcated by the line below the bottom trace of A1/B1 is expanded in A2/B2. Note the change in vertical gain. EEGs are biopolar recordings and were placed according to the Standard International 10-20 System of Electrode Placement with a transverse montage crossing midline. EEG recordings courtesy of Dr. Kevin Graber, Stanford University Epilepsy Center.
Although Loomis et al. (1935) discovered sleep spindles nearly 75 years ago, the functional relevance of these oscillations remains largely unknown – indeed, the function of sleep in general continues to be a source of debate. However, many recent studies have provided compelling evidence for the hypothesis that sleep spindles are important for the consolidation of memories formed during wakefulness. First, spindles are temporally associated with hippocampal activity patterns believed to underlie the transfer of newly acquired information to the cortex (Buzsaki, 2006; Siapas and Wilson, 1998; Sirota et al., 2003; Steriade, 2006). Second, the density of spindles increases in sleep periods that follow intensive learning tasks (Eschenko et al., 2006; Gais et al., 2002; Morin et al., 2008; Peters et al., 2008). Third, long-term synaptic changes are induced in cortical neurons when spindle-like activity is artificially imposed on them (Czarnecki et al., 2007; Rosanova and Ulrich, 2005). Collectively, these studies are beginning to make inroads into putative functions of spindle oscillations. Moreover, they are a harbinger for what is likely to be a major question for the neuroscience community in the coming years – that is, what function, if any, do brain oscillations serve? In this review, however, we only tangentially address the function of sleep-related oscillations. That is, the primary objective of this review is not to describe why the brain oscillates; rather, we will focus on recent advances describing how the brain oscillates. For more detailed commentary on sleep and memory, we suggest several recent review articles and/or books (Born et al., 2006; Buzsaki, 2006; Buzsaki and Draguhn, 2004; Chee and Chuah, 2008; Rasch and Born, 2007; Stickgold and Walker, 2007).
Cellular and Network Underpinnings of Sleep Spindles
Spindles result from interactions between extensively interconnected cortical and thalamic neurons. In attempting to parse out mechanisms mediating spindles, it is instructive to borrow from concepts that have evolved during decades of work on central pattern generators, neural networks that underlie rhythmic motor patterns such as walking, breathing and crustacean feeding behaviors (Grillner, 2006; Hooper and DiCaprio, 2004; Kiehn et al., 2000; Marder, 2000; Marder and Bucher, 2001; Marder and Calabrese, 1996). In this vein, we will independently consider two aspects of network oscillations: (1) factors that initiate rhythmic activity, and (2) factors that sustain/maintain rhythmic activity (i.e. the so-called rhythm generators). As the two are often inter-related, the distinction between rhythm initiation and rhythm maintenance is not always clear. For example, as we discuss below, spindles are intimately associated with a second, slower cortical oscillation, an observation that has motivated the call for considering the two oscillations as unified brain rhythms (Steriade, 2006). If we do so, then the question arises of whether mechanisms underlying spindle oscillations should include mechanisms underlying the generation of the slow cortical oscillation. The answer is probably ‘Yes’. Indeed, a deep understanding of brain function will undoubtedly require knowledge of how activity patterns in different brain structures are coordinated. For practical reasons, however, we will primarily limit our discussion of cortical factors to those that are specifically associated with spindles. We first explore the basis of the spindle rhythm generator.
Spindle Rhythm Generator
A defining feature of a central pattern generator is the capacity to generate rhythmic activity in the absence of extrinsic phasic timing information (Marder and Bucher, 2001). That is, a circuit capable of generating rhythmic activity even when it is isolated from the rest of the nervous system. Therefore, we will define the spindle rhythm generator as the fundamental core circuit that is capable of producing rhythmic spindle-like activity. Several decades of research indicate that circuits intrinsic to the thalamus are endowed with this capacity. First, thalamic neurons continue to generate ∼10 Hz spindle-like activity in decorticated animals (Andersen et al., 1967b; Andersson and Manson, 1971; Morison and Bassett, 1945). Second, spindle activity is relatively generalized, meaning that oscillatory activity can be simultaneously recorded in many anatomically-distinct regions of cortex (see Figure 1A). This feature of generalization has been used to argue that a sub-cortical structure with widespread projections to the cortex likely serves as a central pacemaker that coordinates the activity of many cortical neurons. Because the thalamic nuclei make widespread cortical projections (Jones, 2007) and because recordings made with depth electrodes placed in the thalamus reveal that thalamic neurons oscillate in time with cortical spindles (Andersen et al., 1968; Andersen et al., 1967a), it has long-been hypothesized that the thalamus serves as this pacemaker (Buzsáki, 1991; Fuentealba and Steriade, 2005; Jasper and Kershman, 1941; Penfield and Jasper, 1954; Steriade and Deschenes, 1984; Steriade et al., 1993b). Finally, in vitro brain slice preparations limited to just portions of the thalamus can sustain robust spindle-like network activity (Bal and McCormick, 1996; Jacobsen et al., 2001; Sanchez-Vives and McCormick, 1997; von Krosigk et al., 1993; Warren et al., 1994), indicating that the spindle rhythm generator is located in the thalamus.
The cellular and circuit properties of thalamic neurons that give rise to spindle oscillations have been extensively studied experimentally both in vivo and in brain slice preparations. This work revealed that the spindle-generating circuit is essentially composed of two sets of neurons: inhibitory neurons in the reticular thalamic (RT) nucleus and excitatory thalamocortical (TC) neurons in the dorsal thalamic nuclei (Figure 2A). RT neurons project into the dorsal thalamic nuclei to inhibit TC neurons with the neurotransmitter γ-aminobutyric acid (GABA). TC neurons project their axons to the cortex and, en route, send collaterals into the reticular nucleus where they release glutamate to excite RT neurons. Additionally, both neuron types receive glutamatergic excitatory input from cortical neurons. Because cortical activation of RT neurons is far more powerful than activation of TC neurons, and because RT neurons inhibit TC neurons, cortical activity primarily inhibits TC neurons, an effect mediated by the disynaptic, RT-dominant pathway (Destexhe et al., 1998; Golshani et al., 2001).
Figure 2.

Schematic representation of RT-TC circuitry present in thalamic brain slices. A. Putative spindle generator. Anatomy (left). Shown is one pair of reciprocally-connected RT and TC neurons. RT neurons are GABAergic and inhibit TC neurons via GABAA and GABAB postsynaptic receptors. TC neurons are glutamatergic and excite RT neurons (and cortical neurons). Not shown are the corticothalamic inputs to that are proposed to activate spindle rhythm generator. Oscillations (right). Following RT-mediated inhibition, TC neurons generate post-inhibitory rebound action potential bursts. These TC burst are responsible for recurrent excitation of RT neurons and activation of the subsequent cycle of the oscillation. Both RT and TC bursting activity is mediated by T-type Ca2+ channel activity. Plus signs (+) refer to glutamatergic excitation while minus signs (-) refer to GABAergic inhibition. B. Mechanisms that contribute to the synchronization and desynchronization of RT neuron activity. Electrical coupling among RT neurons is hypothesized to synchronize their activity. RT-to-RT neuron inhibition (i.e. intra-RT inhibition) is proposed to desynchronize RT neuron activity through a burst-shunting mechanism (Sohal and Huguenard, 2003). For example, if RT2 fires slightly before RT1 & RT3, then RT1 & RT3 will not fire bursts of activity. Moreover, if the inhibition is sufficiently strong, then recurrent excitation by TC neurons will be ineffective at triggering a subsequent burst in RT1 & RT3. Also shown is the convergence/divergence of RT-to-TC and TC-to-RT connectivity. Line thickness/synapse size does not represent functional difference in connection strength.
The reciprocally connected RT-TC neuron circuit generates spindle-like oscillatory activity, through mechanisms described in detail elsewhere (Huguenard and McCormick, 2007; McCormick and Bal, 1997; McCormick and Contreras, 2001). Briefly, when a RT neuron fires a burst of action potentials it releases GABA onto TC neurons and activates both GABAA and GABAB receptors. This inhibition hyperpolarizes neurons and deinactivates low-threshold, T-type Ca2+ channels, enabling TC neurons to produce regenerative calcium spikes that can trigger bursts of action potentials (Huguenard, 1996; Llinas and Jahnsen, 1982) (Figure 2A). Thus, due to their ability to generate such a post-inhibitory rebound burst of action potentials, TC neurons are paradoxically activated, albeit with a delay, in response to RT neuron inhibition. The stronger the RT-mediated inhibition, the more robust the resulting TC neuron rebound burst. The burst of TC neuron activity is transmitted back to the reticular nucleus and provides the recurrent excitation necessary to re-activate RT neurons and initiate another cycle of the oscillation. Thus, RT-to-TC neuron inhibition followed by TC-to-RT neuron excitation results in the rhythmic alternation of RT and TC neuron bursting activity and constitutes the core circuitry of the putative spindle rhythm generator. The ∼10 Hz frequency of spindle oscillations reflects the timing properties of RT-to-TC inhibition and TC-mediated recurrent excitation. Because TC neurons also project to the cortex, rhythmic TC neuron activity is relayed to cortical neurons, an action that presumably underlies the oscillatory cortical activity observed during spindles.
The aforementioned post-inhibitory rebound bursting of TC neurons is critical for sustaining oscillatory activity in the thalamus. As inhibition of TC neurons is mediated by RT neurons, much attention has focused on elucidating factors that regulate RT neuron excitability. An important component of thalamic circuitry that largely determines the strength and pattern of RT neuron activity is intra-RT inhibition. That is, in addition to inhibiting TC neurons, RT neurons also inhibit other RT neurons (Bal et al., 1995a; Sanchez-Vives et al., 1997; Deleuze and Huguenard, 2006) (Figure 2B), an action mediated by GABAA receptors. Intra-RT inhibition can limit RT neuron burst duration (Bal et al., 1995; but see Sohal and Huguenard, 2003a) and also limits the number of RT neurons involved in a particular cycle of network activity (Sanchez-Vives et al., 1997; Sohal and Huguenard, 2003a). However, the details regarding how intra-RT inhibition regulates RT neuron bursting are not known. A complete understanding of bursting in RT neurons will likely require a deeper appreciation of how these neurons integrate inhibitory inputs from other RT neurons along with excitatory inputs from both corticothalamic and thalamocortical inputs – that is, how RT neurons integrate inhibitory and excitatory signals. This point is underscored by the observation that blocking inhibition in thalamic slices increases the amplitude and duration of EPSP barrages recorded in RT neurons (Bal et al., 1995). The issue of inhibitory/excitatory integration – and the implications of such integration for thalamic oscillations – is developed more fully in later sections (see Tipping the Balance: Submission of Intra-RT Inhibition).
In this section we limit our discussion to the direct inhibitory mechanisms that regulate RT neuron bursting. Insights into these effects primarily rely on computational approaches. One model proposes that intra-RT inhibition acts to shunt bursting activity in RT neurons (Sohal and Huguenard, 2003; Sohal et al., 2000). To understand this process, one might conceptualize a centrally located RT neuron firing a strong burst of action potentials (see Figure 2B). With intra-RT inhibition in place, this RT neuron will activate a significant GABAA receptor-mediated inhibitory conductance in neighboring RT neurons such that they will not fire – the centrally located RT neuron effectively vetoes burst opportunities in neighboring RT neurons. Moreover, if sufficiently strong, then this conductance will shunt the recurrent excitation provided by TC neurons, thereby reducing the probability that the neighboring RT neurons will fire a burst on the next cycle of the oscillation (Sohal and Huguenard, 2003). This hypothesis is supported by experimental evidence demonstrating that RT neuron burst probability is relatively high when intra-RT inhibition is pharmacologically blocked, and low when pharmacologically enhanced (Sohal and Huguenard, 2003). The effects of a low RT neuron burst probability are two-fold. First, it restricts the number of RT neurons that contribute to any one cycle of the network oscillation, effectively enforcing sparseness in the network. Second, with fewer RT neurons bursting, feed-forward inhibition is weaker and, therefore, less likely to trigger post-inhibitory rebound bursts in TC neurons. Weak TC neuron rebound bursting, in turn, renders recurrent excitation of RT neurons less effective, a condition that cannot sustain robust oscillatory activity. Thus, intra-RT inhibition serves to both desynchronize and limit the duration of thalamic oscillations.
In addition to inhibitory connections, electrical synapses mediated by the gap-junction protein connexin 36 also connect RT neurons. Evidence for the presence of such electrical coupling comes from anatomical (Landisman et al., 2002; Liu and Jones, 2003) and physiological studies (Deleuze and Huguenard, 2006; Lam and Sherman, 2006; Landisman et al., 2002). While intra-RT inhibition serves to desynchronize thalamic circuits, it is thought that electrical coupling among RT neurons does the opposite, although this hypothesis has not been extensively explored in either theoretical or experimental studies.
Spindle Initiation & Modulation
Thus far, we have attempted to dissect the essential circuit features that enable the generation of rhythmic activity associated with spindles. As described above, the evidence to date indicates that the mechanisms of spindle generation are contained within thalamic circuits. What factors, then, activate these circuits? For reasons discussed above, early models of sleep spindles did not incorporate any specific cortical role in the generation of spindles (Andersen et al., 1967b). Now, however, it is generally appreciated that the cortex plays important roles in both initiating and shaping spindle oscillations. Regarding initiation, spindles recorded in vivo are often associated with a slower (<1 Hz) cortical oscillation (Contreras and Steriade, 1995; Steriade et al., 1993a; Steriade et al., 1993d). The slow oscillation is driven by cortical circuits as it survives thalamic lesions (Steriade et al., 1993c) and is absent in the thalamus following cortical lesions (Timofeev and Steriade, 1996). It has been proposed that during the slow cortical oscillation, excitatory neurons projecting from the cortex to the thalamus (i.e. corticothalamic neurons) are rhythmically depolarized and fire action potentials, leading to the excitation of RT neurons and subsequent activation of the thalamic spindle rhythm generator (Steriade et al., 1993a) (Figure 3). Indeed, intracellular recordings of RT neurons in vivo reveal that they receive rhythmic barrages of excitatory postsynaptic potentials (EPSPs) in time with the slow cortical oscillation, and that these EPSPs can trigger spindle-like oscillations (Steriade et al., 1993a). Thus, the correlation between the slow cortical oscillation and thalamic spindles makes a compelling argument for the hypothesis that spindles are evoked by neurons in the cortex. To strengthen this argument, future studies may incorporate strategies involving multi-site recording electrodes placed throughout the cortex and thalamus. By simultaneously recording activity in many areas before and during spindles, one could then more precisely determine the temporal relationships between cortical and thalamic firing and better resolve whether activity in the cortex precedes spindle activity in the thalamus – a condition that must be met if cortical activity initiates spindles. Comparable experiments have been carried out to determine factors that initiate thalamocortical seizure activity (see section entitled Is the Spike-Wave a Hypersynchronous Spindle?).
Figure 3.

Proposed relationship between sleep spindle oscillation and the slow cortical oscillation. A. The slow cortical oscillation is generated in the cortex. The spindle rhythm generator is found in the thalamus. B. As proposed by Contreras and Steriade (1995) and Steriade et al. (1993a,d) cortical activity generated during the slow oscillation is transmitted to the thalamic reticular nucleus and activates the spindle rhythm generator. Spindle activity generated in the thalamus is then relayed back to cortex by thalamocortical neurons. High levels of cortical activity are represented by the plus (+) signs which occur at the peaks of the cortical oscillation (vertical dotted line).
In addition to initiating spindle activity, corticothalamic feedback also appears critical for establishing the high degree of synchrony and coherence associated with cortical spindle activity. That is, the cortex is critical for establishing the spatiotemporal aspects of spindles. Contreras et al. (1996) came to this conclusion after recording spindles in the normal and decorticated cat. They observed that spindles recorded in the intact cortex of anesthetized cats are highly synchronized throughout spatially disparate regions. Next they removed the cortex from one hemisphere and recorded spindles in the remaining intact cortex. They observed that while the spindles persisted, coherence in different cortical regions was lost. This result was subsequently confirmed for spindles observed during natural sleep (Contreras et al., 1997). How, then, do corticothalamic projections promote widespread spindle coherence? For the proposed answer, we once again turn to the slow (<1 Hz) cortical oscillation. This slow oscillation is itself widely synchronized throughout cortex by widespread cortico-cortical connections (Contreras et al., 1996). It is hypothesized that the synchronized nature of the slow oscillation promotes the simultaneous activation of many corticothalamic inputs that, in turn, activate many, spatially disparate regions of the thalamus. This leads to simultaneous activation of multiple thalamic spindle rhythm generators which then behave more or less in unison (although not with the same extent of hypersynchrony associated with seizure activity, as described later). As the synchronized output of these rhythm generators is relayed back to cortex via extensive and divergent thalamocortical fibers, the spindles elicited in the disparate cortical regions occur at the same time. It is noteworthy that this effect does not occur in vitro where, rather than displaying patterns that are generally spatially invariant, thalamic spindles propagate in distinct waves (Kim et al., 1995). However, this apparent inconsistency is not surprising as the vast corticothalamic projections are not maintained in vitro. Indeed, the strong spatiotemporal coherence of thalamic spindle activity recorded in vivo is also lost after decortication (Contreras et al., 1997).
Finally, circuits within the cortex are also hypothesized to amplify spindle activity. This conclusion was derived primarily from current source density (CSD) analyses of cortical regions during spindle activity (Kandel and Buzsaki, 1997). This approach enabled the determination of spatiotemporal profiles of current flux in cortical neurons during spindle oscillations and demonstrated that an early, significant current sink, a presumed site of dendritic current flux, occurred in layer IV of cortex. As thalamocortical neurons primarily project to this layer, this observation is consistent with the hypothesis that thalamic activity mediates at least some aspects of spindle activity in the cortex. However, the authors also observed that neurons throughout all cortical layers were activated during spindles, leading to the suggestion that intra-cortical network activity contributes significantly to the spindle signal. Indeed, the authors conclude that while thalamocortical neurons may trigger a spindle cycle in the cortex, the EPSPs elicited in cortical neurons by thalamocortical neurons likely do not contribute much to the spindle signal. This is corroborated by anatomical evidence indicating that thalamocortical projections into the cortex are numerically small (White, 1979). Thus, according to their model, the relatively small triggering signal from the thalamus is powerfully amplified by cortical circuits, in agreement with the concept of the canonical cortical microcircuit (Douglas and Martin, 2004).
Thalamocortical Dysfunction and Epilepsy
Thus far we have described the mechanisms that contribute to the initiation and generation of a normal, sleep-related thalamocortical oscillation. Thalamocortical networks, however, are also associated with disease, particularly epilepsy. Seizures associated with thalamocortical dysfunction are linked to the idiopathic generalized epilepsies (IGEs). IGEs comprise one-third to one-half of all epilepsies (Loiseau et al., 1991; Wolf and Goosses, 1986) and include epilepsy subtypes characterized by absence seizures, myoclonic jerks and/or generalized tonic-clonic seizures (Beghi et al., 2006). Among the most common IGE subtypes are childhood (CAE) and juvenile (JAE) absence epilepsy, juvenile myclonic epilepsy (JME) and generalized tonic/clonic epilepsy. It is unclear whether the different IGE subtypes constitute independent syndromes, or if they correspond to different points on a broad IGE spectrum (i.e. “IGEs with variable phenotype”), an issue that requires future deliberation according to the organization largely responsible for defining epilepsy subtypes, the International League Against Epilepsy. For example, the cardinal symptoms associated with CAE and JAE are absences, seizures that are behaviorally subtle, primarily consisting of non-convulsive staring episodes that last several seconds, during which the patient is unconscious. Patients with JME, however, often express absence seizures (Hirsch et al., 2008; Panayiotopoulos et al., 1989) despite the fact that this epilepsy syndrome is principally defined by myoclonic seizures (transient muscle twitches).
Regardless of whether the various IGEs are one or many epilepsy syndromes, it is clear that they share two important features. First, the absence seizures associated with the different IGEs include the hallmark synchronized ∼3Hz spike-wave cortical discharges on essentially all scalp recording electrodes (Figure 1B) – indeed, this pattern of activity is a defining feature of absence epilepsy. Similar to the argument outlined above regarding thalamic involvement in spindle generation, this attribute of generalization is often used as evidence that the thalamus plays a critical role in the pathologies underlying the IGEs. The second feature common to the IGE subtypes is that they are considered to be inherited disorders. This is well-illustrated by the high concordance (70-95%) of IGE syndromes within monozygotic twins, and the ∼10-fold higher recurrence risk within first-degree relatives of IGE patients (Sander et al., 2000).
Not surprising for a presumed genetic disease resulting in hyperexcitable neuronal activity, many of the gene mutations associated with the IGEs occur in proteins that regulate the excitability of neurons: i.e. ion channels, transporter proteins and neurotransmitter receptors. With respect to CAE and JAE, recent studies suggest that three types of ion channel and receptor genes are implicated: (1) calcium channels, (2) chloride channels and (3) GABAA receptors. While a fourth gene, EFHC1 (EF-hand domain containing 1), is primarily associated with juvenile myoclonic epilepsy, some patients with mutations in EFHC1 have absence seizures (Suzuki et al., 2004). Interestingly, EFHC1, which is not thought to be neuronal (Suzuki et al., 2008), can augment currents mediated by voltage-dependent R-type calcium channels (Suzuki et al., 2004). However, as very little is known how EFHC1 dysfunction causes absence seizures, we will restrict our discussion below to those mutations associated with CAE and JAE. To better place those mutations in a functional context, we will first highlight the current, more general views regarding the pathophysiologies believed to underlie absence seizures.
Is the Spike-Wave a Hypersynchronous Spindle?
Our understanding of how dysfunctional thalamic circuits promote absence seizures is in its infancy. The hypothesis that human IGEs even result from such circuits has yet to be definitively proven. Nonetheless, one model posits that the circuit responsible for generating absence seizure activity is comparable to the circuit that generates sleep spindles (Huguenard and McCormick, 2007). Indeed, the rhythmic spike-wave discharge has been referred to as a perversion of the spindle oscillation (McCormick and Bal, 1997). For several reasons, this assumption is reasonable. First, qualitative features including duration of spike-wave events and generalization throughout widespread cortical areas are reminiscent of sleep spindles. Both types of network oscillations occur intermittently, last a few seconds and can be recorded in many different cortical regions with EEG electrodes (see Figure 1). Second, recordings from depth electrodes placed in the thalamus of patients with absence epilepsy clearly demonstrate that thalamic activity is oscillatory and phase-locked to the timing of spike-wave discharges recorded in the EEG (Williams, 1953), an observation that is paralleled by the finding that the cortex and thalamus oscillate together during spindles (Andersen et al., 1968; Andersen et al., 1967a). Third, current source density profiles recorded in the cortex are generally similar during spindle oscillations and spike-wave discharges (Kandel and Buzsaki, 1997).
Another commonality between spindles and spike-wave discharges is that both are hypothesized to be initiated by cortical activity. While spike-wave discharges have not been linked to the slow cortical oscillation per se, initiation of the discharge is nonetheless thought to involve the cortex. Specifically, Meeren et al. (2002) recorded from a rat model of genetic absence seizures using multiple electrodes in the cortex and thalamus and performed association analyses. The authors discovered that the cortical focus of epileptic activity (i.e. the site that consistently exhibits the first bouts of activity) preceded any recorded activity in the thalamus by 500 msec, enabling the authors to conclude that cortical activity initiates absence seizures. The assumption guiding this strategy, of course, is that the recording electrodes are placed in the appropriate regions to capture the initiating bouts of activity. Therefore, one could argue that the particular placement of electrodes influenced the authors' conclusion. However, using a similar strategy in another animal model of absence epilepsy, Polack et al. (2007) also found that cortical activity preceded thalamic activity during spike-wave discharges. Comparable results were also found by magnetic resonance imaging (MRI) putative brain activity in a rat model (Nersesyan et al., 2004). Finally, pharmacological inactivation of the cortical focus with lidocaine reduces spike-wave incidence in the entire cortex (Sitnikova and van Luijtelaar, 2004). It could be argued that lidocaine, a known sodium channel blocker, also likely inhibits the activity of thalamocortical afferents, making it formally possible that any initiating thalamic activity propagating to the cortex is likewise silenced. Nonetheless, the accumulated data thus far support the hypothesis that seizure initiation is cortical in nature.
The primary reason that spindles and spike-wave discharges are believed to emerge from similar circuit mechanisms comes from the observation that both types of oscillations can be recorded in the thalamic slice preparation. Moreover, both oscillations can be recorded within the same slice by performing simple pharmacological manipulations. Specifically, application of GABAA receptor antagonists transforms sparse spindle-like activity recorded in the slice into robust 3 Hz epileptiform oscillations (Huguenard and Prince, 1994a; Kim et al., 1997; Sanchez-Vives and McCormick, 1997; von Krosigk et al., 1993), a transformation that occurs within minutes and is observed even when recording neurons at the single cell level. A comparable transformation occurs with infusion of GABAA receptor blockers directly into the thalamus of behaving rats (Castro-Alamancos, 1999; but see Steriade and Contreras, 1998). These results suggest that, as with spindles, the spike-wave rhythm generator is found in the thalamus. Moreover, the rapid transition from normal to epileptiform activity argues that dramatic re-wiring of thalamic circuitry is not required for the expression of spike-wave discharges. In other words, the basic circuitry responsible for the expression of both normal and epileptiform activity is in place.
Switching the Thalamic Circuit
How does GABAA receptor blockade promote the switch between normal and epileptiform activity? The prevailing hypothesis suggests that this blockade eliminates intra-RT inhibition (see Spindle Rhythm Generator), thereby promoting the hypersynchronous firing of thalamic neurons. Specifically, the burst vetoing mechanisms that are hypothesized to occur during normal spindles (see Spindle Rhythm Generator section) are removed when intra-RT inhibition is blocked. Without such vetoing mechanisms, a higher percentage of RT neurons will contribute to the oscillation. Indeed, in contrast to the relative sparseness of thalamocortical activity during spindles, neural activity is more robust and highly synchronized during spike-wave discharges, as evidenced by higher amplitude EEG signals (see Figure 1).
More conclusive evidence for intra-RT inhibition serving as a mechanism that constrains the oscillatory activity in the thalamus was derived from genetic manipulations. GABAA receptors are heteropentamers, with specific subunit composition varying among neurons in different brain areas (Pirker et al., 2000). β3 is one such receptor subunit. GABAA receptors expressed by RT neurons normally contain the β3 subunit while those expressed by TC neurons do not. Thus, genetically removing the β3 subunit from mice essentially eliminates intra-RT inhibition but does not affect RT-to-TC inhibition (Huntsman et al., 1999). The reduced intra-RT inhibition in β3 knockout mice is associated with thalamic oscillations that are significantly more robust and coherent (i.e. epileptiform) than their wild-type counterparts (Huntsman et al., 1999). Importantly, mice lacking the β3 subunit have a phenotype consistent with Angelman Syndrome, including absence-like seizures (DeLorey et al., 1998). Thus, reducing intra-RT inhibition promotes hypersynchrony among RT neurons and likely contributes to absence seizures. Electrical coupling among the RT neurons also likely contributes to RT hypersynchrony during spike-wave discharges (Deleuze and Huguenard, 2006; Slaght et al., 2002). In support of this hypothesis is the finding that carbenoxelone, an inhibitor of gap junctional communication, reduces the number and duration of spike-wave discharges in rat and mouse models of absence epilepsy (Gareri et al., 2005).
If reducing intra-RT inhibition promotes RT hypersynchrony, what then slows the normal 10 Hz oscillation to the pathological 3 Hz oscillation? One hypothesis that has attracted attention is that of differential activation of GABAA and GABAB receptors at the RT-TC synapse. As described above RT neurons inhibit TC neurons, leading to the generation of post-inhibitory rebound activity bursts that are responsible for re-excitation of RT neurons (see Figure 2A). The RT-TC synaptic response is mixed, composed of both fast GABAA receptor-mediated (ionotropic) and slow GABAB receptor-mediated (metabotropic) inhibition. While both types of inhibition can result in post-inhibitory rebound bursting in TC neurons, GABAB receptor-mediated inhibitory postsynaptic potentials (IPSPs) are particularly effective at doing so because they produce long-lasting, strong inhibition that results in substantial deinactivation of T-type calcium channels (Huguenard and Prince, 1994c; von Krosigk et al., 1993). Moreover, because GABAA and GABAB receptor-mediated IPSPs follow different time courses, the latency to rebound burst onset is different in the two forms of inhibition. If the synapse is dominated by GABAA receptor-mediated inhibition, then the latency from synapse activation to rebound burst is relatively brief. On the other hand, if inhibition at the RT-TC synapse is dominated by GABAB receptor activation, then this latency is longer. Thus, the relative contribution of GABAA and GABAB receptor-mediated inhibition defines the latency to TC neuron rebound burst onset and, therefore, dictates when RT neurons will be re-excited by TC neurons. As such, the properties of RT-TC inhibition influence the frequency at which the thalamic circuit oscillates. Therefore, one hypothesis states that the transition from a 10 Hz to 3 Hz oscillation reflects a stronger dependence on GABAB receptor-mediated inhibition at the RT-TC synapse (Kim et al., 1997). The ∼300 msec duration of synaptically-evoked GABAB currents is consistent with this hypothesis. Additionally, GABAB receptor antagonists can eliminate 3 Hz epileptiform activity in both in vitro (Kim et al., 1997) and in vivo (Smith and Fisher, 1996) models of absence epilepsy, and can do so even with direct intrathalamic infusion (Liu et al., 1992). These results suggest a critical role for thalamic GABAB receptors in the generation of 3 Hz spike-wave discharges. It is important to emphasize that this hypothesis does not state that dysfunctional RT-TC synapses mediate absence seizures. Rather, it suggests that factors promoting greater GABAB receptor activation at RT-TC synapses likely underlie the slower frequency of thalamic oscillations associated with absence seizures. Indeed, as significant GABAB receptor activation is primarily achieved only with strong presynaptic activation (Destexhe and Sejnowski, 1995; Dutar and Nicoll, 1988; Huguenard and Prince, 1994b; Isaacson et al., 1993; Kim et al., 1997; Scanziani, 2000), it is hypothesized that any condition/manipulation that facilitates strong, synchronized RT neuron bursting will result in significant GABAB receptor-mediated inhibition of TC neurons and promote slower thalamocortical oscillations. Thus, factors that contribute to RT neuron excitability are actively investigated as potential sources of absence seizure generation.
Tipping the Balance: Submission of Intra-RT Inhibition
Many factors are involved in defining RT neuron excitability. The intra-RT inhibition described above is one factor that limits RT excitability. As such it can be directly targeted, as in the β3 knockout mouse, to promote epileptiform activity. However, perturbations that do not directly modulate intra-RT inhibition can also increase RT neuron excitability. For example, if sufficiently strong, enhanced excitatory drive can overcome intra-RT inhibition such that RT neurons generate synchronized and/or robust bursting activity. Evidence for this comes from in vitro studies in which thalamic oscillations were evoked by electrically stimulating cortical inputs with a variable number of stimuli (Bal et al., 2000; Blumenfeld and McCormick, 2000). These studies demonstrated that oscillations evoked with one/few stimulus shocks were sparse and relatively fast (6-10 Hz). However, when many shocks were delivered to the corticothalamic inputs, the resulting oscillation was much more robust and slower (2-4 Hz). Moreover, under these conditions, i.e. when many shocks were delivered, subsequent application of a GABAB receptor antagonist transformed the responses back to sparse and fast oscillations, like those evoked by one shock. Thus, powerful excitatory feedback onto RT neurons from the cortex can transform spindle oscillations into slow, 3 Hz oscillations, a transition that is dependent on activation of GABAB receptors.
The aforementioned studies by Blumenfeld & McCormick and Bal et al. suggest that increased cortical activity could be one mechanism that triggers the expression of spike-wave discharges. This possibility is entirely plausible. However, as TC neurons also excite RT neurons, it is also possible that enhanced TC neuron activity contributes to RT neuron hyperexcitability. Indeed, several genetic animal models of absence epilepsy are associated with elevated (∼+40%) T-type currents in TC neurons (Song et al., 2004; Tsakiridou et al., 1995; Zhang et al., 2002), an effect that likely leads to more robust post-inhibitory rebound bursting among TC neurons. Interestingly, these studies also show that the kinetics of the T-currents recorded in epileptic animals are not substantially different from those recorded in control animals. This suggests that altered T-type currents in TC neurons are unlikely to directly contribute to the slow oscillatory features of the spike-wave discharges observed in the epileptic animals. However, if TC neuron firing is nonetheless more robust in the epileptic animals, then as a result it is likely that TC-to-RT excitation is likewise increased. If so, then this secondary increase in excitation onto RT neurons might indirectly contribute to slower, more robust oscillations in much the same manner that increased cortical excitation of RT neurons transforms fast spindles into slow and robust oscillations.
A more recent study demonstrates yet another possible means of promoting RT neuron activity; alteration of the synaptic machinery responsible for RT neuron excitation (Beyer et al., 2008). Beyer et al. demonstrate that an inbred mouse prone to absence seizures has a mutation in the gene encoding Gria4, an AMPA-type glutamate receptor subunit that is highly expressed by RT neurons. The Gria4 subunit confers rapid decay kinetics to the AMPA receptor. The authors subsequently knocked out the Gria4 gene and observed that the mutant animals have frequent spike-wave discharges similar to the inbred strain. Consistent with the finding that Gria4-containing AMPA receptors mediate fast excitatory transmission is the observation that EPSPs recorded in RT neurons of Gria4 knockout animals are substantially slower than those in wild type animals. Thus, the authors propose that prolonged excitatory synaptic responses of RT neurons promote RT neuron hyperexcitability and may contribute to the spike-wave discharges observed in the epileptic animals.
A second recent study also probes the question of whether altered synaptic excitation of RT neurons contributes to absence seizures (Menuz and Nicoll, 2008). However, in contrast to the aforementioned studies, these results suggest that decreased RT excitability contributes to seizure activity. The authors examined a mutant mouse called stargazer that has long-been known to have frequent spike-wave discharges (Noebels et al., 1990). In fact, the stargazer mouse is one of the previously described mutant animals associated with substantially elevated T-type currents in TC neurons (Zhang et al., 2002). More recently, it has been shown that stargazin, the protein mutated in stargazer mice, regulates AMPA receptor trafficking (Chen et al., 2000; Milstein and Nicoll, 2008). Thus, the frequency of AMPA receptor-mediated excitatory events is made significantly lower in cultured neurons when stargazin is mutated (Chen et al., 2000). Menuz and Nicoll found a comparable result in RT neurons recorded from thalamic brain slices taken from stargazer mice. Specifically, AMPA receptor-mediated excitatory events were smaller and less frequent in stargazer RT neurons than in wild type RT neurons. No difference was found with excitatory events in TC neurons. The authors propose that the observed decrease in RT neuron excitation promotes the disinhibition of thalamocortical networks and, therefore, promotes the generation absence seizures. Given that most evidence indicates that circuits driving absence epilepsy seem to require strong RT-to-TC inhibition, it is not obvious how disinhibition per se would lead to spike-wave discharges. Thus, the recent characterization of excitatory events in stargazer mice in many ways highlights the challenges we face in attempting to elucidate the mechanisms underlying absence seizures.
The hippocampus and Sleep: Why Do We Ripple?
The hippocampus is located within the medial temporal lobe of the cortex and, similar to the thalamus, is characterized by robust oscillatory network activity. Primary functions associated with hippocampal oscillations include memory formation and spatial navigation. Under normal conditions information enters the hippocampus via the dentate gyrus and then proceeds to area CA3, then to area CA1 and finally to the subiculum (Figure 4). One oscillation associated with both normal and epileptic brains is the sharp wave-ripple complex, a pattern of activity expressed predominantly during sleep that consists of a large, highly synchronized burst of activity in CA3 neurons (sharp wave) followed by the generation of a very fast (∼200 Hz) oscillation in CA1 neurons (ripples) (Figure 4). These events are intermittent, occurring once every few hundred milliseconds to once every 50 seconds (Ylinen et al., 1995). The sharp wave-ripple complex has received considerable attention for its potential role in reactivation, a phenomenon in which hippocampal neurons activated during specific learning tasks exhibit elevated activity patterns once again during sleep (Nadasdy et al., 1999; Pavlides and Winson, 1989; Wilson and McNaughton, 1994). Such reactivation is hypothesized to promote the consolidation of memories within cortical networks (Buzsaki, 2006; Cheng and Frank, 2008; Rasch and Born, 2007; Sutherland and McNaughton, 2000) and is likely to involve sustained changes in synaptic strength such as those observed during long-term potentiation (LTP). Indeed, sharp wave-like stimuli artificially provided to CA3 neurons can induce LTP in CA1 neurons (Buzsáki et al., 1987). Interestingly, the converse is also true: inducing LTP in CA3/CA1 promotes the expression of sharp wave ripple complexes (Behrens et al., 2005; Buzsáki, 1984).
Figure 4.

Schematic representation of the hippocampus and the sharp-wave ripple complex. Left. In general, the flow of information begins in the dentate gyrus, proceeds to CA3, then to CA1 and finally to the subiculum. The sharp wave component of the sharp-wave complex is hypothesized to involve the synchronized busting of many CA3 pyramidal neurons. The excitation provided by CA3 pyramidal neurons is then transmitted to CA1 where subsequent ripple activity is generated. While CA1 ripple activity is hypothesized to emanate from CA1 pyramidal neurons, it is not clear what mechanisms contribute to the generation of the activity. Right. The composite sharp-wave ripple complex in schematic form.
The Sharp Wave-Ripple Generator
In keeping with our previous distinction between rhythm initiation and rhythm generation, we will primarily discuss the sharp wave component in the context of rhythm initiation and the ripple component in the context of rhythm generation. It is generally accepted that the sharp wave component is initiated by a highly synchronized discharge of CA3 pyramidal neurons that results in compound field EPSPs in the dendritic regions of CA1 neurons (Buzsaki et al., 1992; Maier et al., 2003; but see Traub et al., 2001b). After a few milliseconds this activity results in ∼200 Hz rhythmic IPSPs (ripples) in CA1 pyramidal neurons (Ylinen et al., 1995) (Figure 5). Evidence for such a CA3-to-CA1 progression comes from the observation that in both in vivo (Chrobak and Buzsaki, 1996) and in vitro (Maier et al., 2003) preparations, CA3 activity precedes CA1 activity. Furthermore, CA1 sharp wave-ripple complexes are largely suppressed when the fiber pathway connecting CA3 to CA1 (Schaffer collateral) is cut (Maier et al., 2003). Also, both components of sharp wave-ripples are eliminated when glutamate receptor antagonists are applied to in vitro models (Maier et al., 2003), consistent with the hypothesis that CA3-mediated excitation drives these oscillations. While ripple oscillations can be observed in CA3, they are less prominent and more enigmatic than CA1 ripples (Ylinen et al., 1995). For this reason, we primarily focus on CA1 ripples.
Figure 5.
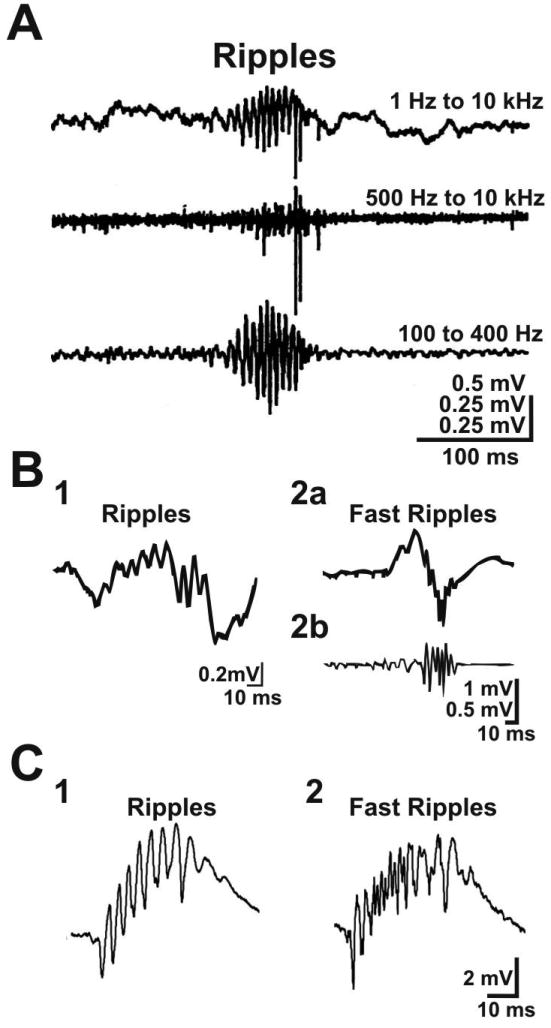
Electrophysiological recordings of ripple and fast ripple activity. A. Ripple oscillation recorded by Buzsaki et al. (1992) in area CA1 of rat hippocampus. The 3 traces correspond to the same recording filtered with 3 different settings. The 200 Hz oscillation is clearly observed when the signal is bandpass filtered between 100 and 400 Hz (bottom trace). B. Ripple (1) and fast ripple (2) oscillations recorded by Staba et al. (2007) with microelectrodes placed in the hippocampus of a human patient with MTLE. Shown are unfiltered (B2a) and bandpass filtered (B2b, filter = 70-600 Hz) fast ripple oscillations. C. Ripple (1) and fast ripple (2) oscillations recorded by Foffani et al. (2007) in hippocampal brain slices of control (1) and epileptic (2) rat. Figure credits. Part A from Buzsaki et al. (1992). Reprinted with permission from AAAS. Part B from Staba et al. (2007) with permission from Wiley. Part C from Foffani et al. (2007) with permission from Elsevier. Original figures were slightly modified.
Factors contributing to CA1 ripples remain unclear. However, most evidence from in vivo studies indicates that ripples arise from circuit properties rather than from intrinsic oscillatory properties of CA1 neurons. Specifically, ripples are hypothesized to reflect rhythmic IPSPs generated in the somatic and perisomatic regions of CA1 pyramidal neurons. Evidence for the dependence of ripple oscillations on synaptic inhibition comes from the observation that spiking activity of CA1 inhibitory neurons significantly increases during ripple activity (Ylinen et al., 1995). Moreover, urethane and ketamine, two anesthetics that modulate GABAergic inhibition, reduce ripple oscillation frequency (Ylinen et al., 1995). The primary question, then, is how do IPSPs occurring at ∼200 Hz in CA1 pyramidal neurons arise? According to the proposed hypothesis CA3 sharp wave excitation activates local inhibitory interneuron circuits in CA1 that function as ripple rhythm generators to produce 200 Hz rhythmic activity (Figure 6). This high frequency rhythmic activity is then imposed on CA1 pyramidal neurons in the form of rhythmic somatic/perisomatic IPSPs – synaptic activity that manifests itself as a ripple oscillation in extracellular recordings of CA1. It is noteworthy that this hypothesis refers to mechanisms of in vivo ripple generation. As discussed below, alternative mechanisms may contribute to ripple oscillations observed in vitro.
Figure 6.

Schematic representation of important experimental observations associated with ripple oscillations. A. Components that contribute to ripple oscillations. Consensus exists that the sharp wave produced by CA3 pyramidal neurons (blue) provides the excitatory drive that initiates the ripple oscillation. Also, it is generally accepted that rhythmic inhibition of CA1 pyramidal neurons (red) underlies ripple oscillations recorded in vivo. What is not clear is how such rhythmic inhibition arises (black box). One hypothesis suggests that rhythmogenic inhibitory networks provide input to CA1 pyramidal neurons (see Figure 7), while another argues that electrically coupled CA1 pyramidal neuron axons form the substrate for rhythmicity (see Figure 8). B. Graphical representation of observed activity patterns during ripple oscillation. Vertical lines placed on horizontal black lines represent extracellularly recorded action potentials of CA3 (blue) and CA1 (red) pyramidal neurons, and CA1 inhibitory neurons (green). The bottom red traces represent synaptic activity of an intracellularly recorded CA1 pyramidal neuron. Before the sharp wave all neurons are weakly active and synaptic activity observed in the CA1 pyramidal neuron is low (left panel). During the sharp wave CA3 pyramidal neurons fire highly synchronized bursts of action potentials, resulting in a moderate elevation of activity in CA1 pyramidal and inhibitory neurons. The sharp wave also causes a CA1 ripple oscillation, which is hypothesized to result from the fast rhythmic inhibition of CA1 pyramidal neurons.
The aforementioned hypothesis is consistent with much of the current dogma stating that inhibitory neurons provide the timing information required to drive coherent network oscillations (Bartos et al., 2007; Fries et al., 2007; Maex and De Schutter, 2005; Mann and Paulsen, 2007). This view has motivated investigators to design computational model networks that utilize synaptic inhibition as a means of generating oscillatory activity. Indeed, many such model networks can generate very fast rhythmic activity, including ripple oscillations. One of the main challenges, however, is to construct a model that captures overall network behavior as well as the physiological activity associated with individual units of the network. With this mind, it is important to appreciate that while the averaged activity of the population of neurons oscillates during hippocampal ripples, the behavior of single neurons that contribute to the population oscillation is likely different. Specifically, intracellular recordings of individual interneurons within the putative ripple network reveal activity that is neither robust nor highly oscillatory, despite the presence of a strong oscillatory signal in the local field potential (LFP, a metric for population activity recorded with extracellular electrodes) (Csicsvari et al., 1998; Csicsvari et al., 1999; Jinno et al., 2007; Klausberger et al., 2003; Klausberger et al., 2004; Le Van Quyen et al., 2008) (Figure 6B). Thus, computational models of ripple-generating networks should capture this emergent property of the oscillation.
The Inhibition Rendition of Ripple Generation
Brunel and Wang (2003) and Geisler et al. (2005) have recently developed model networks that generate robust population-based oscillations that are driven by non-rhythmic constituent neurons. Moreover, these models, which are composed entirely of inhibitory neurons, can sustain network oscillations in the ripple frequency range (200 Hz). This oscillation arises primarily from two assumed features of the network: (1) inhibition is powerful, and (2) synaptic noise – the stochastic fluctuations in a neuron's membrane potential arising from randomly firing synaptic inputs – is high. Individual network neurons fire whenever their membrane potentials are sufficiently depolarized to reach action potential threshold, an event that occurs sporadically with high synaptic noise. As schematized in Figure 7, when the network receives strong excitatory drive from external inputs (e.g. CA3-initiated sharp wave), the firing rate of the population of inhibitory neurons within the network is substantially elevated. However, because recurrent inhibition within the network is strong, the firing rate of individual neurons remains relatively low (i.e. sparse), albeit higher than prior to receiving excitation. As the firing rate increases, recurrent inhibition within the network strengthens. At the time of peak population firing, recurrent circuitry delivers a powerful surge of inhibition throughout the network, resulting in an overall decrease in the population firing rate. The firing rate trough occurs roughly 2.5 msec after the peak, a duration defined primarily by synaptic latencies and rise times. As the inhibitory synaptic events decay, the population firing rate builds up once again to a peak level approximately 2.5 msec later, only to be reduced by the next surge in recurrent inhibition. Thus the activity of the population oscillates between high firing rates during which net excitation is stronger than inhibition and low firing rates during which net inhibition is stronger than excitation (Figure 7A2). As the time lag between two successive cycles of this oscillation is ∼5 msec, the population firing rate oscillates at ∼200 Hz. Importantly, while the population readily oscillates under these conditions, only ∼10% of all network neurons contribute to any cycle of the oscillation, an observation consistent with experimental data. It is important to note that 200 Hz likely represents the upper frequency limit of this inhibitory model network. Specifically, the model can readily oscillate at frequencies greater than 200 Hz but this would require assigning synaptic delay and rise times with unrealistically short durations. Also important to note is the finding that incorporating excitatory neurons within the model network does not necessarily increase oscillation frequency; on the contrary, excitatory neurons can slow the oscillation frequency to ∼80 Hz (Brunel and Wang, 2003; but see Geisler et al., 2005). Nonetheless, it is theoretically possible to build a model network of inhibitory neurons that can generate 200 Hz oscillations. Thus, synthesizing the experimental and modeling data, it is hypothesized that ripples emerge when the CA3 sharp wave provides powerful excitation to inhibitory networks in CA1 that generate 200 Hz oscillations, rhythmic activity that is then imposed on CA1 pyramidal neurons in the form of IPSPs.
Figure 7.

Graphical description of how inhibitory networks may provide the rhythmicity that underlies ripple oscillations. A1. Placing the computational models proposed by Brunel and Wang (2003) and Geisler et al. (2005) in a hippocampal context, the aggregate activity of CA1 inhibitory neurons (green) provides rhythmic drive to CA1 pyramidal neurons (red). As such, local inhibitory networks in CA1 are hypothesized to function as the rhythm generator for ripple oscillations. Excitation provided by CA3 pyramidal neurons (blue) activates CA1 inhibitory networks. A2. Description of the temporal relationship between CA1 network dynamics and ripple oscillations, as represented by a rhythmic 200 Hz local field potential (LFP). Excitation from CA3 activates CA1 inhibitory neurons. CA1 inhibitory neuron activity is low at the LFP trough and progressively increases in response to CA3 excitation. Concomitant with the increase in inhibitory neuron activity is a strengthening of recurrent inhibition within the network. At the LFP peak, recurrent inhibition is strong and overpowers the CA3 excitatory drive. This causes the activity levels among CA1 inhibitory neurons to decrease. The 5 msec period of the LFP is defined by synaptic delays and rise times associated with inhibitory events. B. Graphical representation of neuronal activity patterns according to proposed model. As in Figure 6, vertical lines placed on horizontal time lines represent extracellularly recorded action potentials. The black trace corresponds to the LFP and the red traces represent synaptic activity of an intracellularly recorded CA1 pyramidal neuron. During the sharp wave activity levels of individual CA1 inhibitory neurons are not obviously rhythmic. However, rhythmicity is apparent when summating the activity across all individual neurons (CA1pop.). As described in A2, high-frequency population activity is observed at the LFP peak while low-frequency activity is observed at the LFP trough. CA1 pyramidal neurons receive fast rhythmic inhibition because the activity generated by the inhibitory networks is oscillatory.
Gap Junction: What's Your Function?
The hypothesis that strong synaptic inhibition contributes to the generation of ripple oscillations is not without problems. Specifically, in vitro experiments have shown that ripples persist during GABAA receptor blockade (Maier et al., 2003). Indeed, spontaneous CA1 ripples are observed in the LFP even when all chemical transmission is eliminated (Draguhn et al., 1998). It is noteworthy that both synaptic potentials and action potentials can contribute to the LFP signal, unlike the case with the EEG signals discussed above. Thus, the persistence of ripples during chemical transmission blockade has led to an alternative hypothesis for which evidence is accumulating: ripples are driven by action potential activity that results from direct, gap junction-mediated electrical coupling among the axons of CA1 pyramidal neurons (Draguhn et al., 1998; Traub et al., 2001a). This hypothesis was derived from the observation that ripple oscillations are eliminated by halothane (Draguhn et al., 1998; Ylinen et al., 1995) and carbenoxolone (Maier et al., 2003), two compounds that block gap-junctional communication. Although gap junction blockers are known to have non-specific actions (Leznik and Llinas, 2005; Rouach et al., 2003; Rozental et al., 2001), evidence for the presence of CA1 pyramidal neuron axon-axon electrical coupling comes from both anatomy and physiology experiments. First, when a CA1 pyramidal neuron is filled with a dye that traverses gap junctions, neighboring pyramidal neurons are also labeled, revealing a plexus of interconnected, dye-coupled neurons (Schmitz et al. 2001; Valiente et al., 1995; Church and Baimbridge, 1991; Andrew et al. 1982). When assessing morphological features of the dye-coupled neurons, there is evidence that axons are the coupling sites (Schmitz et al., 2001). Second, when simultaneously recording a pair of CA1 pyramidal cells with intracellular electrodes, one can elicit a carbenoxolone-sensitive, miniature version of an action potential (‘spikelet’) in the axon of one neuron by causing the other neuron to fire an action potential (Schmitz et al., 2001).
How might axon-axon electrical coupling promote ripple rhythm generation? Once again, computational approaches may provide insights. Traub and colleagues (Traub et al., 2001a; Traub and Bibbig, 2000; Traub et al., 1999) have proposed a model (Figure 8) that assumes that (1) under relatively depolarized conditions CA1 axons spontaneously generate action potentials, (2) CA1 axon-axon coupling is relatively sparse (i.e. each axon is coupled to ∼2 others) and (3) axonal gap junctions permit the transfer of action potentials to their coupled neighbors, enabling the activation of coupled axons. If an action potential is spontaneously generated in an axon, then this action potential will propagate to its electrically coupled neighbors. In turn, these secondary action potentials will elicit action potentials in their electrically coupled neighbors and so forth until a wave of action potential-based activity propagates sparsely throughout the CA1 axonal plexus. The duration of this wave of activity is primarily a function of (1) the time required for an action potential to propagate though a gap junction (0.3-0.5 msec) and (2) the size and connectivity of the axonal plexus.
Figure 8.

Graphical description of how electrical coupling among the axons of CA1 pyramidal neurons may provide the rhythmicity that underlies ripple oscillations. A1. As proposed by Traub and colleagues, propagating activity within a plexus of electrically coupled CA1 pyramidal neuron axons forms the basis of the rhythm generator for ripple oscillations. The plexus is activated by CA3 excitatory input. By rhythmically driving CA1 inhibitory neurons (see B), the activated plexus indirectly generates rhythmic IPSPs on somatic regions of CA1 pyramidal neurons. A2. Description of the temporal relationship between CA1 axon plexus dynamics and ripple oscillations, as represented by a rhythmic 200 Hz local field potential (LFP). The plexus is arbitrarily composed of 30 axons (red) that can be subdivided into 10 nodes according to firing times. Numbers above the plexus represent node number. Nodes 1 & 10 contain 1 axon each, nodes 2 & 9 contain 2 axons each, and nodes 3-8 contain 4 axons each. Axons within each node fire action potentials at the same time. The axon in node 1 is electrically coupled to both axons in node 2, which, in turn, are electrically coupled to the axons in node 3, and so forth. Thus, when an action potential is spontaneously generated by the axon in node 1, the activity quickly propagates through the plexus in a wave-like manner (from left to right). The duration of the wave is determined by the time for an action potential to cross a gap junction and the size of the plexus (specifically, the number of nodes through which the wave propagates). For practical reasons, in this example we define the duration for an action potential to propagate through a gap junction to equal 0.5 msec (it is proposed to be slightly briefer). Because the activity propagates through 10 nodes, the duration of the wave is equal to 5 msec (0.5 msec × 10 nodes). Once the wave of activity terminates, another wave begins when the next spontaneously-generated action potential occurs within the plexus (which we arbitrarily set to occur again within node 1). During CA3-mediated excitation, the rate of spontaneously-generated action potentials within the plexus is high, resulting in propagating waves that occur every ∼5 msec (i.e. time-locked to the local field potential, LFP). B. Proposed mechanism for how activity generated by the CA1 pyramidal neuron axon plexus results in rhythmic perisomatic inhibition of CA1 pyramidal neurons. For simplicity, we show only two CA1 pyramidal neurons and two inhibitory neurons. Under normal conditions action potentials generated within the axon plexus propagate orthodromically to trigger the release of glutamate that excites inhibitory neurons. The inhibitory neurons are rhythmically activated because the output of the plexus is rhythmic. Inhibitory neurons, in turn, rhythmically inhibit the perisomatic region of CA1 pyramidal neurons, resulting in ripple oscillations. Under conditions that block chemical neurotransmission the rhythmic waves of activity within the axon plexus persist. However, because communication between pyramidal and inhibitory neurons is abolished, the recorded ripple oscillation is a consequence of action potentials propagating antidromically back to the CA1 pyramidal neuron soma. C. Graphical representation of neuronal activity patterns according to proposed model. As in Figure 6, vertical lines placed on horizontal black lines represent extracellularly recorded action potentials. The two bottom red traces represent synaptic activity of intracellularly recorded CA1 pyramidal neurons and the bottom trace corresponds to the LFP. Population activity within the CA1 axon plexus (red, represented as a histogram) is rhythmic during the CA3 sharp wave (blue). The plexus rhythmically activates a population of CA1 inhibitory neurons (CA1pop., green), resulting in rhythmic IPSPs in the perisomatic region of CA1 pyramidal neurons (bottom red traces).
To understand how activity waves are defined by action potential propagation and axonal plexus size, it is instructive to conceptualize a population of neuronal axons that are sparsely coupled to each other. In Figure 8 we outline an example network with 10 coupled nodes (i.e. groups of electrically coupled axons that fire synchronized action potentials). Clearly, the population of CA1 pyramidal neurons is much larger and, therefore, the corresponding plexus is likely to be much larger. However, the parameter that is critical for oscillatory activity is the portion of the plexus that sustains the wave, which is proposed to be less than 20 coupled nodes (Traub et al., 2002). If it takes 0.5 msec for an action potential from one axon to propagate through a gap junction to its neighbor, and the wave travels through 10 coupled nodes, then the duration of the wave is 5 msec. Once the wave reaches the end of the coupled nodes, it does not propagate in the opposite direction because the action potentials are associated with a refractory period. When the wave of activity has terminated, another wave will initiate when the next action potential in the CA1 axon plexus is generated. If the rate of action potential generation within the plexus is high, then waves will occur every 5 msec (i.e. 200 Hz). Thus, to borrow from a description provided by Traub et al. (2002), the ripple-like activity within the axonal plexus is not an oscillation per se but, rather, a rapid series of propagating waves. What, then, elevates the rate of spontaneous action potential generation such that waves are initiated at ripple frequency? According to the proposed model, the CA3 sharp wave depolarizes CA1 neurons and significantly increases the probability (and, therefore, the rate) that their axons will generate action potentials.
Hypotheses regarding the downstream effects of CA1 axonal waves are thoroughly outlined by Traub et al. (2002). Briefly, under conditions in which chemical transmission is blocked in vitro, these waves propagate antidromically along the axons until they reach their respective somata (Figure 8B). Under more intact conditions, the waves of activity are proposed to propagate orthodromically to provide rhythmic excitatory drive to CA1 inhibitory neurons. In turn, these inhibitory neurons provide feedback onto CA1 pyramidal neurons in somatic/perisomatic regions, resulting in the fast, rhythmic IPSPs presumed to underlie in vivo ripple oscillations.
Recently, a second model based exclusively on electrical coupling among axons has been proposed (Tseng et al., 2008). In this model, fast oscillatory activity results from the reverberation of action potentials within a closed-loop network of electrically coupled axons. Similar to the model proposed by Traub et al., oscillation frequency is largely determined by the size of the coupled network. Interestingly, Tseng et al. also performed experimental work demonstrating that comparable oscillatory mechanisms occur in the network responsible for the tail-flip escape behavior of crayfish. Moreover, the crayfish network, which consists of a relatively small plexus of electrically coupled axons, can oscillate at frequencies greater than 600 Hz. Thus, both computational and biological networks can generate very fast oscillations using axonal electrical coupling as a substrate.
Ascribing a significant role for electrical coupling in the generation of ripples is, however, complicated by the observation that the oscillations persist in mice lacking connexin 36 (Pais et al., 2003), the gap junction protein that predominantly mediates electrical coupling among neurons. Ripples persist in these animals either because gap junctions are not important for the oscillations or because long-term removal of the protein, as occurs in knockout animals, results in the upregulation of other gap junction proteins that can mediate axon-axonal electrical coupling and, therefore, sustain ripple activity (Pais et al., 2003). The inhibition of ripples in the mutant mouse by gap junction blockade (Pais et al., 2003) supports the latter. Future studies using anatomical and physiological approaches comparable to those described above should reveal whether CA1 axons remain electrically coupled in the mutant mouse.
In sum, the mechanisms underlying the ripple rhythm generator remain controversial and rely primarily on insights gleaned from computational techniques. On the one hand, we have a model network driven by powerful synaptic inhibition that must be reconciled with experimental evidence indicating that ripples persist when all chemical synaptic transmission is abolished. On the other hand, we have a model network mediated exclusively by electrical coupling that must be reconciled with the finding that ripples persist in an animal lacking connexin 36, a predominant neuronal gap junction protein. Future studies will ultimately determine which model is closer to reality.
Sharp Wave-Ripple Initiation
As described above, the initiation of sharp wave-ripple oscillations is thought to involve the synchronized burst of many CA3 pyramidal neurons that send projections to CA1. What factors, then, promote large-scale, coordinated CA3 bursting activity? It has long been appreciated that CA3 pyramidal neurons are under inhibitory control and that releasing these neurons from inhibition promotes their synchronous firing of action potential bursts (Miles and Wong, 1987). Moreover, blocking inhibition unmasks significant recurrent excitatory connections among CA3 pyramidal neurons (Miles and Wong, 1987). Therefore, investigators have set out to determine what endogenous factors modulate inhibition within CA3 that might regulate the expression of highly synchronized pyramidal neuron bursting. One illustrative example of a neurotransmitter that regulates such bursting is adenosine, a nucleoside that is present in the extracellular space at relatively high concentrations. Several reports have demonstrated that blocking adenosine receptors induces synchronized bursting of CA3 pyramidal neurons (Alzheimer et al., 1989; Alzheimer et al., 1993; Wu et al., 2009). Moreover, Wu et al. (2009) recently showed that augmenting the actions of adenosine can inhibit spontaneously-occurring CA3 sharp waves in hippocampal slices. Both pre- and postsynaptic mechanisms may regulate CA3 bursting. Inhibiting the actions of adenosine may remove the presynaptic block that the neurotransmitter preferentially places on excitatory terminals (Lambert and Teyler, 1991; Scanziani et al., 1992; Wu and Saggau, 1994), thereby enhancing excitatory neurotransmission. Alternatively, as adenosine can activate postsynaptic potassium channels to hyperpolarize neurons (Pan et al., 1995), blocking adenosine may cause pyramidal neurons to depolarize and make them more likely to burst. Regardless of the mechanism, an intriguing observation is that endogenous extracellular levels of adenosine are reduced by ∼20% during sleep (Basheer et al., 2004) – the time when sharp wave-ripples are primarily expressed.
Hippocampal Dysfunction and Epilepsy
The predominant form of epilepsy associated with the hippocampus is mesial temporal lobe epilepsy (MTLE). Like the idiopathic generalized epilepsies described above, little is known regarding the etiology of MTLE. One dominant hypothesis states that events early in life set in motion a process that ultimately engenders networks in the brain hyperexcitable and capable of generating seizures. As this process can occur slowly, the precipitating event does not necessarily immediately result in MTLE. For example, prolonged febrile seizures (i.e. fever-induced seizures occurring in children) are often associated with later onset MTLE (Falconer, 1971; French et al., 1993; So et al., 1989; Williamson et al., 1993; but see Ellenberg and Nelson, 1978). After the precipitating seizures pass, the brain enters a latent period during which an epileptic circuit “ripens” (Earle and Penfield, 1953). After this latent period, the individual has seizures symptomatic of MTLE, which are commonly characterized spontaneous motor behaviors that often consist of movements of the mouth (e.g. chewing, licking, tooth grinding). Consciousness and/or memory is often impaired during MTLE seizures, rendering patients unaware of their motor activity. While seizures can be treated with anti-epileptic drugs (AEDs), between 60% and 80% of MTLE patients will eventually develop intractable (or refractory) epilepsy (Kwan and Brodie, 2000; Pittau et al., 2009; Semah et al., 1998), a condition in which seizure frequency is high and is not abated by AEDs. In such cases, surgical removal of the affected medial temporal lobe is a common form of treatment.
MTLE is commonly associated with sclerotic tissue in the hippocampus, with neuronal loss most often occurring in the hilar region of the dentate gyrus, CA3 and CA1 (Margerison and Corsellis, 1966; but see Engel et al., 2008). This association was originally made in studies of resected hippocampi from patients with intractable MTLE (Falconer, 1953) and was subsequently supported by high-resolution MRI scans of intractable MTLE patients (Van Paesschen et al., 1997a). However, several recent MRI studies have shown that the hippocampi of between 65% and 90% of newly-diagnosed MTLE patients actually appear normal (Liu et al., 2002; Van Paesschen et al., 1998; Van Paesschen et al., 1997b). Assuming that MRI scans are sufficiently sensitive to assess hippocampal damage, these findings suggest that extensive hippocampus sclerosis is not required to initially cause seizures. This finding has led to the hypothesis that repetitive seizure activity in the hippocampus may itself cause the structural damage that is apparent in intractable MTLE patients (Sutula et al., 2003).
If extensive hippocampal damage does not contribute to MTLE, then how do seizures arise? In a few cases, MTLE is familial, indicating a genetic predisposition for the disease (Berkovic et al., 1996). In 1995, it was discovered that mutations in the leucine rich glioma inactivated (LGI-1) gene are associated with familial forms of MTLE (Ottman et al., 1995). Moreover, the LGI-1 gene is mutated in roughly 50% of all patients with familial MTLE characterized by strong auditory auras (Kalachikov et al., 2002; Ottman et al., 2004). While LGI-1 is highly expressed in the hippocampus (Senechal et al., 2005), it remains unknown how LGI-1 dysfunction contributes to MTLE. However, two recent studies have shed light on how LGI-1 regulates neuronal excitability. First, LGI-1 prevents the inactivation of axonal/presynaptic voltage-gated potassium channels that are critical for shaping action potentials (Schulte et al., 2006). The Kv1.1 and Kvβ1 potassium channel subunits coassemble, with the latter subunit conferring rapid inactivation to the channel. Schulte et al. demonstrated that LGI-1 binds to Kv1.1 and prevents Kvβ1-mediated channel inactivation. Without channel inactivation, the potassium channel is open for longer periods and, therefore, promotes potassium-mediated repolarization of the membrane potential during the action potential, an effect that likely results in short-duration action potentials. Mutant forms of LGI-1 cannot prevent channel inactivation, leading to significantly briefer potassium currents and, presumably, broader action potentials. These experiments were performed in Xenopus oocytes so direct measurements of action potential kinetics were not possible. The results are nonetheless significant because broadened action potentials are hypothesized to prolong the duration that presynaptic terminals are depolarized, thereby enhancing terminal Ca2+ influx and subsequent neurotransmitter release. Indeed, Geiger and Jonas (2000) demonstrated that EPSPs recorded in the hippocampus are larger when triggered by broadened action potentials (also see Shu et al., 2006). These observations are consistent with the finding that mutations in LGI-1 can lead to the hyperexcitable hippocampal networks associated with MTLE.
A second function associated with LGI-1 relates to trafficking of AMPA-subtype glutamate receptors (Fukata et al., 2006). In this context, LGI-1 is believed to encode a secreted protein. These studies demonstrated that exogenous application of LGI-1 to hippocampal slices promotes AMPA receptor-mediated synaptic transmission, an effect attributed to increased surface expression of AMPA receptors. The authors did not perform experiments with mutant LGI-1 so we can only speculate how mutations in the protein might contribute to hyperexcitable networks. If, as might be anticipated, there is less AMPA receptor expression with application of mutant LGI-1, then it is difficult to conceptualize how a reduction in excitatory synaptic transmission might lead to hyperexcitable hippocampal networks. Future studies may elucidate a link. Finally, while these recent studies of LGI-1 provide significant insights into the function of a previously unknown protein, it is important to emphasize that in contrast to the idiopathic generalized epilepsies described above, MTLE is not currently thought to have a strong genetic predisposition. Indeed, a recent study showed that only 7% of all MTLE patients were believed to have a familial form of the disease (Kobayashi et al., 2001).
As there are few leads into the etiology of MTLE, several investigators have turned to animal models. Most of these models are motivated by the aforementioned hypothesis that prolonged neuronal hyperactivity early in life predisposes the more mature hippocampus to generate seizures. In this regard, these animal models are consistent with the hypothesis because they exhibit a latent period during which seizures are not expressed. In these models, animals are induced to have severe prolonged seizures, a condition known as status epilepticus, with chemoconvulsants such as kainic acid or pilocarpine. Following a latent period animals acquire a seizure phenotype consistent with MTLE (Sloviter, 2008; Buckmaster, 2004; White, 2002). Also, animal model pathologies are comparable to those discussed in the human literature such as neuronal degeneration in the dentate gyrus, CA3 and CA1. Of particular relevance to this review is the observation that the electrophysiological activity patterns recorded in these animal models is comparable to activity recorded in human patients.
The Fast Ripple
Both humans with MTLE (Bragin et al., 1999a; Jacobs et al., 2008) and animals with induced MTLE-like seizures (Bragin et al., 1999c) generate a very fast (>200 Hz) oscillation called the fast ripple (Figure 5B,C). Fast ripples have received considerable attention because they appear to occur primarily – if not exclusively – in the epileptic hippocampus (Bragin et al., 1999a; Bragin et al., 1999b). These events can be recorded with depth electrodes placed within the hippocampi of patients with intractable MTLE during pre-operative screening procedures designed to determine the seizure focus for subsequent surgical removal. Interestingly, fast ripples primarily occur in the regions near the seizure onset, leading to the hypothesis that fast ripples reflect hypersynchronous discharges of pyramidal neurons within the epileptogenic region (Bragin et al., 1999b; Bragin et al., 2000). Specifically, while ripples and fast ripples can be recorded in both hippocampi, the incidence of fast ripples is higher in the hippocampus ipsilateral to the seizure onset zone (Staba et al., 2007). Also, epileptic foci are characterized by lower neuronal densities and higher fast ripple-to-ripple discharge ratios. Together, these results suggest that as ripple incidence decreases, the occurrence of fast ripples increases, and that the switch between the two types of oscillations requires a structural reorganization of hippocampal circuits.
Are Fast Ripples Just Fast Ripples?
To begin, it is important to emphasize that current evidence indicates that ripples and fast ripples are distinct phenomena (Bragin et al., 2002). First, the frequency of fast ripples is generally higher than ripples. Second, unlike ripples, fast ripples are only observed in epileptic brains. Third, ripples primarily occur in a unidirectional manner in that they are initiated by CA3 sharp waves and subsequently generated in CA1. With the exception of occurring near epileptic foci, fast ripples, in contrast, do not appear to be localized to specific regions of hippocampus, nor do they propagate in a stereotyped fashion. Finally, fast ripples are not broadly expressed throughout large areas of hippocampus (Bragin et al., 2002). Indeed, Bragin et al. (2002) demonstrated that fast ripples are primarily confined to small clusters near epileptic foci the authors termed pathologically interconnected clusters of neurons (PINs). Ripples, in contrast, are not associated with such PINs. Nonetheless, these differences do not preclude the possibility that the general mechanisms underlying ripples and fast ripples are similar. To conclusively determine whether ripple and fast ripple oscillations are derived from either (1) similar but altered circuit mechanisms or (2) completely different circuit mechanisms obviously requires a deep understanding of how both oscillations are generated. In this review we will attempt to make the argument that comparable circuit mechanisms subserve ripple and fast ripple oscillations. However, as we lack a firm of understanding of how either oscillation is generated, our hypotheses are largely speculative. One piece of evidence that the two oscillations are related comes from the observation that the incidence of ripples and fast ripples appears to be coupled. Specifically, the occurrence of fast ripples increases in the human epileptic hippocampus as the number of observed ripple oscillations decreases. A similar result was found by Foffani et al. (2007) in the pilocarpine animal model of MTLE. Foffani et al. (2007) also found a strong correlation between the peak frequencies of ripple and fast ripple oscillations recorded within slices, suggesting that the two were dynamically connected. Another finding that indicates similar underlying mechanisms is the observation that both oscillation types occur primarily during sleep (Bragin et al., 2000; Pavlides and Winson, 1989). Thus, ripples and fast ripples appear to rely on similar brain states for expression. Additionally, Bragin et al. (2002) demonstrated that fast ripples persist during GABAA receptor blockade, as was shown for ripples (Maier et al., 2003). However, to better assess the possibility that the two oscillations rely on similar mechanisms for generation, it will be important to determine whether fast ripples persist when all chemical transmission is blocked, as was shown for ripples (Draguhn et al., 1998), or whether fast ripples are still present in the connexin-36 knockout animal, as was also shown for ripples (Pais et al., 2003). The results of such experiments will help us determine whether hypotheses regarding ripple generation can be co-opted to explain how fast ripples are produced.
Fast Ripples: Envision Precision
There are relatively few studies that describe the mechanisms underlying fast ripples. Here we highlight two recent studies demonstrating the importance of action potential timing precision in the expression of fast ripples in CA3. It is not clear how well the mechanisms underlying CA3 fast ripples can be extrapolated to oscillations observed in CA1 but as there are few studies on the mechanisms of fast ripples we will discuss their results nonetheless. The first study examined hippocampal slices bathed with solution containing elevated concentrations of potassium ions (Dzhala and Staley, 2004), a manipulation often used to depolarize neurons and make networks hyperexcitable. The authors observed that this manipulation promoted the generation of epileptiform CA3 pyramidal neuron discharges that consisted of fast ripple oscillations. The observation that fast ripple oscillations can be generated in normal slices with acute application of modified bathing solution was subsequently demonstrated by Spampanato and Mody (2007). Dzhala and Staley observed that as high-potassium solution washed into the slice, the burst onset time of two intracellularly recorded neurons gradually became more synchronized. Additionally, elevated potassium levels promoted highly precise spiking activity within individual bursting neurons, as assessed by measuring the variance of the inter-spike interval (ISI). Moreover, pharmacological agents that decreased ISI variance (i.e. increased spike precision) also increased the amplitude of fast ripple oscillations while agents that increased ISI variance decreased fast ripple amplitude. Together, the results of Dzhala and Staley demonstrate that hypersynchrony among CA3 neurons is likely to initiate fast ripple oscillations and that subsequent high-fidelity firing of CA3 neurons contributes fast ripples characterized by high amplitudes.
A recent study further supports the hypothesis that the temporal features of action potentials among bursting CA3 pyramidal neurons is pivotal for the emergence of fast ripples (Foffani et al., 2007). The authors compared ripples and fast ripples in CA3 regions of control and pilocarpine-treated epileptic animals. In slice preparations containing only CA3, the authors observed that the frequency of fast ripples was nearly double that of ripples. Additionally, fast ripples were characterized by greater entropy, indicating that the faster oscillation is more disorganized. To determine the source of network disorganization, they subsequently measured the spontaneous bursting activity of individual CA3 pyramidal neurons. While the average intraburst firing frequency was similar for both groups of animals, the timing of the action potentials was more variable in epileptic animals. Specifically, the 2nd action potential within control bursts occurred at a stereotyped latency following the initial action potential. In epileptic animals, however, the second action potential was associated with more variable latencies (i.e. jitter), an effect attributed to elevated spontaneous synaptic activity in the epileptic animal. So, how does greater spike time variability promote fast ripple activity? The authors propose that such variability in the epileptic hippocampus promotes a phase shift in the oscillatory activity of neighboring ripple-generating networks. To understand this model, one can conceptualize two ripple generating networks operating in close proximity, both of which generate 250 Hz oscillations (see Staley, 2007 for an excellent description of their model). When the two networks are simultaneously activated under normal conditions, as might occur when triggered by a common input, the networks behave in unison due to the high temporal precision of pyramidal cell firing. This results in a high-amplitude 250 Hz oscillation because the activity generated by the two networks summates. In contrast, the poor firing precision of pyramidal neurons in the epileptic hippocampus causes the two ripple-generating networks to fire half a cycle out of phase, resulting in a composite 500 Hz oscillation with lower amplitude. The suggestion, then, is that fast ripples reflect subpopulations of pyramidal neurons firing at ripple frequency but ½ a cycle out of phase with each other. If true, then it is difficult to envision how increased variability in pyramidal firing – variability that is presumably random – can lead to neuronal subpopulations firing exactly ½ a cycle out of phase, a phenomenon that would appear to require a non-random shift in neuronal firing properties. Nevertheless, the authors demonstrated that pharmacologically enhancing spike jitter increases ripple frequency, and that pharmacologically decreasing spike jitter decreases fast ripple frequency.
Several conclusions of Foffani et al. are consistent with those of Dzhala and Staley. First, both studies demonstrate that elevated synaptic activity promotes fast ripple generation. Second, both argue that fast ripples are likely to be initiated by a hypersynchronous burst among CA3 pyramidal neurons. Third, both studies demonstrate that altering the precision of spike-timing changes the frequency of hippocampal oscillations. Finally, the two studies show that pharmacologically promoting spike jitter within neurons results in low amplitude oscillations. However, one major inconsistency appears to exist. Dzhala and Staley argue that highly precise firing is associated with fast ripples, a claim substantiated by the observation that pharmacologically enhancing spike jitter decreases the frequency of fast ripples. In contrast, Foffani et al. argue that imprecise firing is responsible for the expression of fast ripples, a claim substantiated by the observation that pharmacologically reducing spike jitter decreases the frequency of fast ripples. While this apparent discrepancy might arise from different experimental conditions (i.e. acute high potassium solution versus chronic epileptic animal model), these conclusions appear difficult to reconcile.
When Activity is High, Fast Ripples are Nigh
While it is not clear what factors contribute to fast ripples, the aforementioned studies demonstrate that increased neuronal activity promotes the expression of fast ripples. In the animal model study, Foffani et al. suggest that a structural reorganization of the injured area mediates this process because fast ripple expression is positively correlated with CA3 pyramidal layer degeneration. The authors did not observe a significant correlation between fast ripple expression and axonal sprouting. If the increased neuronal activity presumed to underlie fast ripples results from neuronal loss and not sprouting, then it should follow that the neurons that survive in the epileptic tissue exhibit higher spontaneous firing rates. The authors did not discuss whether or how this might arise. One possibility is that potassium regulation by glial cells is altered in the epileptic hippocampus. Extracellular potassium removal is a major function of glial cells (Orkand et al., 1966). Previous work has demonstrated that glia do not efficiently carry out this function in sclerotic human or rat hippocampal tissue (Heinemann et al., 2000). Such reduced buffering capacity may elevate extracellular potassium concentrations and, in turn, increase neuronal activity and fast ripple expression. Indeed, bathing solutions with elevated extracellular potassium concentrations are precisely the manipulation used by Dzhala and Staley use to elicit fast ripples.
So, are the rhythm generators for ripples and fast ripples similar? The answer to this question is not known. However, if they are, we once again must address whether these oscillations are driven by generators whose rhythmicity is based on chemical synaptic transmission or on electrical coupling. The results of Dzhala & Staley and Foffani et al. indicate that neural networks can oscillate at vastly different frequencies when the level of synaptic activity within constituent neurons is modulated. To our knowledge, investigators have not yet designed computational models that recapitulate these experimental observations. Nonetheless the aforementioned computational models of Brunel and Wang (2003) and Geisler et al. (2005) do incorporate features that are consistent with these two studies such as the reliance of fast oscillations on high levels of synaptic activity. Despite this consistency, it remains formally possible that the aforementioned gap junction-based models of ripple rhythmogenesis can also generate oscillations in the fast ripple frequency range. One common property of the two gap junction-based models proposed by Traub et al. (2002) and Tseng et al. (2008) is that oscillation frequency is a function of network size: the fewer the number of coupled axons, the faster the oscillation. This feature is particularly intriguing in light of the observation that expression of fast ripples is highly correlated with neuronal degeneration (Foffani et al., 2007; Staba et al., 2007). If axonal plexus shrinkage accompanies pyramidal neuron degeneration, then this structural change could effectively increase the expression of fast ripples while concurrently decreasing ripple occurrence. The specific features of neuronal death observed in the various animal models are consistent with this hypothesis. In both kainic acid- and pilocarpine-induced status epilepticus, as well as a rodent model of febrile seizures, death among CA3 and CA1 pyramidal neurons is common, with CA3 neurons perhaps more vulnerable (Clifford et al., 1987; Nadler et al., 1978; Pollard et al., 1994; Toth et al., 1998). Moreover, if the damaged area is associated with elevated concentrations of potassium due to reduced glia buffering, then the rate of spontaneously-generated action potentials within the axonal plexus that drive waves of activity would presumably rise as well.
Concluding Remarks
We have outlined above our current understanding of how two different brain circuits generate sleep-related oscillatory patterns. There is evidence that these circuits are co-opted to generate epileptiform activity, yet much work still needs to be done to determine the relationship between sleep- and epilepsy-related circuits. Moreover, it remains to be determined whether the link between these circuits is coincidental, or whether epilepsy is a disease that, as a general rule, takes advantage of sleep circuits. Any potential link is unlikely to do with the behavior of sleep per se. Rather, we argue that both sleep and epilepsy appear to unmask oscillatory circuits that are likely pervasive in the brain. While their function is incompletely understood, these circuits are likely comparable to those described by Douglas and Martin (2004) – i.e. semi-autonomous anatomical microcircuits that process information. If the brain is composed of numerous such microcircuits, and many are rhythmic, then it is not too surprising that a disease characterized by neuronal hyperexcitability manifests itself with oscillatory activity patterns. That is, pathological conditions that render neurons more excitable will channel elevated activity into existing circuitry rather than into completely uncharted pathways. Thus, we argue that the circuitry present in the normal, non-injured brain likely exists as a template upon which pathological changes are placed.
Pathological changes that promote network hyperexcitability include (1) alterations in intrinsic/synaptic excitability, and (2) alterations in anatomical structure (i.e. neuronal death, sprouting, synaptogenesis). Indeed, the pathological activity patterns that are the focus of this review reflect these two possibilities, with thalamocortical spike-wave discharges likely resulting from the former and hippocampal fast ripples likely resulting from the latter. This distinction may provide an explanation for another major difference between spike-wave discharges and fast ripples. Specifically, the thalamocortical circuit appears capable of existing in two distinct stable states, while the hippocampal circuit does not. The justification for this statement stems from the observation that thalamocortical circuits generate both spindle and spike-wave discharges while hippocampal circuits appear to generate either ripples or fast ripples. The lack of apparent bistability in the latter circuit, we believe, is easier to comprehend. If fast ripples (and MTLE seizures in general) are the result of long-term structural changes to the hippocampus, then one might expect that the underlying circuitry will only sustain one form of activity pattern. Indeed, the observation by Staba et al. (2007) that structural reorganization in the epileptic hippocampus promotes fast ripple expression while reducing ripple expression is consistent with the hypothesis that the circuitry permanently exists either in a fast ripple-generating or a ripple-generating configuration, but cannot dynamically switch between the two.
The greater challenge, we believe, is to understand why dynamic bistability exists in the pathological thalamocortical circuit. Indeed, it seems quite surprising that both the appearance and incidence of spindle oscillations are unchanged in patients with absence epilepsy (although we are unaware of any systematic studies). Of course, one explanation for this observation is that the circuits responsible for generating spindles and spike-wave discharges are independent. While possible, this seems unlikely for reasons outlined above. If the two activity patterns do result from the same basic circuit, then what is responsible for the dynamic switch between the two activity patterns? Several avenues of research point to the possibility that the reticular thalamic nucleus provides a switching mechanism. Under normal conditions, strong intra-RT inhibition is difficult to overcome and, therefore, maintains the thalamus in a sparse network configuration that favors the generation of spindles. While intra-RT inhibition in normal brains can be overcome to generate spike-wave discharges, very strong and presumably non-physiological excitation of RT neurons is required to do so (e.g. Blumenfeld and McCormick, 2000; Bal et al. 2000). If genetic mutations elevate the excitability of RT neurons, however, then the threshold required to overcome intra-RT inhibition may be lower, thereby enabling physiological activity patterns to more easily switch thalamic circuits into a hypersynchronous firing mode. Future studies will determine whether it is clinically feasible to keep RT excitation below the threshold for seizure generation, effectively turning off the RT switch for good.
Acknowledgments
This work was supported by NIH/NINDS Grants 34774 and 07280. We would like to thank Drs. Diego Contreras and Roger Traub for insightful comments regarding mechanisms of thalamic and hippocampal oscillations. Drs. Paul Buckmaster and Kevin Graber provided important insights throughout the writing process. Drs. Chris Dulla and Alex Goddard provided helpful commentary on final versions of the manuscript.
References
- Alzheimer C, Sutor B, ten Bruggencate G. Transient and selective blockade of adenosine A1-receptors by 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) causes sustained epileptiform activity in hippocampal CA3 neurons of guinea pigs. Neuroscience Letters. 1989;99:107–112. doi: 10.1016/0304-3940(89)90273-5. [DOI] [PubMed] [Google Scholar]
- Alzheimer C, Sutor B, Ten Bruggencate G. Disinhibition of hippocampal CA3 neurons induced by suppression of an adenosine A1 receptor-mediated inhibitory tonus: Pre- and postsynaptic components. Neuroscience. 1993;57:565–575. doi: 10.1016/0306-4522(93)90006-2. [DOI] [PubMed] [Google Scholar]
- Andersen P, Andersson S, Junge K, Sveen O. Xth Scandinavian EEG meeting: Voksenåsen, Oslo, Norway March 10-11, 1967 Secretary: C. W. Sem-Jacobsen The EEG Research Institute, Gaustad Sykehus, Oslo 3 (Norway) Electroencephalography and Clinical Neurophysiology. 1968;24:87–92. [Google Scholar]
- Andersen P, Andersson SA, Lomo T. Nature of thalamo-cortical relations during spontaneous barbiturate spindle activity. J Physiol. 1967a;192:283–307. doi: 10.1113/jphysiol.1967.sp008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P, Andersson SA, Lomo T. Some factors involved in the thalamic control of spontaneous barbiturate spindles. J Physiol. 1967b;192:257–281. doi: 10.1113/jphysiol.1967.sp008299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson SA, Manson JR. Rhythmic activity in the thalamus of the unanaesthetized decorticate cat. Electroencephalography and Clinical Neurophysiology. 1971;31:21–34. doi: 10.1016/0013-4694(71)90286-0. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Taylor CP, Snow RW, Dudek FE. Coupling in rat hippocampal slices: dye transfer between CA1 pyramidal cells. Brain Res Bull. 1982;8:211–22. doi: 10.1016/0361-9230(82)90048-x. [DOI] [PubMed] [Google Scholar]
- Bal T, Debay D, Destexhe A. Cortical feedback controls the frequency and synchrony of oscillations in the visual thalamus. J Neurosci. 2000;20:7478–7488. doi: 10.1523/JNEUROSCI.20-19-07478.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal T, McCormick DA. What stops synchronized thalamocortical oscillations? Neuron. 1996;17:297–308. doi: 10.1016/s0896-6273(00)80161-0. [DOI] [PubMed] [Google Scholar]
- Bal T, von Krosigk M, McCormick DA. Role of the ferret perigeniculate nucleus in the generation of synchronized oscillations in vitro. J Physiol. 1995;483:665–85. doi: 10.1113/jphysiol.1995.sp020613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Basheer R, Strecker RE, Thakkar MM, McCarley RW. Adenosine and sleep-wake regulation. Progress in Neurobiology. 2004;73:379–396. doi: 10.1016/j.pneurobio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Behrens CJ, van den Boom LP, de Hoz L, Friedman A, Heinemann U. Induction of sharp wave-ripple complexes in vitro and reorganization of hippocampal networks. Nat Neurosci. 2005;8:1560–1567. doi: 10.1038/nn1571. [DOI] [PubMed] [Google Scholar]
- Berkovic S, McIntosh A, Howell R, Mitchell A, Sheffield L, Hopper J. Familial temporal lobe epilepsy: a common disorder identified in twins. Annals of Neurology. 1996;40:227–235. doi: 10.1002/ana.410400214. [DOI] [PubMed] [Google Scholar]
- Beyer B, Deleuze C, Letts VA, Mahaffey CL, Boumil RM, Lew TA, Huguenard JR, Frankel WN. Absence seizures in C3H/HeJ and knockout mice caused by mutation of the AMPA receptor subunit Gria4. Human Molecular Genetics. 2008;17:1738–1749. doi: 10.1093/hmg/ddn064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H, McCormick DA. Corticothalamic inputs control the pattern of activity generated in thalamocortical networks. J Neurosci. 2000;20:5153–5162. doi: 10.1523/JNEUROSCI.20-13-05153.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born J, Rasch B, Gais S. Sleep to remember. Neuroscientist. 2006;12:410–424. doi: 10.1177/1073858406292647. [DOI] [PubMed] [Google Scholar]
- Bragin A, Engel J, Jr, Wilson C, Fried I, Buzsáki G. High-frequency oscillations in human brain. Hippocampus. 1999a;9:137–142. doi: 10.1002/(SICI)1098-1063(1999)9:2<137::AID-HIPO5>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Bragin A, Engel J, Jr, Wilson C, Fried I, Mathern G. Hippocampal and entorhinal cortex high-frequency oscillations (100-500 Hz) in human epileptic brain and in kainic acid-treated rats with chronic seizures. Epilepsia. 1999b;40:127–137. doi: 10.1111/j.1528-1157.1999.tb02065.x. [DOI] [PubMed] [Google Scholar]
- Bragin A, Engel J, Jr, Wilson C, Vizentin E, Mathern G. Electrophysiologic analysis of a chronic seizure model after unilateral hippocampal KA injection. Epilepsia. 1999c;40:1210–1221. doi: 10.1111/j.1528-1157.1999.tb00849.x. [DOI] [PubMed] [Google Scholar]
- Bragin A, Wilson C, Engel J., Jr Chronic epileptogenesis requires development of a network of pathologically interconnected neuron clusters: a hypothesis. Epilepsia. 2000;41:S144–S152. doi: 10.1111/j.1528-1157.2000.tb01573.x. [DOI] [PubMed] [Google Scholar]
- Brunel N, Wang XJ. What determines the frequency of fast network oscillations with irregular neural discharges? I. Synaptic dynamics and excitation-inhibition balance. J Neurophysiol. 2003;90:415–430. doi: 10.1152/jn.01095.2002. [DOI] [PubMed] [Google Scholar]
- Buckmaster P. Laboratory animal models of temporal lobe epilepsy. Comparative Medicine. 2004;54:473–485. [PubMed] [Google Scholar]
- Buzsaki G. Rhythms of the brain. Oxford University Press; 2006. [Google Scholar]
- Buzsáki G. Long-term changes of hippocampal sharp-waves following high frequency afferent activation. Brain Research. 1984;300:179–182. doi: 10.1016/0006-8993(84)91356-8. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. The thalamic clock: Emergent network properties. Neuroscience. 1991;41:351–364. doi: 10.1016/0306-4522(91)90332-i. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–1929. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Haas HL, Anderson EG. Long-term potentiation induced by physiologically relevant stimulus patterns. Brain Research. 1987;435:331–333. doi: 10.1016/0006-8993(87)91618-0. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Horvath Z, Urioste R, Hetke J, Wise K. High-frequency network oscillations in the hippocampus. Science. 1992;256:1025–1027. doi: 10.1126/science.1589772. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA. Neocortical synchronized oscillations induced by thalamic disinhibition in vivo. J Neurosci. 1999;19:RC27 1–7. doi: 10.1523/JNEUROSCI.19-18-j0005.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee M, Chuah L. Functional neuroimaging insights into how sleep and sleep deprivation affect memory and cognition. Current Opinion in Neurology. 2008;21:417–423. doi: 10.1097/WCO.0b013e3283052cf7. [DOI] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Cheng S, Frank LM. New experiences enhance coordinated neural activity in the hippocampus. Neuron. 2008;57:303–313. doi: 10.1016/j.neuron.2007.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrobak JJ, Buzsaki G. High-frequency oscillations in the output networks of the hippocampal-entorhinal axis of the freely behaving rat. J Neurosci. 1996;16:3056–3066. doi: 10.1523/JNEUROSCI.16-09-03056.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church J, Baimbridge KG. Exposure to high-pH medium increases the incidence and extent of dye coupling between rat hippocampal CA1 pyramidal neurons in vitro. J Neurosci. 1991;11:3289–3295. doi: 10.1523/JNEUROSCI.11-10-03289.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford DB, Olney JW, Maniotis A, Collins RC, Zorumski CF. The functional anatomy and pathology of lithium-pilocarpine and high-dose pilocarpine seizures. Neuroscience. 1987;23:953–968. doi: 10.1016/0306-4522(87)90171-0. [DOI] [PubMed] [Google Scholar]
- Contreras D, Destexhe A, Sejnowski TJ, Steriade M. Control of spatiotemporal coherence of a thalamic oscillation by corticothalamic feedback. Science. 1996;274:771–774. doi: 10.1126/science.274.5288.771. [DOI] [PubMed] [Google Scholar]
- Contreras D, Destexhe A, Sejnowski TJ, Steriade M. Spatiotemporal patterns of spindle oscillations in cortex and thalamus. J Neurosci. 1997;17:1179–1196. doi: 10.1523/JNEUROSCI.17-03-01179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D, Steriade M. Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. J Neurosci. 1995;15:604–622. doi: 10.1523/JNEUROSCI.15-01-00604.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurko A, Buzsáki G. Reliability and state dependence of pyramidal cell-interneuron synapses in the hippocampus: an ensemble approach in the behaving rat. Neuron. 1998;21:179–189. doi: 10.1016/s0896-6273(00)80525-5. [DOI] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurko A, Mamiya A, Buzsaki G. Oscillatory coupling of hippocampal pyramidal cells and interneurons in the behaving rat. J Neurosci. 1999;19:274–287. doi: 10.1523/JNEUROSCI.19-01-00274.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnecki A, Birtoli B, Ulrich D. Cellular mechanisms of burst firing-mediated long-term depression in rat neocortical pyramidal cells. J Physiol. 2007;578:471–479. doi: 10.1113/jphysiol.2006.123588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleuze C, Huguenard JR. Distinct Electrical and chemical connectivity maps in the thalamic reticular nucleus: potential roles in synchronization and sensation. J Neurosci. 2006;26:8633–8645. doi: 10.1523/JNEUROSCI.2333-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olsen RW. Mice lacking the beta 3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci. 1998;18:8505–8514. doi: 10.1523/JNEUROSCI.18-20-08505.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M. Mechanisms underlying the synchronizing action of corticothalamic feedback through inhibition of thalamic relay cells. J Neurophysiol. 1998;79:999–1016. doi: 10.1152/jn.1998.79.2.999. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Sejnowski T. G Protein activation kinetics and spillover of {gamma}-aminobutyric acid may account for differences between inhibitory responses in the hippocampus and thalamus. Proc Natl Acad Sci. 1995;92:9515–9519. doi: 10.1073/pnas.92.21.9515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas R, Martin K. Neuronal circuits of the neocortex. Annu Rev Neurosci. 2004;27:419–451. doi: 10.1146/annurev.neuro.27.070203.144152. [DOI] [PubMed] [Google Scholar]
- Draguhn A, Traub RD, Schmitz D, Jefferys JGR. Electrical coupling underlies high-frequency oscillations in the hippocampus in vitro. Nature. 1998;394:189–192. doi: 10.1038/28184. [DOI] [PubMed] [Google Scholar]
- Dutar P, Nicoll RA. A physiological role for GABAB receptors in the central nervous system. Nature. 1988;332:156–158. doi: 10.1038/332156a0. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Staley KJ. Mechanisms of fast Ripples in the hippocampus. J Neurosci. 2004;24:8896–8906. doi: 10.1523/JNEUROSCI.3112-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earle KM, Penfield W. Incisural sclerosis and temporal lobe seizures produced by hippocampal herniation at birth. AMA Arch Neurol Psychiatry. 1953;69:27–42. doi: 10.1001/archneurpsyc.1953.02320250033003. [DOI] [PubMed] [Google Scholar]
- Ellenberg JH, Nelson KB. Febrile seizures and later intellectual performance. Arch Neurol. 1978;35:17–21. doi: 10.1001/archneur.1978.00500250021004. [DOI] [PubMed] [Google Scholar]
- Engel J, Williamson P, Wieser H. Mesial Temporal Lobe Epilepsy With Hippocampal Sclerosis. In: Engel J, Pedley T, editors. Epilepsy: A Comprehensive Textbook. Philadelphia, PA: Lippincott Williams & Wilkins; 2008. pp. 2479–2786. [Google Scholar]
- Eschenko O, Molle M, Born J, Sara SJ. Elevated sleep spindle density after learning or after retrieval in rats. J Neurosci. 2006;26:12914–12920. doi: 10.1523/JNEUROSCI.3175-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer M. Discussion on the surgery of temporal lobe epilepsy: surgical and pathological aspects. Proc R Soc Med. 1953;46:971–974. doi: 10.1177/003591575304601112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer M. Genetic and related aetiological factors in temporal lobe epilepsy. A review. Epilepsia. 1971;12:13–31. doi: 10.1111/j.1528-1157.1971.tb03912.x. [DOI] [PubMed] [Google Scholar]
- Foffani G, Uzcategui YG, Gal B, Menendez de la Prida L. Reduced spike-timing reliability correlates with the emergence of fast ripples in the rat epileptic hippocampus. Neuron. 2007;55:930–941. doi: 10.1016/j.neuron.2007.07.040. [DOI] [PubMed] [Google Scholar]
- French J, Williamson P, Thadani V, Darcey T, Mattson R, Spencer S, Spencer D. Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination. Annals of Neurology. 1993;34:774–780. doi: 10.1002/ana.410340604. [DOI] [PubMed] [Google Scholar]
- Fries P, Nikolic D, Singer W. The gamma cycle. Trends Neurosci. 2007;30:309–316. doi: 10.1016/j.tins.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Fuentealba P, Steriade M. The reticular nucleus revisited: Intrinsic and network properties of a thalamic pacemaker. Progress in Neurobiology. 2005;75:125–141. doi: 10.1016/j.pneurobio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313:1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- Gais S, Molle M, Helms K, Born J. Learning-dependent increases in sleep spindle density. J Neurosci. 2002;22:6830–6834. doi: 10.1523/JNEUROSCI.22-15-06830.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareri P, Condorelli D, Belluardo N, Citraro R, Barresi V, Trovato-Salinato A, Mudò G, Ibbadu GF, Russo E, De Sarro G. Antiabsence effects of carbenoxolone in two genetic animal models of absence epilepsy (WAG/Rij rats and lh/lh mice) Neuropharmacology. 2005;49:551–563. doi: 10.1016/j.neuropharm.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Geiger JRP, Jonas P. Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Geisler C, Brunel N, Wang XJ. Contributions of intrinsic membrane dynamics to fast network oscillations with irregular neuronal discharges. J Neurophysiol. 2005;94:4344–4361. doi: 10.1152/jn.00510.2004. [DOI] [PubMed] [Google Scholar]
- Golshani P, Liu XB, Jones EG. Differences in quantal amplitude reflect GluR4- subunit number at corticothalamic synapses on two populations of thalamic neurons. Proc Natl Acad Sci. 2001;98:4172–4177. doi: 10.1073/pnas.061013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillner S. Biological pattern generation: The cellular and computational logic of networks in motion. Neuron. 2006;52:751–766. doi: 10.1016/j.neuron.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Gabriel S, Jauch R, Schulze K, Kivi A, Eilers A, Kovacs R, Lehmann TN. Alterations of glial cell function in temporal lobe epilepsy. Epilepsia. 2000;41:S185–S189. doi: 10.1111/j.1528-1157.2000.tb01579.x. [DOI] [PubMed] [Google Scholar]
- Herrmann CS, Demiralp T. Human EEG gamma oscillations in neuropsychiatric disorders. Clinical Neurophysiology. 2005;116:2719–2733. doi: 10.1016/j.clinph.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Thomas P, Panayiotopoulos C. Childhood and Juvenile Absence Epilepsies. In: Engel J, Pedley T, editors. Epilepsy: A Comprehensive Textbook. Philadelphia: Lippincott Williams & Wilkins; 2008. pp. 2397–2411. [Google Scholar]
- Hooper SL, DiCaprio RA. Crustacean motor pattern generator networks. Neurosignals. 2004;13:50–69. doi: 10.1159/000076158. [DOI] [PubMed] [Google Scholar]
- Huguenard J, Prince D. Intrathalamic rhythmicity studied in vitro: nominal T-current modulation causes robust antioscillatory effects. J Neurosci. 1994a;14:5485–5502. doi: 10.1523/JNEUROSCI.14-09-05485.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 2007;30:350–356. doi: 10.1016/j.tins.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. Clonazepam suppresses GABAB-mediated inhibition in thalamic relay neurons through effects in nucleus reticularis. J Neurophysiol. 1994b;71:2576–2581. doi: 10.1152/jn.1994.71.6.2576. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. Intrathalamic rhythmicity studied in vitro: nominal T-current modulation causes robust antioscillatory effects. J Neurosci. 1994c;14:5485–5502. doi: 10.1523/JNEUROSCI.14-09-05485.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Jacobs J, LeVan P, Chander R, Hall J, Dubeau F, Gotman J. Interictal high-frequency oscillations (80-500 Hz) are an indicator of seizure onset areas independent of spikes in the human epileptic brain. Epilepsia. 2008;49:1893–1907. doi: 10.1111/j.1528-1167.2008.01656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen RB, Ulrich D, Huguenard JR. GABAB and NMDA receptors contribute to spindle-like oscillations in rat thalamus in vitro. J Neurophysiol. 2001;86:1365–1375. doi: 10.1152/jn.2001.86.3.1365. [DOI] [PubMed] [Google Scholar]
- Jasper H, Kershman J. Electroencephalographic classification of the epilepsies. Arch Neurol Psychiat (Chicago) 1941;45:903–943. [Google Scholar]
- Jinno S, Klausberger T, Marton LF, Dalezios Y, Roberts JDB, Fuentealba P, Bushong EA, Henze D, Buzsaki G, Somogyi P. Neuronal diversity in GABAergic long-range projections from the hippocampus. J Neurosci. 2007;27:8790–8804. doi: 10.1523/JNEUROSCI.1847-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG. The Thalamus. 2nd. Cambridge, UK: Cambridge University Press; 2007. [Google Scholar]
- Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker-Cummings C, Boneschi FM, Choi C, Morozov P, Das K, Teplitskaya E, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335–341. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel A, Buzsaki G. Cellular-synaptic generation of sleep spindles, spike-and-wave discharges, and evoked thalamocortical responses in the neocortex of the rat. J Neurosci. 1997;17:6783–6797. doi: 10.1523/JNEUROSCI.17-17-06783.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehn O, Kjaerulff O, Tresch MC, Harris-Warrick RM. Contributions of intrinsic motor neuron properties to the production of rhythmic motor output in the mammalian spinal cord. Brain Research Bulletin. 2000;53:649–659. doi: 10.1016/s0361-9230(00)00398-1. [DOI] [PubMed] [Google Scholar]
- Kim U, Bal T, McCormick DA. Spindle waves are propagating synchronized oscillations in the ferret LGNd in vitro. J Neurophysiol. 1995;74:1301–1323. doi: 10.1152/jn.1995.74.3.1301. [DOI] [PubMed] [Google Scholar]
- Kim U, Sanchez-Vives MV, McCormick DA. Functional dynamics of GABAergic inhibition in the thalamus. Science. 1997;278:130–134. doi: 10.1126/science.278.5335.130. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Magill PJ, Marton LF, Roberts JDB, Cobden PM, Buzsaki G, Somogyi P. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature. 2003;421:844–848. doi: 10.1038/nature01374. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Marton LF, Baude A, Roberts JDB, Magill PJ, Somogyi P. Spike timing of dendrite-targeting bistratified cells during hippocampal network oscillations in vivo. Nature Neuroscience. 2004;7:41–47. doi: 10.1038/nn1159. [DOI] [PubMed] [Google Scholar]
- Kobayashi E, Lopes-Cendes I, Guerreiro CAM, Sousa SC, Guerreiro MM, Cendes F. Seizure outcome and hippocampal atrophy in familial mesial temporal lobe epilepsy. Neurology. 2001;56:166–172. doi: 10.1212/wnl.56.2.166. [DOI] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–319. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- Lam YW, Sherman SM. Mapping by laser photostimulation of connections between the thalamic reticular and ventral posterior lateral nuclei in the rat. J Neurophysiol. 2006;94:2472–2483. doi: 10.1152/jn.00206.2005. [DOI] [PubMed] [Google Scholar]
- Lambert NA, Teyler TJ. Adenosine depresses excitatory but not fast inhibitory synaptic transmission in area CA1 of the rat hippocampus. Neuroscience Letters. 1991;122:50–52. doi: 10.1016/0304-3940(91)90190-5. [DOI] [PubMed] [Google Scholar]
- Landisman CE, Long MA, Beierlein M, Deans MR, Paul DL, Connors BW. Electrical synapses in the thalamic reticular nucleus. J Neurosci. 2002;22:1002–1009. doi: 10.1523/JNEUROSCI.22-03-01002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Van Quyen M, Bragin A, Staba R, Crepon B, Wilson CL, Engel J., Jr Cell type-specific firing during ripple oscillations in the hippocampal formation of humans. J Neurosci. 2008;28:6104–6110. doi: 10.1523/JNEUROSCI.0437-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leznik E, Llinas R. Role of gap junctions in synchronized neuronal oscillations in the inferior olive. J Neurophysiol. 2005;94:2447–2456. doi: 10.1152/jn.00353.2005. [DOI] [PubMed] [Google Scholar]
- Liu R, Lemieux L, Bell G, Sisodiya S, Bartlett P, Shorvon S, Sander J, Duncan J. The structural consequences of newly diagnosed seizures. Annals of Neurology. 2002;52:573–580. doi: 10.1002/ana.10338. [DOI] [PubMed] [Google Scholar]
- Liu X, Jones E. Fine structural localization of connexin-36 immunoreactivity in mouse cerebral cortex and thalamus. The Journal of Comparative Neurology. 2003;466:457–467. doi: 10.1002/cne.10901. [DOI] [PubMed] [Google Scholar]
- Liu Z, Vergnes M, Depaulis A, Marescaux C. Involvement of intrathalamic GABAb neurotransmission in the control of absence seizures in the rat. Neuroscience. 1992;48:87–93. doi: 10.1016/0306-4522(92)90340-8. [DOI] [PubMed] [Google Scholar]
- Llinas R, Jahnsen H. Electrophysiology of mammalian thalamic neurons in vitro. Nature. 1982;279:406–408. doi: 10.1038/297406a0. [DOI] [PubMed] [Google Scholar]
- Loiseau P, Duche B, Loiseau J. Classification of epilepsies and epileptic syndromes in two different samples of patients. Epilepsia. 1991;32:303–309. doi: 10.1111/j.1528-1157.1991.tb04656.x. [DOI] [PubMed] [Google Scholar]
- Loomis A, Harvey E, Hobart G. Potential rhythms of the cerebral cortex during sleep. Science. 1935;81:597–598. doi: 10.1126/science.81.2111.597. [DOI] [PubMed] [Google Scholar]
- Maex R, De Schutter E. Oscillations in the cerebellar cortex: a prediction of their frequency bands. Progress in Brain Research. In: Cicirata DZ, editor. Creating coordination in the cerebellum. Elsevier; 2005. pp. 181–188. [DOI] [PubMed] [Google Scholar]
- Maier N, Nimmrich V, Draguhn A. Cellular and network mechanisms underlying spontaneous sharp wave-ripple complexes in mouse hippocampal slices. J Physiol. 2003;550:873–887. doi: 10.1113/jphysiol.2003.044602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann EO, Paulsen O. Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci. 2007;30:343–349. doi: 10.1016/j.tins.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Marder E. Motor pattern generation. Curr Opin Neurobiol. 2000;10:691–698. doi: 10.1016/s0959-4388(00)00157-4. [DOI] [PubMed] [Google Scholar]
- Marder E, Bucher D. Central pattern generators and the control of rhythmic movements. Curr Biol. 2001;11:R986–R996. doi: 10.1016/s0960-9822(01)00581-4. [DOI] [PubMed] [Google Scholar]
- Marder E, Calabrese RL. Principles of rhythmic motor pattern generation. Physiol Rev. 1996;76:687–717. doi: 10.1152/physrev.1996.76.3.687. [DOI] [PubMed] [Google Scholar]
- Margerison JH, Corsellis JAN. Epilepsy and the temporal lobes: a clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain. 1966;89:499–530. doi: 10.1093/brain/89.3.499. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Bal T. Sleep and arousal: Thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Contreras D. On the cellular and network bases of epileptic seizures. Annu Rev Physiol. 2001;63:815–846. doi: 10.1146/annurev.physiol.63.1.815. [DOI] [PubMed] [Google Scholar]
- Meeren HKM, Pijn JPM, Van Luijtelaar ELJM, Coenen AML, Lopes da Silva FH. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. 2002;22:1480–1495. doi: 10.1523/JNEUROSCI.22-04-01480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menuz K, Nicoll RA. Loss of inhibitory neuron AMPA receptors contributes to ataxia and epilepsy in stargazer mice. J Neurosci. 2008;28:10599–10603. doi: 10.1523/JNEUROSCI.2732-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Wong RK. Inhibitory control of local excitatory circuits in the guinea-pig hippocampus. J Physiol. 1987;388:611–629. doi: 10.1113/jphysiol.1987.sp016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein AD, Nicoll RA. Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci. 2008;29:333–339. doi: 10.1016/j.tips.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin A, Doyon J, Dostie V, Barakat M, Tahar AH, Korman M, Benali H, Karni A, Ungerleider LG, Carrier J. Motor sequence learning increases sleep spindles and fast frequencies in post-training sleep. Sleep. 2008;31:1149–1156. [PMC free article] [PubMed] [Google Scholar]
- Morison RS, Bassett DL. Electrical activity of the thalamus and basal ganglia in decorticate cats. J Neurophysiol. 1945;8:309–314. [Google Scholar]
- Nadasdy Z, Hirase H, Czurko A, Csicsvari J, Buzsaki G. Replay and time compression of recurring spike sequences in the hippocampus. J Neurosci. 1999;19:9497–9507. doi: 10.1523/JNEUROSCI.19-21-09497.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. 1978;271:676–677. doi: 10.1038/271676a0. [DOI] [PubMed] [Google Scholar]
- Nersesyan H, Hyder F, Rothman D, Blumenfeld H. Dynamic fMRI and EEG recordings during spike-wave seizures and generalized tonic-clonic seizures in WAG/Rij rats. J Cereb Blood Flow Metab. 2004;24:589–599. doi: 10.1097/01.WCB.0000117688.98763.23. [DOI] [PubMed] [Google Scholar]
- Noebels JL, Qiao X, Bronson RT, Spencer C, Davisson MT. Stargazer: a new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Research. 1990;7:129–135. doi: 10.1016/0920-1211(90)90098-g. [DOI] [PubMed] [Google Scholar]
- Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- Ottman R, Risch N, Hauser WA, Pedley TA, Lee JH, Barker-Cummings C, Lustenberger A, Nagle KJ, Lee KS, Scheuer ML, et al. Localization of a gene for partial epilepsy to chromosome 10q. Nat Genet. 1995;10:56–60. doi: 10.1038/ng0595-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottman R, Winawer MR, Kalachikov S, Barker-Cummings C, Gilliam TC, Pedley TA, Hauser WA. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology. 2004;62:1120–1126. doi: 10.1212/01.wnl.0000120098.39231.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pais I, Hormuzdi SG, Monyer H, Traub RD, Wood IC, Buhl EH, Whittington MA, LeBeau FEN. Sharp wave-like activity in the hippocampus in vitro in mice lacking the gap junction protein connexin 36. J Neurophysiol. 2003;89:2046–2054. doi: 10.1152/jn.00549.2002. [DOI] [PubMed] [Google Scholar]
- Pan W, Osmanovic S, Shefner S. Characterization of the adenosine A1 receptor-activated potassium current in rat locus ceruleus neurons. J Pharmacol Exp Ther. 1995;273:537–544. [PubMed] [Google Scholar]
- Panayiotopoulos C, Obeid T, Waheed G. Absences in juvenile myoclonic epilepsy: A clinical and video-electroencephalographic study. Annals of Neurology. 1989;25:391–397. doi: 10.1002/ana.410250411. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Winson J. Influences of hippocampal place cell firing in the awake state on the activity of these cells during subsequent sleep episodes. J Neurosci. 1989;9:2907–2918. doi: 10.1523/JNEUROSCI.09-08-02907.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfield W, Jasper H. Epilepsy and the functional anatomy of the human Brain. Boston: Little Brown; 1954. [Google Scholar]
- Peters K, Ray L, Smith V, Smith C. Changes in the density of stage 2 sleep spindles following motor learning in young and older adults. Journal of Sleep Research. 2008;17:23–33. doi: 10.1111/j.1365-2869.2008.00634.x. [DOI] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- Pittau F, Bisulli F, Mai R, Fares J, Vignatelli L, Labate A, Naldi I, Avoni P, Parmeggiani A, Santucci M, et al. Prognostic factors in patients with mesial temporal lobe epilepsy. Epilepsia. 2009;50:41–44. doi: 10.1111/j.1528-1167.2008.01969.x. [DOI] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 2007;27:6590–6599. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard H, Charriaut-Marlangue C, Cantagrel S, Represa A, Robain O, Moreau J, Ben-Ari Y. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63:7–18. doi: 10.1016/0306-4522(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Rasch B, Born J. Maintaining memories by reactivation. Curr Opin Neurobiol. 2007;17:698–703. doi: 10.1016/j.conb.2007.11.007. [DOI] [PubMed] [Google Scholar]
- Rosanova M, Ulrich D. Pattern-specific associative long-term potentiation induced by a sleep spindle-related spike train. J Neurosci. 2005;25:9398–9405. doi: 10.1523/JNEUROSCI.2149-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Segal M, Koulakaoff A, Giaume C, Avignone E. Carbenoxolone blockade of neuronal network activity in culture is not mediated by an action on gap junctions. J Physiol. 2003;553:729–745. doi: 10.1113/jphysiol.2003.053439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozental R, Srinivas M, Spray DC. How to close a gap junction channel. Efficacies and potencies of uncoupling agents. Methods Mol Biol. 2001;154:447–476. doi: 10.1385/1-59259-043-8:447. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives MV, McCormick DA. Functional properties of perigeniculate inhibition of dorsal lateral geniculate nucleus thalamocortical neurons in vitro. J Neurosci. 1997;17:8880–8893. doi: 10.1523/JNEUROSCI.17-22-08880.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M. GABA spillover activates postsynaptic GABAB receptors to control rhythmic hippocampal activity. Neuron. 2000;25:673–681. doi: 10.1016/s0896-6273(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Capogna M, Gähwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- Schmitz D, Schuchmann S, Fisahn A, Draguhn A, Buhl EH, Petrasch-Parwez E, Dermietzel R, Heinemann U, Traub RD. Axo-axonal coupling: a novel mechanism for ultrafast neuronal communication. Neuron. 2001;31:831–840. doi: 10.1016/s0896-6273(01)00410-x. [DOI] [PubMed] [Google Scholar]
- Schulte U, Thumfart JO, Klöcker N, Sailer CA, Bildl W, Biniossek M, Dehn D, Deller T, Eble S, Abbass K, et al. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kv21. Neuron. 2006;49:697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Semah F, Picot MC, Adam C, Broglin D, Arzimanoglou A, Bazin B, Cavalcanti D, Baulac M. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology. 1998;51:1256–1262. doi: 10.1212/wnl.51.5.1256. [DOI] [PubMed] [Google Scholar]
- Senechal KR, Thaller C, Noebels JL. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet. 2005;14:1613–1620. doi: 10.1093/hmg/ddi169. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, Duque A, Yu Y, McCormick DA. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature. 2006;441:761–765. doi: 10.1038/nature04720. [DOI] [PubMed] [Google Scholar]
- Siapas AG, Wilson MA. Coordinated interactions between hippocampal ripples and cortical spindles during slow-wave sleep. Neuron. 1998;21:1123–1128. doi: 10.1016/s0896-6273(00)80629-7. [DOI] [PubMed] [Google Scholar]
- Sirota A, Csicsvari J, Buhl D, Buzsakii G. Communication between neocortex and hippocampus during sleep in rodents. PNAS. 2003;100:2065–2069. doi: 10.1073/pnas.0437938100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitnikova E, van Luijtelaar G. Cortical control of generalized absence seizures: effect of lidocaine applied to the somatosensory cortex in WAG/Rij rats. Brain Research. 2004;1012:127–137. doi: 10.1016/j.brainres.2004.03.041. [DOI] [PubMed] [Google Scholar]
- Slaght SJ, Leresche N, Deniau JM, Crunelli V, Charpier S. Activity of thalamic reticular neurons during spontaneous genetically determined spike and wave discharges. J Neurosci. 2002;22:2323–2334. doi: 10.1523/JNEUROSCI.22-06-02323.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter R. Hippocampal epileptogenesis in animal models of mesial temporal lobe epilepsy with hippocampal sclerosis: The importance of the “latent period” and other concepts. Epilepsia. 2008;49:85–92. doi: 10.1111/j.1528-1167.2008.01931.x. [DOI] [PubMed] [Google Scholar]
- Smith KA, Fisher RS. The selective GABAB antagonist CGP-35348 blocks spike-wave bursts in the cholesterol synthesis rat absence epilepsy model. Brain Research. 1996;729:147–150. [PubMed] [Google Scholar]
- So N, Olivier A, Andermann F, Gloor P, LQuesney L. Results of surgical treatment in patients with bitemporal epileptiform abnormalities. Annals of Neurology. 1989;25:432–439. doi: 10.1002/ana.410250503. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Huguenard JR. Inhibitory interconnections control burst pattern and emergent network synchrony in reticular thalamus. J Neurosci. 2003;23:8978–8988. doi: 10.1523/JNEUROSCI.23-26-08978.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Huntsman MM, Huguenard JR. Reciprocal inhibitory connections regulate the spatiotemporal properties of intrathalamic oscillations. J Neurosci. 2000;20:1735–1745. doi: 10.1523/JNEUROSCI.20-05-01735.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song I, Kim D, Choi S, Sun M, Kim Y, Shin HS. Role of the {alpha}1G T-type calcium channel in spontaneous absence seizures in mutant mice. J Neurosci. 2004;24:5249–5257. doi: 10.1523/JNEUROSCI.5546-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato J, Mody I. Spike timing of lacunosom-moleculare targeting interneurons and CA3 pyramidal cells during high-frequency network oscillations in vitro. J Neurophysiol. 2007;98:96–104. doi: 10.1152/jn.00188.2007. [DOI] [PubMed] [Google Scholar]
- Staba R, Frighetto L, Behnke E, Mathern G, Fields T, Bragin A, Ogren J, Fried I, Wilson C, Engel J., Jr Increased fast ripple to ripple ratios correlate with reduced hippocampal volumes and neuron loss in temporal lobe epilepsy patients. Epilepsia. 2007;48:2130–2138. doi: 10.1111/j.1528-1167.2007.01225.x. [DOI] [PubMed] [Google Scholar]
- Staba RJ, Wilson CL, Bragin A, Fried I, Engel J., Jr Quantitative analysis of high-frequency oscillations (80-500 Hz) recorded in human epileptic hippocampus and entorhinal cortex. J Neurophysiol. 2002;88:1743–1752. doi: 10.1152/jn.2002.88.4.1743. [DOI] [PubMed] [Google Scholar]
- Staley KJ. Neurons skip a beat during fast ripples. Neuron. 2007;55:828–830. doi: 10.1016/j.neuron.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Steriade M. Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci. 2005;28:317–324. doi: 10.1016/j.tins.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–1106. doi: 10.1016/j.neuroscience.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D. Spike-wave complexes and fast components of cortically generated seizures. I. Role of neocortex and thalamus. J Neurophysiol. 1998;80:1439–1455. doi: 10.1152/jn.1998.80.3.1439. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D, Curro Dossi R, Nunez A. The slow (< 1 Hz) oscillation in reticular thalamic and thalamocortical neurons: scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci. 1993a;13:3284–3299. doi: 10.1523/JNEUROSCI.13-08-03284.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Deschenes M. The thalamus as a neuronal oscillator. Brain Research Reviews. 1984;8:1–63. doi: 10.1016/0165-0173(84)90017-1. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick D, Sejnowski T. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993b;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Steriade M, Nunez A, Amzica F. Intracellular analysis of relations between the slow (< 1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J Neurosci. 1993c;13:3266–3283. doi: 10.1523/JNEUROSCI.13-08-03266.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Nunez A, Amzica F. A novel slow (< 1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993d;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickgold R, Walker MP. Sleep-dependent memory consolidation and reconsolidation. Sleep Medicine Advances in Sleep Medicine. 2007;8:331–343. doi: 10.1016/j.sleep.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland GR, McNaughton B. Memory trace reactivation in hippocampal and neocortical neuronal ensembles. Curr Opin Neurobiol. 2000;10:180–186. doi: 10.1016/s0959-4388(00)00079-9. [DOI] [PubMed] [Google Scholar]
- Sutula T, Hagen J, Pitkänen A. Do epileptic seizures damage the brain? Curr Opin Neurol. 2003;16:189–195. doi: 10.1097/01.wco.0000063770.15877.bc. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Delgado-Escueta AV, Aguan K, Alonso ME, Shi J, Hara Y, Nishida M, Numata T, Medina MT, Takeuchi T, et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004;36:842–849. doi: 10.1038/ng1393. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Inoue I, Yamagata T, Morita N, Furuichi T, Yamakawa K. Sequential expression of Efhc1/myoclonin1 in choroid plexus and ependymal cell cilia. Biochemical and Biophysical Research Communications. 2008;367:226–233. doi: 10.1016/j.bbrc.2007.12.126. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Steriade M. Low-frequency rhythms in the thalamus of intact-cortex and decorticated cats. J Neurophysiol. 1996;76:4152–4168. doi: 10.1152/jn.1996.76.6.4152. [DOI] [PubMed] [Google Scholar]
- Toth Z, Yan XX, Haftoglou S, Ribak CE, Baram TZ. Seizure-induced neuronal injury: Vulnerability to febrile seizures in an immature rat model. J Neurosci. 1998;18:4285–4294. doi: 10.1523/JNEUROSCI.18-11-04285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub R, Draguhn A, Whittington M, Baldeweg T, Bibbig A, Buhl E, Schmitz D. Axonal gap junctions between principal neurons: a novel source of network oscillations, and perhaps epileptogenesis. Rev Neurosci. 2002;13:1–30. doi: 10.1515/revneuro.2002.13.1.1. [DOI] [PubMed] [Google Scholar]
- Traub R, Whittington M, Buhl E, LeBeau F, Bibbig A, Boyd S, Cross H, Baldeweg T. A possible role for gap junctions in generation of very fast EEG oscillations preceding the onset of, and perhaps initiating seizures. Epilepsia. 2001a;42:153–170. doi: 10.1046/j.1528-1157.2001.26900.x. [DOI] [PubMed] [Google Scholar]
- Traub RD, Bibbig A. A model of high-frequency ripples in the hippocampus based on synaptic coupling plus axon-axon gap junctions between pyramidal neurons. J Neurosci. 2000;20:2086–2093. doi: 10.1523/JNEUROSCI.20-06-02086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Bibbig R, Piechotta A, Draguhn R, Schmitz D. Synaptic and nonsynaptic contributions to giant IPSPs and ectopic spikes induced by 4-aminopyridine in the hippocampus in vitro. J Neurophysiol. 2001b;85:1246–1256. doi: 10.1152/jn.2001.85.3.1246. [DOI] [PubMed] [Google Scholar]
- Traub RD, Schmitz D, Jefferys JGR, Draguhn A. High-frequency population oscillations are predicted to occur in hippocampal pyramidal neuronal networks interconnected by axoaxonal gap junctions. Neuroscience. 1999;92:407–426. doi: 10.1016/s0306-4522(98)00755-6. [DOI] [PubMed] [Google Scholar]
- Tsakiridou E, Bertollini L, de Curtis M, Avanzini G, Pape H. Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J Neurosci. 1995;15:3110–3117. doi: 10.1523/JNEUROSCI.15-04-03110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng SH, Tsai LY, Yeh SR. Induction of high-frequency oscillations in a junction-coupled network. J Neurosci. 2008;28:7165–7173. doi: 10.1523/JNEUROSCI.0950-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiante TA, Velazquez JLP, Jahromi SS, Carlen PL. Coupling potentials in CA1 neurons during calcium-free induced field burst activity. J Neurosci. 1995;15:6946–6956. doi: 10.1523/JNEUROSCI.15-10-06946.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Paesschen W, Connelly A, King M, Jackson G, Duncan J. The spectrum of hippocampal sclerosis: a quantitative magnetic resonance imaging study. Annals of Neurology. 1997a;41:41–51. doi: 10.1002/ana.410410109. [DOI] [PubMed] [Google Scholar]
- Van Paesschen W, Duncan JS, Stevens J, Connelly A. Longitudinal quantitative hippocampal magnetic resonance imaging study of adults with newly diagnosed partial seizures: One-year follow-up results. Epilepsia. 1998;39:633–639. doi: 10.1111/j.1528-1157.1998.tb01432.x. [DOI] [PubMed] [Google Scholar]
- Van Paesschen W, Duncan JS, Stevens JM, Connelly A. Etiology and early prognosis of newly diagnosed partial seizures in adults: A quantitative hippocampal MRI study. Neurology. 1997b;49:753–757. doi: 10.1212/wnl.49.3.753. [DOI] [PubMed] [Google Scholar]
- von Krosigk MB, Thierry, McCormick David A. Cellular mechanisms of a synchronized oscillation in the thalamus. Science. 1993:361–364. doi: 10.1126/science.8392750. [DOI] [PubMed] [Google Scholar]
- Warren RA, Agmon A, Jones EG. Oscillatory synaptic interactions between ventroposterior and reticular neurons in mouse thalamus in vitro. J Neurophysiol. 1994;72:1993–2003. doi: 10.1152/jn.1994.72.4.1993. [DOI] [PubMed] [Google Scholar]
- White E. Thalamocortical synaptic relations. A review with emphasis on the projection of specific thalamic nuclei to the primary sensory areas of the neocortex. Brain Res. 1979;180:275–311. doi: 10.1016/0165-0173(79)90008-0. [DOI] [PubMed] [Google Scholar]
- White HS. Animal models of epileptogenesis. Neurology. 2002;59:7S–14. doi: 10.1212/wnl.59.9_suppl_5.s7. [DOI] [PubMed] [Google Scholar]
- Williams D. A study of thalamic and cortical rhythms in petit mal. Brain. 1953;76:50–69. doi: 10.1093/brain/76.1.50. [DOI] [PubMed] [Google Scholar]
- Williamson P, French J, Thadani V, Kim J, Novelly R, Spencer S, Spencer D, Mattson R. Characteristics of medial temporal lobe epilepsy: II. Interictal and ictal scalp electroencephalography, neuropsychological testing, neuroimaging, surgical results, and pathology. Annals of Neurology. 1993;34:781–787. doi: 10.1002/ana.410340605. [DOI] [PubMed] [Google Scholar]
- Wilson MA, McNaughton B. Reactivation of hippocampal ensemble memories during sleep. Science. 1994;265:676–679. doi: 10.1126/science.8036517. [DOI] [PubMed] [Google Scholar]
- Wolf P, Goosses R. Relation of photosensitivity to epileptic syndromes. J Neurol Neurosurg Psychiatry. 1986;49:1386–1391. doi: 10.1136/jnnp.49.12.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Wong T, Wu X, Sheppy E, Zhang L. Adenosine as an endogenous regulating factor of hippocampal sharp waves. Hippocampus. 2009;19:205–220. doi: 10.1002/hipo.20497. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 1994;12:1139–1148. doi: 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]
- Ylinen A, Bragin A, Nadasdy Z, Jando G, Szabo I, Sik A, Buzsaki G. Sharp wave-associated high-frequency oscillation (200 Hz) in the intact hippocampus: network and intracellular mechanisms. J Neurosci. 1995;15:30–46. doi: 10.1523/JNEUROSCI.15-01-00030.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Mori M, Burgess DL, Noebels JL. Mutations in high-voltage-activated calcium channel genes stimulate low-voltage-activated currents in mouse thalamic relay neurons. J Neurosci. 2002;22:6362–6371. doi: 10.1523/JNEUROSCI.22-15-06362.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]