

Abstract
Mycobacterium tuberculosis adenosine 5′-phosphosulfate (APS) reductase is an iron-sulfur protein and a validated target to develop new anti-tubercular agents, particularly for the treatment of latent infection. To facilitate the development of potent and specific inhibitors of APS reductase, we have probed the molecular determinants that underlie binding and specificity through a series of substrate and product analogs. Our study highlights the importance of specific substitutent groups for substrate binding and provides functional evidence for ligand-specific conformational states. An active site model has been developed for M. tuberculosis APS reductase that is in accord with the results presented here as well as prior structural data reported for Pseudomonas aeruginosa APS reductase and related enzymes. This model illustrates the functional features required for the interaction of APS reductase with a ligand and provides a pharmacological road map for the rational design of small-molecules as potential inhibitors of APS reductase present in human pathogens, including M. tuberculosis.
Introduction
Reduced sulfur appears in organic compounds essential to all organisms as constituents of proteins, coenzymes, and cellular metabolites1–3. In the amino acid cysteine, the thiol functional group plays important biological roles in redox chemistry, metal binding, protein structure and catalysis4. In many human pathogens such as Mycobacterium tuberculosis and Pseudomonas aeruginosa, activation of inorganic sulfur for the biosynthesis of cysteine proceeds via adenosine 5′-phosphosulfate (APS)5, 6. This high-energy intermediate is produced by the action of ATP sulfurylase, which condenses sulfate and adenosine 5′-triphosphate (ATP) to form APS1. The iron-sulfur protein, APS reductase catalyzes the first committed step in sulfate reduction and is a validated target to develop new anti-tubercular agents, particularly for the treatment of latent infection7–9.
APS reductase catalyzes the reduction of APS to sulfite (HSO3−) and adenosine 5′-monophosphate (AMP) using reduction potential supplied by the protein cofactor, thioredoxin as shown in Figure 1. Functional and structural studies have been used to investigate the mechanism of APS reductase from sulfate-assimilating bacteria10–12. The proposed mechanism in Figure 2 involves nucleophilic attack by cysteine 2561 on the sulfur atom in APS to form an enzyme S-sulfocysteine intermediate, E-Cys-Sγ–SO3−, which is then reduced through intermolecular thiol-disulfide exchange with thioredoxin. The iron-sulfur cluster in APS reductase is essential for activity; however, it is not involved in redox chemistry and its exact role remains unknown6, 11.
Figure 1.

Sulfate assimilation pathway in M. tuberculosis. The majority of sulfate reducing bacteria use APS as their source of sulfite. In this reaction, APS is reduced to sulfite and adenosine-5′-monophosphate (AMP) by APS reductase. Sulfite, in turn, is reduced by later enzymes in this metabolic pathway, forming first sulfide before incorporation into cysteine and, ultimately, to methionine and other essential reduced sulfur-containing biomolecules.
Figure 2.

Mechanism of sulfonucleotide reduction.
Crystal structure determination at 2.7 Å of P. aeruginosa APS reductase in complex with APS provided the first insight into the molecular basis for substrate recognition (Figure 3)12. M. tuberculosis and P. aeruginosa APS reductases are related by high sequence homology (27.2% of sequence identity and 41.4% of sequence similarity), particularly in the residues that line the active site. The protein monomer folds as a single domain with a central six-stranded β sheet, interleaved with seven α-helices (Figure 3). Opposite the nucleotide at one end of the active site is the [4Fe-4S] cluster. Three additional elements define the active site: the P-loop (residues 60–66), the LDTG motif (residues 85–88) and the Arg-loop (residues 162–173). APS fits into the active site cavity with the phosphosulfate moiety extending toward the protein surface and ten residues interact directly, via hydrogen bonding or hydrophobic interactions with the substrate. The C-terminal segment of residues 250–267, which carries the catalytically essential Cys256, is disordered in this structure, but would be positioned above the active site cleft.
Figure 3.

The structure of P. aeruginosa APS reductase in complex with substrate, APS (PDB 2GOY). The C-terminal segment of residues starting at Glu249 carries the catalytically essential Cys256 and is disordered.
Not all organisms that assimilate sulfate reduce APS as the source of sulfite. Through divergent evolution, some organisms, such as Escherichia coli and Saccharomyces cerevisiae reduce the related metabolite 3′-phosphoadenosine-5′-phosphosulfate (PAPS)10, which is produced by APS kinase from ATP and APS13. PAPS reductases lack the iron-sulfur cofactor, but utilize the same two-step mechanism shown in Figure 210, 11. S. cerevisiae PAPS reductase, crystallized in the presence of the product, 3′-phosphoadenosine 5′-phosphate (PAP)14 has a fold similar to APS reductase (1.6-Å rms deviation over 117 residues). In the structure of yeast PAPS reductase, the Arg-loop and C-terminal segment are folded over the active site and this conformation reveals additional enzyme-ligand contacts, which may also form between APS reductase and its substrate, APS.
Even though M. tuberculosis has plagued humans for millennia, the antibiotic regime is complex and effective drugs that specifically target latent TB infection are yet to be developed15. Novel targets16, 17 and treatment strategies18, 19 are emerging, but new avenues for therapeutic intervention must continue to be explored in order to combat multidrug-resistant strains of TB, which pose a significant threat to global human health20. To this end, APS reductase represents an attractive target for therapeutic intervention because it is essential for mycobacterial survival in the latent phase of TB infection9 and humans do not possess an analogs metabolic pathway. Recently, we have discovered small-molecule inhibitors of APS reductase through virtual ligand screening21. However, the development of more specific and potent inhibitors will be greatly aided through knowledge of the functional importance of interactions between the substrate and enzyme at the active site, which have not yet been experimentally addressed.
Herein, we probe binding determinants of the M. tuberculosis APS reductase active site using synthetic ligand analogs. These studies define chemical groups that are essential for molecular recognition and reveal a network of electrostatic interactions, which play an important role in substrate discrimination. An active site model has been developed for M. tuberculosis APS reductase that is in accord with the results presented here as well as prior structural data reported for P. aeruginosa APS reductase and related enzymes. This model illustrates the functional features required for the interaction of APS reductase with a ligand and provides a pharmacological road map for the rational design of small-molecules as potential inhibitors of APS reductase present in human pathogens, including M. tuberculosis.
Results and Discussion
The substrate and fragments studied and results obtained in these experiments are summarized in Tables 1–3 and Figures 4–9.
Table 1.
Ligand dissociation constants for substrate-fragments with APS reductase.
| Ligand | Structure | Kd [μM][c] | ΔΔG [kcal/mol][d] | pKa |
|---|---|---|---|---|
| APS[a] | 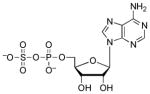 |
0.20 | N/A | ~2 (O)[e] |
| AMP | 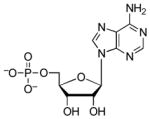 |
5.4 | 2.0 | 6.8 (O)52 |
| 5′-Phosphoribose | 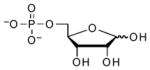 |
93 | 5.1 | ~6.8 (O)[f] |
| Adenosine | 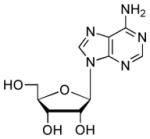 |
3000 | 5.8 | 3.6 (N1), 12.4 (O)54 |
| Phosphate | 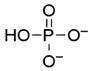 |
66000 | 7.7 | 1.97 (O), 6.82 (O), 12.5 (O)55 |
| Ribose | 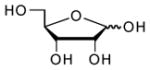 |
680000 | 9.1 | 12.22 (O)56 |
| Adenine[b] | 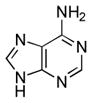 |
≥90000 | ≥7.3 | 4.15 (N1), 9.80 (N9)56 |
| Sulfate[b] | 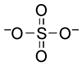 |
≥310000 | ≥8.6 | −3 (O), 1.89 (O)57 |
The Kd of APS was measured under single turnover conditions, in the absence of thioredoxin, as described in the methods section.
Due to the limits of solubility or solution ideality the reported values are lower limits.
For substrate-fragments in this table values of Ki were determined under single turnover conditions from the dependence of the observed rate constant at a given inhibitor concentration under conditions of subsaturating APS, such that Ki is equal to the Kd. Each value reflects the average of at least two independent experiments, and the standard deviation was less than 15% of the value of the mean. Kinetic data were nonlinear-least squares fit to a model of competitive inhibition.
Energetic difference in affinity of APS relative to inhibitor, ΔΔG = −RTln(KdAPS/KdFragment).
pKa estimated from value measured for 2′-deoxy-5′-phosphoribose58.
pKa estimated from value measured for 3′-phospho-5′-adenosinephosphosulfate59.
Table 3.
Ligand dissociation constants for product AMP analogs with APS reductase.
| Ligand | Structure | Kd [μM][a] | ΔΔG [kcal/mol][b] | pKa |
|---|---|---|---|---|
| 5′-AMPS | 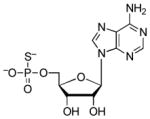 |
3.3 | −0.30 | 5.3 (O)60 |
| 5′-AMPN | 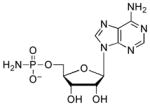 |
100 | 1.8 | ~3.0 (N), ~8.15 (O)[c] |
| 3′,5′-ADP | 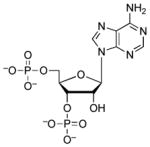 |
770 | 3.0 | 2.49 (O), 8.61(O)65 |
| 3′-AMP | 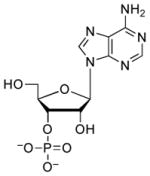 |
3200 | 3.8 | 5.92 (O)52 |
For analogs in this table values of Ki were determined under single turnover conditions from the dependence of the observed rate constant at a given inhibitor concentration under conditions of subsaturating APS, such that Ki is equal to the Kd. Each value reflects the average of at least two independent experiments, and the standard deviation was less than 15% of the value of the mean. Kinetic data were nonlinear-least squares fit to a model of competitive inhibition.
Energetic difference in affinity of AMP relative to inhibitor, ΔΔG = −RTln(KdAMP/KdAnalog).
pKa estimate from value measured for phosphoramidic acid64.
Figure 4.

Effects of cutting the substrate, APS and the product, AMP into fragments on ligand Kd values. The energetic effect that results from connectivity was calculated based on the affinity of the parent ligand ‘XY’, compared with the affinities of its pieces, ‘X’ and ‘Y’: ΔGconn = −RTln(KXY/KXKY), as previously reported23.
Figure 9.

Pharmacophore model of substrate, APS. Chemical structure of APS highlighting key hydrogen bond accepting (HBA) and donating (HBD) interactions, ionic interactions, and van der Waals interaction, based on functional data obtained in the present study.
1. Substrate Affinity
As a starting point to explore the molecular recognition properties of APS reductase, we determined the Kd value of substrate APS for M. tuberculosis APS reductase from the dependence of the observed rate constant for S-sulfocysteine formation (Figure S2), as described in Experimental procedures. The substrate APS binds to APS reductase with a Kd value of 0.2 μM (Table 1), which is ~3-fold lower than the value of the reported substrate Km22.
2. Affinity of Substrate Fragments
To gain further insight into substrate recognition of M. tuberculosis APS reductase, we analyzed the energetic contribution of individual portions of APS to the enzyme-binding interaction. The results obtained in these experiments are summarized in Table 1. The product AMP differs chemically from the natural substrate, APS, by the absence of the sulfate moiety. The loss of sulfate from APS reduced binding to APS reductase about 30-fold (2 kcal/mol), demonstrating that the AMP moiety makes a substantial contribution (7.3 kcal/mol) to the overall binding affinity (9.3 kcal/mol) of APS. Deletion of the adenine or phosphate group from AMP decreased binding to APS reductase by ~170-fold (3.1 kcal/mol) and ~550-fold (3.8 kcal/mol), respectively. Fragments of adenosine – D-ribose and adenine – exhibited weak binding activity toward APS reductase (0.2 and ≤1.5 kcal/mol). The respective free energy of binding to sulfate and phosphate dianions was ≤0.7 and 1.6 kcal/mol. Figure 4 summarizes the binding properties of the substrate, APS and product, AMP for M. tuberculosis APS reductase as compared with those of the fragments obtained by cutting these ligands at several positions, including the glycosidic bond, and at α- or β-positions within the diester moiety. In all cases, the Kd value of APS or AMP was lower than those of its pieces. However, the free energies (ΔGconn) associated with these connectivity effects for APS reductase are modest (≤2.2 kcal/mol), as compared to values that have been determined for adenosine23 and cytidine deaminases24 (~10 kcal/mol).
3. Affinity of Substrate Analogs
i. β-Nucleotide Substitution
The results above suggest that the β-sulfate group plays a modest role (≤2.0 kcal/mol) in molecular recognition of APS. To probe this observation in further detail, we investigated binding affinities for a panel of nucleotide analogs containing systematic modifications at the β-position (Table 2 and Figure 5). A phosphate oxyanion has nearly the same size and shape as a sulfate oxyanion, four atoms arranged tetrahedrally around a sulfur instead of a phosphorous25, 26. However, the overall charge of these analogs differs since the β-sulfate is monoanionic, whereas the β-phosphate is dianionic. Replacement of the β-sulfate moiety with β-phosphate, as in adenosine 5′-diphosphate (ADP), diminished binding to APS reductase about 20-fold (1.8 kcal/mol). To determine whether this decrease in binding affinity is due to additional negative charge at the β-position of ADP, relative to APS, we examined sulfur (ADPβS), fluorine (ADPβF), or amine (AMPPN) substitution of the β-phosphate nonbridging oxygen atom in ADP (Table 2 and Figure 5). Sulfur substitution is considered to be a good mimic of the phosphate moiety since it is isosteric, pseudoisoelectronic, and has a similar charge distribution and similar net charge at physiological pH27, 28. Fluorine substitution replaces an ionizable hydroxyl group, thereby mimicking the protonated nucleotide species in net charge at all pH values29 and, at neutral pH, amine substitution neutralizes the −1 charge at the β-position. Compared to ADP, substitution by sulfur increased binding affinity by 2.0 kcal/mol, fluorine increased binding by less than two-fold (0.3 kcal/mol), and introduction of the amine group decreased the free energy of binding for APS reductase by 2.8 kcal/mol.
Table 2.
Ligand dissociation constants for substrate analogs with APS reductase.
| Ligand | Structure | Kd [μM][a] | ΔΔG [kcal/mol][b] | pKa |
|---|---|---|---|---|
| ADPβS | 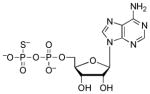 |
0.15 | −0.17 | 5.2 (O)60 |
| ADPαS[c] | 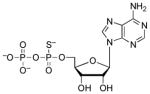 |
0.80 | 0.83 | 6.8 (O)61 |
| ADPβF | 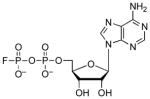 |
2.5 | 1.5 | N/A |
| ADP | 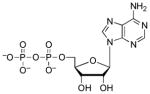 |
4.3 | 1.8 | 6.4 (O)61 |
| AMPCF2P | 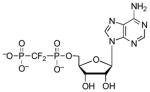 |
13 | 2.5 | ~5.7 (O)[d] |
| AMPCP | 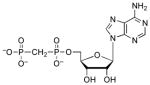 |
27 | 2.9 | 8.1 (O)62 |
| APSαS[c] | 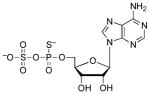 |
43 | 3.2 | ~1.6 (O)[e] |
| AMPNP | 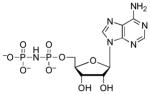 |
260 | 4.3 | 7.7 (O), 8.25 (N)33 |
| AMPPN | 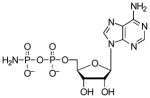 |
410 | 4.6 | 3.0 (O), 8.15 (N)[f] |
| APSβM | 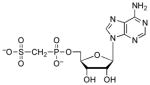 |
700 | 4.9 | ~3.7 (O)[g] |
For substrate analogs in this table values of Ki were determined under single turnover conditions from the dependence of the observed rate constant at a given inhibitor concentration under conditions of subsaturating APS, such that Ki is equal to the Kd. Each value reflects the average of at least two independent experiments, and the standard deviation was less than 15% of the value of the mean. Kinetic data were nonlinear-least squares fit to a model of competitive inhibition.
Energetic difference in affinity of APS relative to inhibitor, ΔΔG = −RTln(KdAPS/KdAnalog).
Kd value for the diastereoisomeric mixture of Rp and Sp isomers.
pKa estimated from value for mono- and difluoro-substituted benzylphosphonic acid63.
pKa estimated from effect of thio-substitution on ADPαS61.
pKa estimated from value measured for acid64.
pestimated phosphoramidic Ka from effect of β-methylene-substitution on AMPCP62.
Figure 5.

Electrostatic potential surfaces of substrate, APS and related nucleotide analogs. Color gradient: red corresponds to most negative and blue corresponds to most positive.
Taken together, the above data suggest that a net charge of −2 correlates with potent enzyme-ligand interactions. The negative charge can be localized entirely to the α-position, as in AMP, or it can be distributed across the diester, as in ADPβF. The similarity of Kd values for AMP and ADP could reflect different modes of nucleotide binding. For example, the α-phosphate of AMP could occupy the same position as the β-phosphate in ADP, thereby establishing key electrostatic interactions with Lys144, Arg242, and Arg245 (Figure 3). However, an upward shift in the position of AMP would likely weaken important enzyme-binding contacts with the adenine ring and ribose sugar of AMP (Figure 3). An alternative possibility is that the C-terminus and Arg-loop of APS reductase could adopt different conformations, depending on whether ADP or AMP is bound at the active site. In other words, binding energy gained through additional charge-charge interactions between the β-phosphate moiety of ADP, Lys144, Arg242 and Arg245 could be cancelled by a decrease in favorable interactions with residues in the C-terminus and the Arg-loop. Evidence in support for the latter proposal is provided in Section 5 below.
Interestingly, substitution of fluorine by an oxyanion had a marginal effect on binding affinity, whereas thiolate substitution increased binding potency by almost 30-fold. The larger size and polarizability of the sulfur atom could enhance binding affinity by: i) shortening the distance to residues that directly contact the ligand, and/or ii) enabling additional electrostatic or stacking interactions with APS reductase30. Finally, we note that neutralizing the −1 charge of the β-phosphate, as in AMPPN, is clearly unfavorable. This significant energetic penalty likely results from repulsive electrostatic interactions between the amine group, which is protonated at physiological pH (Figure 5), and adjacent positively charged residues (Figure 3). In line with this hypothesis, the binding affinity of AMPPN for APS reductase increased at elevated pH (Table S1).
ii. α-β Bridging Oxygen Substitution
Interactions between the 5′-phosphate and two highly conserved residues – Arg171 and His259 – are observed in the structure of S. cerevisiae PAPS reductase bound to PAP14. However, owing to the mobility of the Arg-loop and C-terminal residues, no direct contacts to the α-phosphate are observed in the structure of APS reductase12 (Figure 3). To gain insight into the functional importance of these contacts for M. tuberculosis APS reductase we examined the contribution of the α-β bridging oxygen to binding affinity (Table 2). Replacing the Pα-O-Sβ bridging oxygen in APS with a methylene group (Pα-CH2-Sβ) significantly decreased binding affinity by ~3,500-fold (4.9 kcal/mol). Comparison of ADP with AMPNP (Pα-NH–Pβ), AMPCF2P (Pα-CF2-Pβ), and AMPCP (Pα-CH2-Pβ) shows that imino, difluoromethylene, or methylene substitutions reduced binding potency by approximately 60-fold (2.5 kcal/mol), 3-fold (0.7 kcal/mol), and 6-fold (1.1 kcal/mol), respectively.
The above findings demonstrate that the α-β bridging oxygen does contribute to ligand recognition. Nonetheless, these data pose several questions: Why is the methylene substitution more detrimental for APS binding, relative to the energetic penalty paid for analogs modification of ADP? And, why does AMPNP bind more weakly to APS reductase compared to AMPCF2P? Replacing the bridging oxygen with a methylene group decreases the S/P–X–P bond angle, increases S/P–X bond length, increases the negative charge density on nonbridging oxygens, but makes the phosphate and sulfate groups less acidic31, 32. It is possible that methylene substitution of ADP is less detrimental to binding because i) the phosphonate moiety has more torsional freedom, allowing the nucleotide to adopt an alternative favorable binding mode, and/or ii) conformational differences in the C-terminal and Arg-loop segments minimize unfavorable contacts to the α-β bridging position of AMPCP. NMR studies show that AMPPNP and PNP exist in solution primarily as the imido tautomers33. Thus, the observed decrease in binding affinity could be due to restricted rotation about the Pα-NH–Pβ bonds.
iii. α-Nucleotide Substitution
Next, we examined the effect of sulfur substitution at the α-nonbridging oxygen atom using the analogs, APSαS2 and ADPαS (Table 2). Comparison of APS and APSαS shows that sulfur substitution for oxygen decreased binding by more than 200-fold (3.2 kcal/mol). However, compared to ADP, the binding potency of ADPαS increased by approximately 5-fold (1 kcal/mol). The larger energetic penalty for α-sulfur substitution of APS could result from ligand-related differences in enzyme conformation, analogs to the scenario presented in the subsection above.
4. Affinity of Product AMP Analogs
i. α-Nucleotide Substitution
To probe the molecular binding determinants of APS reductase at the α-position in greater detail, we compared the Kd value for the product, AMP to values measured for related analogs (Table 3). The respective effects of substitution at the α-oxygen by sulfur (AMPS) or amine (AMPN) are −0.3 and +1.8 kcal/mol, compared to AMP. The modest increase in binding energy that results from sulfur modification is similar to the effect observed with ADPαS. The reduction in binding affinity of AMPN also parallels the decrease observed for AMPPN.
In PAPS reductase, residues in the P-loop interact with the 3′-phosphate of PAP14, 34. However, in APS reductase the acidic residue, Asp66, interacts with amide groups of the P-loop and thus, appears to mimic the interaction of the negatively charged 3′-phosphate group. To investigate the role of the 3′-hydroxyl group in ligand discrimination (see also Section 4ii below) we determined the binding affinity for 3′-AMP, which reverses the position of the 5′-phosphate and 3′-hydroxyl groups (Table 3). Switching the position of the phosphate moiety decreased binding by ~600-fold (3.8 kcal/mol) indicating that, while the analog binds poorly, the 3′-phosphate does not impact binding to APS reductase, as compared to adenosine. By contrast, addition of a 3′-phosphate group to AMP, as in PAP, decreased binding affinity by 3.0 kcal/mol. The energetic penalty for 3′-phosphate in PAP, but not 3′-AMP, likely reflects additional binding interactions to the 5′-phosphate, which could decrease conformational freedom and increase unfavorable protein-ligand contacts.
ii. Purine and Ribose Substitution
Next, we analyzed the relative energetic contributions of individual purine and ribose substitutents to the enzyme-binding interaction. Owing to the relative difficulties traditionally associated with the preparation of ADP analogs and the weak binding of adenosine, we determined affinities for a series of compounds derived from the AMP scaffold. As shown in Figure 6, energetic penalties for individual substitutions ranged from 0.6 to 4.7 kcal/mol. First, we probed interactions between the N6 amine and N1 of adenine and Leu85 (Figure 3), the first residue in the conserved LDTG motif. Loss of the N6 amine from AMP reduced the free energy of binding to APS reductase by 1.8 kcal/mol. Replacing hydrogen atoms with methyl groups at the N6 position of adenine decreased binding affinity by 35-fold (2.1 kcal/mol) per substitution. Inverting the hydrogen bond donor and acceptor, as in inosine 5′-monophosphate, was also disfavored (4.7 kcal/mol), presumably due to electrostatic repulsion between the O6-keto and the Leu85 carbonyl, and the N1 by the Leu85 amine. Likewise, introduction of an N1 amine markedly reduced the affinity of this analog for APS reductase (4.7 kcal/mol). In subsequent experiments, we determined the binding potency of AMP analogs with substitutions at the 2, 7 and 8-positions of the purine ring. Methylation at N2 had a detrimental effect on binding (3.4 kcal/mol), likely due to steric clashes with the surrounding adenine-binding pocket (Figure 3). The structure of APS reductase bound to APS shows that C8-H group is directed toward the 5′-phosphosulfate moiety (Figure 3). Not surprisingly then, amine substitution at this position decreased the free energy of binding by 3.7 kcal/mol. No contacts are formed between N7 and APS reductase (Figure 3). Consistent with this observation, replacing N7 with a carbon atom had a relatively minor effect on binding affinity (0.6 kcal/mol). Finally, we investigated the influence of modification at the 2′ and 3′-hydroxyl groups of the ribose sugar. 2′-deoxy and methoxy group substitutions reduced the free energy of binding by a respective 4.0 and 3.6 kcal/mol, whereas 3′-deoxy substitution had only a modest effect on binding affinity (0.9 kcal/mol). This indicates that hydrogen bonds between the 2′-hydroxy group and Ser60 and Gly161 residues (Figure 3) play a vital role in substrate recognition.
Figure 6.

Free energy of binding for purine and ribose-modified analogs of product, AMP. Numbers represent the energetic effect of a given substitution in kcal/mol, relative to AMP (ΔΔG = −RTln(KdAnalog/KdAMP). Positive ΔΔG’s indicate a penalty for substitution.
5. pH Dependence of Ligand Binding
The pH dependence for ligand binding provides information about the relative affinities of the different nucleotide ionization states, and thus provides information about the active site environment. Initially, we investigated the pH dependence for APS binding. However, the substantial increase in reaction rate at higher pH precludes measurement by conventional kinetic methods (data not shown). As an alternative, we determined the affinities of APS reductase for the substrate analog, ADP, and product, AMP, as a function of pH to investigate whether ionizations at α and β-positions are important for binding affinity (Figure 7). The pH dependence for ADP binding is best fit by a pKa of 6.4 ± 0.2, which could reflect ionizations of the free enzyme and ligand (Figure 7A). The most likely candidate for this ionization is the ligand, as the second pKa values of phosphate esters fall in this region35. If this were true, the pH profile would be expected to shift to lower pH for an ADP analog with lower pKa values. To test this, we determined the pH dependence for ADPβS, which differs in its respective pKa value by approximately one pH unit27. The pH dependence for ADPβS binding is best fit by a pKa of 5.8 ± 0.2 (Figure S3A). The dependence of the pKa upon the identity of the ligand suggests that deprotonation of the ligand is responsible for the increase in binding at higher pH.
Figure 7.

pH dependence for ADP (A) and AMP (B) binding. The association equilibrium constant, (Ka = 1/Kd) is plotted as a function of pH. Values of Kd were determined by inhibition of APS reductase (pH 6.0–9.5). Buffers were as follows: MES (pH 6.0–6.5), BisTris (pH 6.5–7.5), TrisHCl (pH 7.5–9.5), CAPS (pH 9–9.5). See Experimental procedures for additional details. (A) The pH dependence for ADP binding. Nonlinear-least-squares fit of the data to a model for a single ionization gave pKa values of 6.4 ± 0.2. (B) The pH dependence for AMP binding. The dashed line represents the best fit of a model for a single ionization and yields a pKa of 7.9 ± 0.1.
The pH dependence for AMP binding is best to best fit by a pKa of 8.1 ± 0.1 (Figure 7B), which could reflect ionizations of the free enzyme and ligand, as described above. The simplest model to explain the weaker binding of AMP below pH 8 is that the dianion binds more tightly than the monoanion. However, the apparent pKa for AMP differs from the expected pKa of 6.8 by more than one unit. The discrepancy between the experimental data and the simplest model is most likely due to concurrent ionization of the enzyme that affects ligand binding, leading to shift in the apparent pKa of AMP. One model that could account for this upward deviation is that an enzymatic group with a pKa of ~6 contributes slightly (~5-fold) to AMP binding when protonated. The most likely residue to exert such an effect on ligand binding is His259, which interacts with the 5′-phosphate of PAP in the structure of S. cerevisiae PAPS reductase14. Since a stimulatory effect is not observed for ADP binding, the C-terminal segment containing His259, and possibly the Arg-loop, may adopt different conformations depending on whether ADP or AMP is bound at the active site. To further confirm that the apparent pKa measured for AMP depends upon the identity of the ligand, we measured the pH dependence for AMPS binding (Figure S3B). The resulting data are best fit by a pKa of 7.7 ± 0.1. The downward shift in apparent pKa suggests that deprotonation of the ligand is responsible for the increase in binding at higher pH.
The observed pH dependence for ADP and AMP binding indicates that the dianion binds more tightly to APS reductase than the monoanion. These data seemingly contradict our earlier comparison between ADP and ADPβF, whose net charges differ by one unit, but bind to APS reductase with similar affinity (Table 2 and Figure 5). Indeed, fluorine modification is often used to determine whether binding of mono- or dianionic phosphate is favored36. However, hydroxyl and fluorine groups are distinguished by unique chemical properties. For example, the high electron density of fluorine gives rise to the ability to act as an acceptor in hydrogen bonds37. By contrast, the hydroxyl group is a strong dipole, with spatially separated partial positive and negative charges that can donate as well as accept hydrogen bonds (Figure 5). Hence, the weaker binding observed for the monoanion may not reflect a loss of charge-charge interactions, but rather an energetic penalty that results from unfavorable charge-dipole interactions with the hydroxyl group.
6. Effect of Mg2+ on Ligand Affinity
In the absence of metal ions, APS binds ~20-times tighter to APS reductase, compared to ADP and AMP. However, the cellular concentrations of these nucleotides are higher than APS38 and thus, raise an important question: How does APS compete against binding of ADP or AMP to the active site of APS reductase in the cell? For the majority of biochemical reactions using ATP and, related nucleotides, the active species is the Mg2+ complex rather than the free nucleotide39. To gain insight into ligand discrimination by APS reductase in vivo, we measured the Kd values for APS, ADP and AMP in the presence of Mg2+. These studies show that the Kd value of APS was independent of Mg2+ concentration (data not shown). This observation is consistent with weak formation constants of APS with Mg2+40 and the general observation that other sulfonucleotide binding enzymes, such sulfokinases and sulfotransferases also do not require Mg2+ as a cofactor11. However, Mg·ADP and Mg·AMP complexes bind approximately 5–6 times weaker to APS reductase, as compared to the free nucleotide (Table S2). The observed reduction in binding energy is most likely due to repelling interactions with multiple positively charged amino acids in the enzyme active site, such as Lys144, Arg242 and Arg245 (Figure 3). Together, these findings indicate that APS reductase discriminates against noncognate adenosine nucleotides through favourable interactions with the sulfate moiety of APS and by disfavouring the binding of Mg2+-nucleotide complexes.
7. Implications for Rational Inhibitor Design
Given that APS reductase is essential for mycobacterial survival during persistent infection9, small-molecule inhibitors of APS reductase might be a source for new drugs to treat latent tuberculosis infection. The increasing number of antibiotic-resistant strains suggests that the availability of such compounds could play an important role in treating the disease and minimizing the negative impact on human health. By defining chemical groups that are essential for molecular recognition, the work described here sets the stage for the development of such drugs. Figure 8 summarizes the network of interactions predicted to occur between APS reductase active site residues and substrate, APS. The structural model was constructed by homology to S. cerevisiae PAPS reductase14 and systematically tested in the present study, from the perspective of the ligand. The total binding energy of APS resulting from these collective interactions is 9.3 kcal/mol and our data indicate several features that are essential for optimized substrate and inhibitor binding. The hydrophobic adenine-binding pocket, pyrimidine ring, 2′-hydroxyl and the α-position are the main determinants for strong target affinity (Figure 9). The significant losses of binding affinity that are found to result from apparently minor structural modifications of ligands at these key positions have encouraging implications for inhibitor design and suggests that, in some cases, the potency of a weak inhibitor might be greatly enhanced by one or two simple modifications. Our studies also suggest that small-molecules that target dynamic elements within the active site – particularly Arg171, Cys256 and His259 – may lead to inhibitors with improved binding affinity. Alternatively, molecules that trap an inactive, ‘open’ conformational state of APS reductase may also represent new opportunities for inhibitor design16.
Figure 8.

APS reductase interactions with substrate, APS inferred from P. aeruginosa APS reductase (PDB deposition 2GOY) and S. cerevisiae PAPS reductase (PDB deposition 2OQ2) structures and functional data obtained in the present study. (A) Summary of proposed active site contacts to APS. (B) Summary of proposed active site contacts to APS, plotted in two dimensions. A total of nine protein residues are shown in proximity around the ligand, with hydrogen bonding interactions shown where detected. Hydrogen bonds are draw as dotted lines with arrows denoting the direction of the bond. Interactions from substrate or the residue backbones of the enzyme are distinguished from the interactions with residue side chains by a solid dot at the end of the interaction line. Active site residues between P. aeruginosa and M. tuberculosis APS reductase are largely conserved, with the exception of residues implicated in hydrophobic interactions (Ser62 to Met67, Ser84 to Phe87, Phe61 to Asn66). The corresponding numbers for residues conserved between M. tuberculosis and P. aeruginosa APS reductase are: Ser65 (Ser60), Leu88 (Leu85), Lys145 (Lys144), Gly162 (Gly161), Arg171 (Arg171), Arg237 (Arg242), Arg240 (Arg245) Cys249 (Cys256), and His252 (His259). See also Figure S1.
Experimental Procedures
Materials
Adenosine, 2-aminoadenosine, 3′-deoxyadenosine, 2′-deoxyadenosine, 1-methyladenosine and inosine-5′-O-monophosphate, were purchased from Sigma. 5′-Phosphoribose, 3′-phosphoadenosine, 3′5′-diphosphoadenosine, adenine and ribose were also purchased from Sigma. 7-Deazaadenosine-5′-O-monophosphate and purine riboside-5′-O-monophosphate were from Biology Life Science Institute. Purchased nucleosides and other analogs were of the highest purity available (≥ 95%) and were used without further purification. Additional reagents and solvents were purchased from Sigma or other commercial sources and were used without further purification.
General Synthetic Methods
Reactions that were moisture sensitive or using anhydrous solvents were performed under a nitrogen or argon atmosphere. Analytical thin layer chromatography (TLC) was performed on pre-coated silica plates obtained from Analtech. Visualization was accomplished with UV light or by staining with ethanolic H2SO4 or ceric ammonium molybdate. Nucleosides were purified by flash chromatography using Merck silica gel (60–200 mesh).
Preparation of Nucleoside and Nucleotide Analogs
N6, N6-Dimethyl-5′-adenosine was prepared from inosine by the procedure described by Veliz and Beal41 and purified by flash chromatography developed in 60:40 ethyl acetate:hexanes. N6-Methyl-5′-adenosine was prepared via reaction of 6-bromoinosine with methylamine as previously described42. 8-Aminoadenosine was prepared by selective bromination of adenosine at the C8 position, exchange of bromine for azide, followed by reductive hydrogenation to afford the amine, as previously described43. 2′-Methoxyadenosine was prepared from adenosine, by reaction with methyl iodide under alkaline conditions as previously described44. Adenosine 5′-O-α,β,-imidodiphosphate (AMPNP) was synthesized by reaction of 5′-tosyladenosine45 with imidodiphosphate salt45, 46. Adenosine 5′-O-α,β-difluoromethylenediphosphate (ADPβF) was prepared by coupling 5′-tosyladenosine to difluoro substituted methylenediphosphonic acid, as previously reported45, 47. Adenosine 5′-O-thiophosphosulfate (APSαS) was synthesized by reacting pyridine-N-sulfonic acid with adenosine 5′-O-thiopshophate (AMPS), as previously described48. Structures and purity (≥ 95%) were confirmed by 1H, 31P NMR and HPLC analysis (data not shown).
Nucleoside Phosphorylation
Nucleotide analogs (2′-deoxy-5′-phosphoadenosine, 2′-methoxy-5′phosphoadenosine, N6, N6-dimethyl-5′-phosphoadenosine, N6-methyl-5′-phosphoadenosine, 8-amino-5′-phosphoadenosine, 1-methyl-5′-phosphoadenosine, and 2-amino-5′-phosphoadenosine) were synthesized by chemical phosphorylation of the corresponding adenosine analog, followed by purification via reversed phase HPLC. Nucleosides were phosphorylated by reaction of nucleoside with POCl3 as described49. The nucleoside (0.15 mmole) was suspended in triethylphosphate at 0 °C (0.65 ml). Water (1 equiv) was added to the reaction. Subsequently, POCl3 (3–5 equivalents, 42 μl–70 μl) was added over a period of 30 min with constant stirring. The suspension was kept stirring for an additional 1.5 h, when the white suspension became a clear solution. Water (1 mL) was added to hydrolyze the phosphoryl chloride and terminate the reaction. The pH was neutralized to ~7 by dropwise addition of 1 M NH4OH. The reaction was passed over a 2 g C18 SPE column (Fisher) that was conditioned in acetonitrile (6 ml) followed by H2O (6 ml). The nucleotide was eluted from the C18 SPE column with H2O. Nucleotides were purified by reversed phase HPLC employing isocratic separation in 20 mM ammonium acetate, pH 7 on a semi-preparative (10 × 25 cm) C18 column and the solvent was removed from the solution by repeated lyophilization. The physical and spectral data for these analogs (confirmed by 1H, 13C and 31P NMR and mass spectrometry) were consistent with those previously reported for these compounds49–51. The purity (≥ 95%) was confirmed by HPLC analysis using the conditions described above. The retention time of each nucleotide analog (N6, N6-Dimethyl-5′-phosphoadenosine, N6-Methyl-5′-phosphoadenosine, 2′-methoxy-5′phosphoadenosine, 2′-deoxy-5′-phosphoadenosine, 8-amino-5′-phosphoadenosine, 1-methyl-5′-phosphoadenosine, and 2-amino-5′-phosphoadenosine) was 19.7 min, 19.6 min, 8.5 min, 6.1 min, 4.4 min, 4 min, 3.5 min, respectively. The concentrations of nucleotide analogs was determined by absorbance at 260 nm, assuming ε260 = 15,400 M−1 cm−152.
Enzyme purification
Purification of APS reductase was carried out as previously described10.
General Kinetic Methods
35S-labeled APS was synthesized and purified as previously described10 with the inclusion of an additional anion exchange purification step on a 5-ml FFQ column (GE Healthcare) eluting with a linear gradient of ammonium bicarbonate, pH 8.0. The reduction of APS to sulfite and AMP was measured in a 35S-based assay as previously described53. Reactions were quenched by the addition of charcoal solution (2% w/v) containing Na2SO3 (20 mM). The suspension was vortexed, clarified by centrifugation and an aliquot of the supernatant containing the radiolabeled sulfite product was counted in scintillation fluid. APS reductase activity was measured in single turnover reactions, with trace amounts of 35S-APS (~1 nM) and excess protein. These reactions can typically be followed to ≥90% completion (Figure S4A), and the reaction time courses fit well to eq 1, in which Frac P is the fraction product, k is the observed rate constant, and t is time:
| (1) |
Unless otherwise specified, the standard reactions conditions were 30 °C with 100 mM bis-tris propane at pH 7.5, DTT (5 mM), and thioredoxin (10 μM). Kinetic data were measured in at least two independent experiments and the standard error was typically less than 15%.
The affinity of APS reductase (E) for APS was determined from the dependence of the observed rate constant for S-sulfocysteine formation on protein concentration according to eq 2:
| (2) |
In this equation, kobs is the observed rate constant at a particular protein concentration, kmax is the maximal rate constant with saturating protein, and K1/2 is the protein concentration that provides half the maximal rate. To ensure that the chemical step was rate-determining, reactions were performed in NaMes (10 mM) at pH 5.5 and control experiments demonstrate that the enzyme is stable under these assay conditions (data not shown). Because the chemical step is rate-determining for S-sulfocysteine formation (kst <, kmax is equal to the rate constant for the reaction of the E·APS complex, and K1/2 is equal to the dissociation constant (Kd) of APS for APS reductase. The concentration of active protein was determined by direct titration with a high concentration of APS (i.e. [APS] ≫ Kd). In theory, the binding affinity of APS could increase at physiological pH. However, several lines of evidence argue against this possibility. First, the pKa of the β-sulfate moiety is less than 2 and thus, at pH 5.5, the sulfonucleotide is completely ionized. Second, the pH dependence of ADP binding (see below) reflects the pKa of this nucleotide in solution. Finally, the Kd measured at pH 5.5 is in line with the apparent Km value measured at pH 8.022.
The affinity of various ligands for APS reductase was determined by inhibition methods. The observed rate constant of the reaction: E + 35S-APS → products (kobs) was determined at varying inhibitor (I) concentrations (Figure S4B), and the [I]-dependence was fit to a simple model for competitive inhibition (eq 3). In eq 3, ko is the rate of the reaction in the absence of analog, and Ki is the inhibition constant of the analog. With subsaturating APS reductase, Ki is equal to the equilibrium dissociation constant (Kd) of the ligand.
| (3) |
pH Dependence of Inhibitor Binding
Values of Ki were determined by inhibition of APS reduction (pH 6–9.5) with [35S-APS] ≪ K1/2, such that Ki is expected to be the Kd. The following buffers were used for the indicated pH ranges: NaMES (6.0–7.0), Bis-Tris (6.5–7.5), Tris (7.5–9.0) and CAPS (9.0–9.5). Reactions were typically carried out with 100 mM buffer and the standard assays and conditions described above were used to monitor kcat/Km for reduction of 35S-APS in the presence and absence of inhibitor. The rate constants at each pH value for multiple reactions were averaged, and the standard deviations were ≤15% of the average. pKa values were determined using eq 4, derived from a model where the binding of the ligand depends on a single ionizable group.
| (4) |
Energetic Contribution of Ligand Substituents to Binding
The energetic contributions of individual ligand substitutents to APS reductase binding were expressed as ΔΔG = −RTln(Kd1/Kd2) in which R is the gas constant, T is the temperature (303 K), and Kd is the equilibrium dissociation constant. A negative value of ΔΔG indicates that a given substitutent contributes to ligand binding by APS reductase.
Electrostatic Surface Potentials
The electrostatic surface potential was calculated using PM3 semi-empirical molecular orbital calculations implemented in SPARTAN software (Wavefunction, Inc) for the fully optimized structure.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM087638 to K.S.C.).
Abbreviations
The following abbreviations have been used in this text:
- S-sulfocysteine
Cys-Sγ–SO3−
- APS
adensoine-5′-phosphosulfate
- Trx
thioredoxin
- HPLC
high-performance liquid chromatography
- TFA
trifluoroacetic acid
- P-loop
phosphate-binding loop
- E
enzyme
- Da
daltons
Footnotes
Residue numbers throughout manuscript correspond to the APS reductase sequence from P. aeruginosa (Figure S1).
In principle, APSαS could be utilized as a substrate by APS reductase. However, under saturating conditions, no evidence for formation of the S-sulfocysteine intermediate has been obtained (data not shown). At present, it is not understood why APS reductase does not effectively reduce APSαS. One possible explanation is that the α-sulfur substitution disrupts contact with residues that could be important for stabilizing charge development in the transition state, such as Arg171 and His259.
Supporting Information Available. Tables S1–S2 and Figures S1–S4. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Schelle MW, Bertozzi CR. Sulfate metabolism in mycobacteria. Chembiochem. 2006;7:1516–1524. doi: 10.1002/cbic.200600224. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell S. Biology of sulfur. In: Mitchell S, editor. Biological Interactions of Sulfur Compounds. Vol. 1. CRC Press; New York: 1996. pp. 20–41. [Google Scholar]
- 3.Kredich NM. Biosynthesis of cysteine. In: Niedhardt FC, Curtiss R, editors. Escherichia coli and Salmonella: Cellular and Molecular Biology. 2. Vol. 1. ASM press; Washigton, D. C.: 1996. pp. 514–527. [Google Scholar]
- 4.Giles NM, Watts AB, Giles GI, Fry FH, Littlechild JA, Jacob C. Metal and redox modulation of cysteine protein function. Chem Biol. 2003;10:677–693. doi: 10.1016/s1074-5521(03)00174-1. [DOI] [PubMed] [Google Scholar]
- 5.Williams SJ, Senaratne RH, Mougous JD, Riley LW, Bertozzi CR. 5′-Adenosinephosphosulfate lies at a metabolic branch point in mycobacteria. J Biol Chem. 2002;277:32606–32615. doi: 10.1074/jbc.M204613200. [DOI] [PubMed] [Google Scholar]
- 6.Kopriva S, Buchert T, Fritz G, Suter M, Benda R, Schunemann V, Koprivova A, Schurmann P, Trautwein AX, Kroneck PM, Brunold C. The presence of an iron-sulfur cluster in adenosine 5′-phosphosulfate reductase separates organisms utilizing adenosine 5′-phosphosulfate and phosphoadenosine 5′-phosphosulfate for sulfate assimilation. J Biol Chem. 2002;277:21786–21791. doi: 10.1074/jbc.M202152200. [DOI] [PubMed] [Google Scholar]
- 7.Bhave DP, Muse WB, 3rd, Carroll KS. Drug targets in mycobacterial sulfur metabolism. Infect Disord Drug Targets. 2007;7:140–158. doi: 10.2174/187152607781001772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mdluli K, Spigelman M. Novel targets for tuberculosis drug discovery. Curr Opin Pharmacol. 2006;6:459–467. doi: 10.1016/j.coph.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Senaratne RH, De Silva AD, Williams SJ, Mougous JD, Reader JR, Zhang T, Chan S, Sidders B, Lee DH, Chan J, Bertozzi CR, Riley LW. 5′-Adenosinephosphosulphate reductase (CysH) protects Mycobacterium tuberculosis against free radicals during chronic infection phase in mice. Mol Microbiol. 2006;59:1744–1753. doi: 10.1111/j.1365-2958.2006.05075.x. [DOI] [PubMed] [Google Scholar]
- 10.Carroll KS, Gao H, Chen H, Stout CD, Leary JA, Bertozzi CR. A conserved mechanism for sulfonucleotide reduction. PLoS Biol. 2005;3:e250. doi: 10.1371/journal.pbio.0030250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carroll KS, Gao H, Chen H, Leary JA, Bertozzi CR. Investigation of the iron-sulfur cluster in Mycobacterium tuberculosis APS reductase: implications for substrate binding and catalysis. Biochemistry. 2005;44:14647–14657. doi: 10.1021/bi051344a. [DOI] [PubMed] [Google Scholar]
- 12.Chartron J, Carroll KS, Shiau C, Gao H, Leary JA, Bertozzi CR, Stout CD. Substrate recognition, protein dynamics, and iron-sulfur cluster in Pseudomonas aeruginosa adenosine 5′-phosphosulfate reductase. J Mol Biol. 2006;364:152–169. doi: 10.1016/j.jmb.2006.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapman E, Best MD, Hanson SR, Wong CH. Sulfotransferases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angew Chem Int Ed. 2004;43:3526–3548. doi: 10.1002/anie.200300631. [DOI] [PubMed] [Google Scholar]
- 14.Yu Z, Lemongello D, Segel IH, Fisher AJ. Crystal structure of Saccharomyces cerevisiae 3′-phosphoadenosine-5′-phosphosulfate reductase complexed with adenosine 3′,5′-bisphosphate. Biochemistry. 2008;47:12777–12786. doi: 10.1021/bi801118f. [DOI] [PubMed] [Google Scholar]
- 15.Young DB, Gideon HP, Wilkinson RJ. Eliminating latent tuberculosis. Trends Microbiol. 2009;17:183–188. doi: 10.1016/j.tim.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Lee GM, Craik CS. Trapping moving targets with small molecules. Science. 2009;324:213–215. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 18.Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE, 3rd, Blanchard JS. Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science. 2009;323:1215–1218. doi: 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palomino JC, Ramos DF, da Silva PA. New anti-tuberculosis drugs: strategies, sources and new molecules. Curr Med Chem. 2009;16:1898–1904. doi: 10.2174/092986709788186066. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen L, Pieters J. Mycobacterial Subversion of Chemotherapeutic Reagents and Host Defense Tactics: Challenges in Tuberculosis Drug Development. Annu Rev Pharmacol Toxicol. 2009;49:427–453. doi: 10.1146/annurev-pharmtox-061008-103123. [DOI] [PubMed] [Google Scholar]
- 21.Cosconati S, Hong JA, Novellino E, Carroll KS, Goodsell DS, Olson AJ. Structure-based virtual screening and biological evaluation of Mycobacterium tuberculosis adenosine 5′-phosphosulfate reductase inhibitors. J Med Chem. 2008;51:6627–6630. doi: 10.1021/jm800571m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun M, Leyh TS. Channeling in sulfate activating complexes. Biochemistry. 2006;45:11304–11311. doi: 10.1021/bi060421e. [DOI] [PubMed] [Google Scholar]
- 23.Dzingeleski GD, Wolfenden R. Hypersensitivity of an enzyme reaction to solvent water. Biochemistry. 1993;32:9143–9147. doi: 10.1021/bi00086a020. [DOI] [PubMed] [Google Scholar]
- 24.Carlow D, Wolfenden R. Substrate connectivity effects in the transition state for cytidine deaminase. Biochemistry. 1998;37:11873–11878. doi: 10.1021/bi980959n. [DOI] [PubMed] [Google Scholar]
- 25.Nikolic-Hughes I, Rees DC, Herschlag D. Do electrostatic interactions with positively charged active site groups tighten the transition state for enzymatic phosphoryl transfer? . J Am Chem Soc. 2004;126:11814–11819. doi: 10.1021/ja0480421. [DOI] [PubMed] [Google Scholar]
- 26.CRC Handbook of Chemistry and Physics. 75. CRC Press; Boca Raton, FL: 1994–1995. pp. 9–12. [Google Scholar]
- 27.Jaffe EK, Cohn M. 31P nuclear magnetic resonance spectra of the thiophosphate analogues of adenine nucleotides; effects of pH and Mg2+ binding. Biochemistry. 1978;17:652–657. doi: 10.1021/bi00597a014. [DOI] [PubMed] [Google Scholar]
- 28.Frey PA, Sammons RD. Bond order and charge localization in nucleoside phosphorothioates. Science. 1985;228:541–545. doi: 10.1126/science.2984773. [DOI] [PubMed] [Google Scholar]
- 29.Satishchandran C, Myers CB, Markham GD. Adenosine-5′-O-(2- fluorodiphosphate) (ADP-β-F), an analog of adenosine-5′-phosphosulfate. Bioorganic Chem. 1992;20:107–114. [Google Scholar]
- 30.Gregoret LM, Rader SD, Fletterick RJ, Cohen FE. Hydrogen bonds involving sulfur atoms in proteins. Proteins. 1991;9:99–107. doi: 10.1002/prot.340090204. [DOI] [PubMed] [Google Scholar]
- 31.Yount RG, Babcock D, Ballantyne W, Ojala D. Adenylyl imidodiphosphate, an adenosine triphosphate analog containing a P--N--P linkage. Biochemistry. 1971;10:2484–2489. doi: 10.1021/bi00789a009. [DOI] [PubMed] [Google Scholar]
- 32.Larsen M, Willett R, Yount RG. Imidodiphosphate and pyrophosphate: possible biological significance of similar structures. Science. 1969;166:1510–1511. doi: 10.1126/science.166.3912.1510. [DOI] [PubMed] [Google Scholar]
- 33.Reynolds MA, Gerlt JA, Demou PC, Oppenheimer NJ, Kenyon GL. N-15 and O-17 NMR-studies of the proton binding-sites in imidodiphosphate, tetraethyl imidodiphosphate, and adenylyl imidodiphosphate. J Am Chem Soc. 1983;105:6475–6481. [Google Scholar]
- 34.Savage H, Montoya G, Svensson C, Schwenn JD, Sinning I. Crystal structure of phosphoadenylyl sulphate (PAPS) reductase: a new family of adenine nucleotide alpha hydrolases. Structure. 1997;5:895–906. doi: 10.1016/s0969-2126(97)00244-x. [DOI] [PubMed] [Google Scholar]
- 35.Cullis PM, Maxwell A, Weiner DP. Energy coupling in DNA gyrase: a thermodynamic limit to the extent of DNA supercoiling. Biochemistry. 1992;31:9642–9646. doi: 10.1021/bi00155a017. [DOI] [PubMed] [Google Scholar]
- 36.Rye CS, Baell JB. Phosphate isosteres in medicinal chemistry. Curr Med Chem. 2005;12:3127–3141. doi: 10.2174/092986705774933452. [DOI] [PubMed] [Google Scholar]
- 37.O’Hagan D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem Soc Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- 38.Harris DA. Cell chemistry and physiology: part II. In: Bittar EE, Bittar N, editors. Principles of Medical Biology. Vol. 4. Elsevier; Conneticut: 1996. p. 27. [Google Scholar]
- 39.Storer AC, Cornish-Bowden A. Concentration of MgATP2− and other ions in solution. Calculation of the true concentrations of species present in mixtures of associating ions. Biochem J. 1976;159:1–5. doi: 10.1042/bj1590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yount RG, Simchuck S, Yu I, Kottke M. Adenosine-5′-sulfatopyrophosphate, an analogue of adenosine triphosphate. I. Preparation, properties, and mode of cleavage by snake venoms. Arch Biochem Biophys. 1966;113:288–295. doi: 10.1016/0003-9861(66)90189-5. [DOI] [PubMed] [Google Scholar]
- 41.Veliz EA, Beal PA. C6 substitution of inosine using hexamethylphosphorous triamide in conjunction with carbon tetrahalide or N-halosuccinimide. Tet Lett. 2000;41:1695–1697. [Google Scholar]
- 42.Veliz EA, Beal PA. 6-Bromopurine nucleosides as reagents for nucleoside analogue synthesis. J Org Chem. 2001;66:8592–8598. doi: 10.1021/jo016078v. [DOI] [PubMed] [Google Scholar]
- 43.Townsend LB, Tipson RS. Nucleic Acid Chemistry:Improved and New Synthetic Procedures, Methods, and Techniques. II Wiley; New York: 1978. [Google Scholar]
- 44.Yano J, Kan LS, Ts’o POP. A simple method for the preparation of adenosine with methyl iodide in anhydrous alkaline medium. Biochim Biophys Acta. 1980;629:178–183. doi: 10.1016/0304-4165(80)90276-7. [DOI] [PubMed] [Google Scholar]
- 45.Davisson VJ, Davis DR, Dixit VM, Poulter CD. Synthesis of nucleotide 5′-diphosphates from 5′-O-tosyl nucleosides. J Org Chem. 1987;52:1794–1801. [Google Scholar]
- 46.Ma Q, Babbitt PC, Kenyon GL. Adenosine 5′-[α,β-imido]triphosphate, a substrate for T7 RNA-polymerase and rabbit muscle creatine-kinase. J Am Chem Soc. 1988;110:4060– 4061. [Google Scholar]
- 47.Mohamady S, Jakeman DL. An improved method for the synthesis of nucleoside triphosphate analogues. J Org Chem. 2005;70:10588–10591. doi: 10.1021/jo0518598. [DOI] [PubMed] [Google Scholar]
- 48.Zhang HP, Leyh TS. alpha-Thio-APS: A stereomechanistic probe of activated sulfate synthesis. J Am Chem Soc. 1999;121:8692–8697. [Google Scholar]
- 49.Yoshikawa M, Kato T, Takenishi T. A novel method for phosphorylation of nucleosides to 5′-nucleotides. Tet Lett. 1967;50:5065–5068. doi: 10.1016/s0040-4039(01)89915-9. [DOI] [PubMed] [Google Scholar]
- 50.Burgess K, Cook D. Syntheses of nucleoside triphosphates. Chem Rev. 2000;100:2047–2059. doi: 10.1021/cr990045m. [DOI] [PubMed] [Google Scholar]
- 51.Freist W, Cramer F. Synthesis of AMP and ATP analogues. In: Townsend LW, Tipson RS, editors. Nucleic Acid Chemistry, Part 2. John Wiley & Sons; New York: 1978. pp. 827–841. [Google Scholar]
- 52.Dawson RMC, Elliott DC, Elliott WH, Jones KM. Data for Biochemical Research. 3. Oxford University Press; Oxford: 1989. [Google Scholar]
- 53.Gao H, Leary J, Carroll KS, Bertozzi CR, Chen HY. Noncovalent complexes of APS reductase from M-tuberculosis: Delineating a mechanistic model using ESI-FTICR MS. J Am Soc Mass Spectrom. 2007;18:167–178. doi: 10.1016/j.jasms.2006.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chargaff E, Davidson JN. The Nucleic Acids I. Academic Press; New York: 1955. [Google Scholar]
- 55.Kumler WD, Eiler JJ. The acid strength of mono and diesters of phosphoric acid. The n-alkyl esters from methyl to butyl, the esters of biological importance, and the natural guanidine phosphoric acids. J Am Chem Soc. 1943;65:2355–2361. [Google Scholar]
- 56.Isbell HS. Carbohydrate in Solution. American Chemical Society; Wachington, DC: 1973. [Google Scholar]
- 57.Chang R. General Chemistry. 8. McGraw-Hill. Inc; New York: 2005. Acids and bases. [Google Scholar]
- 58.Hirota K, Inoue Y, Chujo R. Effect of the base stacking association on the phosphate ionization of 2′-deoxyguanosine 5′-monophosphate. Bull Chem Soc Jpn. 1984;57:247–250. [Google Scholar]
- 59.Falany CN. Enzymology of human cytosolic sulfotransferases. Faseb J. 1997;11:206–216. doi: 10.1096/fasebj.11.4.9068609. [DOI] [PubMed] [Google Scholar]
- 60.Jaffe EK, Cohn M. 31P nuclear magnetic resonance spectra of the thiophosphate analogues of adenine nucleotides; effects of pH and Mg2+ binding. Biochemistry. 1978;17:652–7. doi: 10.1021/bi00597a014. [DOI] [PubMed] [Google Scholar]
- 61.Cullis PM, Maxwell A, Weiner DP. Energy coupling in DNA gyrase: a thermodynamic limit to the extent of DNA supercoiling. Biochemistry. 1992;31:9642–6. doi: 10.1021/bi00155a017. [DOI] [PubMed] [Google Scholar]
- 62.Vogel HJ, Bridger WA. Phosphorus-31 nuclear magnetic resonance studies of the methylene and fluoro analogues of adenine nucleotides. Effects of pH and magnesium ion binding. Biochemistry. 1982;21:394–401. doi: 10.1021/bi00531a029. [DOI] [PubMed] [Google Scholar]
- 63.Rye CS, Baell JB. Phosphate isosteres in medicinal chemistry. Curr Med Chem. 2005;12:3127–41. doi: 10.2174/092986705774933452. [DOI] [PubMed] [Google Scholar]
- 64.Chanley JD, Feageson E. A study of hydrolysis of phosphoramides .2. Solvolysis of phosphoramidic acid and comparison with phosphate esters. J Am Chem Soc. 1963;85:1181–1190. [Google Scholar]
- 65.Singer B. Nucleosides, nucleotides, and nucleic acids. In: Fasman GD, editor. Practical Handbook of Biochemistry and Molecular Biology. 2. Vol. 1. CRC Press; Cleveland: 1989. pp. 392–393. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.