

Abstract
Many lymphocyte functions, such as antigen recognition, take place deep in densely populated lymphoid organs. Because direct in vivo observation was not possible, the dynamics of immune-cell interactions have been inferred or extrapolated from in vitro studies. Two-photon fluorescence excitation uses extremely brief (<1 picosecond) and intense pulses of light to ‘see’ directly into living tissues, to a greater depth and with less phototoxicity than conventional imaging methods. Two-photon microscopy, in combination with newly developed indicator molecules, promises to extend single-cell approaches to the in vivo setting and to reveal in detail the cellular collaborations that underlie the immune response.
Scientific questions drive the development of technology, and new technologies, in turn, influence the type of questions that we pose in an iterative process of discovery. Applied to immunology, this has prompted the innovation and application of powerful techniques for analysing the microscopic world of the immune system. Two methodological lines of investigation have dominated the field: in vivo experiments that examine the behaviour of populations of cells in living animals during an immune response; and in vitro experiments that use individual cells in artificial environments. An immune response is the sum of many complex and dynamic individual cellular behaviours that are shaped by many environmental factors. In vivo experiments maintain this natural environment, but they cannot resolve the behaviours of individual cells. By contrast, in vitro experiments provide information at the subcellular and molecular levels, but they cannot replicate adequately the full repertoire of environmental factors. There is a pressing need for techniques that allow real-time observation of single cells and molecules in intact tissues. Recent developments in imaging technology now make this possible. In this review, we provide a guide to the application of biophotonic techniques to the field of immunology and, in particular, to the use of two-photon laser microscopy. We discuss recent results obtained using this approach and pinpoint some key questions that might be resolved by tissue imaging.
The biophotonics tool kit
Immunologists have long been adept at raiding other disciplines to acquire new research tools. Fluorescence techniques — for example, flow cytometry, video imaging and more exotic imaging modalities, such as FLUORESCENCE RESONANCE ENERGY TRANSFER (FRET) and two-photon microscopy — combined with new probes to track Ca2+ signalling, gene expression, cell division and molecular interactions, form a powerful arsenal of biophotonic techniques to investigate the immune response. Tissue and intravital imaging methods are just beginning to extend the use of these tools to the in vivo setting. Recently, two-photon microscopy has shown enormous potential for the imaging of tissue-dependent, dynamic and asynchronous individual cellular behaviours in intact lymphoid organs, and it can provide information that would be indiscernible in population measurements and completely absent in vitro. This technique promises to contribute to our fundamental understanding of the immune system, from lymphocyte trafficking to antigen presentation and subsequent effector responses.
Imaging T-cell dynamics in vitro
The single-cell approach has intrinsic value, because asynchronous behaviours are not indicated by population measurements. In vitro imaging systems that combine BRIGHT-FIELD MICROSCOPY or NOMARSKI OPTICS with FLUORESCENCE MICROSCOPY have been particularly good at revealing the individual behaviours and interactions of cells in the immune system. Previous in vitro studies that visualized T cells interacting with antigen-presenting cells (APCs), or beads used as surrogate APCs, have shown dynamic changes in T-cell motility, shape, contact requirements, intracellular calcium signalling (through FURA-2 RATIOMETRIC IMAGING) and reporter gene expression1–7 (FIG. 1). These studies showed that T-cell motility is inhibited by the Ca2+ signal, by protein kinase C (PKC) activation and by integrin signalling4,8; this ‘stop signal’ stabilizes the T cell at the site of antigen presentation.
Figure 1. In vitro versus in vivo approaches to immunology.

a | In vitro-imaging approaches allow the visualization of cellular and molecular details — illustrated here by the example of an antigen-presenting cell triggering a calcium signal in an interacting T cell. b | Immunohistological and cytometric in vivo methods have made possible the study of cell populations at discrete time points; however, these techniques do not allow the visualization of the dynamic behaviour of single living cells in their native environment. Images reproduced a | from REF. 4, with permission from Elsevier Science © (1996), and b | by personal communication from J. Cyster and T. Okada.
Studies using DECONVOLUTION MICROSCOPY or CONFOCAL MICROSCOPY on fixed cells stained with antibodies, or living cells that express molecules tagged with green fluorescent protein (GFP), have shown that molecular rearrangements occur at the zone of contact between the T cell and the APC that form what is now known as the immunological synapse9. The intricate choreography of formation and stabilization of the immunological synapse has been reviewed recently10. Some molecules move laterally towards the contact region and become concentrated in a central zone — the central supra-molecular activation complex (cSMAC) — whereas others form a concentric region known as the peripheral supra-molecular activation complex (pSMAC)11. Following the pioneering work of McConnell and colleagues12,13, Dustin and Springer developed a LIPID-BILAYER IMAGING system14,15, which uses dissolved membrane receptors and other proteins to study cell adhesion and motility. This system was used subsequently to define additional requirements and dynamic molecular changes during immunological-synapse formation16. Recently, molecular interactions at the synapse between the T-cell receptor (TCR) and CD4 have been shown using FRET between the GFP variants cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) tagged to CD3ζ and CD4, respectively17. T-cell commitment to activation requires direct contact with antigen-specific APCs, but the nature of this interaction (stable or transient) remains controversial18. The concept of the immunological synapse as a stable organizing element that leads, after a single encounter, to activation was proposed on the basis of the time course of T-cell activation in vitro and the time required for the SMAC to form19. In contrast to previous in vitro observations on a two-dimensional substrate, interactions between T cells and dendritic cells (DCs) in a COLLAGEN GEL MATRIX culture system were shown to be relatively brief (<30 minutes) and intermittent, such that T cells make serial contact with several DCs20. However, the effects of environmental signals on synapse formation are not understood fully21. As a result, there are now two competing models of T-cell–APC interactions, which involve either a single, stable encounter or transient, serial encounters18–20,22.
Studying T-cell responses in animals
Many of us take for granted the availability of instruments that use fluorescence to detect and visualize the distribution of molecules. Flow cytometry, for example, is a method to analyse the expression of specific molecules by particular subsets of cells. Fluorescent antibodies specific for cell-surface markers can be used to identify lymphocyte phenotypes, as well as to assess their activation state and effector functions using methods that include intracellular staining protocols to assay for cytokine expression23,24. Antigen-specific T cells can be identified in vivo using fluorescent peptide–MHC tetramers25. Furthermore, the discovery of CFSE LABELLING has provided a convenient tool for measuring cell division in vivo26. Moreover, because CFSE is non-toxic and bright, it has facilitated the tracking of cells in slice preparations, intact tissues and intravital microscopy27,28. The use of immunofluorescence to stain fixed tissue sections has allowed a high-definition, but static, glimpse inside animal tissues, which has yielded important information about the location, phenotype and physical interactions of various cells that participate in the immune response28. In addition, it has been a useful technique to map the deposition of extracellular matrix and the distribution of chemokines that are thought to direct the organization of lymphoid organs29. The use of GFP and its spectral variants (CFP, YFP and blue fluorescent protein, BFP) as protein tags has been of great benefit to cell biologists, because these proteins are non-toxic, require no labelling step and work equally well in living and fixed tissues. In particular, GFP-transgenic mice have provided markers for T-cell trafficking30,31 and probes for the activation of these cells and their expression of interleukin-2 (IL-2) and IL-4 (REFS 32,33). Other groups have used GFP to track T cells in the high endothelial venules of peripheral lymph nodes34, to study the lipopolysaccharide-induced intraocular recruitment and activation of T cells35, and to follow the interaction of CD8+ T cells with bone-marrow-derived DCs36. Of particular interest is the observation that in the last system, lymphoid cells express the GFP tag strongly, but at different levels, which allows T cells, B cells and DCs to be discriminated.
The need for live-tissue imaging
In vivo approaches have elucidated the basic properties of immune cells and lymphoid tissues, whereas in vitro cell-culture systems have provided a high-definition analysis of the cell-surface receptors and intracellular signalling pathways that lead to an immune response. In vitro culture systems have been invaluable for understanding the single-cell behaviours of lymphocytes, but it is apparent that lymphocyte activation and function are influenced by, and in several cases completely dependent on, factors that are present in the tissue environment18,20,37,38. For example, the structural features of the immunological synapse have been pieced together from in vitro studies — mainly on fixed T cells using specific antibody staining and on the interactions of T cells with planar lipid films. In a few cases, the redistribution of molecules has been observed in living T cells using GFP-tagged proteins17,39–41. Close contacts between T cells and APCs, and the clustering of TCRs near to the site of antigen presentation have been observed also in fixed tissue samples from the lymph nodes of TCR-transgenic mice27,42, but this provides only a static picture of a dynamic event. Different experimental systems have led to fundamentally different views of the T-cell–APC interaction, which has led some immunologists to question the physiological relevance of in vitro findings. Two-photon tissue imaging provides the much-needed methodology to address these questions.
Two-photon microscopy
Fluorescent dyes are standard tools for tracking cell populations in animals by flow cytometry and for the microscopic imaging of individual cells in vitro. However, important challenges arise when attempting to visualize fluorescently labelled cells deep in living tissues, such as lymph nodes. Three main problems limit optical resolution in such a case. First, the high-numerical-aperture objective lenses that are required for fluorescence microscopy have a narrow depth of field. Objects appear in sharp focus over a depth of <1 μm, but with a conventional epi-fluorescence microscope, fluorescent label above and below this plane glows brightly, thereby obscuring the image. Second, most biological tissues scatter light, so that image contrast is lost as the microscope is focused to increasing depths. Finally, the intense excitation light will, over time, bleach the dye and cause photodamage to the cells.
Limitations of confocal microscopy
A partial solution was provided by the introduction of confocal microscopy some 15 years ago43. This technique provides a sharp ‘optical section’ at a given depth in a specimen by rejecting out-of-focus fluorescence that arises from above and below the focal plane. The principle of confocal microscopy is simple (FIGS 2,3). Fluorescence excitation light (usually from a laser) is focused in the specimen by the objective lens, and the fluorescence emitted from that spot is collected through the same lens and refocused to a spot centred on a small aperture (confocal pinhole) placed before a detector. So, in-focus fluorescence passes efficiently to the detector, whereas out-of-focus fluorescence from above or below the imaging plane is largely rejected.
Figure 2. Principles of confocal and two-photon microscopy.

a | Single-photon excitation. Individual photons of high-energy blue light (wavelength, λ = 488 nm) from a krypton-argon (Kr-Argon) laser excite fluorophores in the sample. After an electron in the fluorophore jumps from the energy ground state (S0) to the excited state (S2) (blue arrow), it loses energy rapidly owing to non-radiative relaxation (NR). Subsequently, fluorescence emission occurs at a longer wavelength than the excitation light (Stokes shift) as the electron falls back to the ground state (green arrow). Because excitation involves single photons, fluorescence is emitted along the whole path of a laser beam focused on a sample of fluorescent dye (inset photograph). b | Two-photon excitation. Two infrared photons (λ = 780 nm) from a pulsed titanium-sapphire (Ti-sapphire) laser are absorbed simultaneously (red arrows) to excite the fluorophore; light is emitted in the same manner as for single-photon excitation (green arrow). However, because of the quadratic relationship between excitation intensity and fluorescence emission, light is emitted only at the focal point of the focused laser beam (inset photograph).
Figure 3. Two-photon excitation of fluorophors by spatial and temporal compression of photons.

A | Spatial compression of photons by an objective lens restricts two-photon excitation to the focal spot. The diagram compares one-photon excitation (with blue light) with two-photon excitation (with infrared light). In both cases, the excitation intensity (ex.), which corresponds to the density of photons, is greatest at the focal spot and declines away from this point as the light subtends circles of progressively increasing diameter. For one-photon excitation, green-fluorescence emission (em.) occurs everywhere along the path of the beam, with an intensity that is linearly proportional to that of the excitation light. By contrast, two-photon excitation evokes fluorescence at the focal spot only. The rest of the sample is exposed only to low-energy infrared photons, which fail to excite the fluorophore and can pass through biological tissue with minimal scattering. B | Temporal compression of photons into short packets during femtosecond pulses achieves the high photon densities that are required for two-photon excitation and prevents the sample from being destroyed. In comparison to a continuous laser beam (a), a pulsed laser (b) of the same average power (mean number of photons per second) concentrates photons into brief bursts of much greater instantaneous power. For a mode-locked titanium-sapphire laser, each pulse lasts ~100 femtoseconds (fs), with a gap of 10 nanoseconds (ns) between pulses. So, the temporal compression is by a factor of 105 and, because of the quadratic relationship for two-photon excitation, fluorescence is enhanced by a factor of 1010.
For many applications, confocal microscopy works extremely well, and, by serial acquisition of optical slices taken at increasing depths along the z-axis, it allows subsequent three-dimensional (x–y–z) reconstruction of the sample. However, confocal microscopes are limited in their ability to ‘see’ deep into biological tissues (typically to a depth of only a few tens of μm), because scattering attenuates both the focal spot of laser light and the collection of light from the resulting fluorescent spot. Furthermore, although fluorescence is imaged only from the focal plane, the specimen is exposed to laser light above and below that plane, so that regions suffer from photobleaching and phototoxic effects even when they are not being visualized. So, confocal microscopy has serious limitations for the long-term imaging of live cells.
Principles of two-photon excitation
A solution to these problems is provided by a more recent imaging technique — two-photon microscopy — which, by a completely different principle, provides the same ‘optical-sectioning’ capability, but has the advantages of greater imaging depth and minimal photobleaching and toxicity. Two-photon excitation is a relatively old concept44 and it was used first for biological research more than ten years ago45. However, only in the past few years have improvements in laser technology and the availability of commercial multi-photon microscopes made it a practical tool for biologists who lack specialized expertise in laser optics.
In conventional fluorescence excitation, a fluorophore molecule, such as fluorescein, absorbs energy from a photon and soon after (a few nanoseconds), re-emits most, but not all, of this energy as a second photon. Because the energy of a photon is inversely proportional to its wavelength (λ), the emitted photon has a longer wavelength than the exciting photon (for example, green emission with blue excitation). By contrast, two-photon excitation involves the almost simultaneous absorption by a fluorophore of energy from two photons, each of which contributes one half of the total energy required to induce fluorescence. The emitted fluorescence light is, therefore, of shorter wavelength than the exciting light. For example, a molecule of fluorescein can be excited by two photons of near-infrared light (λ ≈ 800 nm), each of which has approximately half the energy of a single blue photon (λ = 400 nm), and then emit a photon of green light, in the same manner as for standard (one-photon) excitation with blue light. Because excitation depends on the simultaneous absorption of two photons, the resulting fluorescence emission increases in proportion to the square of the excitation intensity. This quadratic relationship gives two-photon microscopy its main advantages. The focusing of light by a microscope objective lens leads to a high density of photons at the focal point, whereas the density falls off rapidly above and below the focal point. So, the probability that a fluorophore might undergo two-photon excitation is highest at the focal point, and drops to nearly zero at distances of less than 1 μm above and below the focal point.
To achieve a useful number of two-photon excitation events, the photon density at the focal spot must be high, because a fluorophore molecule has to absorb two photons within an unimaginably brief time (10−18 seconds) of one another. Under normal conditions, this does not occur — for example, the light intensity of a laser scanning confocal microscope is about one million times too weak. Indeed, an intensity level of ~1012 W cm−2 would be required, which is equivalent to converting the electrical output of 5000 nuclear reactors into light and focusing it on a square centimetre (BOX 1). An obvious question, then, is how a biological specimen can be imaged by two-photon microscopy without vaporizing it. The answer lies in using pulsed lasers, for which the peak power during a pulse is extremely high, but the average laser power is relatively low and not much greater than for a conventional confocal microscope.
Box 1 | Two-photon illumination: brief laser pulses at high power
Average laser power at the specimen = 100 mW, focused on a diffraction-limited spot of 0.5 μm in diameter
Area of the spot = 2 × 10−9 cm2
Average laser power in the spot = 0.1 W × 1/(2 × 10−9 cm2) = 5 × 107 W cm−2
Laser pulse is on for 100 femtoseconds every 10 nanoseconds; therefore, the pulse duration to gap duration ratio = 10−5
Instantaneous power when laser is on = 5 × 1012 W cm−2
Output of a typical nuclear reactor = 1000 MW = 109 W
Therefore, the peak energy density at the specimen during each pulse is equivalent to the output of 5000 nuclear reactors converted to light and focused on a square centimetre.
Such lasers have been available for nearly 20 years, but it is only recently that their design has improved to the point that they can be used as a ‘turn-key’ instrument, requiring no special expertise or maintenance. The most versatile and commonly used system is the mode-locked titanium-sapphire (Ti-sapphire) ‘femto-second’ laser, which provides pulses of infrared light (λ = 700–1000 nm) with a duration of ~80 femto-seconds at a repetition rate of ~80 MHz. An intuitive feel for how brief these pulses are can be obtained by visualizing the laser beam as a series of ‘packets’ of photons, each about 25 μm long, travelling at the speed of light and separated from one another by gaps of about 2 metres. A crucial measure is the ratio of the pulse duration to gap duration — in this case, about 10−5. Therefore, the peak power during each pulse is 105 times greater than the mean laser power, so that the fluorescence emission from a fluorophore excited by two photons is 1010 times greater than would be achieved by a continuous laser of the same mean power.
As described above, focusing the beam from a femto-second laser through a microscope objective results in fluorescence emission restricted to the focal spot. So, an image can be formed simply by measuring the fluorescence intensity while constructing an image by scanning the laser spot point-by-point and line-by-line, in the same manner that a picture is built up on a television screen. Because fluorescence arises only from the focused laser spot, all emitted light carries useful information, even if it has been scattered in the specimen. Unlike for confocal microscopy, there is no need to detect fluorescence through an aperture; instead, a ‘wide-field’ detector (usually a photomultiplier) should be positioned close to the objective lens to collect as much of the emitted fluorescence as possible. Complete two-photon microscope systems are available commercially; alternatively, existing confocal microscopes have been adapted by addition of a femto-second laser, and other investigators have built their systems from scratch.
Lasers
The availability of lasers that produce brief pulses (<1 picosecond) at infrared wavelengths has been the key factor driving the development of two-photon microscopy. Several different types of laser are available, but the wide tuning range and high power output of the mode-locked Ti-sapphire laser make it a near-universal choice. Until a few years ago, large argon-ion lasers were used to pump the Ti-sapphire laser, but newer models use a solid-state pump laser, which requires no maintenance or adjustments, and runs from a standard wall electrical outlet. Another innovation is the availability of broadband optics sets that allow continuous tuning of the laser over a wavelength range of about 720–980 nm. The most versatile configuration is a ‘two-box’ design, with separate Ti-sapphire and pump laser heads coupled on an optical table. This provides the highest power output (2 W with a 10 W pump laser) and greatest tuning range, but it requires some expertise from the user, because tuning and mirror alignment are carried out manually. Alternatively, ‘single-box’ designs combine everything in one laser head and are controlled by a computer.
Objective lenses
The choice of objective lens is crucial to maximize the advantages of two-photon imaging. Most importantly, the NUMERICAL APERTURE (NA) should be as high as possible for two reasons: the amount of emitted fluorescence increases with the square of the NA; and the dimensions of the focal spot decrease (and so resolution increases) with increasing NA. Therefore, dry (air) objectives are unsuitable generally owing to their relatively low NA. Oil-immersion objectives have the highest available NA (up to 1.4), but they have marked drawbacks for imaging deep in live tissue — imaging must be done through a cover glass; the physical working distance of the lens is usually only ~60 μm; and spherical aberration occurs when imaging more than a few μm into aqueous specimens. Instead, water-immersion objectives provide the best compromise; they allow imaging to a depth of hundreds of μm with a NA as great as 1.2. Two basic designs of water-immersion objective are available: those that are corrected to image through a cover glass and ‘dipping’ objectives that are immersed directly in the bathing fluid. The former have a higher NA, but they generally have a working distance of only ~200 μm and they have the further disadvantage for imaging explanted tissues that the cover glass impairs superfusion with oxygenated solution. Dipping objectives have a lower NA (0.80–0.95), but they have the advantage of allowing free access to the specimen with working distances of a few mm. We have obtained the best results with the Olympus 20×, 0.95 NA dipping objective, which provides a unique combination of high NA, low magnification and long (2 mm) working distance.
The objective lenses that are used at present for two-photon imaging were designed originally for conventional (one-photon) applications, and it is probable that improvements will be possible by optimizing lens design for two-photon microscopy. For example, some constraints on lenses for one-photon microscopy (including corrections for chromatic aberration and flatness of field) are eased for two-photon microscopy, and new factors become important (such as good infrared transmission and low dispersion to minimize the broadening of femtosecond pulses).
Advantages of two-photon microscopy
The main advantage of two-photon microscopy is that excitation is confined exclusively to the focal plane, whereas other regions of the specimen are exposed only to the relatively harmless infrared light. Undesirable processes, such as photo-bleaching and photodamage, are not prevented entirely; however, unlike for confocal microscopy, these processes are restricted only to regions where imaging takes place. A secondary, but important, advantage arises from the long wavelengths that are used for excitation. The extent to which light is scattered varies as a steep, inverse power function of wavelength (which is why the sky is blue and sunsets through the Los Angeles smog appear so red). Infrared light penetrates most biological tissues with minimal scattering or absorption, which allows two-photon images to be obtained at much greater depths than is possible with confocal microscopy. As summarized in TABLE 1, the two-photon technique has several advantages over conventional confocal fluorescence imaging, including reduced photobleaching, minimal phototoxicity and the ability to resolve detail at greater depths in living tissues.
Table 1.
Comparison of three-dimensional imaging techniques
| Technique | Three-dimensional resolution | Acquisition speed | Photodamage, photobleaching | Tissue imaging depth |
|---|---|---|---|---|
| Deconvolution microscopy | Submicron resolution, but algorithms are computationally intensive | Depends on camera; fast acquisition, but slow deconvolution | High (depends on excitation wave-length). Whole sample exposed | Superficial (<40 μm), owing to light scattering |
| Confocal microscopy | Submicron resolution with pinhole aperture | Limited by scanhead design (typically two frames per second (2 fps), but 30 fps with resonant mirror or spinning disk) | High (depends on excitation wave-length). Whole sample exposed | Moderate (<80 μm), owing to light scattering |
| Two-photon microscopy | Submicron resolution, but slightly less than for confocal microscopy. Inherent optical sectioning with two-photon effect | Limited by scanhead design (typically 2 fps, but 30 fps with resonant mirror) | Low (long excitation wavelength). Excitation only in focal plane | Hundreds of microns (<500 μm, depends on tissue) |
Probes and applications
The development of fluorescent probes has kept pace with the development of new microscopy techniques to allow the visualization of molecules, cell-signalling events, subcellular organelles and whole cells. Some probes must be loaded into individual cells and have been used mainly for in vitro studies (for example, fura-2 and other Ca2+ indicators). However, CellTracker™ dyes are stable and can be used to track single cells or to follow rounds of cell proliferation by dilution. Genetically encoded indicators (GFP and its variants) can be driven by tissue-specific promoters — such as those for nuclear factor of activated T cells (NFAT), IL-2 and IL-4 — turned into FRET calcium indicators (cameleons and pericams) or used as timers and markers by spontaneous or induced photo-bleaching46. TABLE 2 summarizes several popular probes that have been used in two-photon microscopy. Most probes that are used for conventional fluorescence microscopy will work also for two-photon excitation, but it is difficult to predict how they will behave. Some useful general rules are given in BOX 2.
Table 2.
Common probes for two-photon microscopy and their applications
| Application | Probe | Ex. (nm) | Em. (nm) |
|---|---|---|---|
| Long-term tracking of living cells | CellTracker™ Green CMFDA (CFSE) | 780 | 516 |
| CellTracker™ Orange CMTMR | 820 | 566 | |
| SNARF-1 | 700–810 | 580/640 | |
| Intracellular calcium measurements | Indo-1 free | 700 | 490 |
| Indo-1 with Ca2+ | 700 | 405 | |
| Fura-2 free | 720 | 512 | |
| Fura-2 with Ca2+ | 700 | 505 | |
| Calcium Green-1 with Ca2+ | 725 | 531 | |
| Calcium Orange with Ca2+ | 800 | 576 | |
| Fluo-3 with Ca2+ | 800 | 526 | |
| Yellow cameleon | 780>820 | 535/480 | |
| Fluorescent protein conjugates | FITC | 780>820 | 519 |
| BODIPY | 920 | Variable | |
| TRITC | 800–840 | 572 | |
| Rhodamine B | 840 | 600 | |
| Texas red | 780 | 615 | |
| CY3 | 780 | 565 | |
| CY5 | 780<820 | 670 | |
| Cascade Blue® | 750 | 420 | |
| Genetically encoded protein tags | BFP | 780>820 | 448 |
| eCFP | 860–920 | 470–490 | |
| eGFP | 900–950 | 516 | |
| eYFP | 960 | 525 | |
| DsRed | 960–990 | 583 | |
| Visualization of organelles | Dil (plasma membrane) | 700 | 565 |
| Rhodamine 123 (mitochondria) | 780–860 | 550 | |
| DAPI (nucleus) | 700 | 455 | |
| Hoechst (nucleus) | 780>820 | 478 |
BFP, blue fluorescent protein; CFP, cyan fluorescent protein; GFP, green fluorescent protein; YFP, yellow fluorescent protein. Excitation wavelengths (Ex.) differ from those of conventional one-photon microscopy and are limited by the tuning range of the laser. Emission wavelengths (Em.) are the same for one- and two-photon excitation. See Further information websites for more information.
Box 2 | Guidelines for the use of fluorophores in two-photon microscopy
The optimal wavelength for two-photon excitation is usually shorter than twice the one-photon excitation maximum.
The two-photon excitation spectrum is usually broader than the corresponding one-photon spectrum. This can be advantageous, as it allows the efficient simultaneous excitation of two fluorophores (for example, CFSE and CMTMR) that have clearly distinct emission wavelengths. Furthermore, the wavelength of the femtosecond laser can be tuned to equalize the brightness of the two probes.
The emission spectrum is generally the same for one- and two-photon excitation.
Multi-dimensional imaging
A single, monochrome image of a microscope specimen provides information in only three dimensions (spatial dimensions x and y, and intensity). By contrast, the ability of two-photon microscopy to ‘see’ deep in tissues and to image for long periods of time with minimal damage or bleaching facilitates the acquisition of data sets that are comprised of as many as six dimensions (spatial dimensions x, y and z, time, intensity and wavelength of more than one probe). For example, a typical application would involve the acquisition of a series of colour images taken at increasing depths in the specimen to form a ‘z-stack’, with this process being repeated at fixed time intervals to form a time-lapse movie. Computer-operated microscopes allow the automated acquisition of multi-dimensional image sequences under the control of software packages such as MetaMorph® (Universal Imaging Corporation™), and these packages also include programs for reconstructing and analysing the data. Nevertheless, the large amount of data in multi-dimensional sets creates new technical challenges in terms of storage, management and analysis of the data, and their ultimate representation in two-dimensional print media. For example, a typical 30-minute recording of a three-colour data set (500 × 500 pixel resolution, 20 z-planes and 6 z-stacks per minute) fills >2 GB of disc space. These data sets are not readily portable and require, at the least, a fast personal computer with a large amount of memory and disc-storage capacity to carry out the image processing. Usually, it is possible to analyse only a few of the available dimensions at a given time, and even that might require creative solutions. For example, current computer programs do not allow simultaneous tracking in the dimensions of time, x, y and z, and they provide generally only a maximum-intensity projection in which the image is ‘flattened’ along a chosen dimension. This representation can be misleading. It is easy to confuse two cells superimposed in a maximum-intensity projection as a single cell, even though they might be separated by tens of microns on the z-axis. One solution is to form stereoscopic images, coded as a colour anaglyph that can be viewed through red/green glasses. A more promising approach will be to adapt the virtual-reality technology that is being used for computer video games to allow the rendering of full-colour multi-dimensional data sets. The presentation of such data on the printed page of a scientific publication can be an almost impossible challenge, but most journals now have extensive online sections that host compressed video files as supplementary material.
Imaging lymphocytes in intact tissues
Jenkins and colleagues27,47, among others, have pieced together the time course of the immune response in vivo using a TCR-transgenic mouse system and fluorescence microscopy of fixed tissue sections. However, only recently have attempts been made to image these processes in real time, in intact lymphoid tissues, by two-photon microscopy48–50 and by conventional confocal methodology51. We have established a two-photon imaging method that allows six-dimensional (x, y, z, time, intensity and emission wavelength) imaging of viable T cells, B cells and APCs up to a depth of ~300 μm in intact lymphoid organs (FIG. 4). We used an adoptive-transfer approach to observe the behaviour of individual fluorescently labelled lymphocytes in the peripheral lymph nodes48 and spleen50, whereas Bousso et al.49 studied T-cell positive selection in a reconstituted thymocyte co-culture system. These three reports share the observation that in physiologically relevant three-dimensional substrates, naive T cells are highly motile and have dynamic and heterogeneous responses to antigen. We measured naive T-cell velocities that often exceeded 25 μm min−1, with mean values of 10.2 μm min−1 for BALB/c T cells and 12.2 μm min−1 for ovalbumin-specific TCR-transgenic T cells from DO11.10 mice48. These rapid movements were unexpected, because the lymphoid organs are packed densely with cells and the measured peak velocities are higher than those observed previously in vitro. T cells in lymph nodes or in the re-aggregated thymic culture moved with a ‘stop-and-go’ behaviour and scattered through their environment in a manner that was reminiscent of ants moving in response to disturbance of their anthill. In the x and y dimensions, parallel to the overlying capsule, T cells moved randomly without following any particular paths, although interactions with reticular fibres and other cells were seen clearly. Often, T-cell motion perpendicular to the capsule (z-axis) occurred preferentially at specific locations, through conduits or ‘wormholes’ for transport up and down. By contrast, B cells moved more slowly and followed more circuitous paths in the follicles. These respective behaviours might constitute the default antigen-search programme.
Figure 4. Multi-dimensional two-photon microscopy: tissue imaging and tracking cell proliferation.
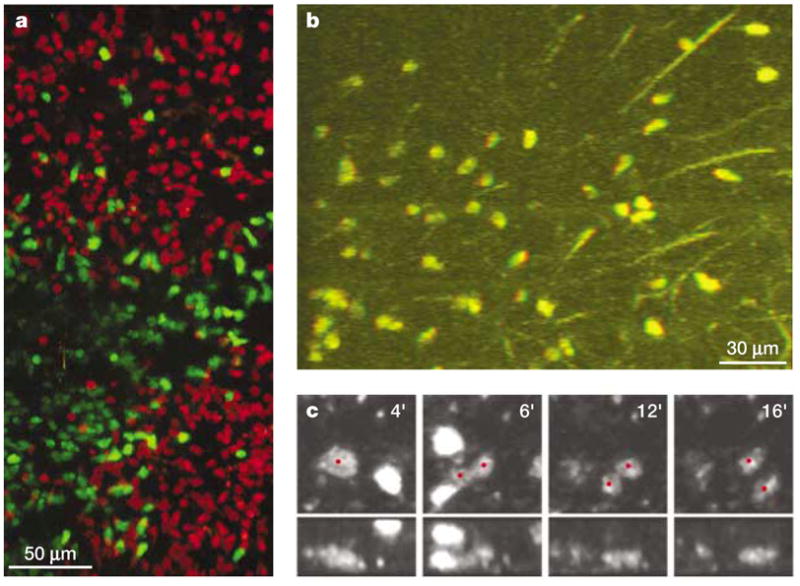
Multi-dimensional imaging data can provide crucial information for understanding the complex behaviour of cells in tissue environments, but they create challenges for analysis and representation in print media. a | Maximum-intensity projection of a 200-μm deep section of an intact lymph node, showing two follicles and an interfollicular space, which contain B cells (red) and T cells (green), respectively. b | A single frame taken from a stereo movie of T cells crawling in a meshwork of reticular fibres (see http://crt.biomol.uci.edu/TCellMovie/stereofiber.mov) that conveys depth information when viewed with red/green stereo glasses. 51-μm thick section. c | An in vivo proliferation event inside the lymph node is verified by inspecting the time course of the event (left to right, in minutes) in two different reconstructions (top and bottom, 0° and 90° rotations of the z-axis). At 4 minutes, the red dot indicates a rounded, enlarged cell just before division. At 5–6 minutes, a furrow develops in the middle of the non-motile dividing cell, followed by complete separation into half-sized cells and full development of motility by 12–16 minutes (see http://crt.biomol.uci.edu/TCellMovie/dividing.mov).
When antigen is introduced, the situation becomes more complex. In intact lymph nodes from antigen-challenged animals and in the thymocyte culture system, both stable clusters and transient interactions of cells were noted48,49. We showed also that after antigen-stimulated cell division, as identified by CFSE dilution by day five, most antigen-specific T cells regained a naive T-cell morphology and pattern of motility48.
In contrast to the two-photon studies described, a similar study using standard confocal methodology concluded that naive T cells in the absence of antigen are non-motile51, and observed some cells moving at a speed of 5–7 μm min−1 in antigen-primed experiments at time points of >37 hours. Furthermore, confocal microscopy showed only stable interactions between T cells and APCs lasting for more than 15 hours. There are several technical differences between these studies that are relevant to understanding these apparently inconsistent observations. First, the issue of physiological oxygen tension in the tissue has been raised34,51, as recent reports have indicated that the lymphoid environment might operate at a relatively low oxygen tension52. Stoll et al.51 superfused their lymph nodes with a 20% oxygen solution, whereas we used a solution saturated with 95% oxygen48, which is standard practice in brain-slice studies, according to the rationale that this would maximize the delivery of oxygen across the fibrous capsule and deep into metabolically active T-cell areas. Another important difference is that Stoll et al.51 used isolated splenic DCs that were primed in vitro and then transferred subcutaneously, whereas we used in situ-primed endogenous DCs. Perhaps, the differences that were observed in terms of the response to antigen are a consequence of the distinct APCs that were used, an idea that is worth testing in future experiments. Alternatively, the discrepancies in cell behaviour might be related to issues associated with the use of single-photon excitation compared with two-photon excitation. Single-photon confocal imaging is restricted to a depth of less than 80 μm below the capsule, whereas two-photon methods allow imaging at a depth of several hundred μm, deep into the T-cell areas of the lymph node. It is possible that T-cell responses to antigen might vary depending on the location at which the T cells encounter antigen in the lymph node. Furthermore, two-photon excitation is less phototoxic, and perhaps the more dynamic cell behaviours observed by this method reflect a level of cell viability that cannot be obtained within the technical limitations of single-photon confocal microscopy.
Pressing questions
Clearly, cell motility and appropriate trafficking are necessary at several stages of the immune response, but many questions remain concerning the dynamics of cell movements and interactions in vivo. For example, there are two models of primary T-cell activation at present that involve either a stable immunological synapse or serial encounters between T cells and APCs. Additional experiments to image APCs and T cells simultaneously and a detailed analysis of the results will be required to characterize the nature of the T-cell–APC antigen-recognition event in vivo. In addition, intravital two-photon microscopy might be the method of choice to investigate the role of chemokines, the extracellular matrix and cell–cell interactions during lymphocyte homing to and trafficking in lymphoid tissues. Furthermore, intravital approaches might be necessary to understand fully the complex environmental interactions that take place in lymphoid tissues during lymphocyte homing, trafficking and antigen recognition in vivo. Intravital techniques have been used for many years to study the homing of lymphocytes and leukocytes34,53,54. Recently, in our laboratory, we have applied two-photon imaging to an intravital preparation of an inguinal lymph node to analyse the homing and migration of T cells in living mice (M.D.C. et al., unpublished observations). Two-photon technology should be applicable to the study of a wide variety of cellular responses in their natural environment.
Conclusions
The ability to see, with one’s own eyes, cells functioning in their native tissues can influence our thinking about cellular processes profoundly and stimulate new questions. It is ironic that despite the wealth of knowledge of the molecular and physiological aspects of the immune system, we do not have a detailed picture of how cells coordinate the initiation or suppression of an immune response. Now, time-lapse video sequences are providing a fresh look at the behaviour of lymphocytes and APCs in living lymphoid organs and have revealed unexpected and exciting findings already. Two-photon microscopy is an elegant solution to elucidate the choreography between T cells, B cells and DCs in these tissues, and it is suited uniquely to reconcile the results obtained using in vitro and in vivo methodologies.
Acknowledgments
We thank J. Cyster and T. Okada for providing the immunohistochemistry picture in FIG. 1b, and S. Schoenberger and M. van Stipdonk for their permission to show FIG. 4b. M.D.C. and I.P. are supported by grants from the National Institutes of Health.
- FLUORESCENCE RESONANCE ENERGY TRANSFER (FRET)
This is used to measure protein–protein interactions microscopically or by a FACS-based method. Proteins fused to cyan, yellow or red fluorescent proteins are expressed and their interactions assessed by measuring the energy transfer between fluors, which can occur only if the proteins interact physically
- BRIGHT-FIELD MICROSCOPY
The original, and most commonly used, form of microscopy, in which the specimen is viewed by transmitted light from a condenser lens
- NOMARSKI OPTICS
One of several implementations of differential interference contrast (DIC) techniques for use with bright-field microscopy. DIC converts optical gradients in a specimen (differences in thickness, slope or refractive index) to differences in intensity, which gives a pseudo three-dimensional appearance to objects that would otherwise appear featureless
- FLUORESCENCE MICROSCOPY
The visualization of fluorescence in a specimen — either natural autofluorescence or after staining with fluorescent probes. Usually, this is achieved using an epi-fluorescence microscope, in which the excitation light is directed by a dichroic mirror through the objective lens, and fluorescence emission is viewed through the same lens after blocking excitation light with a barrier filter
- FURA-2 RATIOMETRIC IMAGING
Calcium imaging using the calcium-sensitive dye fura-2. When excited at short ultraviolet wavelengths (~350 nm), the fluorescence of fura-2 increases with increasing calcium concentration, whereas fluorescence decreases with increasing calcium concentration at longer wavelengths (~380 nm). By forming a ratio of successive images obtained at each wavelength, it is possible to obtain a measure of absolute free calcium concentration
- DECONVOLUTION MICROSCOPY
The use of software to correct partially for diffraction-limited blurring by a microscope and thereby enhance the resolution of three-dimensional images obtained by wide-field or confocal microscopy
- CONFOCAL MICROSCOPY
A form of fluorescence (or reflected light) microscopy in which out-of-focus signals are rejected by measuring through an aperture that restricts all light except that originating from the focused excitation spot
- LIPID-BILAYER IMAGING
This involves phospholipid with protein constituents deposited in a bilayer on a glass surface; it is used to study cell–cell interactions such as the immunological synapse
- COLLAGEN GEL MATRIX
A culture system consisting of a three-dimensional collagen environment that mimics a physiological substrate for the study of immune-cell motility and function
- CFSE LABELLING
CFSE (5,6-carboxyfluorescein diacetate succinimidyl ester) is a green-fluorescent dye that binds covalently to intracellular proteins, providing a stable cell label. When a cell divides, the dye is diluted equally in the daughter cells and fluorescence is halved with each generation, which allows up to 7–10 cell divisions to be detected by flow cytometry
- NUMERICAL APERTURE
(NA). The light-gathering ability of a microscope objective. The NA is equal to the refractive index of the medium multiplied by the sine of half the angular aperture
References
- 1.Donnadieu E, Bismuth G, Trautmann A. Antigen recognition by helper T cells elicits a sequence of distinct changes of their shape and intracellular calcium. Curr Biol. 1994;4:584–595. doi: 10.1016/s0960-9822(00)00130-5. [DOI] [PubMed] [Google Scholar]
- 2.Negulescu PA, Shastri N, Cahalan MD. Intracellular calcium dependence of gene expression in single T lymphocytes. Proc Natl Acad Sci USA. 1994;91:2873–2877. doi: 10.1073/pnas.91.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fanger CM, Hoth M, Crabtree GR, Lewis RS. Characterization of T-cell mutants with defects in capacitative calcium entry: genetic evidence for the physiological roles of CRAC channels. J Cell Biol. 1995;131:655–667. doi: 10.1083/jcb.131.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Negulescu PA, Krasieva TB, Khan A, Kerschbaum HH, Cahalan MD. Polarity of T-cell shape, motility and sensitivity to antigen. Immunity. 1996;4:421–430. doi: 10.1016/s1074-7613(00)80409-4. This study describes the T cell as a motile, polarized antigen sensor, for which Ca2+ functions as a stop signal. [DOI] [PubMed] [Google Scholar]
- 5.Delon J, Bercovici N, Raposo G, Liblau R, Trautmann A. Antigen-dependent and -independent Ca2+ responses triggered in T cells by dendritic cells compared with B cells. J Exp Med. 1998;188:1473–1484. doi: 10.1084/jem.188.8.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 7.Wei X, Tromberg BJ, Cahalan MD. Mapping the sensitivity of T cells with an optical trap: polarity and minimal number of receptors for Ca2+ signaling. Proc Natl Acad Sci USA. 1999;96:8471–8476. doi: 10.1073/pnas.96.15.8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dustin ML, Bromley SK, Kan Z, Peterson DA, Unanue ER. Antigen-receptor engagement delivers a stop signal to migrating T lymphocytes. Proc Natl Acad Sci USA. 1997;94:3909–3913. doi: 10.1073/pnas.94.8.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Merwe PA, Davis SJ, Shaw AS, Dustin ML. Cytoskeletal polarization and redistribution of cell-surface molecules during T-cell antigen recognition. Semin Immunol. 2000;12:5–21. doi: 10.1006/smim.2000.0203. [DOI] [PubMed] [Google Scholar]
- 10.Bromley SK, et al. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 11.Kupfer A, Singer SJ, Janeway CA, Jr, Swain SL. Coclustering of CD4 (L3T4) molecule with the T-cell receptor is induced by specific direct interaction of helper T cells and antigen-presenting cells. Proc Natl Acad Sci USA. 1987;84:5888–5892. doi: 10.1073/pnas.84.16.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McConnell HM, Watts TH, Weis RM, Brian AA. Supported planar membranes in studies of cell–cell recognition in the immune system. Biochim Biophys Acta. 1986;864:95–106. doi: 10.1016/0304-4157(86)90016-x. [DOI] [PubMed] [Google Scholar]
- 13.Watts TH, Gaub HE, McConnell HM. T-cell-mediated association of peptide antigen and major histocompatibility complex protein detected by energy transfer in an evanescent wave-field. Nature. 1986;320:179–181. doi: 10.1038/320179a0. [DOI] [PubMed] [Google Scholar]
- 14.Dustin ML, Springer TA. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature. 1989;341:619–624. doi: 10.1038/341619a0. [DOI] [PubMed] [Google Scholar]
- 15.Dustin ML, Springer TA. Role of lymphocyte adhesion receptors in transient interactions and cell locomotion. Annu Rev Immunol. 1991;9:27–66. doi: 10.1146/annurev.iy.09.040191.000331. [DOI] [PubMed] [Google Scholar]
- 16.Grakoui A, et al. The immunological synapse: a molecular machine controlling T-cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 17.Zal T, Zal MA, Gascoigne NR. Inhibition of T-cell-receptor–coreceptor interactions by antagonist ligands visualized by live FRET imaging of the T-hybridoma immunological synapse. Immunity. 2002;16:521–534. doi: 10.1016/s1074-7613(02)00301-1. [DOI] [PubMed] [Google Scholar]
- 18.Dustin ML, de Fougerolles AR. Reprogramming T cells: the role of extracellular matrix in coordination of T-cell activation and migration. Curr Opin Immunol. 2001;13:286–290. doi: 10.1016/s0952-7915(00)00217-x. [DOI] [PubMed] [Google Scholar]
- 19.Lanzavecchia A, Sallusto F. The instructive role of dendritic cells on T-cell responses: lineages, plasticity and kinetics. Curr Opin Immunol. 2001;13:291–298. doi: 10.1016/s0952-7915(00)00218-1. This paper questions the requirement for stable interactions between T cells and antigen-presenting cells. [DOI] [PubMed] [Google Scholar]
- 20.Gunzer M, et al. Antigen presentation in extracellular matrix: interactions of T cells with dendritic cells are dynamic, short lived and sequential. Immunity. 2000;13:323–332. doi: 10.1016/s1074-7613(00)00032-7. [DOI] [PubMed] [Google Scholar]
- 21.Dustin ML, Allen PM, Shaw AS. Environmental control of immunological synapse formation and duration. Trends Immunol. 2001;22:192–194. doi: 10.1016/s1471-4906(01)01872-5. This article reviews the immunological synapse as a stable organizing element and proposes that the environment might influence the stability of the synapse. [DOI] [PubMed] [Google Scholar]
- 22.Friedl P, Gunzer M. Interaction of T cells with APCs: the serial encounter model. Trends Immunol. 2001;22:187–191. doi: 10.1016/s1471-4906(01)01869-5. [DOI] [PubMed] [Google Scholar]
- 23.Jung T, Schauer U, Heusser C, Neumann C, Rieger C. Detection of intracellular cytokines by flow cytometry. J Immunol Methods. 1993;159:197–207. doi: 10.1016/0022-1759(93)90158-4. [DOI] [PubMed] [Google Scholar]
- 24.Prussin C, Metcalfe DD. Detection of intracytoplasmic cytokine using flow cytometry and directly conjugated anti-cytokine antibodies. J Immunol Methods. 1995;188:117–128. doi: 10.1016/0022-1759(95)00209-x. [DOI] [PubMed] [Google Scholar]
- 25.Altman JD, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 26.Lyons AB, Hasbold J, Hodgkin PD. Flow cytometric analysis of cell-division history using dilution of carboxyfluorescein diacetate succinimidyl ester, a stably integrated fluorescent probe. Methods Cell Biol. 2001;63:375–398. doi: 10.1016/s0091-679x(01)63021-8. [DOI] [PubMed] [Google Scholar]
- 27.Ingulli E, Mondino A, Khoruts A, Jenkins MK. In vivo detection of dendritic-cell antigen presentation to CD4+ T cells. J Exp Med. 1997;185:2133–2141. doi: 10.1084/jem.185.12.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okada T, et al. Chemokine requirements for B-cell entry to lymph nodes and Peyer’s patches. J Exp Med. 2002;196:65–75. doi: 10.1084/jem.20020201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cyster JG. Chemokines and cell migration in secondary lymphoid organs. Science. 1999;286:2098–2102. doi: 10.1126/science.286.5447.2098. [DOI] [PubMed] [Google Scholar]
- 30.Kawakami N, et al. Green fluorescent protein-transgenic mice: immune functions and their application to studies of lymphocyte development. Immunol Lett. 1999;70:165–171. doi: 10.1016/s0165-2478(99)00152-2. [DOI] [PubMed] [Google Scholar]
- 31.Schleicher U, Rollinghoff M, Gessner A. A stable marker for specific T cells: a TCRα/green fluorescent protein (GFP) fusion protein reconstitutes a functionally active TCR complex. J Immunol Methods. 2000;246:165–174. doi: 10.1016/s0022-1759(00)00298-2. [DOI] [PubMed] [Google Scholar]
- 32.Naramura M, Hu RJ, Gu H. Mice with a fluorescent marker for interleukin-2 gene activation. Immunity. 1998;9:209–216. doi: 10.1016/s1074-7613(00)80603-2. [DOI] [PubMed] [Google Scholar]
- 33.Hu-Li J, et al. Regulation of expression of IL-4 alleles: analysis using a chimeric GFP/IL-4 gene. Immunity. 2001;14:1–11. doi: 10.1016/s1074-7613(01)00084-x. [DOI] [PubMed] [Google Scholar]
- 34.von Andrian UH. Immunology. T-cell activation in six dimensions. Science. 2002;296:1815–1817. doi: 10.1126/science.296.5574.1815. [DOI] [PubMed] [Google Scholar]
- 35.Becker MD, et al. Intraocular in vivo imaging of activated T lymphocytes expressing green-fluorescent protein after stimulation with endotoxin. Graefes Arch Clin Exp Ophthalmol. 2001;239:609–612. doi: 10.1007/s004170100320. [DOI] [PubMed] [Google Scholar]
- 36.Kedl RM, et al. T cells compete for access to antigen-bearing antigen-presenting cells. J Exp Med. 2000;192:1105–1113. doi: 10.1084/jem.192.8.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gretz JE, Anderson AO, Shaw S. Cords, channels, corridors and conduits: critical architectural elements facilitating cell interactions in the lymph-node cortex. Immunol Rev. 1997;156:11–24. doi: 10.1111/j.1600-065x.1997.tb00955.x. [DOI] [PubMed] [Google Scholar]
- 38.Gretz JE, Norbury CC, Anderson AO, Proudfoot AE, Shaw S. Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J Exp Med. 2000;192:1425–1440. doi: 10.1084/jem.192.10.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wulfing C, Sjaastad MD, Davis MM. Visualizing the dynamics of T-cell activation: intracellular adhesion molecule 1 migrates rapidly to the T-cell/B-cell interface and acts to sustain calcium levels. Proc Natl Acad Sci USA. 1998;95:6302–6307. doi: 10.1073/pnas.95.11.6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaefer BC, et al. Live cell fluorescence imaging of T-cell MEKK2: redistribution and activation in response to antigen stimulation of the T-cell receptor. Immunity. 1999;11:411–421. doi: 10.1016/s1074-7613(00)80116-8. [DOI] [PubMed] [Google Scholar]
- 41.Krummel MF, Sjaastad MD, Wulfing C, Davis MM. Differential clustering of CD4 and CD3ζ during T-cell recognition. Science. 2000;289:1349–1352. doi: 10.1126/science.289.5483.1349. [DOI] [PubMed] [Google Scholar]
- 42.Reichert P, Reinhardt RL, Ingulli E, Jenkins MK. Cutting edge: in vivo identification of TCR redistribution and polarized IL-2 production by naive CD4 T cells. J Immunol. 2001;166:4278–4281. doi: 10.4049/jimmunol.166.7.4278. [DOI] [PubMed] [Google Scholar]
- 43.Ojcius DM, Niedergang F, Subtil A, Hellio R, Dautry-Varsat A. Immunology and the confocal microscope. Res Immunol. 1996;147:175–188. doi: 10.1016/0923-2494(96)83169-5. [DOI] [PubMed] [Google Scholar]
- 44.Mayer MG. Elementary processes with two-quantum transitions. Ann d Physik. 1931;9:273–294. [Google Scholar]
- 45.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 46.Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- 47.Jenkins MK, et al. In vivo activation of antigen-specific CD4 T cells. Annu Rev Immunol. 2001;19:23–45. doi: 10.1146/annurev.immunol.19.1.23. [DOI] [PubMed] [Google Scholar]
- 48.Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296:1869–1873. doi: 10.1126/science.1070051. The first use of two-photon microscopy to observe the dynamics of T- and B-cell motility in an intact lymphoid organ; T cells were observed with and without antigen challenge. [DOI] [PubMed] [Google Scholar]
- 49.Bousso P, Bhakta NR, Lewis RS, Robey E. Dynamics of thymocyte–stromal cell interactions visualized by two-photon microscopy. Science. 2002;296:1876–1880. doi: 10.1126/science.1070945. Two-photon microscopy was used to image interactions between thymocytes and stromal cells in a reaggregated thymic organ culture system during positive selection. [DOI] [PubMed] [Google Scholar]
- 50.Wei SH, Miller MJ, Cahalan MD, Parker I. Two-photon imaging in intact lymphoid tissue. Adv Exp Med Biol. doi: 10.1007/978-1-4615-0757-4_26. (in the press) [DOI] [PubMed] [Google Scholar]
- 51.Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of T-cell–dendritic cell interactions in lymph nodes. Science. 2002;296:1873–1876. doi: 10.1126/science.1071065. Confocal imaging of T cells interacting with dendritic cells in intact lymph nodes. [DOI] [PubMed] [Google Scholar]
- 52.Caldwell CC, et al. Differential effects of physiologically relevant hypoxic conditions on T-lymphocyte development and effector functions. J Immunol. 2001;167:6140–6149. doi: 10.4049/jimmunol.167.11.6140. [DOI] [PubMed] [Google Scholar]
- 53.Ley K, Pries AR, Gaehtgens P. A versatile intravital microscope design. Int J Microcirc Clin Exp. 1987;6:161–167. [PubMed] [Google Scholar]
- 54.Dunne JL, Ballantyne CM, Beaudet AL, Ley K. Control of leukocyte rolling velocity in TNF-α-induced inflammation by LFA-1 and Mac-1. Blood. 2002;99:336–341. doi: 10.1182/blood.v99.1.336. [DOI] [PubMed] [Google Scholar]