

Abstract
Organisms have the property to adapt to a changing environment and keep certain components within a cell regulated at the same level (homeostasis). “Perfect adaptation” describes an organism's response to an external stepwise perturbation by regulating some of its variables/components precisely to their original preperturbation values. Numerous examples of perfect adaptation/homeostasis have been found, as for example, in bacterial chemotaxis, photoreceptor responses, MAP kinase activities, or in metal-ion homeostasis. Two concepts have evolved to explain how perfect adaptation may be understood: In one approach (robust perfect adaptation), the adaptation is a network property, which is mostly, but not entirely, independent of rate constant values; in the other approach (nonrobust perfect adaptation), a fine-tuning of rate constant values is needed. Here we identify two classes of robust molecular homeostatic mechanisms, which compensate for environmental variations in a controlled variable's inflow or outflow fluxes, and allow for the presence of robust temperature compensation. These two classes of homeostatic mechanisms arise due to the fact that concentrations must have positive values. We show that the concept of integral control (or integral feedback), which leads to robust homeostasis, is associated with a control species that has to work under zero-order flux conditions and does not necessarily require the presence of a physico-chemical feedback structure. There are interesting links between the two identified classes of homeostatic mechanisms and molecular mechanisms found in mammalian iron and calcium homeostasis, indicating that homeostatic mechanisms may underlie similar molecular control structures.
Introduction
Many physiologically important compounds are under tight homeostatic regulation, where internal concentrations are adapted (1) at certain levels, despite environmental disturbances. Two concepts have developed to understand homeostasis: one is related to the intrinsic properties of the network showing that the adaptation response is independent of (most but not all) rate constant values (referred to here as robust (2–4) adaptation/homeostasis), whereas the other concept looks at the homeostasis due to a fine-tuning between rate constants. Perfect adaptation describes an organism's response to an external stepwise perturbation by regulating some of its variables/components precisely to their original preperturbation values. Perfect adaptation has been found, for example, in bacterial chemotaxis (5–8), photoreceptor responses (9), and MAP-kinase regulation (10–12). In this respect, perfect adaptation and homeostasis are closely related and in the following, we look at homeostasis as a perfectly adapted process.
Robust perfect adaptation/homeostasis of a perturbed system can be related to the concept of integral control (13) or integral feedback (14). In this type of control mechanism, the error between the value of the system output (controlled variable, CV) and its setpoint is integrated, and the integral value is fed to the input of the process (the so-called manipulated variable, MV), which results in a robust adaptation of the system output to the setpoint (Fig. 1). Recently, El-Samad et al. (15) have shown that calcium homeostasis under hypocalcemia conditions can be described on the basis of an integral feedback approach, where the error between the calcium setpoint and the actual calcium level is related to the activity of the parathyroid hormone (PTH), an important hormone in calcium regulation.
Figure 1.
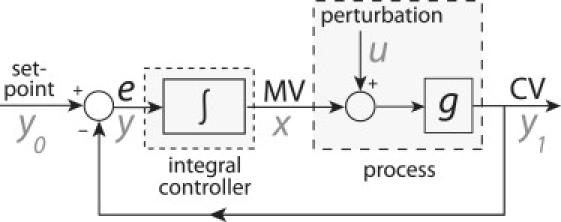
Scheme of integral control/feedback of a perturbed system, where the system output is perfectly adapted to the setpoint (i.e., the error e is robustly controlled to zero). MV and CV are the manipulated and controlled variables, respectively. Symbols in gray denote the notation for integral feedback by Yi et al. (14).
However, the molecular mechanisms behind error-sensing processes are little understood. To investigate the relationship between the integral control/feedback concept and its reaction kinetic realization, we provide here a kinetic analysis. We show that robust perfect adaptation (homeostasis) is associated with a control species working under zero-order flux conditions while acting on another control species in the way of a “control of the controller”. There is an interesting and close analogy between the mechanisms shown here and mechanisms found in mammalian iron and calcium homeostasis, indicating that other homeostatic mechanisms may underlie similar control structures.
Computational methods
Rate equations were solved numerically by using the FORTRAN subroutine LSODE (Livermore Solver of Ordinary Differential Equations) (16) and MATLAB (www.mathworks.com). To make notations simpler, concentrations are denoted by their names without square brackets.
Results
Molecular representation of integral control
Fig. 2 a shows a simple scheme where a homeostatic-regulated intermediate A is being synthesized, transformed, and degraded. Rate constant kpert indicates an environmental perturbation, such as a sudden increase in A. To avoid possible cell damage by excess of A, A has to be homeostatically regulated. A way to achieve this is to use an A-induced enzyme, Eadapt, which clears the cell for excess A. To make the homeostasis perfect, i.e., A adapts always to the same Aset value, the rate in the formation of Eadapt has to be proportional to the difference (i.e., the error) between A and its setpoint, Aset, as indicated by the following equations and shown in Fig. 2, a and b:
| (1) |
| (2) |
Figure 2.
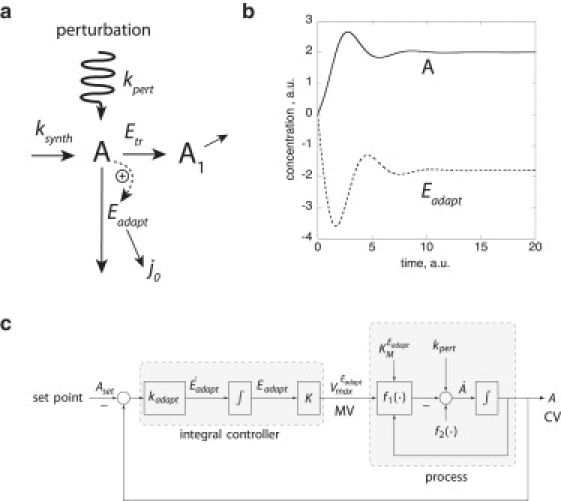
(a) Reaction scheme of system with rate Eqs. 1 and 2. Species A is formed by a zero-order process with rate constant ksynth and then transformed to the product A1. Rate constant kpert is related to a perturbing process (wavy line), which increases the level of A. To remove excess A, A is forming enzyme species Eadapt, which removes A with the flux (indicated by the vertical arrow). To get robust adaptation in A independent of kpert, Eadapt is removed through a zero-order flux j0 = kadaptAset. (b) Calculation showing that negative Eadapt concentrations may arise when Aset is regarded as a fixed setpoint. Initial concentrations of A and Eadapt are zero; kadapt = 5, kpert = ksynth = 0.5, , = 1, = 110, = 100, and Aset = 2. Concentration and timescales are in arbitrary units (a.u.). (c) Scheme of the adaptive process shown in panel a containing the setpoint Aset, the integral controller and the process units. The controlled variable (CV) is A. Eqs. 1 and 2 are written as dA/dt = f2(·) – f1(·) + kpert, dEadapt/dt = kadapt(A – Aset), respectively, with , , and = K · Eadapt. K is the turnover number for Eadapt, i.e., . MV: manipulated variable.
However, writing the rate of formation of Eadapt in proportion to the error (A – Aset) (Eqs. 1 and 2, and Fig. 2 c) does still lack a molecular understanding of how the setpoint Aset is determined. In addition, treating the setpoint Aset as a fixed parameter can lead to the problem that, for certain parameter values, concentrations in Eadapt may become negative (Fig. 2 b).
To avoid negative concentrations, the zero-order term in Eq. 2, j0 = kadaptAset, has to be replaced in a kinetically plausible way. A possibility is the removal of Eadapt by an additional controller/enzymatic species (Eset) working at zero-order conditions. In this case, the set-value Aset is then determined by Eset's maximum velocity, , divided by the A-induced influx rate, which generates Eadapt (Eq. 3). Fig. 3 shows two representations of this mechanism with robust perfect adaptation/homeostasis in A avoiding any negative concentrations. In Fig. 3 a, a fully expanded Michaelis-Menten mechanism is shown, whereas in Fig. 3 b the mechanism is formulated in terms of steady-state or rapid equilibrium assumptions for the individual enzymatic steps. The kinetic equations with rate constants are given in the Appendix. For both cases, the setpoint in A is given by
| (3) |
where is the total concentration of enzyme Eset. Keeping Aset fixed, the mechanism shows robust homeostasis in A even when rate constants of the three enzymatic pathways (Fig. 3 a) are varied by over six orders of magnitude! Fig. 4 shows the A-homeostasis for the scheme shown in Fig. 3 a, using several perturbing and initial conditions (for details, see Appendix and Fig. 3 legend). Fig. 4 a shows the homeostasis in A when kpert is increased from 0.0 to 1.0 a.u. In Fig. 4 b, a large positive excursion in A is observed when kpert is increased from 1.0 to 1 × 103 a.u., which is accompanied by an increased relaxation time in A for reaching Aset. Negative excursions in A are observed when kpert is decreased. This is illustrated in Fig. 4 c when kpert is decreased from 1.0 to 1 × 10−3 a.u.
Figure 3.
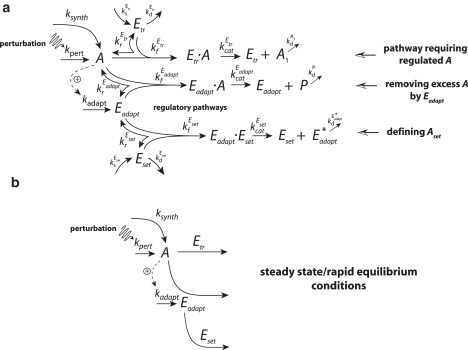
To avoid negative concentrations in Eadapt, j0 (Fig. 2a) is represented as an enzymatic zero-order process. (a) Fully expanded Michaelis-Menten mechanism. The rate equations together with rate constants are shown in the Appendix. To obtain robust homeostasis in A, Eset removes active Eadapt into an inactive form Eadapt∗ under zero-order conditions with Aset given by Eq. 3. (b) Same mechanism as in panel a, but formulating the Michaelis-Menten mechanism under steady-state/rapid equilibrium conditions. Rate equations are given in the Appendix. A zip-archive containing MATLAB and Berkeley Madonna versions of the model shown in Fig. 3 A with instructions and annotation available in the Supporting Material.
Figure 4.
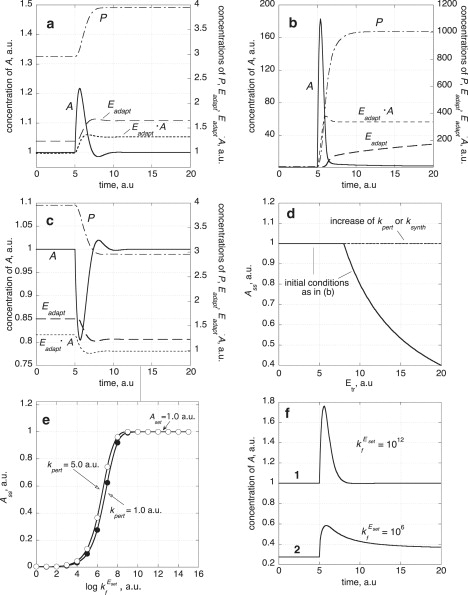
Robust perfect adaptation in A with Aset = 1.0. (a) Model described in Fig. 3a with rate constants as given in the Appendix. At t = 5.0 time units, kpert is increased from 0.0 to 1.0. (b) Initial conditions as given in the Appendix with kpert = 1.0. At t = 5.0 time units, kpert is increased from 1.0 to 103 a.u. (c) Initial conditions as in panel b. At t = 5.0 time units, kpert is decreased from 1.0 to 10−3 a.u. (d) Same initial conditions as in panel b, but Etr is successively increased leading eventually to the breakdown in homeostasis indicated by the decreasing Ass values. This breakdown can be opposed to a certain degree by increasing the values of kpert or ksynth. In the figure, kpert or ksynth were increased from their original values 1.0 and 3.0 to 10.0 and 12.0, respectively, thereby extending the homeostasis to larger Etr values (dashed line). However, at higher Etr concentrations the homeostasis fails again with decreasing Ass values (data not shown). (e) Calculated Ass values for varying with ksynth = 3.0 a.u. and kpert = 1.0 a.u. (solid circles), or with ksynth = 3.0 a.u. and kpert = 5.0 a.u. (open circles). For < 109 a.u., perfect homeostasis in A is lost (indicated by the condition that Ass < Aset), because for decreasing the associated with the removal of Eadapt by Eset increases, which eventually leads to the loss of the zero-order kinetics in the Eadapt degradation. (f) Time profiles in A with two different values. At t = 5.0 time units, kpert is increased from 1.0 to 5.0 a.u. 1 = Perfect homeostasis in A for = 1012 a.u.; 2 = Loss of perfect homeostasis in A when = 106 a.u., which is due to the loss of zero-order kinetics in the degradation of Eadapt.
However, due to the introduction of enzymatic zero-order kinetics (for avoiding negative concentrations in Eadapt), both mechanisms in Fig. 3 show homeostasis only for perturbations, which result in increased or moderate decreased levels in A. When a perturbation removes A too quickly, then the homeostasis in A breaks down. We therefore call this type of homeostatic control for inflow-homeostatic control. In Fig. 4 d such a breakdown in A-homeostasis is illustrated by the A steady-state level (Ass) in relation to the (total) concentration of the A-removing enzyme Etr. When the removal rate in A becomes greater than the total production rate (ksynth + kpert), Ass decreases below Aset and homeostasis in A is lost. This type of homeostatic failure can be avoided by using controllers, which specifically address the removal of A (outflow-homeostasis). A mechanism for calcium homeostasis under outflow conditions (hypocalcemia) was recently suggested by El-Samad et al. (15), but in this mechanism, the problem of zero-order fluxes and their association with negative concentrations was not addressed. Specific examples of other inflow and outflow homeostatic mechanisms are discussed below. Fig. 4 e illustrates the breakdown in A-homeostasis when the kinetics in the removal of Eadapt by Eset is no longer zero-order. For sufficiently large values, the becomes much lower than Eadapt, ensuring zero-order kinetics in the removal of Eadapt and leading to Ass values which are equal to Aset. For lower values, the increases and the zero-order kinetics in the removal of Eadapt are eventually lost leading to Ass values lower than Aset and to the loss in the homeostasis of A. As shown in Fig. 4 e, the Ass values under non-zero-order conditions also depend on kpert. In Fig. 4 f, two A-time profiles are shown for two perturbations, one applied for zero-order conditions (, upper curve), and the other for non-zero-order conditions (, lower curve). In both cases, kpert is increased from 1.0 to 5.0 a.u. at t = 5.0 a.u. Clearly, zero-order kinetics in the removal of Eadapt is required to ensure robust homeostasis in A.
Robust perfect temperature compensation
Temperature is an important environmental parameter, which influences each reaction step in a reaction kinetic network. Van 't Hoff's rule states that the velocity of a chemical or biochemical process increases generally by a factor between 2 and 3 (the so-called Q10) when the temperature is increased by 10°C. A Q10 of 2 corresponds to an activation energy of ∼50 kJ/mol (17). In general, the concentration of a chemical component, a flux within a kinetic network, or the period length of an oscillatory network, can show temperature compensation/adaptation near a given reference temperature, Tref, when the following balancing equation, here written for the concentration in A, is satisfied (18–21):
| (4) |
Here is the control coefficient (22,23) describing how sensitive concentration A is with respect to variations to the network's rate constants ki. The values R, T, and Ei describe the gas constant, the temperature (in Kelvin), and activation energy (in J/mol) of the process indexed by i, respectively. The balancing equation (Eq. 4) requires a fine-tuning between the control coefficients and activation energies. In general, the resulting temperature compensation in A is not robust, i.e., temperature compensation is only observed within a local region around Tref (see, for example, (24)). Considering the network in Fig. 3, we have 21 rate constants with associated activation energies, and in general, temperature compensation in A is given by Eq. 4 including all 21 terms.
However, this situation changes dramatically when one assumes that Eadapt is removed by Eset under saturating (zero-order kinetics) conditions and that Eset's turnover is negligible compared to the other fluxes associated with Eset. In this case, most of the control coefficients become zero, except for two, which are related to the rate constants kadapt and . Together with the concentration of Eset, kadapt and define the setpoint for A (Eq. 3). Due to the concentration summation theorem (22,25,26),
| (5) |
and have the same magnitude but opposite signs. This indicates that the network can show robust temperature compensation in the level of A when the activation energies for kadapt and are equal. In fact, when all activation energies are equal, say each reaction step has an activation energy Ea, then all concentrations of the reaction intermediates in the network, Ij, become robust perfectly adapted, as seen by Eq. 6:
| (6) |
Fig. 5, a and b, shows this situation for 5°C and 100°C. When activation energies are different (except for the activation energies of kadapt and ), then only A shows robust temperature compensation, whereas the concentrations of the other intermediates are no longer invariant. This is indicated in Fig. 5, c and d, for temperature changes between 5°C and 100°C.
Figure 5.
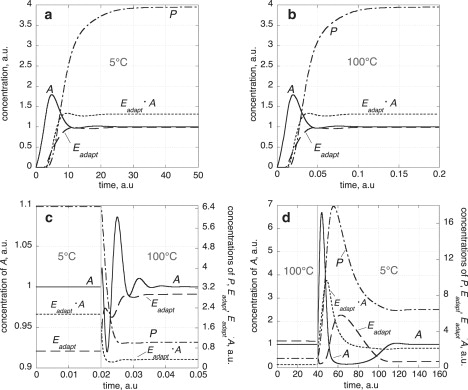
Robust perfect temperature compensation of the model described in Fig. 3a. Rate constants (Appendix) refer to 25°C with Aset = 1. (a) All activation energies are 50 kJ/mol and temperature is 5°C. (b) All activation energies are 50 kJ/mol and temperature is 100°C. Please note the much shorter timescale compared to panel a needed for the system to approach the same steady state. (c) Activation energies are as given in the Appendix. The system is initially at its steady state at 5°C. At 0.02 time units, the temperature is changed to 100°C showing perfect homeostasis only in A. (d) Activation energies as in panel c. The system is initially at its steady state at 100°C. At 40.0 time units the temperature is changed to 5°C showing perfect homeostasis only in A.
Discussion
Zero-order kinetics, integral feedback, and homeostatic breakdown
Integral feedback (14) or integral control (13) is a concept from control theory assuring that the output (the CV, Fig. 1) of a perturbed process is kept at a certain setpoint by integrating the associated error e such that e approaches zero (Fig. 1).
To keep the level of A homeostatic-regulated by integral control/feedback, the rate of formation of an additional species (Eadapt) has to be linked to the error, integrated, and then fed into the production rate of A. Integrated A is subtracted from Aset and the error e is recalculated (Fig. 2 c). Essential for this approach is the definition of the error e through Eq. 2, which provides the actual condition that A approaches Aset when the system's steady state is reached. Critically in this respect is the kinetic interpretation of the term kadaptAset. To avoid unrealistic situations such as negative concentrations (Fig. 2 b), the zero-order flux needs to be put into a proper mechanistic perspective. To achieve this, the mechanism shown in Fig. 3 includes an additional enzymatic species (Eset) leading to zero-order degradation/inactivation in Eadapt. This step is essential to obtain robust homeostasis. It requires that the level of Eset is kept constant and that the ratio between kadapt and remains unchanged. The latter condition is similar to that found by Levchenko and Iglesias for a model of eukaryotic chemotaxis (27) and a model by Ingalls et al. for a fast excitation-slow inhibition mechanism (Fig. 12.7 in (28)), where activation and inhibition steps are simultaneously activated by a common environmental signal. It may be noted that such a control is, principally, still based on balancing. In our model (Fig. 3 a), the balancing between 21 components has been effectively reduced to three parameters, as indicated by Eq. 3.
Interestingly, the kinetic restriction that concentrations must be positive leads to the breakdown of homeostasis for the mechanism in Fig. 3 at high removal/outflow rates in A. Whereas the homeostasis in Fig. 2 is robust for both high inflow and high outflow rates in A (leading sometimes to unrealistic negative concentrations in Eadapt), the chemically realistic mechanism shown in Fig. 3 works only for (high) inflow and moderate outflow rates in A. To address the situation of A-homeostasis at higher outflow rates (outflow-homeostasis), another homeostatic mechanism is necessary. Fig. 6 shows four motifs of homeostatic control mechanisms, two addressing inflow-homeostasis (Fig. 6, a and b) and two addressing outflow-homeostasis (Fig. 6, c and d). Each of these mechanisms work properly when the perturbing inflow and outflow conditions in A match their appropriate working conditions, but will fail otherwise, i.e., when total outflow in A becomes too large for an inflow-homeostatic controller or when total inflow in A becomes too large in an outflow-homeostatic controller. Thus, biochemical homeostasis will, in general, require at least two types of mechanisms, i.e., one addressing inflow-homeostasis and another addressing outflow-homeostasis.
Figure 6.
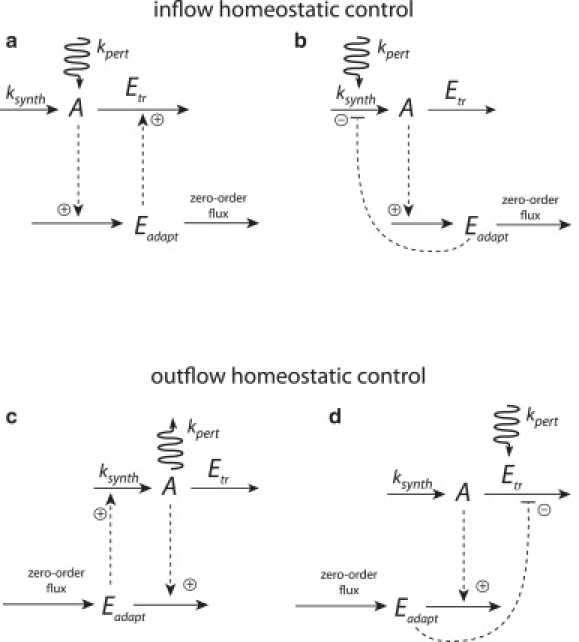
Homeostatic control motifs. Due to the kinetic restriction that concentrations need to be positive, two classes of homeostatic controllers arise: 1), inflow homeostatic controllers leading to homeostasis in the concentration of A for increasing (and moderate decreasing) perturbations in A (panels a and b); and 2), outflow homeostatic controllers leading to homeostasis in A for decreasing (and moderate increasing) perturbations in A (panels c and d). Rate equations with example parameter values are given in the Appendix. Note that many of the parameter values may be changed within certain limits (besides changing kpert) without affecting the homeostasis. (a) Schematic representation of the two (inflow) homeostatic models shown in Fig. 3. Robust homeostasis is due to the zero-order kinetic removal of Eadapt. (b) Inflow homeostatic model where Eadapt inhibits the inflow of A through ksynth. To maintain homeostasis the perturbation needs to be applied to the same reaction channel as ksynth. The integral feedback is due to the zero-order removal of Eadapt and is not related to the physico-chemical negative feedback from Eadapt to (ksynth + kpert). (c) Outflow homeostatic controller by removing Eadapt through A. (d) Outflow homeostatic controller by inhibiting Etr through Eadapt. Similar to panel b, homeostasis is obtained when the perturbation increases the outflow of A through the same reaction channel that is used by enzyme Etr.
Fig. 6 a shows an outline of the inflow-control mechanisms described in Fig. 3. The inflow-control mechanism in Fig. 6 b shows a related scheme suggested by Yi et al., including a zero order reaction step (14), where instead of the increased removal of A the formation of A is inhibited by a molecular feedback loop.
Fig. 6 c shows an outflow-homeostatic mechanisms closely related to the scheme by El-Samad et al. (15), but avoiding negative concentrations, as shown in their Fig. 8. In our Fig. 6 d, outflow homeostatic control is achieved by inhibiting the outflow of A through Eadapt. In the Appendix we show kinetic representations of these four mechanisms.
Robust perfect adaptation can be related to the concept of integral control or integral feedback (13,14), which involves a negative feedback in the control-theoretic formulation of the system as indicated in Fig. 1 or Fig. 2 c. Although some schemes, as in Fig. 6, b and d, or in the literature (14,29), contain molecular feedback inhibitions (molecular negative feedbacks), the presence of robust perfect adaptation, i.e., the behavior of a control-theoretic negative feedback, does not necessarily require molecular negative feedbacks. An example of robust perfect adaptation with integral feedback behavior but without molecular feedback loops is given by a consecutive reaction such as → A → B →, where B (or the flux forming B) can show robust perfect adaptation for any stepwise change in the rate constant forming intermediate B (11,30,31), as long as A is formed by zero-order kinetics. Thus, the essential part to get robust homeostasis in the mechanisms shown in Fig. 6, and as illustrated in Fig. 4, e and f, is the presence of the zero-order kinetic term (i.e., “control of the controller”).
Possible regulation points in homeostatic mechanisms
Iron homeostasis
Iron is an essential element for all mammalian cells, but gets toxic when in excess. Special transport and regulatory processes are therefore needed to ensure iron homeostasis within the organism as a whole as well as in individual cells (32). Ferroportin (33) and hepcidin (34) have been suggested to be two key players in iron homeostasis. Ferroportin is an iron exporter, which transports iron from cells such as macrophages or intestinal or liver cells into the blood plasma. Hepcidin, a liver-produced hormone, is a negative regulator of iron absorption with antimicrobial properties, which itself is under homeostatic regulation. An interesting regulatory aspect, which relates to the models in Fig. 3, is that hepcidin binds to ferroportin and leads to its degradation in a similar way as Eset removes Eadapt. Considering the interaction between ferroportin and hepcidin, the mechanism in Fig. 3 suggests that under iron inflow conditions, hepcidin may serve as a setpoint controller for cell-internal iron concentrations with ferroportin having the role as Eadapt, i.e., removing iron (A) out of the cell. The binding between ferroportin (Eadapt) and hepcidin (Eset), which leads to the degradation of ferroportin (Eadapt) (34), may thus provide a mechanism of how hepcidin acts as a “control of the controller” and leads to potential robust homeostasis. Hepcidin works at concentrations as low as 10 nM (34) and can efficiently reduce upregulated ferroportin levels when iron influx into the cell is high (35). It is not known whether the removal of ferroportin by hepcidin at normal iron concentrations is a zero-order process.
Calcium homeostasis
Fig. 7 shows a scheme of calcium homeostasis in humans. Calcitonin (CT), parathyroid hormone (PTH), and the active form of vitamin D (calcitriol) are important (but not the only) factors involved in the regulation of Ca2+ and bone metabolism (36). PTH increases bone resorption and plasma Ca2+ levels. Calcitriol increases intestinal Ca2+ absorption, bone resorption, and plasma Ca2+. Calcitonin (CT) decreases bone resorption and plasma Ca2+. CaSR denotes the calcium-sensing receptor in the nephron, which appears to mediate effects of hypercalcemia on calcium excretion (37). In case of low calcium levels or when the outflow of calcium needs to be compensated for, an outflow-control mechanism like that indicated in Fig. 6 c may come into play. The mechanism is similar to that suggested by El-Samad et al. (15) for hypocalcemia. In this mechanism, Eadapt plays the role of PTH. The level of PTH is decreased by increased calcium levels. Robust calcium homeostasis is obtained due to a zero kinetic formation rate of Eadapt (PTH) and its downregulation by calcium. In the case of high calcium levels, an inflow-control mechanism like that shown in Fig. 6 a appears to be operative. High calcium (A) levels activate CT and CaSR, which are responsible for the removal of plasma calcium by transporting it into the bone and/or by excretion through the urine. Homeostatic control may be achieved by zero-order kinetic inactivation of CT and/or CaSR.
Figure 7.
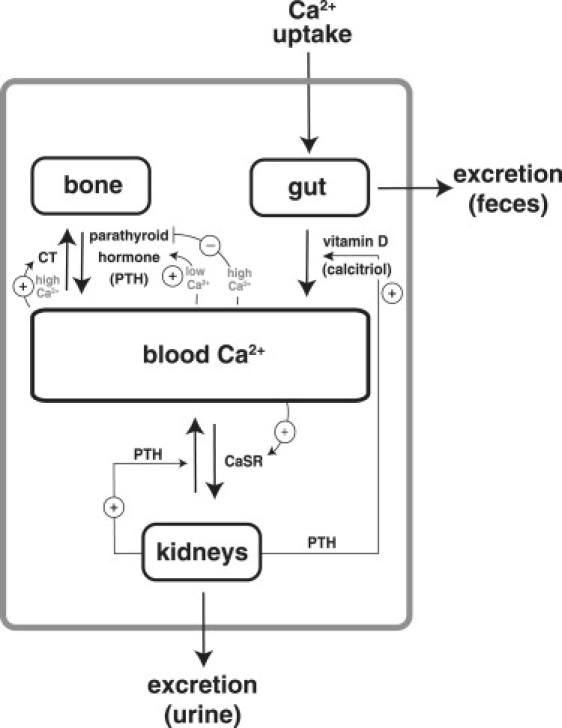
Schematic representation of blood calcium homeostasis in humans. Important regulators are parathyroid hormone (PTH), calcitonin (CT), vitamin D, and the calcium-sensing receptor in the nephron. For a discussion of how these regulators may participate in inflow- and outflow mechanisms, see main text.
Robust temperature compensation
It is interesting that the occurrence of robust (activation-energy-independent) temperature compensation for a certain intermediate is closely associated with a robust homeostatic control of that intermediate. This indicates that calcium, iron, and other homeostatic mechanisms may be capable of showing temperature compensation. Unfortunately, there have been few studies in this direction. Herrera et al. (38) studied the temperature dependence of calcium homeostasis in rat pachytene spermatocytes and rat round spermatids in suspension without external calcium concentration. The pachytene spermatocytes showed practically unchanged calcium levels at 10 nmol/L when the temperature was varied (increased) between 16°C and 33°C. Above 33°C, the internal calcium levels quickly increased, reaching levels at 120 nmol/L at 40°C. In the rat round spermatids, the internal calcium levels did not show any temperature compensation, but a monotonic increase from 30 nmol/L to ∼150 nmol/L was observed when the temperature was varied from 5°C to 40°C. Interestingly, the temperature compensation in the calcium homeostasis in the pachytene spermatocytes appears to be due to a balance between two opposing reactions, i.e., between uptake and leakage to and from the cell's internal calcium stores, with determined activation energies of 62 kJ/mol and 55 kJ/mol, respectively.
Although robust homeostatic and adaptation mechanisms appear to be attractive concepts, it is still unclear to what extent temperature compensation (of oscillatory or nonoscillatory processes) is due to a balancing between individual reaction steps (18,21) or due to mechanisms as outlined in Fig. 5, where the balancing is reduced to a few parameters (39). Characteristic to all physiological and chemical temperature compensated systems (20,24,38,40–48) is that the compensation mechanism operates at a local (for the organism) important temperature range and not globally over the whole temperature range such as shown in Fig. 5. However, this does not necessarily invalidate homeostatic control structures as those shown in Figs. 3 and 6. The controllers (for example, Eadapt and Eset) have to be seen in the context of the dynamics of the whole cell and the whole organism (2), a systems (biology) perspective (49–51), where the controllers themselves are controlled and influenced by factors important for other cellular purposes.
Appendix
Rate equations, rate constants, and activation energies for the mechanism in Fig. 3a.
Rate equations
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
| (16) |
Rate constants with activation energies
The following rate constants and activation energies (given in parenthesis) have been used unless otherwise stated in the text. Rate constant values refer to 25°C. The Arrhenius equation ki = Ai·exp(−Ei/(RT)) has been used to calculate the rate constant ki at other temperatures (Ai: preexponential factor, assumed to be temperature-independent; Ei, activation energy; R, gas constant; and T, temperature in Kelvin). All rate constants are given in arbitrary units (a.u.):
Rate equations for the mechanism in Fig. 3b/Fig. 6a
| (17) |
| (18) |
The following rate constants and initial concentrations give perfect homeostasis with Aset = 1.0, ksynth = 1.0, and kpert ≥ 0: = 1.0; = 2.0; kadapt = 3.0; = 6.0e+6; = 1.0e−6; = 0.01; and = 5.0, where ; ; and .
Initial concentrations: A = 1.0; Eadapt = 0.01; = 5.0e−7; and = 0.1. Concentrations of and are kept constant.
Rate equations for the mechanism in Fig. 6b
| (19) |
| (20) |
The following rate constants with zero initial concentrations (both a.u.) give perfect homeostasis in A with Aset = 1.0, when varying kpert (≥0): ksynth = 10.0; = 0.1; = 40; = 1.0; kadapt = 1.0; = 1.0; and = 1.0e−6.
Rate equations for the mechanism in Fig. 6c
| (21) |
| (22) |
The following rate constants with zero initial concentrations (both a.u.) give perfect homeostasis in A with Aset = 1.0 when varying kpert (≥0.1): ksynth = 1.0; k = 1.0; and = 1; = 0.1; zero-order flux j0 = 1.0; = 1.0; and = 1.0e−6.
Rate equations for the mechanism in Fig. 6d
| (23) |
| (24) |
The following rate constants with zero initial concentrations (both a.u.) give perfect homeostasis in A with Aset = 1.0 when varying kpert (≥0): ksynth = 1.0; = 10; = 1.0; and = 1.0; zero-order flux j0 = 1.0; = 1.0; and = 1.0e−6.
Supporting Material
Supporting Material files are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01166-7.
Supporting Material
Acknowledgments
This research was supported by the Norwegian Research Council under FUGE Systems Biology (SysMO) grant No. 183085/S10.
References
- 1.Asthagiri A.R., Lauffenburger D.A. Bioengineering models of cell signaling. Annu. Rev. Biomed. Eng. 2000;2:31–53. doi: 10.1146/annurev.bioeng.2.1.31. [DOI] [PubMed] [Google Scholar]
- 2.Carlson J.M., Doyle J. Complexity and robustness. Proc. Natl. Acad. Sci. USA. 2002;99(Suppl 1):2538–2545. doi: 10.1073/pnas.012582499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stelling J., Sauer U., Szallasi Z., Doyle F.J., Doyle J. Robustness of cellular functions. Cell. 2004;118:675–685. doi: 10.1016/j.cell.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Kitano H. Towards a theory of biological robustness. Mol. Syst. Biol. 2007;3:1–7. doi: 10.1038/msb4100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berg H.C., Tedesco P.M. Transient response to chemotactic stimuli in Escherichia coli. Proc. Natl. Acad. Sci. USA. 1975;72:3235–3239. doi: 10.1073/pnas.72.8.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barkai N., Leibler S. Robustness in simple biochemical networks. Nature. 1997;387:913–917. doi: 10.1038/43199. [DOI] [PubMed] [Google Scholar]
- 7.Alon U., Surette M.G., Barkai N., Leibler S. Robustness in bacterial chemotaxis. Nature. 1999;397:168–171. doi: 10.1038/16483. [DOI] [PubMed] [Google Scholar]
- 8.Hansen C.H., Endres R.G., Wingreen N.S. Chemotaxis in Escherichia coli: a molecular model for robust precise adaptation. PLoS Comput. Biol. 2008;4:0014–0027. doi: 10.1371/journal.pcbi.0040001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ratliff F., Hartline H.K., Miller W.H. Spatial and temporal aspects of retinal inhibitory interaction. J. Opt. Soc. Am. 1963;53:110–120. doi: 10.1364/josa.53.000110. [DOI] [PubMed] [Google Scholar]
- 10.Asthagiri A.R., Nelson C.M., Horwitz A.F., Lauffenburger D.A. Quantitative relationship among integrin-ligand binding, adhesion, and signaling via focal adhesion kinase and extracellular signal-regulated kinase 2. J. Biol. Chem. 1999;274:27119–27127. doi: 10.1074/jbc.274.38.27119. [DOI] [PubMed] [Google Scholar]
- 11.Hao N., Behar M., Elston T.C., Dohlman H.G. Systems biology analysis of G protein and MAP kinase signaling in yeast. Oncogene. 2007;26:3254–3266. doi: 10.1038/sj.onc.1210416. [DOI] [PubMed] [Google Scholar]
- 12.Mettetal J.T., Muzzey D., Gómez-Uribe C., van Oudenaarden A. The frequency dependence of osmo-adaptation in Saccharomyces cerevisiae. Science. 2008;319:482–484. doi: 10.1126/science.1151582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilkie J., Johnson M., Reza K. Palgrave; New York: 2002. Control Engineering. An Introductory Course. [Google Scholar]
- 14.Yi T.M., Huang Y., Simon M.I., Doyle J. Robust perfect adaptation in bacterial chemotaxis through integral feedback control. Proc. Natl. Acad. Sci. USA. 2000;97:4649–4653. doi: 10.1073/pnas.97.9.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Samad H., Goff J.P., Khammash M. Calcium homeostasis and parturient hypocalcemia: an integral feedback perspective. J. Theor. Biol. 2002;214:17–29. doi: 10.1006/jtbi.2001.2422. [DOI] [PubMed] [Google Scholar]
- 16.Radhakrishnan, K., and A. C. Hindmarsh. 1993. Description and Use of LSODE, the Livermore Solver for Ordinary Differential Equations. NASA Reference Publication 1327, Lawrence Livermore National Laboratory Report UCRL-ID-113855. National Aeronautics and Space Administration, Lewis Research Center, Cleveland, OH.
- 17.Ruoff P., Vinsjevik M., Rensing L. Temperature compensation in biological oscillators: a challenge for joint experimental and theoretical analysis. Comments Theor. Biol. 2000;5:361–382. [Google Scholar]
- 18.Ruoff P. Introducing temperature-compensation in any reaction kinetic oscillator model. J. Interdiscipl. Cycle Res. 1992;23:92–99. [Google Scholar]
- 19.Ruoff P., Christensen M.K., Wolf J., Heinrich R. Temperature dependency and temperature compensation in a model of yeast glycolytic oscillations. Biophys. Chem. 2003;106:179–192. doi: 10.1016/s0301-4622(03)00191-1. [DOI] [PubMed] [Google Scholar]
- 20.Ruoff P., Loros J.J., Dunlap J.C. The relationship between FRQ-protein stability and temperature compensation in the Neurospora circadian clock. Proc. Natl. Acad. Sci. USA. 2005;102:17681–17686. doi: 10.1073/pnas.0505137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruoff P., Zakhartsev M., Westerhoff H.V. Temperature compensation through systems biology. FEBS J. 2007;274:940–950. doi: 10.1111/j.1742-4658.2007.05641.x. [DOI] [PubMed] [Google Scholar]
- 22.Heinrich R., Schuster S. Chapman and Hall; New York: 1996. The Regulation of Cellular Systems. [Google Scholar]
- 23.Fell D. Portland Press; London and Miami: 1997. Understanding the Control of Metabolism. [Google Scholar]
- 24.Aase S.O., Ruoff P. Semi-algebraic optimization of temperature compensation in a general switch-type negative feedback model of circadian clocks. J. Math. Biol. 2008;56:279–292. doi: 10.1007/s00285-007-0115-5. [DOI] [PubMed] [Google Scholar]
- 25.Kacser H., Burns J.A. Molecular democracy: who shares the controls? Biochem. Soc. Trans. 1979;7:1149–1160. doi: 10.1042/bst0071149. [DOI] [PubMed] [Google Scholar]
- 26.Burns J.A., Cornish-Bowden A., Groen A.K., Heinrich R., Kacser H. Control analysis of metabolic systems. Trends Biochem. Sci. 1985;19:16. [Google Scholar]
- 27.Levchenko A., Iglesias P.A. Models of eukaryotic gradient sensing: application to chemotaxis of amoebae and neutrophils. Biophys. J. 2002;82:50–63. doi: 10.1016/S0006-3495(02)75373-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ingalls B.P., Yi T.-M., Iglesias P.A. Using control theory to study biology. In: Szallasi Z., Stelling J., Periwal V., editors. System Modeling in Cellular Biology. MIT Press; Cambridge, MA: 2006. [Google Scholar]
- 29.Alon U. Chapman & Hall; New York: 2006. An Introduction to Systems Biology: Design Principles of Biological Circuits. [Google Scholar]
- 30.Csikasz-Nagy A., Soyer O.S. Adaptive dynamics with a single two-state protein. J.R. Soc. Interface. 2008;5(Suppl 1):S41–S47. doi: 10.1098/rsif.2008.0099.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drengstig T., Ueda H.R., Ruoff P. Predicting perfect adaptation motifs in reaction kinetic networks. J. Phys. Chem. B. 2008;112:16752–16758. doi: 10.1021/jp806818c. [DOI] [PubMed] [Google Scholar]
- 32.Andrews N.C., Schmidt P.J. Iron homeostasis. Annu. Rev. Physiol. 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- 33.Donovan A., Lima C.A., Pinkus J.L., Pinkus G.S., Zon L.I. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Nemeth E., Tuttle M.S., Powelson J., Vaughn M.B., Donovan A. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 35.Delaby C., Pilard N., Goncalves A.S., Beaumont C., Canonne-Hergaux F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood. 2005;106:3979–3984. doi: 10.1182/blood-2005-06-2398. [DOI] [PubMed] [Google Scholar]
- 36.Molina P.E. McGraw-Hill; Blacklick, OH: 2006. Endocrine Physiology. [Google Scholar]
- 37.Friedman P.A. Calcium transport in the kidney. Curr. Opin. Nephrol. Hypertens. 1999;8:589–595. doi: 10.1097/00041552-199909000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Herrera E., Salas K., Lagos N., Benos D.J., Reyes J.G. Temperature dependence of intracellular Ca2+ homeostasis in rat meiotic and postmeiotic spermatogenic cells. Reproduction. 2001;122:545–551. doi: 10.1530/rep.0.1220545. [DOI] [PubMed] [Google Scholar]
- 39.Hong C.I., Conrad E.D., Tyson J.J. A proposal for robust temperature compensation of circadian rhythms. Proc. Natl. Acad. Sci. USA. 2007;104:1195–1200. doi: 10.1073/pnas.0601378104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hastings J.W., Sweeney B.M. On the mechanism of temperature independence in a biological clock. Proc. Natl. Acad. Sci. USA. 1957;43:804–811. doi: 10.1073/pnas.43.9.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bünning E. Springer-Verlag; Berlin: 1963. The Physiological Clock. [Google Scholar]
- 42.Zimmerman W.F., Pittendrigh C.S., Pavlidis T. Temperature compensation of the circadian oscillation in Drosophila pseudoobscura and its entrainment by temperature cycles. J. Insect Physiol. 1968;14:669–684. doi: 10.1016/0022-1910(68)90226-6. [DOI] [PubMed] [Google Scholar]
- 43.Hazel J.R., Prosser C.L. Molecular mechanisms of temperature compensation in poikilotherms. Physiol. Rev. 1974;54:620–677. doi: 10.1152/physrev.1974.54.3.620. [DOI] [PubMed] [Google Scholar]
- 44.Aronson B.D., Johnson K.A., Dunlap J.C. Circadian clock locus frequency: protein encoded by a single open reading frame defines period length and temperature compensation. Proc. Natl. Acad. Sci. USA. 1994;91:7683–7687. doi: 10.1073/pnas.91.16.7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rábai G., Hanazaki I. Temperature compensation in the oscillatory hydrogen peroxide-thiosulfate-sulfite flow system. Chem. Comm. 1999;19:1965–1966. [Google Scholar]
- 46.Kóvacs K.M., Rábai G. Temperature-compensation in pH-oscillators. Phys. Chem. Chem. Phys. 2002;4:5265–5269. doi: 10.1021/jp0538159. [DOI] [PubMed] [Google Scholar]
- 47.Edwards K.D., Lynn J.R., Gyula P., Nagy F., Millar A.J. Natural allelic variation in the temperature-compensation mechanisms of the Arabidopsis thaliana circadian clock. Genetics. 2005;170:387–400. doi: 10.1534/genetics.104.035238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gould P.D., Locke J.C., Larue C., Southern M., Davis S.J. The molecular basis of temperature compensation in the Arabidopsis circadian clock. Plant Cell. 2006;18:1177–1187. doi: 10.1105/tpc.105.039990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.von Bertalanffy L. George Braziller; New York: 1975. Perspectives on General System Theory. [Google Scholar]
- 50.Sontag E.D. Some new directions in control theory inspired by systems biology. Syst. Biol. (Stevenage) 2004;1:9–18. doi: 10.1049/sb:20045006. [DOI] [PubMed] [Google Scholar]
- 51.Wolkenhauer O., Sreenath S.N., Wellstead P., Ullah M., Cho K.H. A systems- and signal-oriented approach to intracellular dynamics. Biochem. Soc. Trans. 2005;33:507–515. doi: 10.1042/BST0330507. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.