

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive motor neuron degenerative disease, and the inherited form, familial ALS (fALS), has been linked to over 100 different point mutations scattered throughout the Cu-Zn superoxide dismutase protein (SOD1). The disease is likely due to a toxic gain of function caused by the misfolding, oligomerization, and eventual aggregation of mutant SOD1, but it is not yet understood how the structurally diverse mutations result in a common disease phenotype. The behavior of the apo-monomer fALS-associated mutant protein A4V was explored using molecular-dynamics simulations to elucidate characteristic structural changes to the protein that may allow the mutant form to improperly associate with other monomer subunits. Simulations showed that the mutant protein is less stable than the WT protein overall, with shifts in residue-residue contacts that lead to destabilization of the dimer and metal-binding sites, and stabilization of nonnative contacts that leads to a misfolded state. These findings provide a unifying explanation for disparate experimental observations, allow a better understanding of alterations of residue contacts that accompany loss of SOD1 structural integrity, and suggest sites where compensatory changes may stabilize the mutant structure.
Introduction
A subset of familial amyotrophic lateral sclerosis (fALS) cases has been tied to mutations in the Cu-Zn superoxide dismutase protein (SOD1). Though the mechanism of toxicity has been under intense scrutiny for 15 years, it remains largely undetermined. One current model involves the misfolding of the mutant protein (1), which results in a new structural species that directly confers toxicity and/or sequesters cellular machinery that is important for protein homeostasis, such as chaperones or proteasome components.
SOD1 is normally responsible for the disproportionation of superoxide to molecular oxygen and hydrogen peroxide, in which one superoxide molecule is oxidized and then another is reduced by copper in the active site of the enzyme. Superoxide is a naturally occurring byproduct of respiration. Although SOD1 is abundant and ubiquitous in the cytoplasm, it has also been found in the mitochondrial intermembrane space. The concentrations are especially high in certain subcellular locations, such as motor neuron axons (2).
Only minimal changes have been observed in the crystal structure of fully metallated disease-associated mutant forms of SOD1 in comparison to wild-type (WT) SOD1. Many SOD1 mutants display lower melting temperatures than their WT counterparts, regardless of the metallation state (3), and they unfold more easily with urea or guanidine-HCL (4). However, some of the metal-binding mutants have higher melting temperatures than WT (5). Both sporadic and familial forms of ALS show evidence of cytoplasmic inclusion bodies—deposits that are characteristic of many neurodegenerative disorders, including Huntington's disease, Alzheimer's disease, and Parkinson's disease (6)—and aggregates have been found in mouse models of the disease (7–9). The familial ALS aggregates contain SOD1, neurofilament proteins, ubiquitin, and a host of other cellular components, but it is not clear whether zinc or copper is present (10). The current thinking is that the aggregation is a cellular protective mechanism and the most toxic form is either a misfolded monomer or a soluble oligomeric species or protofibril (6). Similar models have been proposed for other neurodegenerative diseases (11).
The unfolding and aggregation pathway of the protein involves the dissociation of the dimer and loss of metal binding, followed by the subsequent oligomeric assembly of the protein (12–14). It is also possible that the aggregates are favored when the amount of misfolded protein reaches a point where the ubiquitin proteolytic machinery becomes unable to handle the load (15,16). Although the aforementioned experimental approaches have been enlightening, there are other opportunities to investigate the effects of mutation on the protein. An atomic-level look at the protein dynamics through molecular-dynamics (MD) simulations can be informative in determining the effect the mutations have on the structure of the protein, allowing investigation of how changing one residue to another can create a ripple effect through the protein that eventually affects dimerization, metal binding, and/or overall protein stability.
SOD1 conforms to the Greek key β-barrel folding topology, and each monomer subunit of the homodimer binds one copper and one zinc ion. Although the crystal structures of several ALS-causing mutants have been solved, the structure of WT SOD1 in solution differs from the crystal structure (17). These average structures are very informative; however, proteins are dynamic and important conformational states may have low sampling rates.
Previous MD studies have included short (<1 ns) simulations of WT bovine and human SOD1 dimers (18–20), as well as comparisons of simulations of the WT monomer and dimer (21,22). In addition, 5 ns simulations of WT and mutant (A4V, G37R, and H46R) dimers and monomers (23) have provided the first direct comparison of mutant and WT MD simulations. These results indicate that altered long-range communication within the protein structure could be an underlying cause of aggregation of mutant SOD1 in fALS. Also, a recent study examined several 100 ns simulations of WT apo and holo monomers and dimers, and the effects of disulfide bonds on SOD1 stability (24).
Here we report findings from multiple long (60 ns) simulations of WT and A4V apo (demetallated) monomers to explore the dominant patterns of structural changes upon mutation in the monomer after loss of metallation. A4V is very close to the dimer interface and represents one of the most common and most lethal mutations, with rapid disease progression and death occurring on average at 1.4 years vs. 3–5 years for other mutants (25). These simulations reveal significant changes in the mutant protein structure, which is discussed in terms of potential effects on dimer destabilization, loss of metallation, and the aberrant oligomerization of the misfolded monomer.
Materials And Methods
Models
The starting structure for WT SOD1 monomer was obtained from a 1.8 Å crystal structure of human SOD1 (1HL5 Chain A) (26). The A4V SOD1 starting structure was obtained from a 1.9 Å crystal structure of the A4V mutant of human SOD1 (1UXM Chain A) (17). Before energy minimization was performed, the Cu2+ and Zn2+ ions were removed and H63 was left unprotonated. The ionization states of the amino acids corresponded to neutral pH (Asp−, Glu−, His0, Lys+, and Arg+). The C57-C146 disulfide bond was intact to match in vitro studies.
The holo 1HL5 structure was chosen over the apo 1HL4 structure for a variety of reasons. In brief, of the four SOD1 proteins available in the 1HL4 tetramer, chains C and D are missing many residues (26 and 28 out of 153, respectively). Chains A and B are missing many heavy atoms (18 and 12 atoms, respectively) and they also have bound zinc. The model-building required to render these structures suitable for simulation would be quite extensive, and the resultant starting structure would be inferior to the 1HL5 structure used.
MD simulations
Simulations of apo WT and A4V SOD1 were performed with the in lucem Molecular Mechanics (ilmm) (27) simulation software using protocols described elsewhere (27–32). In brief, the simulations included all hydrogens and explicit flexible three-center waters (29). The protein was solvated in a box extending at least 10 Å from any protein atom, with the solvent density set to the experimental value at 310 K of 0.993 g/mL (33). Periodic boundary conditions were employed to minimize edge effects. The microcanonical constant number of molecules, volume, and energy (NVE) ensemble was employed. A 10 Å force-shifted cutoff range was used for all nonbonded atom interactions, and the interaction pair list was updated every three steps. The potential energy functions were used to propagate MD trajectories with a 2 fs time step, and structures were saved every 1 ps for analysis. Three 60 ns simulations were run for each WT and A4V apoprotein at 310 K, yielding ∼180,000 structures each for analysis. The long simulation time allows the different simulations to reach a “converged” state for each monomer in solution with enough sampling of the state to be statistically sound.
Analyses
Except where noted, all analyses were performed over all structures. The Define Secondary Structure of Proteins (DSSP) algorithm (34) was used to assign secondary structure based on hydrogen-bond energies. The DSSP data used to generate the graphs were sampled at 1 ps intervals; however, conformational time-based populations from DSSP were calculated at 100 ps intervals.
The CONGENEAL structural dissimilarity scores (35) for each simulation were calculated at each time point based on the minimized structure of the protein. CONGENEAL is based on the weighted distance maps of two structures, such that the weight is higher for atoms that are closer together. Each element of the matrix is calculated using the equation wi,j = di,j−p, where w is the weight, d is the Cα distance between the two residues, and p = 2.
Differences between the total contact time of residue pairs in the WT and A4V simulations were considered to be statistically significant if the difference in the averages of the two different types of simulations was greater than the combined standard deviation (SD) of those values. Atoms were considered to be in contact if the heavy atom distance was <4.6 Å, except for C-C, where the cutoff was 5.4 Å. Protein images were produced using UCSF Chimera (36).
Results And Discussion
Simulations of the fALS-associated SOD1 mutant A4V showed the effects of the mutation on the kinetic stability of the protein when compared with WT under similar conditions (neutral pH, 310 K, apo, monomer). For each WT and mutant protein, three simulations were run for 60.5–63.0 ns and then analyzed. The differences between the two sequences may be illustrated in a number of ways to provide clues for the previously reported effects of this mutation on dimer stability (14) and metal binding (37,38).
Overall protein instability
The simulations reveal that during the course of simulation, the A4V mutant protein undergoes larger structural changes than WT. An examination of the Cα root mean-square deviation (RMSD) (see Fig. S1, A and B, in the Supporting Material), total solvent accessible surface area (SASA) (Fig. S1, C and D), radius of gyration (Fig. S1, E and F), and CONGENEAL structural dissimilarity score (Fig. S1, G and H) for the simulations shows a general increase in all of these measures across the simulations for the mutant versus WT. The smaller changes in the SASA and radius of gyration data indicate that the mutant protein sample conformations are somewhat different but still fairly compact. A breakdown of the various components that comprise the total SASA of the protein reveals that each of the components of the total SASA is generally higher in the mutant than in the WT (data not shown). Overall, larger changes in the structure are observed for A4V than for WT.
Perturbations of residues normally at the dimer interface
Dimerization of the individual monomer subunits increases the stability of the SOD1 enzyme (39), and thus mutations that destabilize the dimer may decrease the overall protein stability. Dimer destabilization may be viewed in two ways: instability in the dimer itself, or a decreased affinity for dimerization by the individual monomer subunits. Perturbations of the native dimer interface residues may either prevent the initial dimerization or decrease the lifetime of the dimer. Although the effects of the mutation on the dimer would be better studied using dimer simulations, our intent was to study the A4V-associated structural changes in the monomer that occur after the known loss of dimerization, which may have implications for the ability of the dimer to reform.
One way to measure perturbations of the protein in MD simulations is to examine the changes in contacts between residues over time. A minimum increase or decrease in total contact time of 50% over the entire simulation between any residue pair in the protein was used as a cutoff, allowing for the creation of a map showing the major changes in intramonomer residue-residue contacts as the effects propagated from one point of interest to another (Fig. 1 A). There is an overall increase in the total contact time of the dimer interface residues (50–53, 114, and 148–153) (17) with other residues in the A4V simulations in comparison with WT, although there is a significant decrease (51%) in total contact time between residues I149 and F20 (Fig. 1, B and C). These intramonomer contacts serve to bring the N- and C-termini of the protein into tighter association in the mutant simulations, potentially impairing the ability of the A4V mutant to maintain dimerization.
Figure 1.
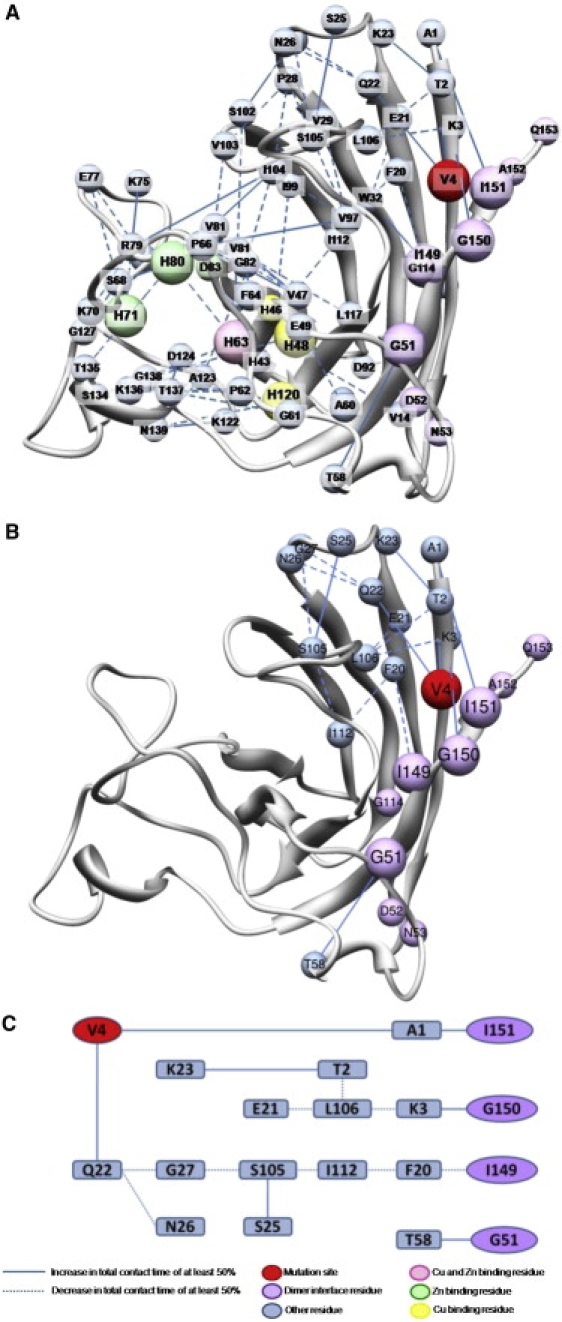
Contact differences between WT and A4V simulations. (A) All changes in residue-residue contact time > 50% of the total simulation time, relative to the WT simulations. (B) Changes in contact time between various residues mapped onto the minimized crystal structure of the mutant protein. (C) Changes in contact time between various residues displayed for clarity.
Residues S105 and C111 form an isolated β-bridge structure at the beginning of all simulations (Fig. 2, A and B). A bridge is essentially defined as an elementary hydrogen-bonding pattern that is a building block for α- or β-sheet. In the mutant runs, this β-bridge is unstable and each simulation adopts a fairly stable π-helix comprised of residues L106–H110 after some period of time (Fig. 2 B). The time it takes to form this helix varies from one simulation to the next, and it is variably stable after it forms. In contrast, no helix is found in this region during any of the WT simulations, and the β-bridge is retained to varying degrees in the three runs (Fig. 2 A). Although they are not directly involved in the dimer interface, these residues have an impact on it through the contacts formed or lost during the course of the simulations. For instance, there is a large loss of contacts between L106 and K3, and an increase in contacts between K3 and dimer interface residue G150, as described above. Additionally, there is a loss of contacts between nearby S105 and I112, I112 with F20, and F20 with dimer interface residue I149 (Fig. 1, B and C). These structural changes near the dimer interface provide a reasonable mechanism to explain the substantial destabilization of the A4V mutant SOD1 dimer (17), which is the first step of the proposed oligomerization pathway.
Figure 2.
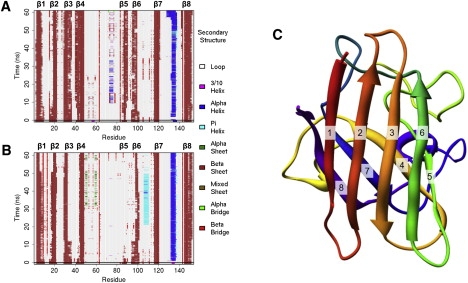
Protein secondary structure. Hydrogen-bond analysis and secondary structure assignment of representative (A) WT and (B) A4V simulations using DSSP. (C) The starting structure of the WT protein with the β-strands labeled.
Perturbations of the metal-binding sites
Copper-binding site
The residues involved in coordinating the metal (Fig. S2) are distal to the A4V mutation, yet they show greater movement during simulation in the mutant than in WT. The Cα root mean-square fluctuation (RMSF) (Fig. 3, A and B) and Cα RMSD (not shown) plots show higher values for these residues, although the distances between the Cu2+ ion-binding atoms are not significantly different between the WT and mutant proteins (Fig. 3 C). This is not surprising given the known role of the Cu2+ ion in the enzymatic reaction, and the normal level of catalytic activity of this mutant (37,40).
Figure 3.
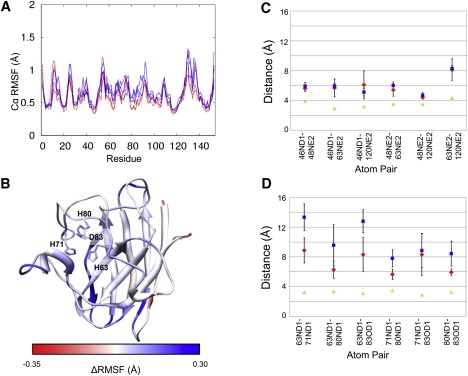
Changes to metal-binding residues. (A) Cα-RMSF by residue for three each WT (light gray, shown in red online) and A4V (dark gray, blue online) simulation. (B) Differences in Cα-RMSF are mapped onto the protein structure. Regions with a higher RMSF in the mutant than in the WT are shown in dark gray (blue online), and regions with higher RMSF in the WT than in the mutant are shown in light gray (red online). White indicates no difference. The four residues displayed are the Zn2+-binding residues. H71, H80, and D83 each show higher RMSF and RMSD (not shown) in the A4V mutant simulations than in the WT. (C) Average pairwise distances between the Cu2+-binding atoms for three simulations of WT (diamonds) and A4V (squares) proteins. In each case the error bars represent the SD of the three simulations, and in each case the error bars overlap. The WT crystal structure distances are shown as triangles. Mutant crystal structure distances differ from WT by <0.2 Å in all cases, including those shown in panel D (not shown). (D) Average pairwise distances between the Zn2+-binding atoms for three simulations of WT (diamonds) and A4V (squares) proteins. In each case the error bars represent the SD of the three simulations. There are significant differences between the distances of the pairs: 63ND1-71ND1, 63ND1-83OD1, 71ND1-80ND1, and 80ND1-83OD1. Triangles are as in panel C.
Motion of the electrostatic loop in WT apo dimer simulations was previously reported (22) and we also observe this motion in our monomer simulations. However, the helix in the electrostatic loop near the copper-binding site is more stable and has less apparent motion in the WT form (Fig. S3 A) than in the mutant (Fig. S3B). The rotation of this helix may be explained by a large decrease in the contacts between the residues of the helix and the body of the protein, specifically residue H63, which coordinates both Zn2+ and Cu2+ binding. Table S1 shows that H63 has greatly reduced total contact times with S134 (57% reduction), T135 (43%), T137 (27%), and G138 (58%), and H120 has less contact with G138 (57%) and N139 (75%) during the mutant simulations compared to WT. Residues in this region (residues 122–140) that differ by >50% in total contact time between the WT and A4V simulations are shown in Fig. 4. Toward the end of the third WT simulation, the helix moves into a similar position as seen in the mutant, which may mean that this motion is not an important effect of the mutation. However, the apo form of the protein is less stable (3,5,39,41), and this may simply be a consequence of the lower stability. The mutant protein spends far more time with the helix in the new position than does WT. There is also greater movement of the C-terminal residues in WT than in the mutant, as discussed above.
Figure 4.
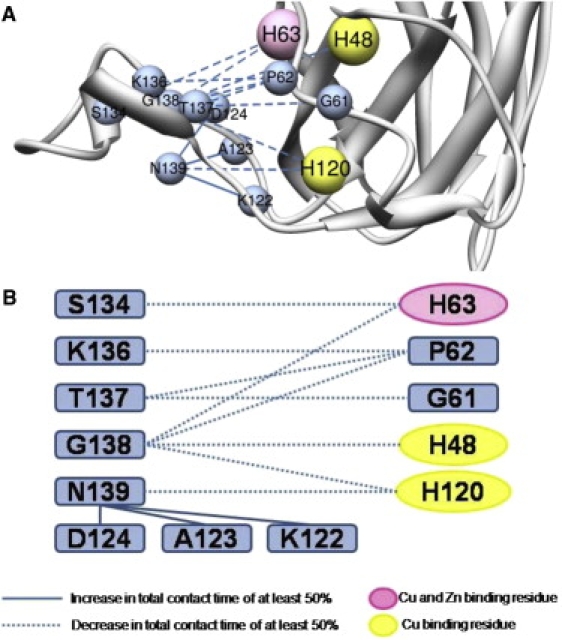
Contact differences of the helix near the Cu2+-binding site. All changes are relative to WT simulations. (A) Changes in contact time between various residues mapped onto the minimized crystal structure of the mutant protein. (B) Changes in contact time between various residues displayed for clarity.
The native α-helix in the electrostatic loop, comprised of residues E132–T137 (Fig. 5 A), is somewhat more stable in the WT simulations than in the mutant runs. Toward the end of one WT run at ∼56 ns, the N-terminus of the helix extends to include neighboring residues through K128. In another WT run, the helix converts to π-helix intermittently from ∼7 to 23.5 ns, spending ∼57% of this time in a π-helical structure before returning more stably to the α-helix structure. The mutant simulations are somewhat different. In one case, the α-helix in this region is very stable. In another, a fairly stable π-helix forms that lasts from ∼27.5 ns until the end of the run (60 ns). In the third simulation, the helix falls apart after only 3.5 ns. It returns stably for over 20 ns beginning around 27 ns into the run, but after that it comes apart again and does not refold before the end of the simulation.
Figure 5.
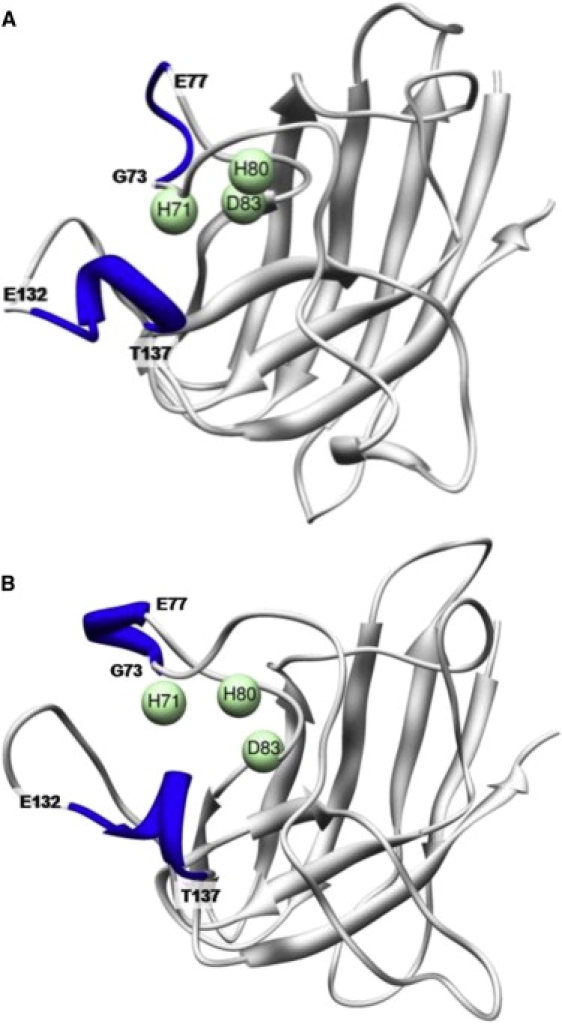
Helix near the Zn2+-binding residues. Locations of the native (E132-T137) and nonnative (G73-E77) helix are shown in WT (A) in the starting structure and (B) at 11.6 ns. Three of the zinc-binding residues are shown as spheres.
Although the A4V mutant has roughly WT levels of dismutase activity per copper, it also has only 50% as much copper bound (37). The destabilization of the electrostatic loop observed by MD may affect the ability of the copper chaperone to adequately load the copper ion into the binding site. Alternatively, the deformations of the loop may affect the protein's ability to hold onto the metal, accounting for the drop in observed copper binding.
Zinc-binding site
The change in average distance between some of the zinc-binding atoms is significant in the mutant simulations (Fig. 3 D). Although there is no difference in the distance between atoms 63ND1 and 80ND1 or between 71ND1 and 83OD1, there is a difference in the distances between 63ND1 and both 71ND1 and 83OD1, as well as between 80ND1 and both 71ND1 and 83OD1. These results indicate that residues H63 and H80 move together in the mutant, as do H71 and D83. This motion and the increased atom-atom distance may prevent the mutant from properly binding Zn2+, consistent with the experimentally observed 30-fold decrease in zinc affinity in the A4V mutant (38). Zinc binding is known to stabilize the protein (3,5,41), and loss of metal binding precedes oligomerization. Recent evidence suggests that loss of zinc binding occurs simultaneously with dimer dissociation on the millisecond timescale of the experimental work (42).
A nonnative helix is formed around residues 73–77 in WT (Fig. 5 B), a stretch that is unstructured in the crystal structure. Although these residues are not themselves involved with zinc binding, they are close to the zinc-binding residues H71 and H80. The different WT runs show a helical structure forming in this region for varying amounts of time. The nature of the helix also varies, alternating between α-helix, π-helix, and 3/10 helix; however, the formation of some helical structure at this location is consistent across all three simulations, whereas no significant amount of helix is found in this location during any of the mutant simulations. Structural stability in this region may be increased by the helix in the WT simulations, accounting for the smaller changes in the zinc-coordinating atom distances compared to the mutant simulations. Movements of the zinc-binding loop were noted in a previous work (22), but no mention was made of a helix in this area.
Contact network analysis revealed a potential propagation pathway for structural changes across the protein, which may also lead to the observed differences in the pairwise distances of the Zn2+-binding residues (Fig. 6 A). A maximum of seven contacts are needed for the four Zn2+ residues in the map (H63, H71, H80, and D83) at a 45% cutoff. The three Cu2+-binding residues (H48, H63, and H120) required a maximum of nine contacts, whereas the fourth residue (H46) could not be included at that cutoff.
Figure 6.
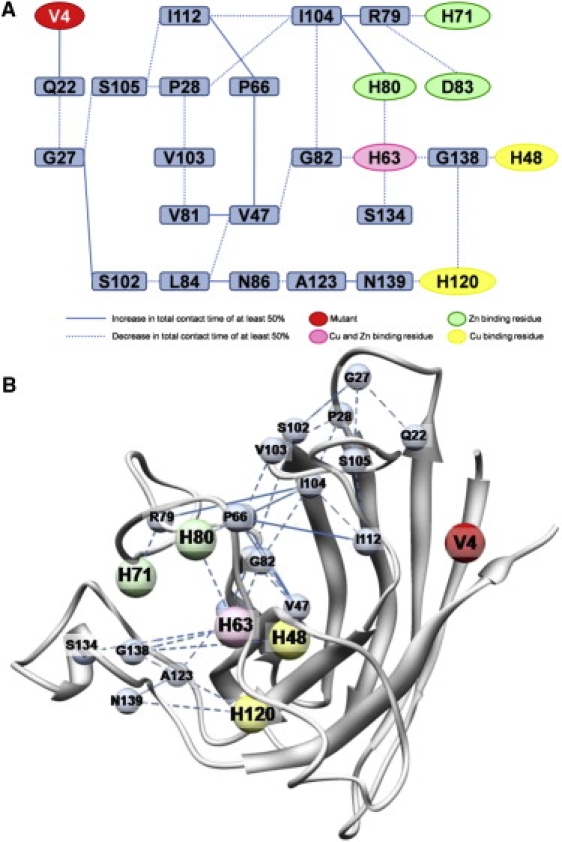
Contact differences between WT and A4V simulations. All changes shown represent a minimum of a 45% absolute change in contact time relative to WT simulations. (A) Map of the changes in contact time displayed for clarity. (B) The starting structure of the A4V mutant shown with selected changes in contact between residues. The changes displayed represent the shortest pathways of change between the mutation site and the metal-binding residues. Metal-binding residue not shown (Cu2+: H46) did not meet the threshold of 45% change in absolute contact time between WT and A4V simulations.
In Fig. 6 B this contact network is mapped onto the structure to illustrate how the A4V mutation causes a ripple effect through the protein. The contacts shift between residues, and the changes propagate throughout the protein. For example, G27 loses contact with S105 and moves to S102. The loss of contact between G27 and S105 causes S105 to lose contact with residues I112 and P28, both of which then lose contact with residue I104. Loss of contact between residues I112 and I104 causes residue I112 to come into contact with residue P66, a contact that almost never occurs in the WT simulations (2% ± 3.46%). Residue I104 then loses contact with G82 and has a significant increase in contact with residues H80 and R79, and R79 loses contact with H71. Note that H80 and H71 are zinc-binding residues. A list of the contacts gained and lost during the simulations, as reflected in Fig. 6, is provided in Table S1.
Through this type of analysis, we are able to identify key residues between the mutation site and the site of interest, such as residue I104, which serves as a branch point on the map leading to all three of the Zn2+-binding residues. This residue could be mutated to halt the propagation of the changes and, it is hoped, eliminate the phenotypes caused by the A4V mutation. However, even though I104 is highly conserved (43), there is a known fALS mutation at this residue: I104F. If I104 serves as a key residue in maintaining SOD1 stability, mutation at this site may only further exacerbate the instability of the monomer.
Potential effects on oligomeric assembly
Exposure of cysteines
Previous research indicates that C6 and C111 play a role in SOD1 aggregation, although reports vary. It is not clear whether both Cys residues are important for oligomerization (44,45), C111 is important but C6 is not (46), or C6 and C111 play some role but are not the only important factors (47).
Our results indicate that C111 has a ∼20% higher average solvent exposure in A4V compared to WT (69.1 Å2 for A4V vs. 57.1 Å2 for WT), and the close association of β8 with β1 in the mutant simulations effectively prevents a significant change in solvent accessibility for C6 (1.1 Å2 for A4V vs. 1.5 Å2 for WT). The increase in solvent accessibility for C111 was predicted via Carr-Purcell-Meiboom-Gill 15N nuclear spin relaxation dispersion measurements by K. Teilum, M. H. Smith, E. Schulz, G. Solomentsev, M. Oliveberg, and M. Akke (unpublished). This solvent exposure of the free cysteine C111 may allow for nonnative monomer-monomer interactions.
β-Sheet instability
The starting structures of both the WT (Fig. 2 C) and mutant proteins contain β-strand comprising residues G85–A89 (strand β5) and V94–D101 (strand β6). β5 is an edge strand of the sheet formed with strands β4, β7, and β8, whereas strand β6 is an edge strand of the sheet formed with strands β1, β2, and β3. Strands β5 and β6 are more stable in WT than in mutant simulations (Fig. 2, A and B). Previous MD simulations also found destabilization of β5 and β6 (23).
In the first two WT runs, the β-sheets are largely retained at β5 and β6, with some loss of strand from the N-terminal end of strand β6. In the third simulation, strand β6 loses most hydrogen bonds with β3 by 26 ns, although some β-bridge is retained through the end of the simulation. In the A4V simulations, it is β5 that is less stable. In the first mutant run, the hydrogen bonds formed with β4 have largely disappeared after only 2.2 ns, although some β-bridges appear infrequently. Strand β6 is much more stable, retaining some β-sheet contacts for ∼97% of the simulation and at a minimum some β-bridges for >99% of the 60 ns simulation. The second simulation reveals a loss of β-sheet beginning with β5 (residues G85–A89), which loses most of the β-structure after only a few nanoseconds. Some degree of β-structure is retained in this region for nearly 40 ns, however, alternating between fragments of β-sheet and β-bridges during that time. After 40 ns, what little structure is found in this area is α in nature, mostly α-bridges formed between residues N86 on β5 and H43 on β4. Strand β6 also begins to lose structure early in the simulation; however, the ends of the strand remain in contact with β3 for much of the first 40 ns of simulation. By 52.5 ns, there is a complete loss of β-structure in β6, but by 44.6 ns residue D96 begins to form frequent α-bridge contact with S34 on neighboring β3. This contact is present ∼94% of the time through the end of the simulation. The third mutant simulation also rapidly loses β-sheet contacts involving β5, as well as a slower but substantial loss of β-sheet contacts from β6. Strand β5 loses most of its β-structure by 4 ns, whereas β6 loses β-structure at ∼30 ns. In each of the mutant simulations, the loss of β-sheet contacts in β5 and β6 are accompanied by some loss of contacts in neighboring strands β4 and β3, respectively; however, each simulation retains β-sheet in these regions through the end of the runs. The observed destabilization of β5 and β6 is consistent with previous MD studies (23).
The typical forms of edge β-strands protect proteins from edge-edge aggregation (48). The perturbation and loss of the native edge strands in the mutant simulations provide another potential mechanism by which the mutant monomers can improperly interact with other monomers. The β-barrel, specifically β3 and β4 (49) and β5 and β6, was previously identified experimentally as a potential hotspot for local unfolding leading to aggregation in fALS cases (50), and our results are in good agreement with recent findings of β-strand destabilization of apo SOD1 monomers (24).
α-Strand and α-bridges
A hydrogen-bond analysis of the first A4V run shows nonhelical local α-structure beginning after 31 ns and continuing at some level through the end of the simulation. This local α-structure, when alternating between αL and αR, can give rise to strands similar to β-strands, but in this case the main chain carbonyl oxygens are aligned on one side and the amide hydrogens along the other, rather than alternating. We refer to this structure as α-sheet. From 31.1 ns to 51.3 ns, α-strand or bridges are present in the area of residues F50–G51 and S59–A60, near the dimer interface, for ∼44% of the time. At this point there is a ∼6 ns period during which there is no sampling of this structure; sampling then resumes at ∼57.6 ns and continues for roughly 64% of the time through the end of the run. A representative structure (32.8 ns) was chosen for further analysis of the α-sheet hydrogen-bond energies, particularly between residues 49–52 and 58–61 (Fig. 7). Note that residues G51 and D52 are dimer interface residues.
Figure 7.
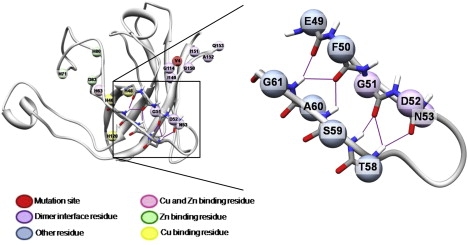
α-Sheet in the mutant simulations. A representative structure of the A4V simulations shows one location of α-sheet formation. In the right panel, the background structure and atoms have been removed for clarity.
In the second mutant simulation, intermittent α-bridges form between residues G51 and A60, H43 and N86, and S34 and D96, beginning at different time points but continuing through the end of the simulation. The α-bridge between residues G51 and A60 forms first, initially appearing at ∼6.6 ns into the simulation and persisting for ∼44% of the time. The α-bridge between residues H43 and N86 forms next at ∼40 ns of the run and is present ∼40% of the time through the end of the run. Finally, the α-bridge between residues S34 and D96 does not form until ∼45 ns into the run, but it is present ∼94% of the time thereafter. None of these α-bridges require either of the others to be present for formation. In the third A4V run, α-sheet again forms between residues F50–G51 and S59–A60, much like in the first mutant run. There is an α-bridge between residues G51 and A60 beginning around 4 ns of the simulation and continuing nearly 73% of the time through 8.4 ns of the simulation. At this point, it breaks and no significant amount of α-bridge or α-sheet appears until after nearly 27 ns of simulation time. After this time, there is an α-bridge or α-sheet present in this region >83% of the time through the end of the run.
In contrast to A4V, the α-bridge is barely populated (<5%) in the second WT simulation, and none is found in either of the other two WT runs. When we examine the data by (φ, ψ) angles, we can see that there is no α-strand comprised of four or more consecutive residues in any of the three neutral pH WT simulations that were run (data not shown); however, it is present in all three of the A4V simulations. In the first A4V simulation, this strand is predominantly found from residues T88 to D92, forming in the loop between β5 and β6. The other two simulations form α-strand in different locations, specifically residues L42–F45 in the second simulation (in strand β4) and at a lower level in the third simulation residues E40–H43 (beginning in the loop between β3 and β4 and continuing into β4). Some α-strand is also formed at residues V103–L106 in the second simulation, which is part of the β-barrel crossover loop between strands β6 and β7. This change may be permitted due to the significant changes in the contacts of residues V103–S105 in all of the mutant simulations, as discussed above.
It is not clear what role, if any, the presence of α-bridge or α-sheet may play in protein aggregation, although it has been observed in simulations of amyloidogenic proteins (32,51–53), leading to the suggestion that aggregation can be mediated by α-sheet (54). This structure is frequently seen in the A4V mutant SOD1 simulations and conspicuously absent from the WT simulations. As such, α-sheet could provide a catalyst for aberrant oligomerization.
Conclusions
MD simulations of the fALS-associated SOD1 mutant A4V in comparison with WT simulations indicate significant structural differences. We found lower overall stability in the mutant by a variety of measures, as well as perturbations of important dimer interface residues that explain the reduced dimerization observed experimentally. The zinc-binding site was disrupted, which could account for the reported 30-fold decrease in zinc-binding affinity in the mutant. This may be caused by a failure of the mutant to form a stabilizing α-helix in the zinc-binding loop. The helix in the electrostatic loop also undergoes greater conformational changes in the mutant simulations. It may be that the movement of this helix and the lack of helix in the zinc-binding loop contribute to the loss of β-strand observed in the mutant simulations. We also observed increased solvent exposure of a free cysteine residue and the presence of α-sheet in the A4V simulations, but not in the WT simulations. Taken together, these results provide possible explanations for dimer destabilization, loss of metallation, and oligomerization of the mutant protein.
Acknowledgments
This study was supported by a Genetic Approaches to Aging Training Grant from the Nathan Shock Center of Excellence in the Basic Biology of Aging (AG 00057 to T.S.) and a grant from the National Institutes of Health (GM 81407 to V.D.).
Supporting Material
References
- 1.Rakhit R., Chakrabartty A. Structure, folding, and misfolding of Cu,Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim. Biophys. Acta. 2006;1762:1025–1037. doi: 10.1016/j.bbadis.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 2.Pardo C.A., Xu Z., Borchelt D.R., Price D.L., Sisodia S.S. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc. Natl. Acad. Sci. USA. 1995;92:954–958. doi: 10.1073/pnas.92.4.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodriguez J.A., Valentine J.S., Eggers D.K., Roe J.A., Tiwari A. Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J. Biol. Chem. 2002;277:15932–15937. doi: 10.1074/jbc.M112088200. [DOI] [PubMed] [Google Scholar]
- 4.Lindberg M.J., Tibell L., Oliveberg M. Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo state. Proc. Natl. Acad. Sci. USA. 2002;99:16607–16612. doi: 10.1073/pnas.262527099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez J.A., Shaw B.F., Durazo A., Sohn S.H., Doucette P.A. Destabilization of apoprotein is insufficient to explain Cu,Zn-superoxide dismutase-linked ALS pathogenesis. Proc. Natl. Acad. Sci. USA. 2005;102:10516–10521. doi: 10.1073/pnas.0502515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross C.A., Poirier M.A. Opinion: what is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 7.Bruijn L.I., Houseweart M.K., Kato S., Anderson K.L., Anderson S.D. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 8.Durham H.D., Roy J., Dong L., Figlewicz D.A. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J. Neuropathol. Exp. Neurol. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Johnston J.A., Dalton M.J., Gurney M.E., Kopito R.R. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valentine J.S., Doucette P.A., Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- 11.Walsh D.M., Klyubin I., Fadeeva J.V., Rowan M.J., Selkoe D.J. Amyloid-β oligomers: their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- 12.Khare S.D., Caplow M., Dokholyan N.V. The rate and equilibrium constants for a multistep reaction sequence for the aggregation of superoxide dismutase in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA. 2004;101:15094–15099. doi: 10.1073/pnas.0406650101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rakhit R., Crow J.P., Lepock J.R., Kondejewski L.H., Cashman N.R. Monomeric Cu,Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J. Biol. Chem. 2004;279:15499–15504. doi: 10.1074/jbc.M313295200. [DOI] [PubMed] [Google Scholar]
- 14.Ray S.S., Nowak R.J., Strokovich K., Brown R.H., Jr., Walz T. An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry. 2004;43:4899–4905. doi: 10.1021/bi030246r. [DOI] [PubMed] [Google Scholar]
- 15.Johnston J.A., Madura K. Rings, chains and ladders: ubiquitin goes to work in the neuron. Prog. Neurobiol. 2004;73:227–257. doi: 10.1016/j.pneurobio.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Sherman M.Y., Goldberg A.L. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 17.Hough M.A., Grossmann J.G., Antonyuk S.V., Strange R.W., Doucette P.A. Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc. Natl. Acad. Sci. USA. 2004;101:5976–5981. doi: 10.1073/pnas.0305143101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chillemi G., Falconi M., Amadei A., Zimatore G., Desideri A. The essential dynamics of Cu, Zn superoxide dismutase: suggestion of intersubunit communication. Biophys. J. 1997;73:1007–1018. doi: 10.1016/S0006-3495(97)78134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Falconi M., Gallimbeni R., Paci E. Dimer asymmetry in superoxide dismutase studied by molecular dynamics simulation. J. Comput. Aided Mol. Des. 1996;10:490–498. doi: 10.1007/BF00124478. [DOI] [PubMed] [Google Scholar]
- 20.Khare S.D., Wilcox K.C., Gong P., Dokholyan N.V. Sequence and structural determinants of Cu, Zn superoxide dismutase aggregation. Proteins. 2005;61:617–632. doi: 10.1002/prot.20629. [DOI] [PubMed] [Google Scholar]
- 21.Khare S.D., Ding F., Dokholyan N.V. Folding of Cu, Zn superoxide dismutase and familial amyotrophic lateral sclerosis. J. Mol. Biol. 2003;334:515–525. doi: 10.1016/j.jmb.2003.09.069. [DOI] [PubMed] [Google Scholar]
- 22.Strange R.W., Yong C.W., Smith W., Hasnain S.S. Molecular dynamics using atomic-resolution structure reveal structural fluctuations that may lead to polymerization of human Cu-Zn superoxide dismutase. Proc. Natl. Acad. Sci. USA. 2007;104:10040–10044. doi: 10.1073/pnas.0703857104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khare S.D., Dokholyan N.V. Common dynamical signatures of familial amyotrophic lateral sclerosis-associated structurally diverse Cu, Zn superoxide dismutase mutants. Proc. Natl. Acad. Sci. USA. 2006;103:3147–3152. doi: 10.1073/pnas.0511266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding F., Dokholyan N.V. Dynamical roles of metal ions and the disulfide bond in Cu, Zn superoxide dismutase folding and aggregation. Proc. Natl. Acad. Sci. USA. 2008;105:19696–19701. doi: 10.1073/pnas.0803266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broom W.J., Johnson D.V., Auwarter K.E., Iafrate A.J., Russ C. SOD1A4V-mediated ALS: absence of a closely linked modifier gene and origination in Asia. Neurosci. Lett. 2008;430:241–245. doi: 10.1016/j.neulet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 26.Strange R.W., Antonyuk S., Hough M.A., Doucette P.A., Rodriguez J.A. The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J. Mol. Biol. 2003;328:877–891. doi: 10.1016/s0022-2836(03)00355-3. [DOI] [PubMed] [Google Scholar]
- 27.Beck, D.A.C., Alonso, D.O.V., Daggett, V. 2000–2008. in lucem Molecular Mechanics (ilmm). University of Washington, Seattle, WA.
- 28.Levitt M., Hirshberg M., Sharon R., Daggett V. Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution. Comput. Phys. Commun. 1995;91:215–231. [Google Scholar]
- 29.Levitt M., Hirshberg M., Sharon R., Laidig K.E., Daggett V. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. J. Phys. Chem. B. 1997;101:5051–5061. [Google Scholar]
- 30.Beck D.A., Armen R.S., Daggett V. Cutoff size need not strongly influence molecular dynamics results for solvated polypeptides. Biochemistry. 2005;44:609–616. doi: 10.1021/bi0486381. [DOI] [PubMed] [Google Scholar]
- 31.Beck D.A., Daggett V. Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods. 2004;34:112–120. doi: 10.1016/j.ymeth.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Armen R.S., Bernard B.M., Day R., Alonso D.O., Daggett V. Characterization of a possible amyloidogenic precursor in glutamine-repeat neurodegenerative diseases. Proc. Natl. Acad. Sci. USA. 2005;102:13433–13438. doi: 10.1073/pnas.0502068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kell G.S. Precise representation of volume properties of water at one atmosphere. J. Chem. Eng. Data. 1967;12:66–69. [Google Scholar]
- 34.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 35.Yee D.P., Dill K.A. Families and the structural relatedness among globular proteins. Protein Sci. 1993;2:884–899. doi: 10.1002/pro.5560020603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Hayward L.J., Rodriguez J.A., Kim J.W., Tiwari A., Goto J.J. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 2002;277:15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- 38.Crow J.P., Sampson J.B., Zhuang Y., Thompson J.A., Beckman J.S. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- 39.Doucette P.A., Whitson L.J., Cao X., Schirf V., Demeler B. Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant. Insights into the molecular basis for dimer stability. J. Biol. Chem. 2004;279:54558–54566. doi: 10.1074/jbc.M409744200. [DOI] [PubMed] [Google Scholar]
- 40.Borchelt D.R., Lee M.K., Slunt H.S., Guarnieri M., Xu Z.S. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. USA. 1994;91:8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Potter S.Z., Zhu H., Shaw B.F., Rodriguez J.A., Doucette P.A. Binding of a single zinc ion to one subunit of copper-zinc superoxide dismutase apoprotein substantially influences the structure and stability of the entire homodimeric protein. J. Am. Chem. Soc. 2007;129:4575–4583. doi: 10.1021/ja066690+. [DOI] [PubMed] [Google Scholar]
- 42.Mulligan V.K., Kerman A., Ho S., Chakrabartty A. Denaturational stress induces formation of zinc-deficient monomers of cu,zn superoxide dismutase: implications for pathogenesis in amyotrophic lateral sclerosis. J. Mol. Biol. 2008;383:424–436. doi: 10.1016/j.jmb.2008.08.024. [DOI] [PubMed] [Google Scholar]
- 43.Ikeda M., Abe K., Aoki M., Sahara M., Watanabe M. Variable clinical symptoms in familial amyotrophic lateral sclerosis with a novel point mutation in the Cu/Zn superoxide dismutase gene. Neurology. 1995;45:2038–2042. doi: 10.1212/wnl.45.11.2038. [DOI] [PubMed] [Google Scholar]
- 44.Niwa J., Yamada S., Ishigaki S., Sone J., Takahashi M. Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1. J. Biol. Chem. 2007;282:28087–28095. doi: 10.1074/jbc.M704465200. [DOI] [PubMed] [Google Scholar]
- 45.Banci L., Bertini I., Durazo A., Girotto S., Gralla E.B. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: a possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. USA. 2007;104:11263–11267. doi: 10.1073/pnas.0704307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cozzolino M., Amori I., Pesaresi M.G., Ferri A., Nencini M. Cysteine 111 affects aggregation and cytotoxicity of mutant Cu,Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 2008;283:866–874. doi: 10.1074/jbc.M705657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karch C.M., Borchelt D.R. A limited role for disulfide cross-linking in the aggregation of mutant SOD1 linked to familial amyotrophic lateral sclerosis. J. Biol. Chem. 2008;283:13528–13537. doi: 10.1074/jbc.M800564200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richardson J.S., Richardson D.C. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaw B.F., Durazo A., Nersissian A.M., Whitelegge J.P., Faull K.F. Local unfolding in a destabilized, pathogenic variant of superoxide dismutase 1 observed with H/D exchange and mass spectrometry. J. Biol. Chem. 2006;281:18167–18176. doi: 10.1074/jbc.M600623200. [DOI] [PubMed] [Google Scholar]
- 50.Nordlund A., Oliveberg M. Folding of Cu/Zn superoxide dismutase suggests structural hotspots for gain of neurotoxic function in ALS: parallels to precursors in amyloid disease. Proc. Natl. Acad. Sci. USA. 2006;103:10218–10223. doi: 10.1073/pnas.0601696103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Armen R.S., Daggett V. Characterization of two distinct β2-microglobulin unfolding intermediates that may lead to amyloid fibrils of different morphology. Biochemistry. 2005;44:16098–16107. doi: 10.1021/bi050731h. [DOI] [PubMed] [Google Scholar]
- 52.Armen R.S., DeMarco M.L., Alonso D.O., Daggett V. Pauling and Corey's α-pleated sheet structure may define the prefibrillar amyloidogenic intermediate in amyloid disease. Proc. Natl. Acad. Sci. USA. 2004;101:11622–11627. doi: 10.1073/pnas.0401781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Armen R.S., Alonso D.O., Daggett V. Anatomy of an amyloidogenic intermediate: conversion of β-sheet to α-sheet structure in transthyretin at acidic pH. Structure. 2004;12:1847–1863. doi: 10.1016/j.str.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 54.Daggett V. α-Sheet: the toxic conformer in amyloid diseases? Acc. Chem. Res. 2006;39:594–602. doi: 10.1021/ar0500719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.