

Abstract
 Potassium azidoaryltrifluoroborates have been prepared from the corresponding haloaryltrifluoroborates in 73–98% yields. Also, we successfully cross-coupled the azido-functionalized organotrifluoroborates and carried out a one-pot sequential cross-coupling/1,3-dipolar cycloaddition and a one-pot cross-coupling/azide reduction process.
Potassium azidoaryltrifluoroborates have been prepared from the corresponding haloaryltrifluoroborates in 73–98% yields. Also, we successfully cross-coupled the azido-functionalized organotrifluoroborates and carried out a one-pot sequential cross-coupling/1,3-dipolar cycloaddition and a one-pot cross-coupling/azide reduction process.
The azide functional group has been used as an important moiety for the formation of nitrogen-containing compounds in fields ranging from synthetic organic chemistry, to pharmaceutical chemistry, materials science, and biology. 1 Alkyl and aryl azides have gained prominence in particular because they may be used for the preparation of [1,2,3]-triazoles by Cu-catalyzed 1,3-dipolar cycloadditions onto terminal alkynes (via “Click” chemistry). 2 Unfortunately, the preparation of certain classes of organic azides has presented considerable challenges. Moreover, to the best of our knowledge, the Suzuki-Miyaura cross-coupling reaction with boron reagents bearing the azide functional group has not been reported, perhaps because of the inherent difficulty in preparing such bifunctional molecules and the perceived instability of the azide under cross-coupling reaction conditions.
Recently, organotrifluoroborate salts have been used as important synthetic reagents in the Suzuki-Miyaura cross-coupling reaction, providing many advantages over the corresponding boronic acids or boronate esters. 3 The organotrifluoroborates are air- and moisture-stable, crystalline solids that are inert to various nucleophilic reagents owing to the tetracoordinate nature of the boron.4
Consequently, it seemed likely that they would be tolerant of conditions allowing the incorporation of the azide functional group. Herein, we describe the first preparation of azidoaryltrifluoroborates from the corresponding haloaryltrifluoroborates and reaction conditions permitting Suzuki-Miyaura cross-coupling reactions of the azidoaryltrifluoroborates thus generated.
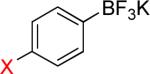
As a starting point, potassium haloaryltrifluoroborates were generated via the one-pot synthesis of aryl dihalides and B(OiPr)3 using 1.0 equiv of n-BuLi (Table 1). When dibromo or diiodobenzenes were used as starting materials, the target compounds were obtained in good yields except when 1,2-diiodobenzene was used. Interestingly, the reaction of dibromo pyridines gave the corresponding organotrifluoroborates in excellent yields. The reaction of 2,5-dibromo-p-xylene was also problematic, providing the desired product in much lower yield (Table 1, 5a 47%) under the same reaction conditions.
Table 1.
One-Pot Preparation of Potassium Haloaryltrifluoroborates from Various Aryl Dihalidesa
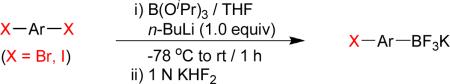 | |||||
|---|---|---|---|---|---|
| entry | X–Ar–X | product | yield | ||
| 1 | 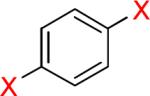 |
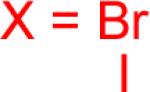 |
 |
1a-Br 1a-I |
86% 90% |
| 2 | 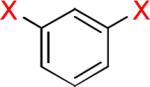 |
 |
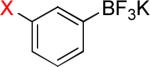 |
2a-Br 2a-I |
56% 85% |
| 3 | 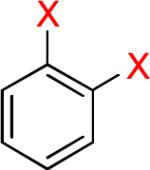 |
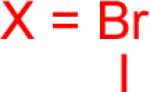 |
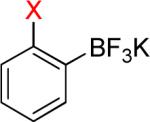 |
3a – |
52% – |
| 4 | 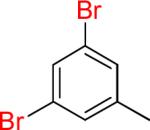 |
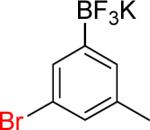 |
4a | 81% | |
| 5 | 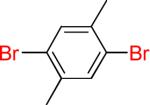 |
 |
5a | 47% | |
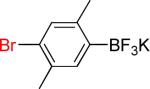
| 6 | 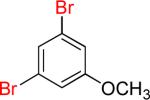 |
 |
6a | 82% | |
| 7 | 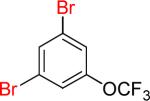 |
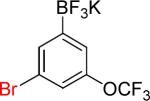 |
7a | 82% | |
| 8 | 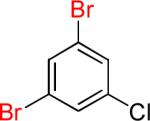 |
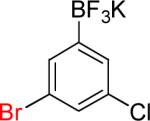 |
8a | 72% | |
| 9 | 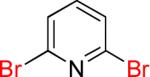 |
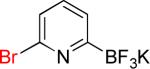 |
9a | 90% | |
| 10 | 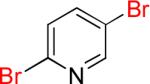 |
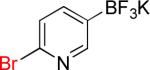 |
10a | 87% | |
| 11 | 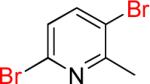 |
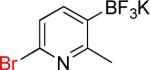 |
11a | 90% | |
| 12 | 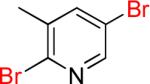 |
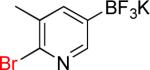 |
12a | 85% | |
| 13 | 13a | 93% | |||
| 14 | 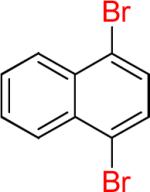 |
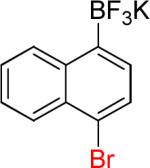 |
14a | 94% | |
| 15 | 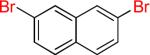 |
15a | 90% | ||
| 16 | 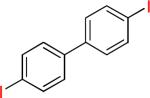 |
 |
16a | 80% | |
Reaction conditions: aryl dihalide (2.0 mmol), triisopropyl borate (2.0 mmol), and n-BuLi (2.0 mmol) at −78 °C to rt under N2 and then quenched with 1 N KHF2.
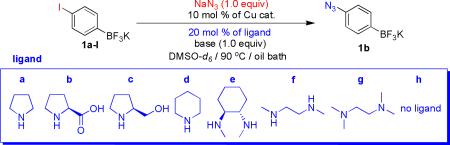
Next, using conditions previously developed for the preparation of aryl azides with aryl halide and Cu/amine-ligand,1, 5 we attempted the formation of azidoaryltrifluoroborates (Table 2).
Table 2.
Optimization of Reaction Conditions for the Preparation of Potassium 4-Azidophenyltrifluoroborate (1b)a
 | |||||
|---|---|---|---|---|---|
| entry | CuX | ligand | base | reaction time (h) | conversion yield (%)b |
| 1 | CuI | a | K2CO3 | 24 | 66 |
| 2 | CuI | b | K2CO3 | 5 | 98 |
| 3 | CuI | c | K2CO3 | 16 | 100 |
| 4 | CuI | d | K2CO3 | 24 | 81 |
| 5 | CuI | e | K2CO3 | 15 | 100 |
| 6 | CuI | f | K2CO3 | 1 | 100 (88)c |
| 7 | CuI | g | K2CO3 | 24 | trace |
| 8 | CuI | h | K2CO3 | 48 | 68 |
| 9 | CuId | f | K2CO3 | 2 | 100 (89)c |
| 10 | CuBr | f | K2CO3 | 1 | 100 (92)c |
| 11 | CuBr | f | Cs2CO3 | 0.5 | 100 (94)c |
| 12e | CuBr | f | Cs2CO3 | 2.5 | 100 |
All reactions were performed on a 0.05 mmol scale in 0.5 mL of DMSO-d6 in an NMR tube.
Percent conversions were determined by 1H NMR of the reaction mixtures. The conversion yield was based on the integration of peaks at 7.14 (1a-I) ppm and 6.85 (1b) ppm, respectively.
Reactions were performed on a 0.1 mmol scale and isolated yields are reported.
5 mol % of CuI was used.
Reaction was performed in DMF-d7.
We first carried out the reaction of potassium 4-azidophenyltrifluoroborate (1b) generated in situ by treatment of 1a-I with 1.0 equiv of NaN3 in the presence of 10 mol % of Cu(I) and various amine ligands.
A number of different amine ligands were screened for their efficacy in promoting the azidation, and it was found that N,N’-dimethylethylenediamine (ligand f) provided the fastest reaction time and the highest converted yield (Table 2, entry 6). Although both CuI and CuBr catalysts generated the target compound 1b under the same reaction conditions (Table 2, entries 6 and 10), the isolated yield and purity of compound 1b using CuBr were better than those of using CuI. A decrease in the catalyst loading from 10 to 5 mol % effectively doubled the reaction time (Table 2, entries 6 and 9).
When Cs2CO3 was used as a base instead of K2CO3, the reaction time decreased from 1 h to 30 min (Table 2, entries 10 and 11), and DMSO-d6 appeared to be a better solvent than DMF-d7 (Table 2, entries 11 and 12). Using these conditions (Table 2, entry 11), the azidation of various potassium haloaryltrifluoroborates was examined (Table 3). As a general rule, isolated yields and reaction rates of the corresponding azidophenyltrifluoroborates increased in the order para > meta > ortho under the same conditions (Table 3, 1b– 3b). Both mono- and dimethyl-substituted aryltrifluoroborates afford the corresponding azidoaryltrifluoroborates in satisfactory yields (Table 3, 4b– 5b). Surprisingly, when organotrifluoroborates 7a, 8a, and 13a were used as starting materials, aminoaryltrifluoroborates were generated in good yields instead of azidoaryltrifluoroborates.
Table 3.
One-Pot Preparation of Potassium Azidoaryltrifluoroboratesa
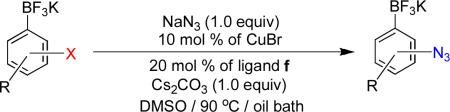 | ||||||
|---|---|---|---|---|---|---|
| entry | X–Ar–BF3K | reaction time (h) | product | yield | ||
| 1 |  |
1a-Br 1a-I |
7 1 |
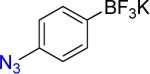 |
1b | 95% 95%b |
| 2 | 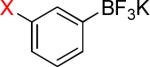 |
2a-Br 2a-I |
9 6 |
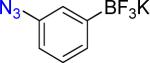 |
2b | 90% 93% |
| 3 | 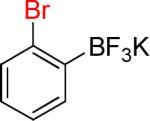 |
3a | 24 | 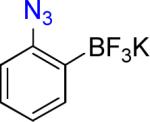 |
3b | 73% |
| 4 | 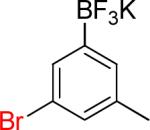 |
4a | 8 | 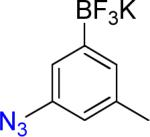 |
4b | 93% |
| 5 | 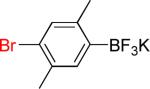 |
5a | 12 | 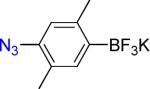 |
5b | 90% |
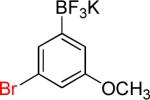
| 6 | 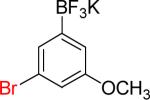 |
6a | 4 | 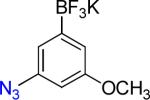 |
6b | 92%c |
| 7 | 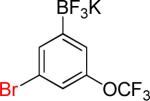 |
7a | 12 | 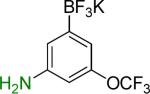 |
7b | 83%c |
| 8 | 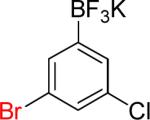 |
8a | 12 | 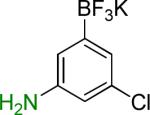 |
8b | 91%c |
| 9 | 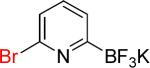 |
9a | 1 | 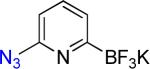 |
9b | 98% |
| 10 | 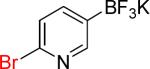 |
10a | 1 | 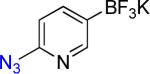 |
10b | 98% |
| 11 | 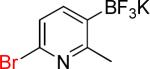 |
11a | 3 | 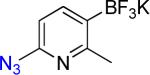 |
11b | 95% |
| 12 | 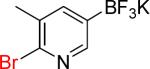 |
12a | 3 | 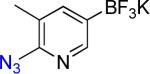 |
12b | 95% |
| 13 | 13a | 12 | 13b | 90% | ||
| 14 | 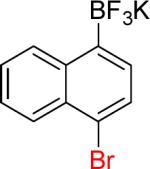 |
14a | 10 | 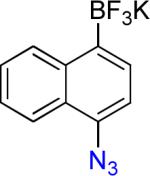 |
14b | (82%)c,d |
| 15 | 15a | 13 | 15b | (82%)e | ||
| 16 | 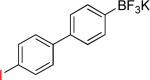 |
16a | 5 min | 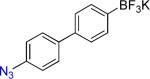 |
16b | 96% |
All reactions were performed on a 1.0 mmol scale in 4 mL of DMSO and monitored by 1H NMR in D2O. Yields are given for isolated products.
Reaction was performed on a 3.0 mmol scale.
Products were obtained as amorphous solids.
Product was contaminated with about 10% of 14a.
Product was contaminated with about 15% of the azide product.
On the other hand, the azidation reactions of bromopyridinyl and iodonaphthyl organotrifluoroborates proceeded readily to give the desired azide compounds in excellent yields (Table 3, 9b– 12b and 16b). Naphthyl organotrifluoroborates were not suitable for this azidation because the resulting products were contaminated with the starting material or mixtures of azido- and aminoaryltrifluoroborates (Table 3, 14b and 15b).
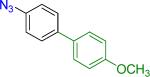
We next examined the Suzuki-Miyaura cross-coupling reaction of 4-azidophenyltrifluoroborate (1b) and various aryl halides in the presence of Pd catalyst and 3.0 equiv of Cs2CO3 (Table 4). As expected, the coupling of aryl and heteroaryl bromides led to the corresponding target products in good yields. Aryl chlorides were generally ineffective as coupling partners.
Table 4.
Cross-Coupling Reactionsa
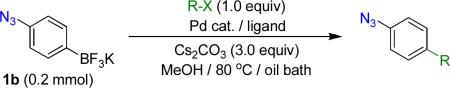 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R-X | reaction conditions | reaction time (h) | product | yield | ||
| 1 | 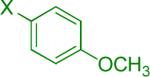 |
A B |
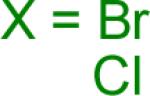 |
0.5 6 |
 |
17 | 85% – |
| 2 | 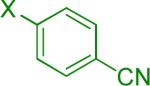 |
A B |
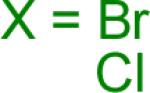 |
1 6 |
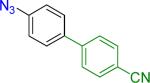 |
18 | 83% 48% |
| 3 | 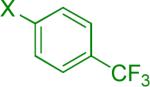 |
A B |
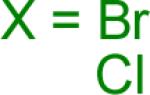 |
0.5 3 |
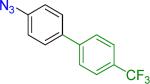 |
19 | 58% 45% |
| 4 | 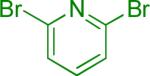 |
A | 3 | 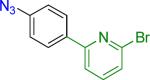 |
20 | 81% | |
Reaction conditions A: potassium 4-azidophenyltrifluoroborate (1b, 0.2 mmol), aryl bromide (0.2 mmol), 10 mol % of PdCl2(dppf)·CH2Cl2, Cs2CO3 (0.6 mmol), MeOH (1.5 mL), 80 °C.; B: 1b (0.2 mmol), aryl chloride (0.2 mmol), 3 mol % of Pd(OAc)2, 6 mol % of XPhos, Cs2CO3(0.6 mmol), 1,4-dioxane/H2O (10/1, 1.5 mL), 100 °C. Yields are given for isolated products.
Finally, we examined one-pot sequential reactions that incorporated cross-coupling followed by 1,3-dipolar cycloaddition or NaBH4 reduction to the amine (eqs 1 and 2).6 These processes provided the desired compounds in good overall yields.
 |
In summary, we have developed a new synthetic method for the preparation of potassium azidoarytrifluoroborates from the corresponding haloaryltrifluoroborates. Additionally, we successfully cross-coupled the azido-functionalized organotrifluoroborates and carried out a one-pot sequential cross-coupling/1,3-dipolar cycloaddition and a one-pot cross-coupling/azide reduction process. Further investigations on transformations of azido-substituted trifluoroborates are currently underway in our laboratory.
Supplementary Material
Acknowledgment
This research was supported by KIST Institutional Program (2Z03270) and a grant from Marine Biotechnology Program funded by Ministry of Land, Transport and Maritime Affairs, Republic of Korea. Support from the NIH General Medical Sciences Institute (USA) is also acknowledged.
Footnotes
Supporting Information Available: General experimental procedures, compound characterization data, and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Sheradsky T. In: Chemistry of the Azido Group. Patai S, editor. Wiley; New York: 1971. [Google Scholar]; b Scriven EFV, Turnbull K. Chem. Rev. 1988;88:297. [Google Scholar]; c Bräse S, Gil C, Knepper K, Zimmermann V. Angew. Chem., Int. Ed. 2005;44:5188. doi: 10.1002/anie.200400657. and references therein. [DOI] [PubMed] [Google Scholar]
- 2.a Huisgen R. In: 1,3-Dipolar Cycloaddition Chemistry. Chapter 1. Padwa A, editor. Wiley; New York: 1984. p. 1. [Google Scholar]; b Padwa A. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 4. Pergamon; Oxford: 1991. p. 1069. [Google Scholar]; c Gothelf KV, Jørgensen KA. Chem. Rev. 1998;98:863. doi: 10.1021/cr970324e. [DOI] [PubMed] [Google Scholar]; d Kolb HC, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; e Gil MV, Arévalo MJ, Lopez Ó. Synthesis. 2007;11:1589. [Google Scholar]; f Meldal M, Tornoe CW. Chem. Rev. 2008;108:2952. doi: 10.1021/cr0783479. and references therein. [DOI] [PubMed] [Google Scholar]
- 3.a Molander GA, Figueroa R. Aldrichimica Acta. 2005;38:49. [Google Scholar]; b Stefani HA, Cella R, Vieira AS. Tetrahedron. 2007;63:3623. [Google Scholar]; c Molander GA, Ellis N. Acc. Chem. Res. 2007;40:275. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]; d Darses S, Genet J-P. Chem. Rev. 2008;108:288. doi: 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]
- 4.a Molander GA, Ham J. Org. Lett. 2006;8:2031. doi: 10.1021/ol060375a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Molander GA, Sandrock DL. Org. Lett. 2007;9:1597. doi: 10.1021/ol070543e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Molander GA, Canturk B. Org. Lett. 2008;10:2135. doi: 10.1021/ol800532p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ahn HR, Cho YA, Kim D-S, Chin J, Gyoung Y-S, Lee S, Kang H, Ham J. Org. Lett. 2009;11:361. doi: 10.1021/ol8025999. [DOI] [PubMed] [Google Scholar]
- 5.a Zhu W, Ma D. Chem. Commun. 2004:888. doi: 10.1039/b400878b. [DOI] [PubMed] [Google Scholar]; b Andersen J, Madsen U, Björkling F, Liang X. Synlett. 2005;14:2209. and references therein. [Google Scholar]
- 6.For detailed procedures, see Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.