

Summary
Purine nucleoside phosphorylase (PNP) catalyzes the phosphorolysis of 6-oxy-purine nucleosides to the corresponding purine base and α-d-ribose 1-phosphate. Its genetic loss causes a lethal T-cell deficiency. The highly reactive ribocation transition state of human PNP is protected from solvent by hydrophobic residues that sequester the catalytic site. The catalytic-site was enlarged by replacing individual catalytic site amino acids with glycine. Reactivity of the ribocation transition state was tested for capture by water and other nucleophiles. In the absence of phosphate, inosine is hydrolyzed by native, Y88G, F159G, H257G, and F200G enzymes. Phosphorolysis but not hydrolysis is detected when phosphate is bound. An unprecedented N9-to-N3 isomerization of inosine is catalyzed by H257G and F200G in the presence of phosphate and by all PNPs in the absence of phosphate. These results establish a ribocation lifetime too short to permit capture by water. An enlarged catalytic site permits ribocation formation with relaxed geometric constraints, permitting nucleophilic rebound and N3-inosine isomerization.
Introduction
Purine nucleoside phosphorylase (PNP) catalyzes the reversible phosphorolysis of 6-oxo-purine (deoxy)ribonucleosides to the corresponding purine base and α-d-ribose 1-phosphate (Figure 1, path b) (Schramm, 2005; Stoeckler, et al., 1978; Stoeckler, et al., 1980). This enzyme is a current target for clinical trials involving T-cell cancers and autoimmune disease (BioCryst, 2009). Kinetic isotope effects have characterized the reaction catalyzed by human PNP as proceeding via a fully-dissociated ribocation transition state (TS) with complete leaving-group departure prior to ribosyl migration to the phosphate nucleophile (Lewandowicz and Schramm, 2004). Despite the high amino acid sequence identity (87%) with human PNP, bovine PNP forms an earlier ribooxacarbenium-ion TS featuring a C1′–N9 bond length of ∼1.8 Å (Kline and Schramm, 1993). Bovine PNP catalyzes the slow hydrolysis of inosine to hypoxanthine and ribose in the absence of phosphate with a turnover number of 1.3 × 10-4 s-1 (Figure 1, path c) (Kline and Schramm, 1992; Kline and Schramm, 1995). Inosine hydrolysis has not been previously reported for human PNP.
Figure 1. Reactions catalyzed by purine nucleoside phosphorylase.

Depending on reaction conditions, the ribooxacarbenium-ion transition state can partition between (a) return to inosine [1], (b) reaction with phosphate to yield hypoxanthine [2] and α-d-ribose 1-phosphate [4], (c) reaction with water to yield hypoxanthine [2] and d-ribose [3], or (d) reaction with N3 of hypoxanthine to yield N3-isoinosine [5].
Crystal structures of human PNP complexed with substrate and TS analogues have defined the active site residues in contact with the purine nucleoside and the phosphate nucleophile (de Azevedo, et al., 2003; Koellner, et al., 1997; Rinaldo-Matthis, et al., 2008; Shi, et al., 2004). Human PNP is a homotrimer, and the catalytic site of PNP is located near the subunit-subunit interfaces. The catalytic site conforms closely to bound inosine with residues N243, E201, H257, F200, Y88, M219, and F159′ (from the adjacent subunit) as contacts for the nucleoside. The side chains of N243 and E201 are involved in leaving-group activation by forming hydrogen-bond contacts with the purine base (N7H and N1H, respectively) (Ghanem, et al., 2008b; Nunez, et al., 2006). Residues F159′, H257, F200, and Y88 form a hydrophobic cover to the catalytic site and are proposed to shield reactants in the catalytic site from bulk solvent (Figure 2) (Saen-Oon, et al., 2008a). The phenyl group of F200 is perpendicular to the purine base, and residues Y88, F159′, H257 and the bound phosphate nucleophile provide contacts to the ribose. Residue F159′ from a mobile loop in the adjacent subunit covers the top surface of the ribosyl group of the purine nucleoside (Figure 2). Quantum mechanical simulations and laser-induced temperature-jump studies establish that dynamic motion of this residue is closely associated with TS formation (Ghanem, et al., 2009; Saen-Oon, et al., 2008a). H257 is a key residue in positioning the “oxygen stack” (H257:Nδ–O5′–O4′–OP) that contributes to catalysis through a vibrational promoting mode of the three oxygen atoms to provide electron density changes to destabilize the ribosyl group, form the carbocation TS and thereby enhance departure of the purine-base leaving group (Murkin, et al., 2007; Saen-Oon, et al., 2008a). The crystal structure of human PNP in complex with a TS analogue (Immucillin-H) and phosphate shows structural water molecules near O6 (2.6 Å) and another within 4.4 Å of the ribosyl anomeric carbon (Figure 2), and these structural waters are also found in the catalytic site of bovine PNP (Fedorov, et al., 2001).
Figure 2. Views of the subunit-subunit interface of human PNP in complex with a TS analogue and phosphate (PO4).
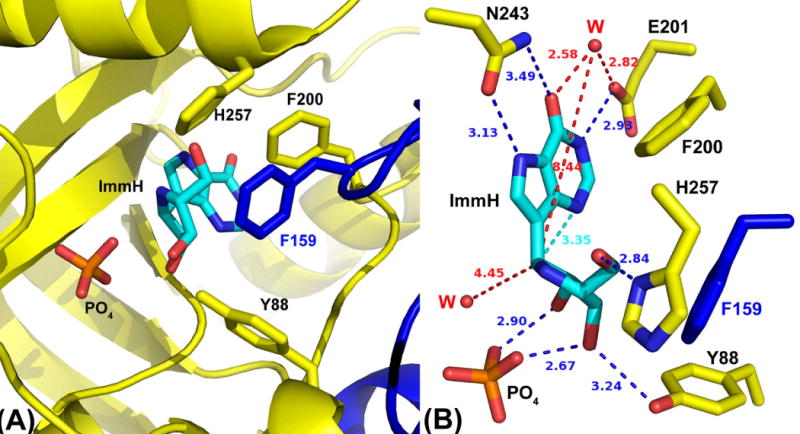
(A) PNP catalytic-site contact residues surrounding Immucillin-H (ImmH) (PDB 1RR6). Adjacent subunits in the trimeric protein have been colored yellow and blue.
(B) Contact residues map, with distances in Å units. The two water molecules (W) closest to ImmH have been included.
We explored two possibilities for water capture of the highly reactive ribocation: 1) water molecules located within the catalytic site prior to ribocation formation or 2) water molecules that diffuse from bulk solvent into the catalytic site as a consequence of catalytic-site mutations. Water capture of the ribocation in the presence of phosphate would partition the pentose between ribose and α-d-ribose 1-phosphate products. This approach provides a test of ribocation reactivity, its lifetime relative to water diffusion, and allows an estimate of the relative proclivity of the ribocation to react with water, phosphate, the hypoxanthine leaving group or adventitious catalytic site nucleophiles. This work contributes substantially to our understanding of the ribocation transition state.
13C and 1H NMR spectroscopy were used to monitor the hydrolysis and phosphorolysis reactions with [1′-13C]inosine as substrate. Native PNP and catalytic-site glycine mutants of human PNP were analyzed. Four different glycine mutants of residues that are proposed to shield the catalytic site from bulk solvent were engineered in attempts to introduce a solvent leak. Partition of products between d-ribose and α-d-ribose 1-phosphate permit estimation of ribocation lifetime, based on the diffusion rate of water and the distance of the ribocation from bulk solvent in a leaky catalytic site.
Results
Steady-State Kinetic Properties
The steady-state kinetic parameters of four catalytic-site glycine mutants were determined with inosine as substrate and compared to those for the native enzyme. All catalytic-site glycine mutants showed a decrease in their kcat values ranging from 2-(Y88G) to 40-fold (F200G) compared to native PNP (Table S1). The catalytic efficiencies (kcat/Km) of those mutants were reduced by ∼8-fold (Y88G) and up to 11,000-fold (F200G) relative to native PNP (Table S1). Purified PNPs showed sufficient catalytic stability in the reaction mixtures at 25 °C to permit collection of 13C or 1H NMR spectra of actively equilibrating enzyme mixtures for 24 h.
13C and 1H NMR Spectroscopy and Assignments
The 13C and 1H NMR spectra of reaction mixtures containing PNPs (native, Y88G, F159G, H257G and F200G) with [1′-13C]inosine in the presence or absence of phosphate were acquired and compared with the reference spectra of free [1′-13C]inosine and [1-13C]ribose (Figure 3 and Table S2). The 13C NMR spectrum of free [1′-13C]inosine [1] is a singlet with a chemical shift of 88.68 ppm (Chenon, et al., 1975a; Ebrahimi, et al., 2001). The downfield region of the 1H NMR spectrum of free [1′-13C]inosine shows two singlets for H8 and H2 with chemical shifts of 8.33 and 8.21 ppm, respectively, and a doublet of doublets for H-1′ (J1′,2′ = 5 Hz, JH,C = 170 Hz), with a chemical shift of 6.07 ppm (Figure 3A and Table S2). The peak assignments were based on the previously reported chemical shifts for purine nucleosides (Chenon, et al., 1975a; Chenon, et al., 1975b). The 13C NMR spectrum of [1-13C]ribose [3], obtained through the irreversible arsenolysis of [1′-13C]inosine in the presence of PNP [5], shows four singlets with chemical shifts of 93.94, 94.22, 96.68 and 101.35 ppm (Figure 3B and Table S2), corresponding to the four major isomers of d-ribose. The chemical shifts and ratio of the four isomer singlets agree with previously determined values of 95.1 (20%), 95.4 (60%), 97.8 (7%) and 102.5 ppm (13%) for α-d-ribopyranose, β-d-ribopyranose, α-d-ribofuranose and β-d-ribofuranose, respectively (Kenneth N. Drew, 1998; Ryu, et al., 2004). The 1H NMR spectrum of free hypoxanthine [2] obtained through the irreversible arsenolysis of labeled inosine shows two downfield singlets for H-8 and H-2, with chemical shifts of 8.19 and 8.17 ppm, respectively (Figure 3B).
Figure 3. 13C NMR (I) and 1H NMR (II) spectra of [1′-13C]inosine, [1-13C]ribose and hypoxanthine.
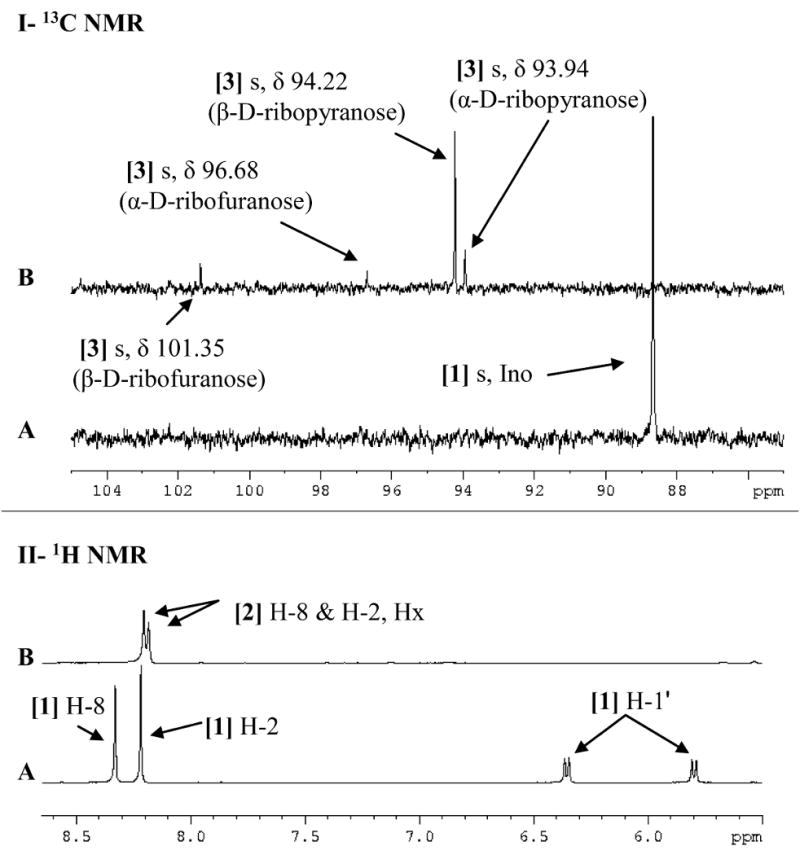
Spectra are of (A) 5.3 mM free [1′-13C]inosine and (B) [1-13C]ribose and hypoxanthine obtained through the irreversible arsenolysis of 5.3 mM [1′-13C]inosine in the presence of human PNP and 50 mM Na2HAsO4 (pH 7.4). Upfield signals have been omitted to highlight the downfield regions of interest. All spectra were acquired at 25 °C in 10% D2O. [1] Ino, inosine; [2] Hx, hypoxanthine.
Phosphate-free PNP Reactions and Inosine Isomerization
In the absence of phosphate, the time-course (to 24 h) 13C NMR spectra of the inosine reaction mixtures for F159G, Y88G, H257G and F200G showed no spectral changes (data not shown). Native PNP with [1′-13C]inosine showed the formation of a new singlet with a chemical shift of 90.59 ppm (Figure 4-I). This chemical shift does not match those for ribose isomers (Figure 3B). The time-course (0 to 24 h) 1H NMR spectra of mixtures of PNPs (native, F159G, Y88G, H257G and F200G) with [1′-13C]inosine were also acquired. The 1H NMR spectra of [1′-13C]inosine with native PNP showed two new downfield singlets with chemical shifts of 8.55 and 8.18 ppm and another doublet of doublets with a chemical shift of 6.28 ppm between ∼3-6 and 24 h (Figure 4-II). The chemical shifts resemble those reported for the chemically synthesized N3-isomer of inosine (3-β-d-ribofuranosyl hypoxanthine or N3-isoinosine) [5] (s δ 8.67 or 8.59, s δ 8.32 or 8.25 and d δ 5.98 or 5.87 for H-8, H-2 and H-1′, respectively, in d6, DMSO) (Lehikoinen, et al., 1989; Tindall, et al., 1972). The rate of isomerization was approximately 1 × 10-3 s-1 (Table 1). In comparison, the four catalytic-site glycine mutants did not form any detectable isomerized product during the time-course of 1H NMR observation (Figure 4-III, as an example for F200G).
Figure 4. 13C and 1H NMR spectra of [1′-13C]inosine with human PNP in the absence of phosphate.
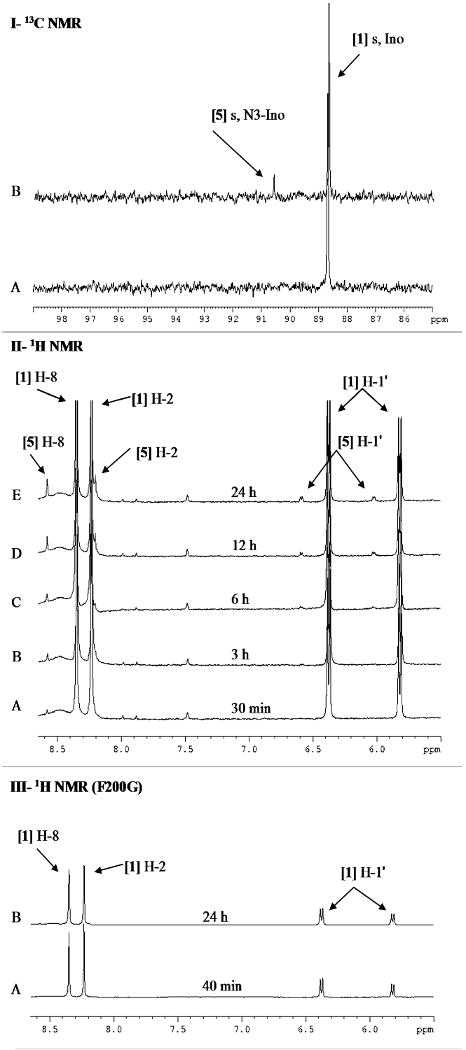
Panel I, 13C NMR spectra of [1′-13C]inosine with native human PNP after (A) 30 min and (B) 24 h.
Panel II, 1H NMR spectra of [1′-13C]inosine with native human PNP after (A) 30 min, (B) 3 h, (C) 6 h, (D) 12 h, and (E) 24 h.
Panel III, 1H NMR spectra of [1′-13C]inosine with F200G after (A) 40 min and (B) 24 h.
All 13C and 1H spectra were acquired with 5.3 mM [1′-13C]inosine and 6 μM PNP in the absence of phosphate at 25 °C in 10% D2O. [1] Ino, inosine; [5] N3-Ino, N3-inosine.
Table 1.
Comparison of the N3 isomerization and single-turnover hydrolysis reactions for PNPs with inosine as substrate.
| Isomerization kineticsa | Hydrolysis kineticsb | ||
|---|---|---|---|
| PNP | kiso (s-1), no phosphatec | kiso (s-1), with phosphatec | khyd (s-1) c |
| BtPNPd | not determined | not determined | 2.0 × 10-3 |
| Native | 1 × 10-3 | undetectablee | (7.5 ± 1.6) × 10-4 |
| Y88G | undetectablee | undetectablee | (1.7 ± 0.5) × 10-3 |
| F159G | undetectablee | undetectablee | (3.0 ± 0.8) × 10-3 |
| H257G | undetectablee | 7.0 × 10-5 | (4.8 ± 0.4) × 10-4 |
| F200G | undetectablee | 4.2 × 10-4 | (3.6 ± 1.2) × 10-3 |
Isomerization assays were performed with 5.3 mM inosine in 20 mM Tris-HCl (pH 7.4) in the presence or absence of 50 mM KH2PO4 (pH 7.4). PNPs were added to give a phosphorolysis Vmax of 2.6 × 10-4 M s-1 (kcat × [E]T).
Hydrolysis assays were performed with equal amounts of inosine and PNP (80 μM) in 20 mM Tris-Cl, pH 7.4, at 25 °C. Reactions were quenched with 1 N HCl, and the ratio between substrate (inosine) and product (hypoxanthine) was quantified by reversed-phase HPLC.
kiso and khyd were calculated as indicated in the Supplemental Data.
From ref 7, at 30 °C.
N3-Isoinosine was not detectable by 1H or 13C NMR during the time-course; the limit for detection was estimated to be ∼25 μM (0.5% of the initial [inosine]).
The identity of the isomerized product as N3-isoinosine was determined by several independent criteria. Resolution from inosine and hypoxanthine by HPLC established a distinct chemical entity. The ESI-MS mass value (m/z = 270, M+H+ for the 1′-13C substituted species) corresponds to [1′-13C]N3-isoinosine (but does not distinguish it from [1′-13C]inosine or [1′-13C]N7-isoinosine). UV-spectral properties at pH 1, 7, and 11 matched published findings for N3-isoinosine and are distinct from those reported for inosine and N7-isoinosine (Table S3) (Lehikoinen, et al., 1989; Montgomery and Thomas, 1969; Tindall, et al., 1972; Wolfenden, et al., 1966). The 1H NMR spectrum of this isolated product agreed with the published chemical shift data for authentic N3-isoinosine but not for inosine or N7-isoinosine (Table S3) (Chenon, et al., 1975a; Lehikoinen, et al., 1989; Tindall, et al., 1972). The 1′-13C chemical shift is consistent with [1′-13C]N3-isoinosine but not inosine or N7-isoinosine (Table S3) (Chenon, et al., 1975a).
PNP Reaction Fidelity with Phosphate
Conditions were selected to permit 2 × 107 catalytic turnovers of PNP at chemical equilibrium in the presence of phosphate and inosine. The 13C and 1H NMR spectra of reaction mixtures with native, Y88G and F159G PNPs showed only the formation of hypoxanthine [2] and α-d-[1-13C]ribose 1-phosphate [4] (Figure 5). The 13C and 1H NMR spectra for H257G and F200G PNPs also showed formation of N3-isoinosine [5] but not ribose (Figure 5-I and II, E and F). N3-Isoinosine is apparent after ∼3 × 106 catalytic turnovers in the phosphorolysis reactions catalyzed by both H257G and F200G. After the entire inosine sample had passed through the catalytic site approximately 4,000 times (∼24 h), the N3-isoinosine peak was integrated to be ∼5% and ∼10% for H257G and F200G, respectively, relative to the initial [1′-13C]inosine peak (Figure 5); thus, the probability for N3 capture of the ribocation is 1:80,000 for H257G and 1:40,000 for F200G. In contrast, the phosphorolysis reaction of native human PNP showed no detectable N3-isoinosine or ribose formation after more than 107 transits through the catalytic site.
Figure 5. 13C NMR (I) and 1H NMR (II) spectra of [1′-13C]inosine with human PNP in the presence of phosphate.
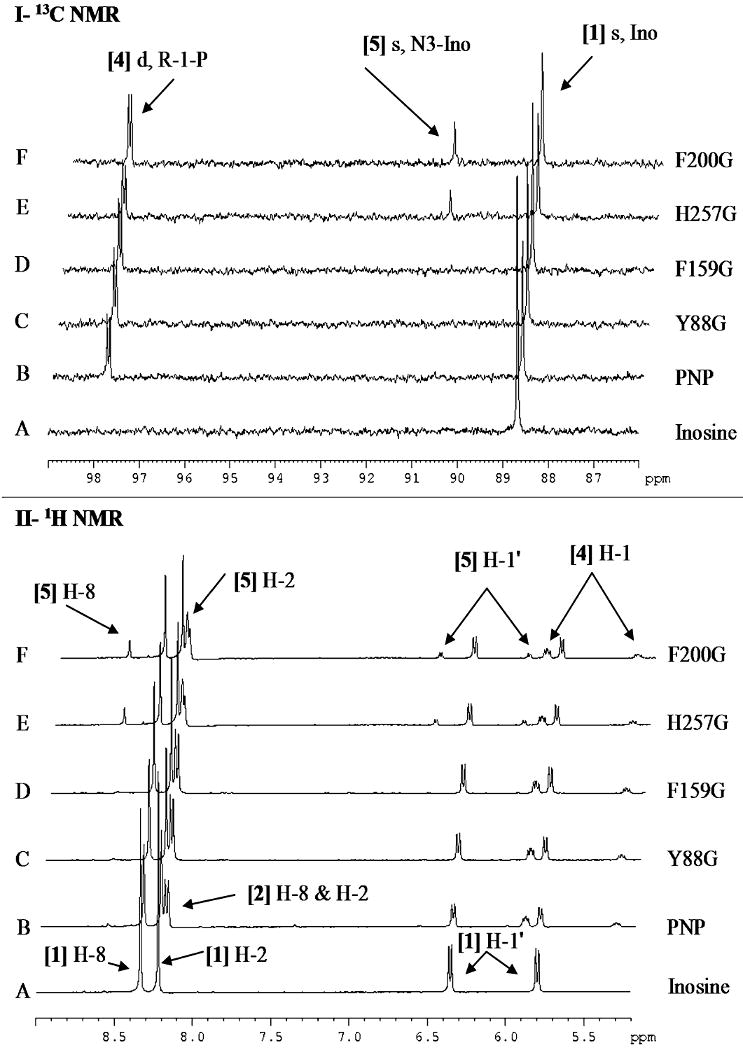
[1′-13C]Inosine (5.3 mM) was incubated (A) alone or with (B) 6 μM native human PNP, (C) 14 μM Y88G, (D) 33 μM F159G, (E) 151 μM H257G, or (F) 264 μM F200G. All spectra were acquired in the presence of 50 mM KH2PO4, pH 7.4, at 25 °C in 10% D2O and after ∼2 × 107 enzymatic turnovers (20-24 h). [1] Ino, inosine; [2] Hx, hypoxanthine; [4] R-1-P, α-d-ribose 1-phosphate; [5] N3-Ino, N3-isoinosine.
Single-Turnover Hydrolysis
Rates of single-turnover inosine hydrolysis catalyzed by native and catalytic-site glycine mutants of human PNP were compared to the previously reported values for bovine PNP (Table 1). The single-turnover hydrolysis rates for native human PNP were found to be ∼3-fold greater than those for the bovine enzyme (Table 1, Figure 6A). The introduction of solvent leaks into the active site of human PNP through the four engineered glycine mutants caused increased rates of inosine hydrolysis compared to native PNP (Table 1 and Figure 6A). The rate of hydrolysis for Y88G increased 2.3-fold, for F159G increased by 4-fold, for H257G decreased by 1.6-fold and for F200G increased by 4.8-fold, relative to native PNP (Table 1 and Figure 6A).
Figure 6. Hydrolysis reactions under high enzyme concentrations.
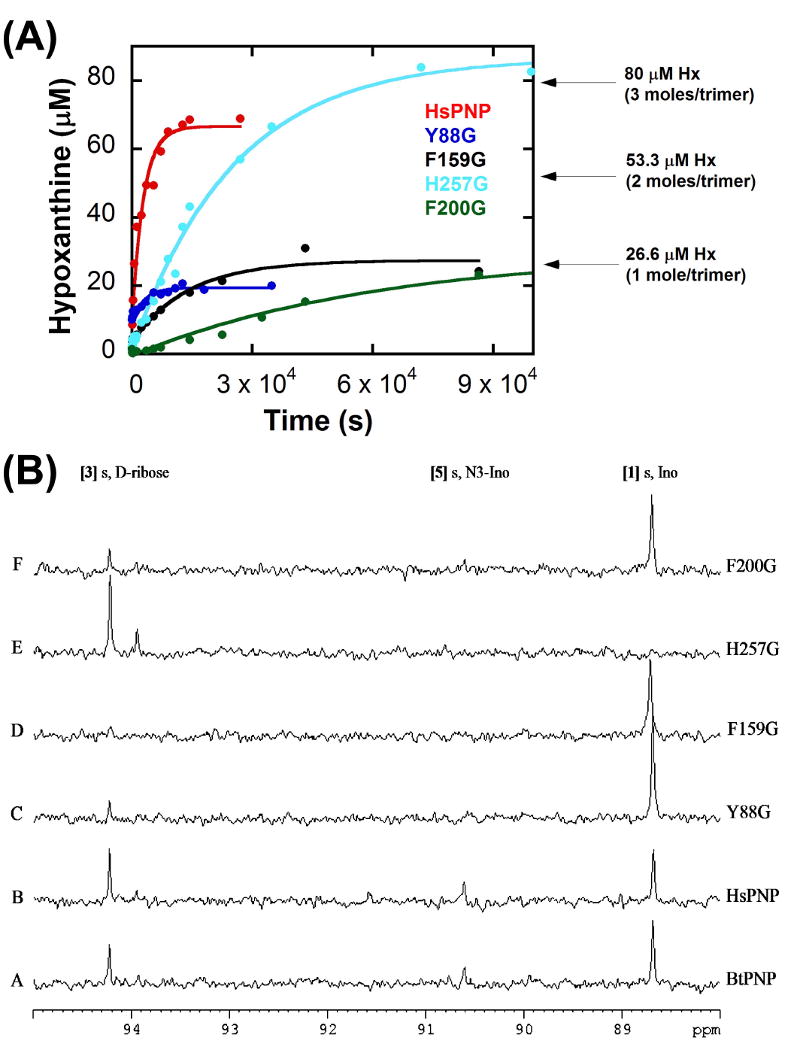
(A) Single-turnover hydrolysis reaction kinetics, plotting the concentration of hypoxanthine (isolated on a C18 analytical column by reversed-phase HPLC) formed during the hydrolysis reaction of native ( ), Y88G (
), Y88G ( ), F159G (
), F159G ( ), H257G (
), H257G ( ), and F200G human PNP (
), and F200G human PNP ( ) as a function of mixing time prior to acid quenching. Data were fit to equation (1).
) as a function of mixing time prior to acid quenching. Data were fit to equation (1).
(B) 13C NMR spectra of the PNP hydrolysis reactions. [1′-13C]inosine (900 μM) with (A) native bovine PNP (BtPNP), (B) native human PNP (HsPNP), (C) Y88G, (D) F159G, (E) H257G, and (F) F200G. All spectra were acquired after 20 h at 25 °C with 200 μM PNP in 10% D2O. [1] Ino, inosine; [3], d-ribose; [5] N3-Ino, N3-isoinosine.
13C NMR Detection of Hydrolysis under High Enzyme Concentrations
To analyze the PNP reaction for slow hydrolysis, 0.9 mM of [1′-13C]inosine, 0.2 mM (monomers) of PNPs, and increased acquisition times were used. These conditions enable 13C-NMR detection of ribose resulting from the hydrolysis of a single equivalent of inosine, even if subsequent turnovers are hindered by rate-limiting hypoxanthine release, as previously found for bovine PNP (Kline and Schramm, 1995). The 13C NMR spectra with [1′-13C]inosine under these conditions showed the appearance of d-ribose [3] (β-d-ribopyranose, δ 94.22) (Figure 6B). After 20 h, H257G had converted all of the 0.9 mM inosine to d-ribose [3] (β- and α-d-ribopyranose) (Figure 6B, spectrum E). Although inosine hydrolysis by H257G is 1.6-fold slower than native PNP, it formed hypoxanthine and ribose without any detectable isomerization to N3-inosine. Native human, bovine and the other three catalytic-site glycine mutants of PNP showed incomplete hydrolysis of [1′-13C]inosine [1] and accumulation of d-ribose [3] (Figure 6B). N3-Isoinosine [5] was also readily detected with native (human or bovine) PNPs and to a lesser extent with F200G (Figure 6B). 13C NMR spectra of similar experiments for Y88G, F159G and F200G recorded after 48 h showed significant accumulations of ribose and N3-isoinosine at approximately equal concentrations (Figure S2, as an example for F159G). Attempts to perform similar solvolysis experiments with aqueous mixtures of methanol (up to 20% v/v) failed to yield any detectable methyl riboside by 13C or 1H NMR, even after prolonged incubations (> 48 h).
Discussion
Inosine Hydrolysis by Human PNP
Without phosphate, solvent H2O is expected to fill the relatively large phosphate binding site of PNP. From its position in the phosphate site, water acts as an inefficient nucleophile to capture the ribocation. In contrast, bound phosphate is known to be highly polarized and activated at the catalytic site (Deng, et al., 2004). Phosphate also participates in ribocation formation and geometric stabilization by forming an ion pair with the developing ribocation and H-bonds to the ribosyl 2′- and 3′-hydroxyls (Figure 2). Our results suggest that when these interactions are missing, the ribosyl group may not be highly constrained in the catalytic site, and once it forms, it can rebound to form inosine (Figure 1, path a), react with water (path c), or react with N3 of hypoxanthine to form N3-isoinosine (path d; see below). Interestingly, Richard and coworkers have also characterized the partitioning of an enzyme-generated oxacarbenium ion among reactant reformation, capture by various nucleophiles and capture by solvent water (Richard, et al., 1996).
Human PNP hydrolyzes inosine 2.7-fold more slowly than the bovine enzyme (Kline and Schramm, 1992; Kline and Schramm, 1995). The hydrolytic reaction rate of 7.5 × 10-4 s-1 for native HsPNP is approximately 5 × 10-6 of the on-enzyme chemical turnover rate of the phosphorolytic reaction (154 s-1) (Saen-Oon, et al., 2008a). Bound phosphate therefore contributes a factor of approximately 105 to TS formation.
Ribocation Reactivity
Transition state lifetimes of nonenzymatic chemical reactions have been shown to be ∼10-13 s, as determined with the use of femtosecond time-resolved spectroscopy (Baumert, et al., 2001). Postulates that enzymes preferentially stabilize their transition states suggest that their lifetimes may be substantially longer. Kinetic isotope effect studies indicate that the TS of human PNP is a fully-dissociated ribocation with complete leaving-group departure (C1′–N9 distance = 3 Å) and a similar distance to the phosphate nucleophile (i.e., an SN1 mechanism) (Lewandowicz and Schramm, 2004; Schramm, 2005). The TS lifetime for human PNP has been estimated by computational transition path sampling to be ∼10 fs (10-14 s) with reaction coordinate motion occurring over ∼70 fsec (Saen-Oon, et al., 2008b). On-enzyme chemistry occurs over a much longer timescale (154 s-1, once every 6 × 10-3 s), reflecting the necessity for multiple geometric alignments between the protein and reactants to permit formation of the TS (Saen-Oon, et al., 2008a; Saen-Oon, et al., 2008b). Since the lifetime of the ribocation is between 10-14 (the computed TS lifetime) and 6 × 10-3 s, we designed an experimental test to determine if the ribocation is stabilized (near 6 × 10-3 s) or has a lifetime near the time scale of a single bond vibration lifetime (near 10-14 s). The lifetime of the ribocation can be estimated by catalytic-site mutations that are designed to permit water access to the reactive ribocation.
Undetectable inosine hydrolysis in the presence of phosphate establishes steric or temporal exclusion for water capture of the ribocation. Transition state analysis (Lewandowicz and Schramm, 2004) and X-ray structures of PNP complexed with TS analogues (Figure 2) reveal a >3-Å separation between the bound phosphate and C-1′, a distance that is expected to permit the diffusional approach of water at assay temperatures. Based on the X-ray structures, phosphate is poised more closely to the ribocation than structurally observed catalytic site waters. If the ribocation is an intermediate and has a lifetime permitting the diffusion of solvent or catalytic site waters to the ribocation, some ribose formation would be observed. The lack of hydrolysis is consistent with a ribocation lifetime that is too short to allow diffusion of a water molecule to within reactive distance of the ribocation C-1′.
Times for water diffusion are a function of distance and environment. The shortest trajectory from solvent water to C1′ of the ribocation is approximately 10 Å by way of a wide channel located at the interface of the two adjacent subunits, surrounded by H257, F159′, F200, and Y88. Solvent water must diffuse around the ribosyl ring, a path that is partially obstructed by phosphate and enzyme residues. An alternative consideration is water bound within the catalytic site, as is presumed to exist in the absence of phosphate. X-ray structures of phosphate-bound PNPs reveal the existence of a structural water within 4.4 Å of C-1′ (Figure 2B). Diffusion of approximately 3.0 Å would permit formation of ribose. The upper rate of water diffusion is approximately 1 Å ps-1 (based on the water diffusion coefficient of 3.01 × 10-5 cm2·s-1 at 25 °C (Wang, 1951)), which sets lower limits of 10 and 3 ps, respectively, for the time of diffusion of solvent water and the catalytic-site water. The lower limit on the rate of diffusion, however, is not defined for the specific environment in the catalytic site of PNP. If we assume it is slowed by a factor of five, we can, with this assumption, set upper limits of 50 and 15 ps, respectively, for these water molecules. The lifetimes of carbohydrate oxacarbenium ions in water are estimated to be ∼1 ps (Amyes and Jencks, 1989). Based on our upper estimates of diffusion time, we can propose that the ribocation formed at the TS of PNP has a lifetime less than the 15 to 50 ps required for diffusional capture by catalytic-site or solvent water.
Formation of N3-Isoinosine
A control for the ribocation capture experiment is the rate of inosine solvolysis in the absence of phosphate. Removal of phosphate permits solvent access to the catalytic site and the unequivocal formation of ribose. Thus, phosphate-free PNP forms the ribocation, and without phosphate, most is captured by water to give ribose. In addition to the hydrolytic reaction, a novel product, N3-isoinosine, was also formed. The mechanism proposed for N3-isoinosine formation involves ribocation formation in a catalytic site where mutations weaken the geometric alignment of the purine base and/or the ribocation (Figure 2). Without tight geometric constraints on the ribocation and displaced base, nucleophilic rebound can occur to both N9 (the normal reaction) and, by misalignment, to N3, causing isomerization. Phosphate is directly involved in this alignment by its H-bond contacts with the 2′- and 3′-hydroxyl groups of ribose (Figure 2). In the presence of phosphate, the alignment of N9-hypoxanthine with the ribocation fails less than once in a million transfers through the catalytic site, as evidenced by the failure to detect any N3-isomer in multiple-turnover NMR studies. In the absence of phosphate, the purine leaving-group interactions assist in ribocation formation, but the poor reactivity of the disordered waters in the phosphate-binding site allows the ribocation to wobble on its nucleophilic rebound.
The slow formation of N3-isoinosine in the absence of phosphate is consistent with the full formation of the ribocation. With water molecules in the phosphate site, ribocation capture occurs with N9 (to reform inosine), N3 to form isoinosine, and solvent to form ribose. In the presence of phosphate, an alternative mechanism for isomerization involving N3 attack upon the ribose 1-phosphate product cannot be excluded, but more complex mechanisms depart from known features of the physiological PNP mechanism.
N3-Isoinosine is a byproduct in the chemical synthesis of inosine (Lehikoinen, et al., 1989; Ryu, et al., 2004; Wolfenden, et al., 1966) and is an alternative substrate for PNPs (Bzowska, et al., 1996; Lehikoinen, et al., 1989), but it has never been reported as a product in any enzymatic reactions. Nucleoside deoxyribosyltransferases have been demonstrated to form 2′-deoxynucleosides with N3- and N7-fused purine derivatives (Huang, et al., 1983; Steenkamp, 1991); however, unlike with PNP, these isomers are likely formed via the covalent deoxyribosylated enzyme intermediate that is known to form during their function with natural substrates (Smar, et al., 1991).
The lifetime of PNP intermediates can be compared with a nonenzymatic isomerization reaction for cyclopropane stereomutation (Hrovat, et al., 1997). The mechanism of isomerization proceeds via C–C bond homolysis to generate a trimethylene biradical, which rotates about the linearized bonds before closing. Their calculations estimated a biradical lifetime of 140 fs, within the range of the 10- to 50,000-fs lifetime for the PNP-generated ribocation estimated in our computational (Saen-Oon, et al., 2008b) and solvolysis experiments, respectively.
Inosine Hydrolysis and Isomerization by Catalytic-Site Mutants of Human PNP
Four glycine mutants of the residues that are proposed to shield the catalytic site from solvent were prepared to create a water leak into the catalytic site and thereby introduce a hydrolytic branch point in the reaction mechanism (Figure 1, path c). The hydrolytic rate is a measure of the water access and reactivity as well as the lifetime of the highly reactive ribocation. The mutants are reduced in kcat by up to 44-fold relative to the native enzyme. All enzymes were capable of inosine hydrolysis with rates similar to or faster than native PNP.
The formation of N3-isoinosine in the absence of phosphate is consistent with the formation of a ribocation TS that reacts with hypoxanthine at N9 (to form inosine) or N3 (yielding N3-isoinosine) or with water (yielding ribose). Unlike with the native enzyme, isomerization to N3-isoinosine was not observed with any of the mutants under the conditions of the hydrolysis experiments. In the presence of phosphate, the PNP species with relatively high kcat/Km values (i.e., native, Y88G and F159G; Table S1) efficiently transfer the ribocation to phosphate without any detectable side reactions. Less efficient PNP mutants (i.e., H257G and F200G) occasionally misdirect the electrophile, resulting in formation of N3-isoinosine. Reaction with N3 is rare – only 1 out of 40,000 (F200G) or 80,000 (H257G) catalytic cycles generates N3-inosine, based on its rate of formation. H257 anchors the 5′-hydroxyl group of the ribosyl moiety, and its removal provides the ribosyl cation sufficient positional flexibility to permit slow ribocation capture by N3. The crystal structure of human PNP complexed with a TS-analogue inhibitor (Immucillin-H) and phosphate indicates that the phenyl-group side chain of residue F200 is involved in a herringbone-type interaction with the purine base (Figure 2) (de Azevedo, et al., 2003; Ghanem, et al., 2009). In F200G, the positional stability of the purine base is compromised. In this mutant, the ribocation contacts are unchanged from native PNP, but weakened interactions to hypoxanthine permit both N9 and N3 to react with the ribocation.
Significance
Native human PNP and catalytic-site glycine mutants slowly hydrolyze inosine when phosphate is absent but not in the presence of phosphate. These mutants were designed to permit solvent leaks; however, the lifetime of the ribocation is too short to permit solvent capture of the ribocation in competition with phosphate. In the absence of phosphate, human PNP catalyzes the capture of the ribocation by N3 of the purine group, resulting in isomerization of the substrate. The TS of human PNP involves a fully-dissociated ribooxacarbenium ion (i.e., SN1 character). The chemical properties of a ribocation are consistent with the isomerization at N3, in which hypoxanthine departure is required to permit N9 → N3 isomerization. The chemical reactivity of PNP and its catalytic site mutants provide experimental evidence for a ribocation lifetime too short to permit efficient solvent capture of the ribocation. Amino acids that stabilize both the ribocation (e.g., H257) and hypoxanthine (e.g., F200) are required for fidelity of product formation. Atomic freedom of motion around the TS results in the formation of N3-isoinosine during phosphorolysis by these mutants.
Experimental Procedures
Steady-State Enzyme Assays
Activity assays for native and mutant PNPs with inosine as a substrate were carried out by monitoring the conversion of hypoxanthine to uric acid (ε293 = 12.9 mM-1 cm-1) (Kim, et al., 1968) in a coupled assay containing 60 milliunits of xanthine oxidase and variable concentrations of inosine in 50 mM KH2PO4, pH 7.4 at 25 °C (Lewandowicz, et al., 2003). Substrate concentration (inosine or [1′-13C]inosine) was determined spectrophotometrically using the published millimolar extinction coefficient of 12.3 at 248.5 nm, pH 7 (Dawson, et al., 1986).
[1′-13C]Inosine
[1′-13C]Adenosine (Omicron Biochemicals, Inc.) was converted to [1′-13C]inosine by treatment with calf spleen adenosine deaminase (Sigma) (Pfrogner, 1967a; Pfrogner, 1967b), with spectrophotometric monitoring at 267 nm (Δε267 = 6.5 mM-1 cm-1) (Schramm and Baker, 1985) at pH 7. [1′-13C]Inosine was purified by reversed-phase HPLC (5% methanol in 50 mM triethylammonium acetate, pH 5.0, at 1 mL min-1) using an analytical C18 column (Waters Deltapak, 15 μm, 300 Å, 300 × 3.9 mm).
Steady-State NMR Detection for Hydrolysis, Phosphorolysis, and Isomerization
The 13C and 1H NMR spectra of reaction mixtures with PNP and labeled inosine were obtained by mixing 6 μM of native human PNP with 5.3 mM of [1′-13C]inosine in 20 mM Tris-HCl (pH 7.4) and 10% D2O, in the presence or the absence of 50 mM KH2PO4, pH 7.4, for ∼2 × 107 enzymatic turnovers (∼20 to 24 h). Native human PNP has a chemical turnover of 154 s-1 and steady-state rate of 44 s-1, limited by the rate of hypoxanthine release (Ghanem, et al., 2008a). During 24 h, the entire inosine sample passes through the catalytic site approximately 15,000 times (according to chemical turnovers) in this freely reversible reaction. Thus, a hydrolytic reaction rate of once in every 300,000 phosphorolytic reactions (5 × 10-4 s-1) would cause 5% of the inosine to accumulate as ribose and would be readily detected by NMR. For mutant PNPs, enzyme was added at concentrations that yield the same steady-state phosphorolytic rate as native PNP (e.g., [H257G] = [Native PNP] × kcat,native ÷ kcat,H257G = 6 μM × 44 s-1 ÷ 1.75 s-1 = 150 μM). Standard references included free [1′-13C]inosine and [1-13C]ribose obtained through the irreversible arsenolysis of [1′-13C]inosine in the presence of PNP and 50 mM Na2HAsO4, pH 7.4. All spectra were acquired using a Bruker 300 MHz spectrometer with 5 mm BBI probe at 298 K. 1H-decoupled 13C NMR spectra were collected with 256 scans of 64 K points (∼ 15 min/spectrum), a recycle delay of 2 s and a carbon sweep width of 315 ppm with the carrier set to 126 ppm. 1H NMR spectra were collected with 64 scans of 16 K points, a recycle delay of 1.3 s and a proton sweep width of 14 ppm with the carrier set to 4.7 ppm. Spectra were processed with an exponential window function of 1 Hz. Spectra were assigned as described previously (Dorman and Roberts, 1970; Teijeira, et al., 1997; Tindall, et al., 1972).
13C NMR Detection for Hydrolysis under High Enzyme Concentrations
13C NMR spectra of reaction mixtures with high molar ratios of PNP to inosine were obtained by mixing 200 μM (monomers) of PNP with 900 μM of [1′-13C]inosine in 20 mM Tris-HCl (pH 7.4) and 10% D2O, for ∼20 to 24 h. 1H-decoupled 13C NMR spectra were collected with 1024 scans of 64 K points (∼58 min), a recycle delay of 2 s and a carbon sweep width of 315 ppm with the carrier set to 126 ppm using a Bruker 300 MHz spectrometer with 5 mm BBI probe at 298 K. See Supplemental Data for calculation of kiso.
Single-Turnover Hydrolysis
Equal amounts of inosine and enzyme (80 μM final) were incubated at 25 °C for 30 s and up to 72 h, followed by quenching with HCl (1 N final) and removal of the denatured protein by centrifugation. Hypoxanthine (retention time 4.3 min) and inosine (retention time 7.3 min) were quantitated by UV detection at 249 nm using reversed-phase HPLC on a Waters Deltapak C18 column (15 μm, 300 Å, 300 × 3.9 mm), equilibrated with 5% methanol in 50 mM triethylammonium acetate (pH 5.0) at 1 mL min-1. See Supplemental Data for calculation of khyd.
Characterization of N3-Isoinosine
N3-Isoinosine was isolated from incubations of inosine with human PNP in the absence of phosphate by reversed-phase HPLC using an analytical C18 column (Phenomenex Luna 5 μm, 100 Å, 150 × 4.6 mm), equilibrated with an ion-pair buffer (100 mM KH2PO4, 8 mM Bu4NHSO4, pH 6.0) at 1 mL min-1, with monitoring by a Waters 996 Photodiode Array Detector. Under these conditions, hypoxanthine, inosine, and N3-isoinosine eluted with retention times of 3.2, 8.07, and 8.8 min, respectively (Figure S1). The N3-isomer peak was collected and reduced to 400 μL in a Speedvac concentrator, and this sample was desalted by application to a second C18 column (Waters Deltapak 15 μm, 300 Å, 300 × 3.9 mm), equilibrated with water at 1.5 mL min-1. N3-Isoinosine was collected, concentrated to dryness in a Speedvac concentrator, redissolved in water, and analyzed by ESI-MS and by UV/Vis spectroscopy at pH 1, 7, and 11.
Supplementary Material
Acknowledgments
We acknowledge the US National Institutes of Health grants GM41916 and GM068036 for funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amyes TL, Jencks WP. Lifetimes of oxocarbenium ions in aqueous solution from common ion inhibition of the solvolysis of .alpha.-azido ethers by added azide ion. J Am Chem Soc. 1989;111:7888–7900. [Google Scholar]
- Baumert T, Frohnmeyer T, Kiefer B, Niklaus P, Strehle M, Gerber G, Zewail AH. Femtosecond transition state dynamics of cis-stillbene. Appl Phys B. 2001;72:105–108. [Google Scholar]
- BioCryst Pharmaceuticals Inc. 2009 http://www.biocryst.com/pipeline.htm.
- Bzowska A, Kulikowska E, Poopeiko NE, Shugar D. Kinetics of phosphorolysis of 3-(beta-D-ribofuranosyl)adenine and 3-(beta-D-ribofuranosyl)hypoxanthine, non-conventional substrates of purine-nucleoside phosphorylase. Eur J Biochem. 1996;239:229–234. doi: 10.1111/j.1432-1033.1996.0229u.x. [DOI] [PubMed] [Google Scholar]
- Chenon MT, Pugmire RJ, Grant DM, Panzica RP, Townsend LB. Carbon-13 magnetic resonance. XXV. A basic set of parameters for the investigation of tautomerism im purines. Established from carbon-13 magnetic resonance studies using certain purines and pyrrolo[2,3-d]pyrimidines. J Am Chem Soc. 1975a;97:4627–4636. doi: 10.1021/ja00849a027. [DOI] [PubMed] [Google Scholar]
- Chenon MT, Pugmire RJ, Grant DM, Panzica RP, Townsend LB. Carbon-13 magnetic resonance. XXVI. A quantitative determination of the tautomeric populations of certain purines. J Am Chem Soc. 1975b;97:4636–4642. doi: 10.1021/ja00849a028. [DOI] [PubMed] [Google Scholar]
- Dawson RMC, Elliott D, Elliott WH, Jones KM. Spectral data and pKa values for purines, pyrimidines, nucleosides, and nucleotides. In: Dawson RMC, editor. Data for Biochemical Research. 3rd. New York: Oxford Univ. Press; 1986. pp. 103–114. [Google Scholar]
- de Azevedo WF, Jr, Canduri F, dos Santos DM, Silva RG, de Oliveira JS, de Carvalho LP, Basso LA, Mendes MA, Palma MS, et al. Crystal structure of human purine nucleoside phosphorylase at 2.3 A resolution. Biochem Biophys Res Commun. 2003;308:545–552. doi: 10.1016/s0006-291x(03)01431-1. [DOI] [PubMed] [Google Scholar]
- Deng H, Lewandowicz A, Schramm VL, Callender R. Activating the phosphate nucleophile at the catalytic site of purine nucleoside phosphorylase: a vibrational spectroscopic study. J Am Chem Soc. 2004;126:9516–9517. doi: 10.1021/ja049296p. [DOI] [PubMed] [Google Scholar]
- Dorman DE, Roberts JD. Nuclear magnetic resonance spectroscopy: 13C spectra of some common nucleotides. Proc Natl Acad Sci U S A. 1970;65:19–26. doi: 10.1073/pnas.65.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi M, Rossi P, Rogers C, Harbison GS. Dependence of 13C NMR chemical shifts on conformations of rna nucleosides and nucleotides. J Magn Reson. 2001;150:1–9. doi: 10.1006/jmre.2001.2314. [DOI] [PubMed] [Google Scholar]
- Fedorov A, Shi W, Kicska G, Fedorov E, Tyler PC, Furneaux RH, Hanson JC, Gainsford GJ, Larese JZ, et al. Transition state structure of purine nucleoside phosphorylase and principles of atomic motion in enzymatic catalysis. Biochemistry. 2001;40:853–860. doi: 10.1021/bi002499f. [DOI] [PubMed] [Google Scholar]
- Ghanem M, Li L, Wing C, Schramm VL. Altered thermodynamics from remote mutations altering human toward bovine purine nucleoside phosphorylase. Biochemistry. 2008a;47:2559–2564. doi: 10.1021/bi702132e. [DOI] [PubMed] [Google Scholar]
- Ghanem M, Saen-oon S, Zhadin N, Wing C, Cahill SM, Schwartz SD, Callender R, Schramm VL. Tryptophan-free human PNP reveals catalytic site interactions. Biochemistry. 2008b;47:3202–3215. doi: 10.1021/bi702491d. [DOI] [PubMed] [Google Scholar]
- Ghanem M, Zhadin N, Callender R, Schramm VL. Loop-tryptophan human PNP reveals submillisecond protein dynamics. Biochemistry. 2009 doi: 10.1021/bi802339c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrovat DA, Fang S, Borden WT, Carpenter BK. Investigation of Cyclopropane Stereomutation by Quasiclassical Trajectories on an Analytical Potential Energy Surface. J Am Chem Soc. 1997;119:5253–5254. [Google Scholar]
- Huang MC, Montgomery JA, Thorpe MC, Stewart EL, Secrist JA, 3rd, Blakley RL. Formation of 3-(2′-deoxyribofuranosyl) and 9-(2′-deoxyribofuranosyl) nucleosides of 8-substituted purines by nucleoside deoxyribosyltransferase. Arch Biochem Biophys. 1983;222:133–144. doi: 10.1016/0003-9861(83)90510-6. [DOI] [PubMed] [Google Scholar]
- Drew Kenneth N, Z J, Bondo Gail, Bose Bidisha, Serianni Anthony S. 13C-labeled aldopentoses: detection and quantitation of cyclic and acyclic forms by heteronuclear 1D and 2D NMR spectroscopy. Carbohydr Res. 1998;307:199–209. [Google Scholar]
- Kim BK, Cha S, Parks RE., Jr Purine nucleoside phosphorylase from human erythroyctes. II. Kinetic analysis and substrate-binding studies. J Biol Chem. 1968;243:1771–1776. [PubMed] [Google Scholar]
- Kline PC, Schramm VL. Purine nucleoside phosphorylase. Inosine hydrolysis, tight binding of the hypoxanthine intermediate, and third-the-sites reactivity. Biochemistry. 1992;31:5964–5973. doi: 10.1021/bi00141a003. [DOI] [PubMed] [Google Scholar]
- Kline PC, Schramm VL. Purine nucleoside phosphorylase. Catalytic mechanism and transition-state analysis of the arsenolysis reaction. Biochemistry. 1993;32:13212–13219. doi: 10.1021/bi00211a033. [DOI] [PubMed] [Google Scholar]
- Kline PC, Schramm VL. Pre-steady-state transition-state analysis of the hydrolytic reaction catalyzed by purine nucleoside phosphorylase. Biochemistry. 1995;34:1153–1162. doi: 10.1021/bi00004a008. [DOI] [PubMed] [Google Scholar]
- Koellner G, Luic M, Shugar D, Saenger W, Bzowska A. Crystal structure of calf spleen purine nucleoside phosphorylase in a complex with hypoxanthine at 2.15 A resolution. J Mol Biol. 1997;265:202–216. doi: 10.1006/jmbi.1996.0730. [DOI] [PubMed] [Google Scholar]
- Lehikoinen PK, Sinnott ML, Krenitsky TA. Investigation of α-deuterium kinetic isotope effects on the purine nucleoside phosphorylase reaction by the equilibrium-perturbation technique. Biochem J. 1989;257:355–359. doi: 10.1042/bj2570355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowicz A, Schramm VL. Transition state analysis for human and Plasmodium falciparum purine nucleoside phosphorylases. Biochemistry. 2004;43:1458–1468. doi: 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- Lewandowicz A, Shi W, Evans GB, Tyler PC, Furneaux RH, Basso LA, Santos DS, Almo SC, Schramm VL. Over-the-barrier transition state analogues and crystal structure with Mycobacterium tuberculosis purine nucleoside phosphorylase. Biochemistry. 2003;42:6057–6066. doi: 10.1021/bi0343830. [DOI] [PubMed] [Google Scholar]
- Montgomery JA, Thomas HJ. Ribosyl derivatives of hypoxanthine. J Org Chem. 1969;34:2646–2650. doi: 10.1021/jo01261a034. [DOI] [PubMed] [Google Scholar]
- Murkin AS, Birck MR, Rinaldo-Matthis A, Shi W, Taylor EA, Almo SC, Schramm VL. Neighboring group participation in the transition state of human purine nucleoside phosphorylase. Biochemistry. 2007;46:5038–5049. doi: 10.1021/bi700147b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez S, Wing C, Antoniou D, Schramm VL, Schwartz SD. Insight into catalytically relevant correlated motions in human purine nucleoside phosphorylase. J Phys Chem A. 2006;110:463–472. doi: 10.1021/jp051277u. [DOI] [PubMed] [Google Scholar]
- Pfrogner N. Adenosine deaminase from calf spleen. I. Purificiation. Arch Biochem Biophys. 1967a;119:141–146. doi: 10.1016/0003-9861(67)90439-0. [DOI] [PubMed] [Google Scholar]
- Pfrogner N. Adenosine deaminase from calf spleen. II. Chemical and enzymological properties. Arch Biochem Biophys. 1967b;119:147–154. doi: 10.1016/0003-9861(67)90440-7. [DOI] [PubMed] [Google Scholar]
- Richard JP, Huber RE, Heo C, Amyes TL, Lin S. Structure-reactivity relationships for beta-galactosidase (Escherichia coli, lac Z). 4. Mechanism for reaction of nucleophiles with the galactosyl-enzyme intermediates of E461G and E461Q beta-galactosidases. Biochemistry. 1996;35:12387–12401. doi: 10.1021/bi961029b. [DOI] [PubMed] [Google Scholar]
- Rinaldo-Matthis A, Murkin AS, Ramagopal UA, Clinch K, Mee SP, Evans GB, Tyler PC, Furneaux RH, Almo SC, et al. L-Enantiomers of transition state analogue inhibitors bound to human purine nucleoside phosphorylase. J Am Chem Soc. 2008;130:842–844. doi: 10.1021/ja710733g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KS, Kim C, Park C, Choi BS. NMR analysis of enzyme-catalyzed and free-equilibrium mutarotation kinetics of monosaccharides. J Am Chem Soc. 2004;126:9180–9181. doi: 10.1021/ja047911j. [DOI] [PubMed] [Google Scholar]
- Saen-Oon S, Ghanem M, Schramm VL, Schwartz SD. Remote mutations and active site dynamics correlate with catalytic properties of purine nucleoside phosphorylase. Biophys J. 2008a;94:4078–4088. doi: 10.1529/biophysj.107.121913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saen-Oon S, Quaytman-Machleder S, Schramm VL, Schwartz SD. Atomic detail of chemical transformation at the transition state of an enzymatic reaction. Proc Natl Acad Sci U S A. 2008b;105:16543–16548. doi: 10.1073/pnas.0808413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm VL. Enzymatic transition states: thermodynamics, dynamics and analogue design. Arch Biochem Biophys. 2005;433:13–26. doi: 10.1016/j.abb.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Schramm VL, Baker DC. Spontaneous epimerization of (S)-deoxycoformycin and interaction of (R)-deoxycoformycin, (S)-deoxycoformycin, and 8-ketodeoxycoformycin with adenosine deaminase. Biochemistry. 1985;24:641–646. doi: 10.1021/bi00324a016. [DOI] [PubMed] [Google Scholar]
- Shi W, Ting LM, Kicska GA, Lewandowicz A, Tyler PC, Evans GB, Furneaux RH, Kim K, Almo SC, et al. Plasmodium falciparum purine nucleoside phosphorylase: crystal structures, Immucillin inhibitors, and dual catalytic function. J Biol Chem. 2004;279:18103–18106. doi: 10.1074/jbc.C400068200. [DOI] [PubMed] [Google Scholar]
- Smar M, Short SA, Wolfenden R. Lyase activity of nucleoside 2-deoxyribosyltransferase: transient generation of ribal and its use in the synthesis of 2′-deoxynucleosides. Biochemistry. 1991;30:7908–7912. doi: 10.1021/bi00246a006. [DOI] [PubMed] [Google Scholar]
- Steenkamp DJ. The purine-2-deoxyribonucleosidase from Crithidia luciliae. Purification and trans-N-deoxyribosylase activity. Eur J Biochem. 1991;197:431–439. doi: 10.1111/j.1432-1033.1991.tb15929.x. [DOI] [PubMed] [Google Scholar]
- Stoeckler JD, Agarwal RP, Agarwal KC, Parks RE., Jr Purine nucleoside phosphorylase from human erythrocytes. Methods Enzymol. 1978;51:530–538. doi: 10.1016/s0076-6879(78)51074-4. [DOI] [PubMed] [Google Scholar]
- Stoeckler JD, Cambor C, Parks RE., Jr Human erythrocytic purine nucleoside phosphorylase: reaction with sugar-modified nucleoside substrates. Biochemistry. 1980;19:102–107. doi: 10.1021/bi00542a016. [DOI] [PubMed] [Google Scholar]
- Teijeira M, Santana L, Uriarte E. Assignment of the 13C NMR spectra of some adenine, hypoxanthine and guanine carbonucleosides. Magn Reson Chem. 1997;35:806–807. [Google Scholar]
- Tindall CG, Jr, Robins RK, Tolman RL, Hutzenlaub W. Directed glycosylation of 8-bromoadenine. Synthesis and reactions of 8-substituted 3-glycosyladenine derivatives. J Org Chem. 1972;37:3985–3989. doi: 10.1021/jo00798a003. [DOI] [PubMed] [Google Scholar]
- Wang JH. Self-diffusion and structure of liquid water. II. Measurement of self-diffusion of liquid water with O18 as tracer. J Am Chem Soc. 1951;73:4181–4183. [Google Scholar]
- Wolfenden R, Sharpless TK, Ragade IS, Leonard NJ. Enzymatic and chemical deamination of 3-(β-D-ribofuranosyl)adenine. J Am Chem Soc. 1966;88:185–186. doi: 10.1021/ja00953a047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.