

Abstract
For almost five decades, antibiotics have been used successfully to control infectious diseases caused by bacterial pathogens. More recently, however, two-thirds of bacterial pathogens exhibit resistance and are continually evolving new resistance mechanisms against almost every clinically used antibiotic. Novel efforts are required for the development of new drugs or drug leads to combat these infectious diseases. A number of antibiotics target the bacterial aminoacyl-tRNA site (A site) of 16S ribosomal RNA (rRNA). Mutations in the A-site region are known to cause antibiotic resistance. In this study, a bacterial (E. coli) A-site rRNA model was chosen as a target to screen for peptide binders. Two heptapeptides, HPVHHYQ and LPLTPLP, were selected through M13 phage display. Both peptides display selective binding to the A-site 16S rRNA with on-bead fluorescence assays. Dissociation constants (Kds) of the amidated peptide HPVHHYQ-NH2 to various A-site RNA constructs were determined by using enzymatic footprinting, electrospray ionization mass spectrometry (ESI-MS), and isothermal titration calorimetry (ITC) under a variety of buffer and solution conditions. HPVHHYQ-NH2 exhibits moderate affinity for the A-site RNA, with an average Kd value of 16 μM. In addition, enzymatic footprinting assays and competition ESI-MS with a known A-site binder (paromomycin) revealed that peptide binding occurs near the asymmetric bulge at positions U1495 and G1494 and leads to increased exposure of residues A1492 and A1493.
It has been half a century since the first antibiotic penicillin was discovered and successfully applied to the treatment of infectious diseases; however, infectious diseases still remain the third-leading cause of death in the United States and the second-leading cause of death worldwide (1). Widespread and intensive use of antibiotics in hospitals and agriculture has led to significant levels of resistance (2, 3). The emergence of antibiotic resistance, especially multidrug resistance, is a growing threat to human health (4). Therefore, a significant driving force exists for the development of new antimicrobial agents, especially those to combat multidrug-resistant pathogens.
The majority of antibiotics used in the clinic target protein synthesis, which occurs at the core of the ribosome (5). High-resolution X-ray crystal structures of rRNA fragments, 30S and 50S subunits, and whole ribosomes, either free or complexed with antibiotics, have provided important information that will help in the design of new anti-infective compounds (6–10). More specifically, X-ray crystal structures of the 30S subunit revealed the role of the aminoacyl-tRNA site (A site) of the small subunit 16S ribosomal RNA (rRNA) in translation fidelity (11). This dynamic region plays a discriminatory role by switching from a closed conformation (with two universally conserved adenine residues, A1492 and A1493, stacked inside of the internal loop) in the absence of cognate tRNA/mRNA to an open conformation (in which A1492 and A1493 flip out of helix 44 to interact with the mRNA-tRNA minihelix) (6, 7, 11). The aminoglycoside class of antibiotics binds readily to rRNA because they are highly positively charged and flexible (12). Aminoglycosides such as paromomycin and kanamycin bind to the A-site rRNA and interfere with protein synthesis by reducing translation fidelity (11). The binding of the aminoglycosides to the A site leads to displacement of A1492 and A1493 and stabilizes the open conformation, therefore impeding discrimination (13). Furthermore, mutations in the A site of 16S rRNA are known to lead to antibiotic resistance due to impaired drug interactions (14–18).
The A site is more than 95% conserved among bacteria (19), but the sequence differs in the human rRNA variant (Figure 1). Small RNA models containing the minimal bacterial ribosomal A site have been developed previously in order to facilitate the investigation of aminoglycoside binding mechanisms (20, 21). These models have similar patterns of chemical modification as the whole ribosome (22). Therefore, the A-site-containing rRNA models are believed to be validated drug targets for the development of new antimicrobial agents that can possibly overcome resistance caused by gene mutation. By using such systems, it may be possible to develop new broad-spectrum antibiotics with high selectivity for the bacterial A-site RNAs.
Figure 1.

The sequence of the E. coli 16S rRNA decoding region is shown. The A site is highlighted by the gray box. Model RNAs containing the A site of bacterial and human rRNAs are compared (hp-AS and human-AP-AS, respectively). The nucleotides that differ between bacteria and human are highlighted in white.
The intrinsic variability of peptide sequences (20 natural amino acids) and their ability to recognize RNA in a specific manner, as well as their ease of synthesis, render them attractive moieties for drug-lead design. The goal of this study was to screen for peptides that can bind tightly to the A-site RNA, and then to characterize the binding interactions between selected peptides and the A-site RNA. Ultimately, understanding the interactions between the functional group of the selected peptides and the A-site RNA can be used in the design of new drug leads or potential antibiotic reagents. Furthermore, peptides can be easily manipulated in order to optimize for binding to mutant forms of the target A-site RNA.
In this study, phage display was used to screen for peptides with affinity for the A-site RNA. Phage display is a powerful tool that has been used in the discovery of peptide ligands as drugs (23–27). Phage display has several key advantages, such as a high diversity of variant peptides in the library (as high as 109), a direct connection between the nucleotide sequence and sequence of the peptide displayed on the phage surface, replicative capacity of the phage, and applicability to high-throughput screening (28, 29). Furthermore, several phage-display libraries are commercially available. In this study, we used Ph.D.-7™ (New England Biolabs), an M13 phage-display library with a random heptapeptide sequence fused to the N-terminus of the pIII minor coat protein. The M13 phage is non-lytic, minimizing the potential for RNA target degradation, and only five copies of the peptide are displayed on each phage surface, therefore minimizing possible multivalent interactions.
Utilizing the phage-display technology, two peptides were selected and demonstrated to have specific binding to the A-site RNA target (Figure 2A). Further investigation of the binding interaction (Figure 2B) revealed that one peptide exhibits moderately high binding affinity (with a dissociation constant in the μM range) for the A-site RNA. Isothermal titration calorimetry (ITC) was carried out to determine the dissociation constant, as well as the thermodynamic parameters, of the binding interaction. Electrospray ionization mass spectrometry (ESI-MS) was used to determine the relative dissociation constants and binding stoichiometries at various pH conditions, compare binding to both related and unrelated RNAs, and examine competition with a known A-site-binding ligand. Enzymatic footprinting revealed the peptide-binding site on the target RNA. Overall, the knowledge gained from this work regarding RNA-peptide interactions will provide useful guidelines for the design of small molecules that can specifically target the ribosomal A site and for future development of novel anti-infective compounds.
Figure 2.

A schematic diagram shows the phage-display strategy to identify A-site RNA-binding peptides (A) and to analyze for binding (B). The peptides displayed on phage, on bead, or free in solution are represented as various colored symbols (diamonds, triangles, etc.), the target A-site RNA is shown as a blue ¾ circle attached to the streptavidin surface, competitor tRNA is in black, and competitor hp-C RNA is shown as a red hairpin. In panel B, the full circles represent TentaGel beads decorated with selected, synthetic peptides, and F represents the 5′-fluorescein tag on A-site RNA.
MATERIAL AND METHODS
Materials
The Ph.D.-7™ phage-display peptide library kit, T4 polynucleotide kinase (T4 PNK), T4 RNA ligase, and T7 RNA polymerase were purchased from New England Biolabs (Beverly, MA). Taq DNA polymerase, calf intestinal alkaline phosphatase (CIAP), DNA polymerase I large (Klenow) fragment, T4 DNA ligase, RNase A, restriction endonucleases, E. coli S30 extract system for circular DNA and pBESTluc plasmid, plasmid pUC19, E. coli DH5α, 5-bromo-4-chloro-3-indolyl-phosphate (BCIP) were bought from Promega (Madison, WI). Dynabeads® M-270 streptavidin were from Invitrogen (Carlsbad, CA). The pfu DNA polymerase was from Stratagene (La Jolla, CA). Centricon ultracell YM-3 membranes were from Millipore (Billerica, MA). All Fmoc-protected amino acids, 1-hydroxybenzotriazole (HOBt), and Rink-amide resin were purchased from Novabiochem (San Diego, CA). Dimethylformamide (analysis grade) was purchased from Acros Organic (Geel, Belgium). Diisopropylcarbodiimide, trifluoroacetic acid, anisole, thioanisole, and triisopropyl silane were from Acros (Morris Plains, NJ), and paromomycin, E. coli tRNAPhe, and goat anti-biotin-alkaline phosphatase antibody were obtained from Sigma-Aldrich (St. Louis, MO). TentaGel S-NH2 resin was from Rapp Polymer (Tübingen, Germany). Superblock buffer was from Pierce (Rockford, IL). The 5′-biotin-labeled DNA (5′-biotin-AGCAATCGCC ATCCGCACC-3′) was synthesized by IDT (Coralville, IA). The unlabeled RNA (5′-GGCGUCACAC CUUCGGGUGA AGUCGCC-3′, hp-AS) and fluorescein-labeled RNAs (5′-fluorescein-GGCGUCACAC CUUCGGGUGA AGUCGCC-3′, F-hp-AS; 5′-fluorescein-GGCGUCGCUA CUUCGGUAAA AGUCGCC-3′, F-human-hp-AS; 5′-fluorescein-GGCGUCACAC CGGUGAAGUC GCC-3′, F-ds-AS; 5′-fluorescein-GGCAGAUCUC AGCCUGGGAG CUGUCUGCC-3′, F-TAR) were obtained from Dharmacon Research, Inc. (Lafayette, CO) (generated by chemical synthesis). The 3-hydroxypicolinic acid (MALDI matrix) was from Fluka (St. Louis, MO). The Sequitherm EXCEL II DNA sequencing kit was from Epicentre Biotechnologies (Madison, WI). The [5′-32P] pCp (cytidine 3′,5′-bisphosphate, 10 μCi/μl) was purchased from PerkinElmer (Waltham, MA). All other chemicals and organic solvents were from Fisher Scientific (Pittsburgh, PA).
Plasmid Construction
The primers used for cloning the A-site RNA sequence into plasmid DNA are listed in Table 1. The primers P1 and P2 were first 5′-phosphorylated with T4 PNK and then annealed by heating to 95 °C for 3 min followed by slowly cooling down to 25 °C over 4 h. The annealed duplex was ligated into BclI/XbaI linearized plasmid pWK122 (a gift from Prof. Philip Cunningham, Department of Biological Sciences, Wayne State University) to generate plasmid pWK-hp-AS (2782 bp). Similarly, primers P3 and P4 were 5′ phosphorylated with T4 PNK before being annealed to form a duplex as described above. Subsequently, the annealed duplex was extended to double-stranded DNA with blunt ends by Klenow DNA polymerase. Ligation of the annealed duplex and CIAP dephosphorylated linear plasmid pUC19 (SmaI) resulted in plasmid pUC-hp-C. The desired recombinants in DH5α were selected on IPTG/X-Gal LB plates containing ampicillin (100 μg/ml). Both plasmids were sequenced to confirm the correct nucleotide composition.
Table 1.
Primers used for cloning and PCR
| Primer | Sequence | Appplication |
|---|---|---|
| P1 | 5′GATCAGGcgtcacaccttcgggtgaagtcgCCTGATCAAACTCTTCAATCTCCCGGTGCGGATGGCGATTGCT3′ | pWK-hp-AS |
| P2 | 5′CTAGAGCAATCGCCATCCGCACCGGGAGATTGAAGAGTTTGATCAggcgacttcacccgaaggtgtgacgCCT3′ | pWK-hp-AS |
| P3 | 5′AAATTAATACGACTCACTATAggcgccaccttcgggtggcgcc3′ | pUC-hp-C |
| P4 | 5′AGCAATCGCCATCCGCACCggcgccacccgaaggtggcgcc3′ | pUC-hp-C |
| P5 | 5′AAATTAATACGACTCACTATAgg3′ | PCR |
| P6 | 5′AGCAATCGCCATCCGCACCgg3′ | PCR |
The A-site DNA sequence and non-specific competitor DNA sequence are in bold lowercase. The 3′ tail sequence is in bold uppercase. The T7 promoter is highlighted in gray with the +1 position in italics and underlined.
RNA Preparation
The hp-AS-pwk RNA (Figure 3) was synthesized by in vitro transcription with plasmid pWK-hp-AS as a template through the following steps. Prior to transcription, plasmid pWK-hp-AS was cleaved with XbaI followed by desalting. The transcription was carried out at 37 °C for 4 h in 50 μl 1× transcription buffer (40 mM Tris-HCl, pH 7.5, 6 mM MgCl2, 10 mM NaCl, 2 mM spermidine, 0.01% Triton X-100), 7 mM NTPs, 10 mM dithiothreitol, 1.5 μg DNA template (linear pWK-hp-AS), and 20 U of T7 RNA polymerase. The hp-C RNA transcript was prepared in a similar way with PCR product as the DNA template, which was amplified by using primers P5 and P6 (Table 1), pfu DNA polymerase, and plasmid pUC-hp-C. The control RNA helix 17 (h17; 5′-GGCGGGGAGG AAGGGAGUAA AGUUAAUACC UUUGCUCAUU GACGUUACCC GCCGGUGCGG AUGGCGAUUG CU-3′) was generated the same way. It also contained a 3′ tail that was complementary to a 5′-biotinylated DNA.
Figure 3.

The RNAs used in the phage-display experiments are shown. A) Target A-site RNAs used for selection are given. The A-site motif is shown in grey and boxed (hp-AS-pwk). The modified A-site is shown in black (hp-C). Note that hp-AS-pwk is hybridized to a complementary 5′-biotinylated DNA sequence through the 3′-tail region. The DNA portion is underlined. B) The DNA sequences for the A-site RNAs on recombinant pWK and pUC19 plasmids are shown. The promoter region is boxed and +1 position is labeled. The repeating A-site sequence is in bold uppercase. The extra sequence is in lowercase and the 3′-tail region is in uppercase and underlined.
The transcribed RNAs were purified on 15% denaturing polyacrylamide gels containing 29:1 acrylamide:bisacrylamide, 0.5× TBE buffer (45 mM Tris, 45 mM boric acid, 0.5 mM EDTA, pH 8.2) and 7 M urea. Electrophoresis was performed at 400 V for ~2 h. The RNA band was visualized by UV shadowing, excised, and subjected to electroelution using an Amicon Centrilutor and Centricon™ YM-3 devices in 0.5× TBE buffer at 200 V for 2.5 h. The sample tube was centrifuged at 3000 rpm at 4 °C overnight to remove buffer and washed with water to desalt.
The size of transcribed RNA was confirmed with MALDI-TOF mass spectrometry (Ultraflex-Tof™, Bruker-Daltonics, Billerica, MA) under negative ion mode and linear acquisition operation mode. The synthetic RNA constructs (from Dharmacon Research, Inc.) were deprotected according to the manufacturer’s instructions (RNA was dissolved in 400 μl of deprotection buffer (100 mM acetic acid, pH 3.8 with TEMED) and incubated at 60 °C for 30 min prior to lyophilization) and purified as described above.
Screening of the Ph.D.7 Phage-Display Peptide Library
For screening, the hp-AS-pwk RNA was refolded and hybridized with the 5′-biotin-DNA by mixing in a 1:1 molar ratio of RNA to DNA in buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.2) and heating for 5 min at 95 °C, followed by slow cooling to room temperature. Hybridization was verified on an 8% non-denaturing polyacrylamide gel (19:1 acrylamide/bisacrylamide, 1× TBE). All other RNAs (hp-C, F-TAR, F-human-hp-AS, F-ds-AS, F-hp-AS, and h17) were refolded or annealed to 5′-biotinylated DNA in a similar manner for binding, counter-selection, or ELISA assays.
On-bead, Two-step Selection
The M-270 streptavidin-coated beads were employed for biopanning to screen for peptides binding to the A-site RNA. The beads were first subjected to preselection to remove streptavidin-binding phage (Figure 2). Pre-selection was conducted by incubation of the original Ph.D.-7™ library (10 μl, 2 × 1013 pfu/ml) with pre-washed beads (1 mg, 1 mg/100 μl) in 100 μl buffer A (20 mM HEPES-KOH, pH 7.5, 100 mM KCl, 5 mM MgCl2, 5% glycerol, 0.1 mg/ml BSA) overnight at 4 °C. The supernatant (pre-selected library) was titered to determine the phage concentration and used for the first round of biopanning. In the first round of selection, the pre-selected phage library was incubated with 30 pmol RNA (hp-AS-pwk)/biotin-DNA complex for 2 h at room temperature with gentle agitation. Next, the RNA-phage complexes were captured by mixing with 0.1 mg pre-washed beads for 30 min at room temperature. After washing, the bound phage were removed by either specific or non-specific elution. For non-specific elution, 100 μl buffer B (0.2 M glycine-HCl, pH 2.2, 1 mg/ml BSA) was added to the on-bead target and incubated for exactly 9 min followed by neutralization with 15 μl of buffer C (1 M Tris-HCl, pH 9.1). For specific elution, buffer A (100 μl) containing 300 pmol of non-biotin-labeled target RNA (hp-AS) was added and incubated for 1 h at room temperature. The eluted phage were amplified and titered to determine the phage concentration, and then subjected to the next round of selection. Four rounds of biopanning were carried out. Each round of biopanning was conducted with increasing stringency as summarized in Table 2. In round four, a non-specific competitor RNA (hp-C) was employed for counter-selection (Figure 2).
Table 2.
Biopanning using M-270 streptavidin-coated magnetic beads and hp-AS-pwk*
| Round | Washing | Competitor RNA (pmol) | Binding time (hr) | Elution |
|---|---|---|---|---|
| 1 | 3 times, 0.3% Triton | 30 (tRNA) | 2 | Non-SP SP |
| 2 | 3 times, 0.3% Triton | 30 (tRNA) | 2 | Non-SP SP |
| 3 | 8 times, 0.5% Triton | 60 (tRNA) 90 (tRNA) |
1 | Non-SP SP |
| 4 | 10 times, 0.5% Triton | 90 (tRNA) 30 (hp-C) |
0.5 | Non-SP SP |
The library was preselected against streptavidin beads only. The washing buffer is identical to the binding buffer (20 mM HEPES-KOH, pH 7.5, 100 mM KCl, 5 mM MgCl2, 5% glycerol, 0.1 mg/ml BSA) with addition of different concentrations of Triton X-100. The amount of A-site RNA (hp-AS-pwk) used for each round of selection was 30 pmol (hp-C: competitor A-site RNA; Non-SP: non-specific; SP: specific).
Phage DNA Purification and Sequencing
To sequence DNAs that encode for the selected 7-mer peptides, DNA templates were prepared either by isolation and purification of the phage genome DNAs or by direct PCR amplification of the phage DNAs from individual plaques. Phage genome DNAs were prepared by the following steps. An overnight culture of ER2378 in LB-Tet was prepared from a single colony and diluted in 1:100 with LB media. A blue plaque was inoculated in 1 ml of diluted culture. The culture was incubated at 37 °C with vigorous shaking for 4.5–5 h followed by centrifugation at 10,000 rpm for 5 min. The supernatant (800 μl) was transferred to a new tube with addition of 20% PEG/2.5 M NaCl (400 μl) and thoroughly mixed by vortexing. After incubation at room temperature for 10 min, the mixture was centrifuged at 10,000 rpm for 10 min. The pellet was resuspended in 70 μl of TE buffer (10 mM Tris-HCl, pH 7.3, 1 mM EDTA). An equilibrated phenol solution (70 μl, pH >8) was added and the mixture was vortexed, followed by centrifugation at 10,000 rpm for 2 min. The supernatant (aqueous) was mixed with CHCl3 (70 μl) and centrifuged in the same manner. The supernatant was mixed with 3 M sodium acetate (NaOAc) (7 μl) and cold 100% ethanol (175 μl), then stored at −20 °C overnight. After centrifugation at 15,000 rpm for 15 min, the resulting pellet was washed with cold 70% ethanol (500 μl), dried under vacuum, and redissolved in dH2O (30 μl).
PCR amplification of the phage DNA from an individual plaque was carried out in a 10 μl reaction volume containing 10× Taq buffer (1 μl, 50 mM Tris-HCl, 50 mM NaCl, 5 mM MgCl2, 0.1 mg/ml BSA, provided with the Taq DNA polymerase), 2.5 mM dNTPs (0.8 μl), 50 μM 110-G3 (forward primer, 5′-GCAAGCTGAT AAACCGATAC AAT-3′, 0.2 μl), 50 μM 96-G3 (reverse primer, 5′-CCCTCATAGT TAGCGTAACG-3′, 0.2 μl), dH2O (2.75 μl), Taq DNA polymerase (0.05 μl, 0.25 U) and 5 μl plaque solution. The plaque solution was prepared by picking up a single blue plaque with a sterile pipette tip and mixing in 5 μl dH2O. The PCR reaction was carried out as follows: 1 cycle of 98 °C for 1 min, 29 cycles of 98 °C for 10 sec, 55 °C for 30 sec and 72 °C for 30 sec, and 1 cycle of 72 °C for 1 min.
DNA sequencing was performed on a LI-COR sequencer (Lincoln, Nebraska) by following the recommended protocol provided by the manufacturer. The primer 700 M13-96 rev (LI-COR) was used for sequencing.
Peptide Synthesis
The peptides were synthesized manually following the standard Merrifield solid-phase peptide synthesis protocol (30, 31). The free peptides (~300 mg scale) were cleaved from the resin by using a mixture of trifluoroacetic acid, anisole, thioanisole, and triisopropyl silane (7 ml of a 94:2:2:2 ratio). The cleaved peptides were precipitated in 2×25 ml of cold (−80 °C) diethylether and washed twice with 15 ml diethylether, then resuspended in 10 ml dH2O and lyophilized. The heptapeptides were purified on a C18 reverse-phase HPLC column (Phenomenex, Torrance, CA) with a gradient of acetonitrile/deionized water and 0.1% trifluoroacetic acid. The desired fractions were collected, diluted to 50% with water, and lyophilized. The peptide masses were verified by ESI and MALDI-TOF mass spectrometry (Figure S1, Supplementary Information). The peptide powder was resuspended in water or the appropriate buffer for the chosen biophysical method. The concentration of the peptide was calculated by A280 in the presence of tyrosine, using the Beer-Lambert law, A= ε• C•l, in which l is the pathlength (cm), C the concentration (mol•L−1), and ε the extinction coefficient (M−1•cm−1). The extinction coefficient for tyrosine (εY280) is 1,300 M−1•cm−1 (32).
For on-bead, peptide-binding analyses, selected peptides were synthesized manually by solid-phase peptide synthesis on micro TentaGel S-NH2 beads following standard Fmoc solid-phase peptide synthesis chemistry (33). The beads containing the peptides were washed thoroughly with DMF and stored in DMF at 4 °C.
Peptide Bead Fluorescence Binding Assay
The peptide beads (5 μl) were washed with dH2O (6 × 1 ml) to remove DMF and then blocked with 200 μl of SuperBlock buffer (Pierce, Rockford, IL) overnight at 4 °C. Following a wash with dH2O (6 × 1 ml), the peptide beads were incubated with 10 μl of 1 μM fluorescein-labeled A-site RNAs (F-hp-AS, F-ds-AS, F-human-hp-AS) or an unrelated RNA (F-TAR) in binding buffer A (20 mM HEPES-KOH, pH 7.5, 100 mM KCl, 5 mM MgCl2, 5% glycerol, and 0.1 mg/ml BSA) for 3 days at 4 °C with gently shaking. Before incubation with the beads, the RNAs were heat denatured followed by slow cooling to room temperature to refold or reanneal. Ten μl of 1 μM fresh fluorescein-labeled RNA was added daily. Peptide beads incubated with 3 × 10 μl of 1 μM free fluorescein (in the same manner as F-AS) were used as the background control. After 72 h, the beads were washed with buffer A (6 × 1 ml) and visualized under a fluorescence microscope (Zeiss fluorescent microscope; 490 nm excitation wavelength, 525 nm emission wavelength for fluorescein) equipped with a CCD camera and image-processing software.
Peptide Bead ELISA Assay
The peptide beads (5 μl) were washed and blocked as described above and then incubated with 10 μl of 1 μM biotin-tagged A-site RNA (hp-AS-pwk annealed to 5′-biotin-DNA) for 4 days at 4 °C with daily addition of fresh 5 μl of 1 μM biotin-tagged RNAs. An unrelated biotin-tagged helix 17 (h17) was used as a control and incubated with beads in the same manner. Beads incubated with buffer were used as the background control. Following washes with buffer A (6 × 1 ml), the beads were incubated with goat anti-biotin-alkaline phosphatase antibody at 1:8000 dilution in antibody binding buffer (50 mM Tris-HCl, pH7.5, 138 mM NaCl, 27 nM KCl, 0.05% Tween-20) for 1 h at room temperature. The beads were first washed with antibody binding buffer (2 × 0.5 ml, each for 5 min) and then with alkaline phosphatase buffer (100 mM Tris-HCl pH 9.5, 100 mM NaCl, 1 mM MgCl2) (1 × 0.5 ml, 5 min). Finally, the beads were incubated with 50 μl of alkaline phosphatase buffer containing 5-bromo-4-chloro-3-indolyl-phosphate (BCIP, 0.165 μg/μl) at 4 °C overnight and then washed with dH2O (6 × 1 ml).
Electrospray Ionization Mass Spectrometry (ESI-MS)
Mass spectrometric measurements were carried out on a Micromass Quattro-LC double quadrupole mass spectrometer (Micromass, Manchester, UK). The electrospray source was operated in the negative ion mode. The samples were injected via a syringe pump at 6 μl/min (34). The capillary ion spray voltage was 2500 V, sampling cone 50 V, and extractor 2 V. The sample was scanned over the mass range m/z 1000 to 2500 with a scope gain of 78, scan time of 5.0 sec/each scan, interscan time of 0.1 sec, and scan for 7.0 min. The spectra were collected using the MCA (multi-channel analysis) method. The spectra were analyzed using Masslynx™ 4.0 (Micromass, Manchester, UK).
To determine apparent dissociation constants for peptide-RNA complexes, the gel-purified, desalted RNA (hp-AS) was renatured in 100 mM NH4OAc (pH 6.7) prior to binding with peptide. Next, 1 μM RNA was incubated with varying concentrations of peptide (0 – 375 μM) at room temperature for 10 min in a 50 μl reaction volume containing 50% 2-propanol (IPA) and 100 mM NH4OAc (pH 6.7). The bound fraction of RNA (Fr) was the ratio of peak intensity of free RNA over the peak intensity of total RNA (the sum of free RNA and peptide-complexed RNA). In this analysis, it is assumed that the peak intensity is proportional to the concentration of the RNA in solution (35). The apparent dissociation constants were obtained by plotting Fr vs. peptide concentration and using non-linear curve fitting with a quadratic equation (Equation 1) (35, 36).
| Equation 1 |
In Equation 1, [R]o is the total concentration of RNA, [P]o is the total concentration of peptide, ΣRPn− is the total intensity of RNA-peptide complexes, and ΣRn− is the total intensity of free RNA. In the plots, fraction bound, or Fr, is equal to (ΣRPn−/(ΣRn− + ΣRPn−)).
The salt concentration effect on peptide-RNA complex formation was investigated by analyzing 1 μM RNA incubated with 34 μM peptide at room temperature for 10 min in solution containing 50% IPA and increasing concentrations of NH4OAc (30, 50, 100, 150, 200 mM) at pH 6.7. Similarly, the pH effect on binding was determined in the same binding mixtures with 100 mM NH4OAc at pH 5.0, 6.0, 6.7, 6.9, 7.4, and 7.6. Competitive binding with paromomycin was conducted by pre-incubating 1 μM RNA with 34 μM peptide at 4 °C overnight in 100 mM NH4OAc at pH 6.7, followed by incubation with 1 or 4 μM paromomycin at room temperature for 30 min.
3′-32P RNA Labeling
The RNA was 3′-32P labeled in a 30 μl reaction volume containing 25 pmol of gel-purified RNA (27 nt, hp-AS), 1× T4 RNA ligase buffer (50 mM Tris-HCl, 10 mM MgCl2, 1 mM ATP, 10 mM dithiothreitol, pH 7.8 at 25 °C), 10% dimethyl sulfoxide, 10 mM ATP, 20 μCi pmol [5′-32P] pCp (Perkin-Elmer, Waltham, MA), and 1 μl T4 RNA ligase (20 U/μl). The mixture was incubated at 4 °C for 4 h followed by addition of 25 μl cold 4 M ammonium acetate (NH4OAc), 5 μg tRNAPhe, and 300 μl cold 100% ethanol. After incubation on dry ice for 1 h, the labeled RNA was centrifuged at 12,000 × g at 4 °C for 20 min. The RNA pellet was resuspended and repelleted by incubation with 200 μl of 0.5 M NH4OAc and 750 μl cold ethanol on dry ice for 1 h, and centrifuged as in the previous step. Finally, the RNA pellet was washed with 500 μl cold (−20 °C) ethanol and dried on a rotary evaporator.
Enzymatic Footprinting
Enzymatic footprinting was carried out by incubating 1 μl 3′-32P-labeled hp-AS (250,000 cpm) with 1 μl of various peptide concentrations (0–120 μM) in buffer containing 10 mM Tris-HCl, pH 7.2, 15 mM NaCl, and 1 mM EDTA at 37 °C for 1.5 h, followed by addition of 1 μl RNase A (0.002 U/μl) into the RNA/peptide mixture to a final volume of 5 μl and incubated at 37 °C for 30 min. The reaction was then quenched on dry ice and all samples were denatured by boiling for 1 min with 2 μl of formamide loading buffer (0.1% bromophenol blue, 0.1% xylene cyanole, 80% formamide, 20% glycerol) prior to loading onto the gel. An RNA ladder was generated by boiling 1 μl labeled RNA in 3 μl of alkaline hydrolysis buffer (50 mM sodium carbonate, 1 mM EDTA, pH 9.2) for 5 min. The reaction samples (50,000 cpm/each) were loaded onto a pre-run 20% denaturing polyacrylamide gel (7 M urea, 1× TBE buffer) and run at 1800 V for 3 h. The gel was exposed overnight at −20 °C to a storage phosphor screen (Amersham Biosciences, Piscataway, NJ), which was then scanned on a Phosphorimager Storm 860 (Molecular Dynamics, Sunnyvale, CA). The gel image was analyzed by using the ImageQuant™ program (Amersham Biosciences). Each band intensity was determined by dividing the specific band intensity by the whole lane intensity and multiplying by 100. The fraction of RNA bound (Fr) was calculated by dividing the normalized intensity of the specific band (corresponding to cleaved RNA) by that of the control, assuming that the maximum cleavage efficiency corresponds to 0% bound and the minimum cleavage efficiency corresponds to 100% bound. In some cases, enhancement rather than protection of cleavage was observed. The Fr values were plotted as function of peptide concentration. The Kd value was achieved through curve fitting using Equation 1.
Isothermal Titration Calorimetry
Isothermal titration calorimetry experiments were carried out using a VP-ITC instrument from Microcal, Inc. (Northampton, MA). The sample cell was passivated by 1× phosphate buffer (10 mM potassium phosphate, 100 mM KCl, pH 7.2) for 2 to 16 h and the reference cell was filled with degassed water. The RNA and the peptide were extensively dialyzed against water, dried, and resuspended in 1× phosphate buffer as above. The RNA and peptide samples were degassed in a Thermovac (Northhampton, MA) at 5 °C for 7 min. Typically, the titrations were carried out with a preliminary injection of 2 μl followed by injections of 20 μl of peptide solution (250–600 μM) into the sample solution of RNA (20–40 μM) with 300 s spacing between each. All experiments were carried out at 10 °C. Before analysis, data from the preliminary 2 μl injection were discarded, and heats of dilution of the peptide into buffer (in the absence of RNA) were subtracted from the peptide into RNA experiments. The heat of dilution of the peptide was found to be endothermic (data not shown). The corrected data were integrated and plotted as a function of the molar ratio, and the binding isotherms obtained were fitted to the Origin one-site model (Origin X.0, Microcal) (37).
In Vitro Translation Inhibition
The E. coli S30 extract for circular DNA system was used for in vitro translation inhibition assay. The reporter gene in plasmid pBESTluc was the eukaryotic firefly luciferase gene (Promega, Madison, WI). The experiment was carried out by incubating the translation mixture (2 μl of pBESTluc DNA (1 μg/μl), 5 μl of complete amino acid mixture (1 mM), 20 μl of S30 premix without amino acids, 15 μl of S30 extract for circular DNA, for a final volume of 50 μl) with increasing concentrations of peptide at 37 °C for 1–2 h. The reaction was quenched on ice for 5 min, followed by mixing with 5 μl luciferase assay reagent (Promega). The samples were transferred into a 96-well plate and the intensity of luminescence was monitored using a Luminometer Genios Plus (Tecan, CA) with plate reader (the background level was determined using a sample without any DNA template). Neomycin and neamine were used as positive controls (38).
RESULTS
RNA Target Design
The transcribed RNA (hp-AS-pwk) used for peptide screening had a 19-nucleotide (nt) 3′-tail region complementary to a 5′-biotin-tagged DNA (biotin probe) (Figure 3A), which allowed for immobilization onto streptavidin-coated beads and facilitated isolation of the bound phage. The hp-AS-pwk was transcribed under control of the T7 promoter of modified plasmid pWK122, which is located upstream of the BclI site (Figure 3B). To allow for stable secondary structure formation of hp-AS-pwk, 22 additional nts were added at the 5′ end of the transcribed RNA, which produced a 90-nt-long RNA transcript with a long double-stranded stem region (Figure 3A). A competitor RNA (hp-C) was designed to be just downstream of the T7 promoter of plasmid pUC19 and transcription was initiated at the first residue. For this RNA, the artificial UUCG tetraloop was included, but no extra residues were present in the helical region and the A-bulge motif was removed (Figure 3A).
Screening a Heptapeptide Phage Library on Streptavidin-coated Beads
The Ph.D.-7 library from New England Biolabs was pre-screened against beads to remove streptavidin-binding phage (Figure 2A). Increasing amounts of competitor tRNA (30 to 90 pmoles, tRNA or hp-C) and Triton (0.3 to 0.5%), decreased binding times (2 to 0.5 h), and increasing numbers of washes were used in each round. The competitor RNA (hp-C) (Figure 3), which is missing the triple A bulge at nucleotides 1492, 1493, and 1408, was used in a counter-selection in the 4th round (Figure 2A). By using this approach, we anticipated removing tRNA and hp-C binders from the pool and yielding peptides with specificity for the A-site (triple A bulge) motif. Furthermore, both specific and non-specific elution conditions were employed in each round (see Material and Methods section for details).
The yields (percent of phage recovery) for the four rounds of screening showed similar trends for both specific and nonspecific elution, with increasing amounts of output phage relative to input phage for each round (Figure 4). After the 3rd round of biopanning, several broad groups of peptide sequences were observed under both specific and non-specific elution conditions (Table 3). The LPLTPLP peptide and several related sequences were observed (LVPPSFS and LPLTALP). In round 4, two peptides, LPLTPLP (and related LPVTPLP) and HPVHHYQ, were predominant and the LPLTPLP peptide appeared with the highest frequency. In addition, the yield dropped significantly in the final round, suggesting that the selected peptides had deleterious effects on bacterial cell growth. The peptide TIGAITS appeared in rounds 3 and 4 of specific elution and round 4 of non-specific elution. The specific and non-specific elution methods yielded similar results, but the non-specific elution generated a wider variety of sequences in round 4. The observed differences between the non-specific and specific elution experiments are not surprising. For the specific elution, only a 10-fold higher concentration of free target RNA (hp-AS) over bound target RNA (hp-AS-pwk) was used for the elution. These conditions may not have been sufficient to elute the stronger-binding peptides. As a consequence, the stronger binders may be lost during each round of selection when using specific elution, whereas all of the peptides are expected to be removed when using non-specific elution conditions. The relative affinities of the two major peptides, HPVHHYQ and LPLTPLP, will be discussed in the following section.
Figure 4.
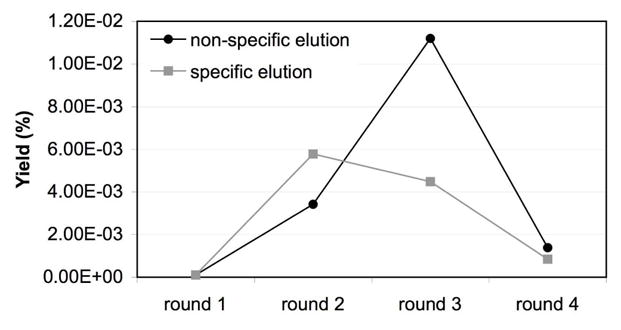
The yields are plotted for four rounds of biopanning against A-site RNAs using streptavidin beads. The yield corresponds to the ratio of input phage (plaque-forming units, pfu) to output phage (pfu) × 100.
Table 3.
Peptide sequences obtained from screening on streptavidin-coated beads.
| Round 3 | Round 4 | ||
|---|---|---|---|
| Non-specific elution | specific elution | Non-specific elution | specific elution |
| (2) LVPPSFS | (1) AQATALP | (4) LPLTPLP | (7) LPLTPLP |
| (2) LPLTALP | (2) LPLTALA | (1) LPVTPLP | (1) LPLTTLH |
| (2) LPLTPLP | (1) LPLTPLA | (2) HPVHHYQ | (1) QLPTTLP |
| (1) LPGIMSL | (2) LPLTPLP | (1) NQDVPLF | (1) TIGAITS |
| (2) HPVHHYQ | (1) HPVHHYQ | (1) VSSGPHW | |
| (1) HKVVAYY | (1) TLHPAHP | (1) TIGAITS | |
| (1) HSNTGYP | (1) TIGAITS | (2) SAKLMGH | |
| (1) TIGLITS | (1) TQSLASR | ||
| (1) TSGLASR | (1) GSWPSLL | ||
| (1) FPLLNML | (1) NWSSLY | ||
| (1) NNLLPPY | (1) NAFHSHI | ||
| (1) SIVRLQV | (1) SHIMPPN | ||
| (1) SISVIQE | (1) QPTSEGL | ||
| (1) QPKQFFQ | (1) QCWSPSL | ||
| (1) AQCLRIP | (1) QSTLNPT | ||
The numbers in parentheses indicate the number of times the peptides appeared in the sequenced pool. Specific vs. non-specific elution conditions are given in the Materials and Methods section (see also Table 2).
Selections against the same or alternative A-site model RNAs with 7-mer or 12-mer peptide phage-display libraries were also carried out, leading to predominant peptide sequences with similar motifs as the ones identified from the 7-mer peptide library selection against hp-AS-pwk (data not shown), demonstrating that the selection method is repeatable.
Detection of the Interactions between Selected Peptides and the A-Site RNA
The predominant peptides, HPVHHYQ and LPLTPLP, obtained from screening the A-site RNA construct against the heptapeptide phage library, were chosen for binding studies. The peptides were synthesized with the C terminus covalently attached to beads and then incubated with fluorescein-tagged A-site RNAs (Figure 2B) (F-ds-AS and F-hp-AS), as well as several control RNAs (F-TAR and F-human-hp-AS, Figure 5). To validate the methodology, two known HIV-1 TAR RNA binding peptides (33) with different relative affinities, KL-KD-NL (high affinity, Kd = 420 nM) and RL-KD-VD (moderate affinity, Kd = 10 μM), were also synthesized and tested against their target using fluorescein-labeled F-TAR (data not shown). The optimized conditions for the F-TAR system were used to monitor binding of peptides HPVHHYQ and LPLTPLP to F-ds-AS and F-hp-AS RNAs. Both RNAs contain the A-site motif, but differ in the number of potential binding sites, and only one contains the UUCG tetraloop motif. Both sets of beads containing the selected peptides HPVHHYQ and LPLTPLP showed strong fluorescence upon incubation with the fluorescent A-site RNAs, indicating a strong binding interaction (Figure 5A). In contrast, no binding was detected when those beads were incubated with an unrelated RNA (F-TAR) or a closely related human A-site RNA (F-human-hp-AS), indicating specific binding of the selected peptides to the bacterial A-site RNA. This result also suggested preferred binding to the A-site region (asymmetric A bulge) rather than the artificial UUCG tetraloop motif.
Figure 5.

The results of fluorescence binding assays on peptide beads are shown. A) The peptide sequences displayed on the beads are indicated on the left. The beads were incubated with fluorescein-tagged RNAs (from left to right, F-TAR, F-human-hp-AS, F-ds-AS, and F-hp-AS). B) The fluorescein-labeled RNAs used for the bead binding assays are shown with the A-site motif highlighted by the gray box. The nucleotides that differ between bacteria and human are highlighted in white.
To further validate the results obtained with the peptide beads and fluorescent RNAs, a bead ELISA assay was carried out. The HPVHHYQ peptide beads showed a much stronger response than LPLTPLP, indicating stronger binding of HPVHHYQ for the A-site RNA (Figure 6). No binding was observed between the two peptides and an unrelated helix 17 (h17) RNA from 16S rRNA. During the course of this study, a similar selection was carried out with a different rRNA target and the same peptide sequence, LPLTPLP, was identified. Our preliminary in vitro solution binding studies indicated that this peptide binds to other RNA targets, therefore further studies with LPLTPLP were not carried out.
Figure 6.

The results of the peptide bead ELISA assay are shown. “RNA−” represents the control without addition of target RNA and “RNA+” represents the peptide beads treated with target RNA (hp-AS-pwk). A control RNA, h17, is also used for comparison.
Binding Studies with Isothermal Calorimetry: Relative Contributions of Entropy and Enthalpy to Binding
The binding of HPVHHYQ-NH2 to hp-AS was examined by using ITC in phosphate buffer containing 100 mM KCl. The peptide HPVHHYQ-NH2 was synthesized with a free N-terminus to mimic the orientation on the phage, whereas the C-terminus was amidated to mimic fusion to the pIII protein on phage and to avoid negative charge repulsion with the RNA-phosphate backbone (Figure 7). Complex formation between the HPVHHYQ-NH2 peptide and the AS-RNA hairpin is an exothermic process (Figure 8). The thermodynamic parameters, enthalpy (ΔH) and entropy (ΔS), show that the binding is both enthalpically (ΔH = −0.56 ± 0.05 kcal/mol) and entropically (−TΔS = −6.5 ± 0.3 kcal/mol) favorable. The thermodynamic parameters that govern the interaction provide insights about the specificity of the binding. In particular, the favorable enthalpy value implies that the binding is mainly due to specific interactions between the RNA and the peptide, i.e., through van der Waals interactions and/or formation of hydrogen bonds. On the other hand, the small but favorable entropy value relates to non-specific hydrophobic interactions occurring in the binding event (displacement of water molecules) and loss of degrees of freedom. The dissociation constant that was obtained from this data is 2.0 ± 0.4 μM (in 110 mM K+, pH 7.2 buffer, 10 °C).
Figure 7.

The chemical structure of the peptide HPVHHYQ-NH2 is shown.
Figure 8.
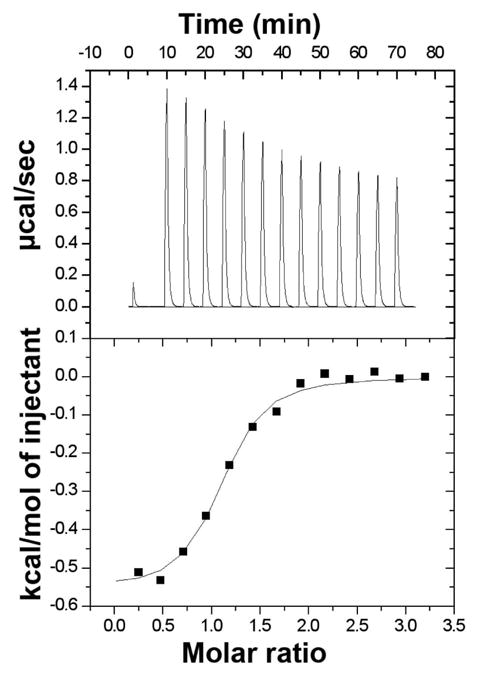
The results of the calorimetric titration of HPVHHYQ-NH2 into hp-AS in 10 mM potassium phosphate and 100 mM KCl at 10 °C are shown. The upper panel shows the zeroed raw data, and the lower panel represents the integration of the reference-subtracted peaks. The data were integrated and plotted as a function of the molar ratio and the resulting binding isotherm was fit using the Origin one-site model (Origin X.0, Microcal). The dissociation constant, stoichiometry, and thermodynamic parameters obtained are: Kd = 2.0 ± 0.4 μM; n= 0.9 ± 0.1; ΔH = −0.56 ± 0.05 kcal•mol−1; −TΔS = −6.5 ± 0.3 kcal•mol−1 (average of three independent experiments).
Binding Studies Using Electrospray Ionization Mass Spectrometry
Electrospray ionization mass spectrometry (ESI-MS) was used to further verify binding of peptide HPVHHYQ-NH2 to the A-site RNA hairpin model, hp-AS (Figure 1). It was also desirable to establish a binding assay that 1) does not require much RNA or peptide, 2) can be used for rapid screening of peptide or RNA variants, and 3) can be used in competition assays (39). The apparent dissociation constant, Kd, of HPVHHYQ-NH2 binding to the A-site RNA was determined by increasing the peptide concentration (0 to 375 μM) and monitoring complex formation (Figure 9A). The ESI-MS peak intensities of RNA ions and RNA-Na+ or RNA-K+ adducts in the −4, −5 and −6 charge states were summed and represent the free RNA. Likewise, the peak intensities of RNA-peptide complexes (including 1:1 and 1:2 complexes) and their ion adducts in −4, −5 and −6 charge states were added summed and represent the complexed RNA. The resulting apparent Kd value is 29 ± 2 μM (χ2 = 0.0068, in 100 mM NH4+, pH 6.7 buffer, 25 °C) (Figure 9B). The fact that this number varies slightly from the ITC value is not surprising since different buffers and temperatures were employed.
Figure 9.


A) ESI-MS spectra of hp-AS RNA binding to peptide HPVHHYQ-NH2 with increasing concentrations (0–375 μM) after incubation at room temperature for 10 min. Circles (●) represent free hp-AS RNA (−4, −5, and −6 charge states), stars (*) represent RNA-peptide complexes (1:1), triangles (▼;) represent RNA-peptide complexes (1:2), and squares (■) and diamonds (◆) represent peptide dimers and aggregates that occur at high peptide concentrations. B) Plot of fraction RNA bound against peptide (ligand) concentration. A non-linear least squares fitting to Equation 1 give an apparent dissociation constant, Kd, of 29 ± 2 μM (χ2 = 0.0068; average of three independent experiments).
The salt concentration is an important factor that could affect the RNA-peptide interaction. The salt might also affect the RNA ionization efficiency and possibly lead to either signal enhancement or repression when using ESI-MS. Thus, RNA ionization and RNA-peptide complex formation were monitored at different ammonium acetate (NH4OAc) concentrations. Both free RNA and RNA-peptide complexes showed fewer charged states at higher salt concentrations (the main charge state at 150 mM NH4OAc was −5) without affecting complex formation. Also, higher salt concentrations reduced formation of peptide-peptide dimer at m/z = 1831 (gas phase aggregate or clusters). In comparison, lower salt concentrations (30 mM NH4OAc) led to peaks at four different charge states: −4, −5, −6 and −7. (Figure S2 and S3, Supporting Information). As the concentration of NH4OAc increased from 30 to 100 mM, the fraction of bound RNA decreased from 52 to 30%, then increased slightly to 34% at 150 and 200 mM NH4OAc. These data indicate that the Kd values determined by ESI-MS could vary by as much as 20 to 25%, depending on the salt concentration. This information is important for future comparative assays with mutant RNAs or variant peptides, in which the buffer conditions should be carefully controlled.
The specificity of the free peptide for the A-site RNA target was assessed by employing ESI-MS competition experiments (data not shown). In ESI-MS, the selected peptide HPVHHYQ-NH2 displayed a five-fold preference for the A-site RNA over TAR RNA. This difference in binding is consistent with the results shown in Figure 5. The relatively low selectivity is not surprising given the fact that TAR is a flexible, promiscuous RNA with a tendency to undergo conformational changes and bind to numerous ligands, including the A-site-binding aminoglycoside antibiotics (33, 41, 42).
The selected peptide HPVHHYQ-NH2 contains three histidine residues; therefore, the effect of pH on peptide binding to the negatively charged RNA was also investigated. When comparing the RNA-peptide complex formation at pH values between 5.0 and 7.6, no significant change in the ratio of free RNA to RNA-peptide complexes was observed (data not shown). Therefore, pH does not appear to affect the binding of peptide HPVHHYQ-NH2 to the A-site RNA.
Binding Stoichiometry of the Peptide for the AS-RNA Hairpin
The binding of ligands to RNA often occurs at regions displaying flexibility such as loops or internal bulges, since the major groove is then widened and easier to access (40–42). The UUCG loop of the AS-RNA hairpin is artificial – it is not part of the natural 16S rRNA decoding region – and could be an alternate site of peptide interaction, as observed with other small molecule ligands (43). Since the peptide was selected for the internal asymmetric A bulge by using a simplified hairpin RNA containing the tetraloop in a counter-selection (hp-C), binding was anticipated to occur only in the bulge region. The interaction of the peptide with the AS-RNA hairpin has been found by ESI-MS to be predominately a 1:1 interaction (>98%) (Figure 9). This finding is confirmed by ITC, in which the binding was found to be a single exothermic event, with a stoichiometry of 1:1 (n = 0.9, Figure 8).
Competitive Binding with Paromomycin
Paromomycin is an aminoglycoside antibiotic and a known A-site binder with Kds ranging from 0.1 to 10 μM (20, 35, 44–48), depending on the method and solution conditions employed. In order to demonstrate that the binding of selected peptide HPVHHYQ-NH2 to the A-site RNA is a specific interaction rather than non-specific binding or the result of aggregation in the gas phase, paromomycin was used for competitive binding to the A-site RNA. As shown in Figure 10, upon addition of 1 μM paromomycin, the peak intensity corresponding to RNA-peptide complexes decreases, whereas the peak intensities corresponding to the RNA-paromomycin complex increase. When the paromomycin concentration reached 4 μM, all of the RNA-peptide complexes disappeared. Thus, paromomycin is able to displace peptide HPVHHYQ-NH2 from the RNA-peptide complex, indicating that the binding site of HPVHHYQ-NH2 is similar to or overlapping with that of paromomycin. Some additional peaks corresponding to a 1:1:1 interaction between RNA, peptide, and aminoglycoside are observed, which we believe arises from a secondary binding site of paromomycin on the A-site RNA. In support of this statement, a 1:2 complex of RNA:paromomycin is observed in Figure 10, whereas there is little evidence for a 1:2 complex between RNA and peptide.
Figure 10.

Competitive binding between peptide HPVHHYQ-NH2 and aminoglycoside paromomycin for the A-site RNA (R) is shown. A) The free A-site RNA (hp-AS) peaks (R) corresponding to the −5 and −6 charge states are shown (●). B) The A-site RNA and peptide (P) (17 μM) form a 1:1 complex in the −5 and −6 charge states (*). C) and D) The 1:1 RNA-peptide complex peak (*) diminishes with increasing concentrations of paromomycin (A) (1 and 4 μM, respectively). The 1:1 RNA-paromomycin (■), 1:2 RNA-paromomycin (◆), and 1:1:1 RNA-paromomycin-peptide (▼;) complexes in −5 and −6 charge states are observed.
Binding Site Determination and Quantitative RNase Footprinting
ESI-MS has been used previously to characterize aminoglycoside interactions with RNA, yielding results similar to those of other methods (35, 44–46); however, peptides have different chemical properties than the aminoglycosides, such as greater hydrophobicity, reduced electrostatic character, or fewer hydrogen-bonding moieties. Therefore, an additional method was employed to examine the peptide-RNA interactions. Enzymatic footprinting experiments were carried out using RNase A, radiolabeled hp-AS, and HPVHHYQ-NH2. The results reveal that residues G1494 and U1495 are protected by the peptide, with a reduced cleavage intensity at these two residues upon peptide binding (Figure 11A). In contrast, cleavage by RNase A is enhanced at residues A1492 and A1493 of hp-AS. These results indicate that the binding site of peptide HPVHHYQ-NH2 is located near the internal loop region of the A-site RNA, as indicated on the secondary structure map of hp-AS in Figure 11A. Based on quantification of the decreased cleavage intensities at residues G1494, and enhanced cleavage intensity at A1493, the average Kd value is 16 ± 4 μM (range 14 – 18 μM; 15 mM Na+, pH 7.2 buffer, 37 °C). The binding curve for residue G1494 is shown in Figure 11B (the binding curve for A1493 is shown in Figure S4, Supplementary Information). The slight variability in Kd values at different residues suggests that the hp-AS RNA is undergoing conformational changes as the peptide binds, such that not all sites are behaving equally. Alternatively, a slightly higher level of error exists using the enzyme-based method. Nonetheless, the peptide-binding site (A bulge motif) that was identified by using the footprinting approach is consistent with the results obtained from the ESI-MS and fluorescent-bead assays. Moreover, the apparent dissociation constant determined using footprinting (16 μM) is in good agreement with that determined using ESI-MS (29 μM) and ITC (2 μM) under different buffer and temperature conditions.
Figure 11.

Enzymatic footprinting analysis of the A-site RNA/peptide (HPVHHYQ-NH2) complex is shown. A) On the right, an autoradiogram of the 20% denaturing polyacrylamide gel reveals the RNase A cleavage sites on 32P-labeled hp-AS. Lane 1: RNA control; Lane 2: alkaline hydrolysis ladder; Lane 3: RNase A cleavage in the absence of peptide; Lanes 4–13: RNase A cleavage in the presence of 5 to 120 μM peptide. The A-site RNA model is shown to the left. The residues being protected are labeled with stars (*), and residues with enhanced cleavage are labeled with closed triangles (▲). B) A plot of the fraction RNA bound vs. peptide (ligand) concentration based on relative cleavage by RNase A at residue G1494 gives an apparent Kd of 18 ± 4 μM (χ2 = 0.0512, average of two independent experiments)..
In Vitro Translation Inhibition
After screening, selection, and characterization of peptide binding affinity, specificity, and binding site on the target RNA, the next step was to carry out functional analyses in vitro to determine whether or not the ligand can inhibit protein synthesis. An E. coli S30 extract translation system was used to test the affect of peptide HPVHHYQ-NH2 on in vitro protein synthesis. When the peptide concentration was 34 μM, no inhibition was observed; however, when the peptide concentration was increased to 620 μM, approximately 50% enzyme activity was observed (Figure 12). Based on this data, the IC50 of HPVHHYQ-NH2 is approximately 600 μM. This value is high compared with the known antibiotic neomycin; however, we are encouraged by the fact that the peptide displays an effect on the biological function of the rRNA.
Figure 12.

The results of in vitro translation inhibition by peptide HPVHHYQ-NH2 are shown. The relative luciferase activity is the ratio of luciferin intensity of each sample to the control in absence of peptide. Neomycin and neamine were used as controls.
DISCUSSION
In this study, we were able to select peptides that bind to the A-site RNA from a random linear heptapeptide phage-display library. Among the selected peptides, amidated HPVHHYQ displayed low micromolar dissociation constants for its target RNA. Thus, we succeeded in finding a peptide from screening with relatively good binding affinity for the target A-site RNA and detectable activity in a protein synthesis inhibition assay.
The binding of A-site rRNA to peptides HPVHHYQ and LPLTPLP on beads was observed via fluorescence and enzyme-linked assays. It was also demonstrated that the binding is specific, since unrelated RNAs such as TAR RNA and helix 17 did not bind to the immobilized peptides. Although both methods demonstrated that the two peptides bind to the A-site RNA, the bead ELISA method was more reliable in distinguishing the relative binding abilities of the peptides. The fluorescence method could only reveal these differences at very low concentrations of fluorescein-labeled RNA and was not very practical due to the long incubation periods and high number of binding and washing steps that were required. Furthermore, the affect of the linker or bead attachment to the overall affinity of peptide to RNA could not be determined. Therefore, a variety of solution assays with free (off-bead) amidated peptide HPVHHYQ-NH2 and hp-AS RNA were utilized as well. We chose methods that have been widely established previously to examine RNA-aminoglycoside interactions, namely, ESI-MS (35), ITC (46), and enzymatic footprinting (22).
It has been shown in previous studies that the specificity of the interactions between the aminoglycoside antibiotics and the 16S A-site RNA are governed by the three-dimensional structure of the rRNA, as well as electrostatic, hydrogen-bonding, and base-specific interactions (14, 20, 21). The RNA-binding affinities of the aminoglycosides decrease with increasing pH and/or salt concentrations (46). In this study, the binding affinity of HPVHHYQ-NH2 for the A-site RNA in the presence of varying salt concentrations and pH values was explored by using ESI-MS. This method is somewhat limited to ammonium acetate buffer because cations such as Na+ and Mg2+ form numerous RNA adducts, which make the RNA spectra difficult to interpret. In order to stabilize the RNA and to assist with sample transfer into the gas phase, organic solvents such as isopropanol were also employed. Based on a number of ESI-MS experiments, we determined that the A-site RNA, HPVHHYQ-NH2, and the A-site RNA/peptide complex tolerate organic solvents and high concentrations of NH4OAc. Our salt-dependence results (i.e., minimal salt effect on the relative binding affinity) also suggested that the binding interaction between A-site RNA and HPVHHYQ-NH2 is governed mainly by stacking and hydrogen-bonding, rather than electrostatic interactions. This conclusion is further supported by the ITC results, in which the thermodynamic parameters show that the binding is enthalpically driven, suggesting the involvement of specific interactions such as hydrogen bonding and van der Waals contacts. Work is currently in progress to determine the roles of the individual histidines in the binding interaction; however, the results presented here are suggestive of stacking interactions with those particular residues, because pH did not significantly alter the binding interaction with the A-site RNA.
A similar range of Kd values for amidated peptide HPVHHYQ-NH2 interactions with A-site RNA were observed, even under very different buffer systems. The affinity determined by ITC is in the low micromolar range, with a Kd of 2 μM. The dissociation constant, while in the same range, is slightly higher when determined by other methods (15 to 29 μM). Of note, however, is the fact that ITC determines the affinity directly – not through an enzyme reaction or in the presence of chemical tags on the RNA or peptide – and takes place completely in solution, therefore the slightest changes in environment can be detected. Unfortunately, the ITC method requires large quantities of RNA and peptide and is not really suitable for further screening assays. The apparent Kd value for the peptide-RNA interaction using ESI-MS under optimized conditions is consistent with that achieved in solution phase by ITC or enzymatic footprinting. Therefore, ESI-MS is a more efficient and relatively easy way to detect RNA-peptide complexes and to determine the stoichiometry of binding as well as relative binding affinities. In addition, ESI-MS competition studies with paromomycin and peptide indicate that their binding regions on the A-site RNA are overlapping or similar. Therefore, even though ESI-MS is not as reliable as ITC for determining absolute binding affinities, it is a valuable method for screening for peptides or compounds with higher A-site RNA affinities than HPVHHYQ-NH2, as well as determining relative binding affinities of related molecules.
Enzymatic footprinting revealed an average apparent Kd value of 16 ± 4 μM. This value was in good agreement with those achieved using ESI-MS. As with ESI-MS, the footprinting method does not give absolute binding affinities, but is highly useful for comparative studies and for providing key information about ligand binding sites on the RNA target. The footprinting assay revealed that the peptide-binding site is located at the A-site asymmetrical bulge, with protection of U1495 and G1494, which are residues adjacent to the internal loop. These data also strongly support the results obtained from fluorescent-bead assays and ESI-MS competition binding experiments that indicated the A-bulge motif as a binding determinant for HPVHHYQ-NH2. The results from the footprinting experiment also indicate that binding of peptide to the A-site RNA at sites G1494 and U1495 induces a conformational change that leads to enhanced cleavage at A1493 and A1492. The enhanced exposure towards RNase A cleavage is suggestive of possible base flipping of the two adenine residues. Such an induced conformational change upon binding of aminoglycosides to the A-site RNA has been observed previously in model RNAs (14, 47–49) and in X-ray crystal structures and molecular modeling studies of the complete ribosome (7, 11, 50, 51). The binding mode of HPVHHYQ-NH2, with respect to base flipping, may be similar to the antibiotics such as paromomycin, but with a reduced electrostatic component of binding. Such an affect would be significant with respect to new antibiotic development. NMR structure studies that are currently in progress should provide further information regarding peptide-induced conformational changes of the A-site RNA.
Of note, the two phage-selected peptides appeared to have higher A-site RNA affinity and a higher level of selectivity on the beads. It is possible that C-terminal attachment influences the binding behavior of the peptides. The role of peptide amidation versus other types of C-terminal modifications was not explored in this study, but will be a topic of interest for future studies. Such experiments will help to determine whether C-terminal sequences, such as those found on the phage-displayed peptides, induce peptide folding, and therefore enhance the binding affinity and/or selectivity for the RNA target.
The ideal drug lead or ligand needs to have several key properties, such as strong binding affinity and high specificity to its target. However, the most important factor is to have a ligand that performs certain functions in vitro and in vivo. Our ultimate goal is to interfere with the biological function of bacterial 16S rRNA, which participates in protein synthesis, and is specifically involved in base flipping and contacts with both mRNA and tRNA. Although the IC50 was much higher than neomycin, some level of translation inhibition was detected. Of course, even very tight binders may not have the desired functional effects in vivo. Another caveat is that in this experiment, a circular DNA template was used rather than mRNA. Therefore, further experiments need to be conducted in order to determine whether inhibition occurs at the translational or transcriptional levels. In addition, although the A-site RNA used in this study has been shown to be a validated model system for ligand interactions that occur in the ribosome (12, 13), further studies with whole ribosomes and the selected peptides still need to be carried out.
We conclude that using the phage–display library can quickly lead to peptide ligands that bind to a specific RNA target at the desired site or motif with moderately high binding affinity (low μM range). Related studies that were reported recently reveal similar results (52). The limitation of using phage display to screen peptides that specifically interfere with protein synthesis of bacteria is that the strongest binders may not actually be selected because the host E. coli would not survive, therefore failing to amplify the phage bearing the infused peptide. We also observed that the pool of peptides after screening tend to be hydrophobic. This may be due to the process of maturation of M13 phage. Therefore, the peptide sequences must be optimized through alanine scanning and other synthetic/screening methods. Overall, this study provides information regarding the binding mode of the peptide by using a variety of methods. Further studies on the RNA-peptide interactions using other methods such as NMR spectroscopy and alanine scanning, as well as in vivo functional investigations using already established genetic systems (53), will give us more insight into this new peptide-RNA interaction. Future studies will involve peptide screening against mutant A-site RNAs that could potentially lead to antibiotic resistance. The knowledge gained from this work regarding A-site-RNA-peptide interactions will provide useful guidelines for the design of new small molecules that can specifically target the ribosomal decoding region and ultimately lead to novel anti–infective compounds.
Supplementary Material
Acknowledgments
We thank P. Cunningham and K. Baker for providing plasmid pWK122 and for assistance with DNA sequencing, A. Feig and N. Salim for training, helpful discussion, and providing instrumentation for the ITC experiments, P.-W. Chao and B. Shay for ESI-MS training, T. Morii and M. Fukuda for assistance with RNA construct design and helpful discussion on phage-display techniques, and M. Friedrich and K. Honn for providing access to the fluorescence microscope and plate reader.
Abbreviations
- A site
aminoacyl-tRNA site
- rRNA
ribosomal RNA
- ITC
isothermal titration calorimetry
- ESI-MS
electrospray ionization mass spectrometry
- MALDI-TOF
matrix assisted laser desorption/ionization – time of flight
- [5′-32P] pCp
cytidine-3′, 5′-bisphosphate
- CIAP
calf intestinal alkaline phosphatase
- BCIP
5-bromo-4-chloro-3-indolyl-phosphate
- HOBt
1-hydroxybenzotriazole
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- X-Gal
5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside
- PNK
polynucleotide kinase
- TEMED
N,N,N′,N′-tetramethyl-ethane-1,2-diamine
- PEG
polyethylene glycol
- BSA
bovine serum albumin
- LB-Tet
Luria Bertani medium containing 40 mg/ml tetracycline
- PCR
polymerase chain reaction
- NTP
nucleotide triphosphate
- dNTP
deoxyribonucleotide triphosphate
- DMF
dimethylformamide
- pfu
plaque-forming unit
- nt
nucleotide
Footnotes
This work was supported by NIH grant AI061192.
SUPPORTING INFORMATION AVAILABLE
Figures S1–S4 show MALDI-TOF characterization of HPVHHYQ-NH2, ESI-MS data (salt dependence), and binding curve for RNase footprinting data (A1493). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Spellberg B, Powers JH, Brass EP, Miller LG, Edwards JE., Jr Trends in antimicrobial drug development: implications for the future. Clin Infect Dis. 2004;38:1279–1286. doi: 10.1086/420937. [DOI] [PubMed] [Google Scholar]
- 2.Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5:175–186. doi: 10.1038/nrmicro1614. [DOI] [PubMed] [Google Scholar]
- 3.Anderson AD, Nelson JM, Rossiter S, Angulo FJ. Public health consequences of use of antimicrobial agents in food animals in the United States. Microb Drug Resist. 2003;9:373–379. doi: 10.1089/107662903322762815. [DOI] [PubMed] [Google Scholar]
- 4.Tomasz A. Multiple-antibiotic-resistant pathogenic bacteria. A report on the Rockefeller University Workshop. N Engl J Med. 1994;330:1247–1251. doi: 10.1056/NEJM199404283301725. [DOI] [PubMed] [Google Scholar]
- 5.Sutcliffe JA. Improving on nature: antibiotics that target the ribosome. Curr Opin Microbiol. 2005;8:534–542. doi: 10.1016/j.mib.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 7.Ogle JM, Brodersen DE, Clemons WM, Jr, Tarry MJ, Carter AP, Ramakrishnan V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001;292:897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- 8.Lynch SR, Gonzalez RL, Puglisi JD. Comparison of X-ray crystal structure of the 30S subunit-antibiotic complex with NMR structure of decoding site oligonucleotide-paromomycin complex. Structure. 2003;11:43–53. doi: 10.1016/s0969-2126(02)00934-6. [DOI] [PubMed] [Google Scholar]
- 9.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 10.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 11.Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell. 2002;111:721–732. doi: 10.1016/s0092-8674(02)01086-3. [DOI] [PubMed] [Google Scholar]
- 12.Lynch SR, Puglisi JD. Structural origins of aminoglycoside specificity for prokaryotic ribosomes. J Mol Biol. 2001;306:1037–1058. doi: 10.1006/jmbi.2000.4420. [DOI] [PubMed] [Google Scholar]
- 13.Yoshizawa S, Fourmy D, Puglisi JD. Recognition of the codon-anticodon helix by ribosomal RNA. Science. 1999;285:1722–1725. doi: 10.1126/science.285.5434.1722. [DOI] [PubMed] [Google Scholar]
- 14.Pfister P, Hobbie S, Vicens Q, Böttger EC, Westhof E. The molecular basis for A-site mutations conferring aminoglycoside resistance: relationship between ribosomal susceptibility and X-ray crystal structures. Chembiochem. 2003;4:1078–1088. doi: 10.1002/cbic.200300657. [DOI] [PubMed] [Google Scholar]
- 15.Pfister P, Hobbie S, Brüll C, Corti N, Vasella A, Westhof E, Böttger EC. Mutagenesis of 16S rRNA C1409-G1491 base-pair differentiates between 6′OH and 6′NH3+ aminoglycosides. J Mol Biol. 2005;346:467–475. doi: 10.1016/j.jmb.2004.11.073. [DOI] [PubMed] [Google Scholar]
- 16.Beauclerk AA, Cundliffe E. Sites of action of two ribosomal RNA methylases responsible for resistance to aminoglycosides. J Mol Biol. 1987;193:661–671. doi: 10.1016/0022-2836(87)90349-4. [DOI] [PubMed] [Google Scholar]
- 17.Thompson J, Skeggs PA, Cundliffe E. Methylation of 16S ribosomal RNA and resistance to the aminoglycoside antibiotics gentamicin and kanamycin determined by DNA from the gentamicin-producer Micromonospora purpurea. Mol Gen Genet. 1985;201:168–173. doi: 10.1007/BF00425655. [DOI] [PubMed] [Google Scholar]
- 18.Gregory ST, Carr JF, Dahlberg AE. A mutation in the decoding center of Thermus thermophilus 16S rRNA suggests a novel mechanism of streptomycin resistance. J Bacteriol. 2005;187:2200–2202. doi: 10.1128/JB.187.6.2200-2202.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gutell RR. Collection of small subunit (16S- and 16S-like) ribosomal RNA structures: 1994. Nucleic Acids Res. 1994;22:3502–3507. doi: 10.1093/nar/22.17.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science. 1996;274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 21.Vicens Q, Westhof E. Crystal structure of paromomycin docked into the eubacterial ribosomal decoding A site. Structure. 2001;9:647–658. doi: 10.1016/s0969-2126(01)00629-3. [DOI] [PubMed] [Google Scholar]
- 22.Recht MI, Fourmy D, Blanchard SC, Dahlquist KD, Puglisi JD. RNA sequence determinants for aminoglycoside binding to an A-site rRNA model oligonucleotide. J Mol Biol. 1996;262:421–436. doi: 10.1006/jmbi.1996.0526. [DOI] [PubMed] [Google Scholar]
- 23.Pierce HH, Adey N, Kay BK. Identification of cyclized calmodulin antagonists from a phage display random peptide library. Mol Divers. 1996;1:259–265. doi: 10.1007/BF01715530. [DOI] [PubMed] [Google Scholar]
- 24.Ramanujam P, Tan WS, Nathan S, Yusoff K. Novel peptides that inhibit the propagation of Newcastle disease virus. Arch Virol. 2002;147:981–993. doi: 10.1007/s00705-001-0778-y. [DOI] [PubMed] [Google Scholar]
- 25.Ho KL, Yusoff K, Seow HF, Tan WS. Selection of high affinity ligands to hepatitis B core antigen from a phage-displayed cyclic peptide library. J Med Virol. 2003;69:27–32. doi: 10.1002/jmv.10266. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura GR, Reynolds ME, Chen YM, Starovasnik MA, Lowman HB. Stable “zeta” peptides that act as potent antagonists of the high-affinity IgE receptor. Proc Natl Acad Sci USA. 2002;99:1303–1308. doi: 10.1073/pnas.022635599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popa I, Ishikawa D, Tanaka M, Ogino K, Portoukalian J, Taki T. GD3-replica peptides selected from a phage peptide library induce a GD3 ganglioside antibody response. FEBS Lett. 2006;580:1398–1404. doi: 10.1016/j.febslet.2006.01.063. [DOI] [PubMed] [Google Scholar]
- 28.Smith GP, Petrenko VA. Phage display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 29.Willats WG. Phage display: practicalities and prospects. Plant Mol Biol. 2002;50:837–854. doi: 10.1023/a:1021215516430. [DOI] [PubMed] [Google Scholar]
- 30.Gooch BD, Krishnamurthy M, Shadid M, Beal PA. Binding of helix-threading peptides to E. coli 16S ribosomal RNA and inhibition of the S15–16S complex. Chembiochem. 2005;6:2247–2254. doi: 10.1002/cbic.200500285. [DOI] [PubMed] [Google Scholar]
- 31.Atherton E, Sheppard RC. Solid Phase peptide synthesis: a practical approach. IRL Press; Oxford, England: 1989. [Google Scholar]
- 32.Goodwin TW, Morton RA. The spectrophotometric determination of tyrosine and tryptophan in proteins. Biochem J. 1946;40:628–632. doi: 10.1042/bj0400628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hwang S, Tamilarasu N, Ryan K, Huq I, Richter S, Still WC, Rana TM. Inhibition of gene expression in human cells through small molecule-RNA interactions. Proc Natl Acad Sci USA. 1999;96:12997–13002. doi: 10.1073/pnas.96.23.12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kieltyka JW, Chow CS. Probing RNA hairpins with cobalt(III)hexammine and electrospray ionization mass spectrometry. J Am Soc Mass Spectrom. 2006;17:1376–1382. doi: 10.1016/j.jasms.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 35.Sannes-Lowery KA, Griffey RH, Hofstadler SA. Measuring dissociation constants of RNA and aminoglycoside antibiotics by electrospray ionization mass spectrometry. Anal Biochem. 2000;280:264–271. doi: 10.1006/abio.2000.4550. [DOI] [PubMed] [Google Scholar]
- 36.Bligh SW, Haley T, Lowe PN. Measurement of dissociation constants of inhibitors binding to Src SH2 domain protein by non-covalent electrospray ionization mass spectrometry. J Mol Recognit. 2003;16:139–148. doi: 10.1002/jmr.622. [DOI] [PubMed] [Google Scholar]
- 37.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 38.Davies J, Gorini L, Davis BD. Misreading of RNA codewords induced by aminoglycoside antibiotics. Mol Pharmacol. 1965;1:93–106. [PubMed] [Google Scholar]
- 39.Swayze EE, Jefferson EA, Sannes-Lowery KA, Blyn LB, Risen LM, Arakawa S, Osgood SA, Hofstadler SA, Griffey RH. SAR by MS: a ligand based technique for drug lead discovery against structured RNA targets. J Med Chem. 2002;45:3816–3819. doi: 10.1021/jm0255466. [DOI] [PubMed] [Google Scholar]
- 40.Weeks KM, Crothers DM. Major groove accessibility of RNA. Science. 1993;261:1574–1577. doi: 10.1126/science.7690496. [DOI] [PubMed] [Google Scholar]
- 41.Chow CS, Bogdan FM. A structural basis for RNA-ligand interactions. Chem Rev. 1997;97:1489–1514. doi: 10.1021/cr960415w. [DOI] [PubMed] [Google Scholar]
- 42.Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem Rev. 2008;108:1171–1224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 43.Thomas JR, Liu X, Hergenrother PJ. Biochemical and thermodynamic characterization of compounds that bind to RNA hairpin loops: towards an understanding of selectivity. Biochemistry. 2006;45:10928–10938. doi: 10.1021/bi0607296. [DOI] [PubMed] [Google Scholar]
- 44.Llano-Sotelo B, Azucena EF, Jr, Kotra LP, Mobashery S, Chow CS. Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem Biol. 2002;9:455–463. doi: 10.1016/s1074-5521(02)00125-4. [DOI] [PubMed] [Google Scholar]
- 45.Wong CH, Hendrix M, Priestley ES, Greenberg WA. Specificity of aminoglycoside antibiotics for the A-site of the decoding region of ribosomal RNA. Chem Biol. 1998;5:397–406. doi: 10.1016/s1074-5521(98)90073-4. [DOI] [PubMed] [Google Scholar]
- 46.Kaul M, Pilch DS. Thermodynamics of aminoglycoside-rRNA recognition: the binding of neomycin-class aminoglycosides to the A site of 16S rRNA. Biochemistry. 2002;41:7695–7706. doi: 10.1021/bi020130f. [DOI] [PubMed] [Google Scholar]
- 47.Shandrick S, Zhao Q, Han Q, Ayida BK, Takahashi M, Winters GC, Simonsen KB, Vourloumis D, Hermann T. Monitoring molecular recognition of the ribosomal decoding site. Angew Chem Int Ed Engl. 2004;43:3177–3182. doi: 10.1002/anie.200454217. [DOI] [PubMed] [Google Scholar]
- 48.Kaul M, Barbieri CM, Pilch DS. Fluorescence-based approach for detecting and characterizing antibiotic-induced conformational changes in ribosomal RNA: comparing aminoglycoside binding to prokaryotic and eukaryotic ribosomal RNA sequences. J Am Chem Soc. 2004;126:3447–3453. doi: 10.1021/ja030568i. [DOI] [PubMed] [Google Scholar]
- 49.Chao PW, Chow CS. Monitoring aminoglycoside-induced conformational changes in 16S rRNA through acrylamide quenching. Bioorg Med Chem. 2008;15:3825–3831. doi: 10.1016/j.bmc.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meroueh SO, Mobashery S. Conformational transition in the aminoacyl t-RNA site of the bacterial ribosome both in the presence and absence of an aminoglycoside antibiotic. Chem Biol Drug Des. 2007;69:291–297. doi: 10.1111/j.1747-0285.2007.00505.x. [DOI] [PubMed] [Google Scholar]
- 51.Vaiana AC, Sanbonmatsu KY. Stochastic gating and drug-ribosome interactions. J Mol Biol. 2009;386:648–661. doi: 10.1016/j.jmb.2008.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Llano-Sotelo B, Klepacki D, Mankin A. Selection of Small Peptides, Inhibitors of Translation. J Mol Biol. 2009 doi: 10.1016/j.jmb.2009.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laios E, Waddington M, Saraiya AA, Baker KA, O’Connor E, Pamarathy D, Cunningham PR. Combinatorial genetic technology for the development of new anti-infectives. Arch Pathol Lab Med. 2004;128:1351–1359. doi: 10.5858/2004-128-1351-CGTFTD. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.