

von Willebrand Factor (VWF) is secreted as hyperactive, ultralarge multimers that are cleaved by the metalloprotease ADAMTS13 to smaller multimers. Cleaved VWF is activated by hydrodynamic forces found in arteriolar bleeding to promote hemostasis, whereas uncleaved VWF is activated at lower, physiologic shear stresses and causes thrombosis. ADAMTS13 cleaves a specific peptide bond in the VWF A2 domain. Here we use single molecule experiments to test the hypothesis that tensile force, applied to the N and C-termini of the A2 domain, acts as a cofactor for its enzymatic cleavage. Unfolding of the A2 domain occurs in a physiologically relevant range, with an intermediate unfolded state. The unfolded, but not folded A2 domain is cleaved by ADAMTS13. The lifetime of the unfolded state of 1.9 s in absence of force provides a limited time window for cleavage. The single molecule KM is lower than for peptide substrates, suggesting that A2 unfolded by force is the physiologic substrate. In shear flow, force on a VWF multimer goes up with the square of multimer length and is highest at the middle, providing an efficient mechanism for homeostatic regulation in vivo of the size distribution of VWF multimers by force-induced A2 unfolding and cleavage by ADAMTS13.
VWF is the key shear-sensing protein in hemostasis, and is especially important in arterial bleeding where shear is high (1). VWF is biosynthesized and stored in the Weibel-Palade bodies of endothelial cells in an ultralarge form (ULVWF). The VWF 240,000 Mr monomer (Fig. 1a) is concatenated through specific disulfide bonds at both its N and C- termini into multimers of up to ∼50 × 106 Mr in ULVWF (1, 2). ULVWF is secreted in response to thrombogenic stimuli. A portion of secreted ULVWF is bound locally to endothelial cells from which it is released, and also through its A3 domain to collagen at sites of tissue injury. Vessel wall-bound VWF multimers, as well as multimers free in the bloodstream, are extended to a length of up to 15 μm by the hydrodynamic forces in shear flow (2). These forces induce a conformational change in VWF that exposes a binding site in the A1 domain for the gpIb molecule of platelets, enabling formation of a hemostatic platelet plug (1, 3).
Figure 1. A2 domain unfolding and refolding with laser tweezers.

a. Domain organization of VWF. Cysteines and disulfide bonds are shown beneath, and N- and O-linked sites above as filled and open lollipops, respectively. b. Experimental setup. A2 domain (enlarged in inset with ADAMTS13 cleavage site arrowed) is coupled to double-stranded DNA handles, which are bound through tags at their other ends to beads held by a laser trap and a translatable (arrows) micropipette. c. Force on a molecular tether during representative cycles of force increase, decrease, and clamping at a constant low level. d. Force-extension traces during force loading in cycles ii and iii from panel b.
Within two hours after release from endothelium into the circulation, ULVWF is converted by ADAMTS13 to smaller multimers with a wide range of size distributions that are characteristic of the circulating pool of VWF (4). Since the length of VWF multimers strongly correlates with hemostatic potential, cleavage by ADAMTS13 is an important regulatory mechanism. Absence of ADAMTS13 results in increased thrombogenic potential of VWF, and thrombotic thrombocytopenic purpura, a life threatening disease caused by uncontrolled microvascular thrombosis (5). On the other hand, mutations in the A2 domain that presumably destabilize it cause excessive cleavage by ADAMTS13, and a shift in the distribution of VWF multimers to lower Mr. The smaller VWF multimers have less hemostatic potential, and result in the bleeding disorder known as type 2A von Willebrand disease (3, 6).
VWF is cleaved by ADAMTS13 within the A2 domain at its Tyr1605-Met1606 bond (1, 3, 5, 7, 8). Cleavage is activated by shear when A2 is present in large VWF concatamers, but not when present as the much smaller, isolated domain (5-7). Presumably, this is because the forces acting on proteins in shear flow scale with protein size and length (9). Shear flow elongates VWF (2), and tensile force exerted on the concatamer is thought to cause conformational changes in A2 domains that enable cleavage (3, 5, 8). The nature of this conformational change is unknown. The A2 domain of VWF is cleaved specifically. It is thought that the scissile bond would be buried in the native state (10, 11). Therefore, partial or complete unfolding may be the mechanism for substrate activation (6). Here, by directly applying force with laser tweezers (12, 13) to a single A2 domain, we test the hypothesis that unfolding and folding of the A2 domain may occur at forces capable of being experienced by VWF in its transit through the circulation or at sites of hemostasis and thrombosis, and that force acts as a cofactor to unfold A2 for cleavage by ADAMTS13.
Single, N-glycosylated A2 domains coupled to DNA handles through N and C-terminal Cys tags (Fig. S1) were suspended between beads held in a laser trap and micropipette (Fig. 1b). A2 domains were subjected to cycles of force increase, force decrease, and clamping at a low force to enable refolding prior to the next cycle (Fig. 1c). A2 domain unfolding was marked by abrupt tether extension events (Fig. 1c inset and 1d, cycle ii). The increase in length at different forces was fit to the worm-like chain (WLC) model (14) (Fig. 2a), yielding an A2 contour length of 57 nm ± 5 nm and a persistence length of 1.1 nm ± 0.4 nm. A2 N-terminal and C-terminal residues Met1495 and Ser1671 are 1 nm apart in the folded state (15). The total length of 58 ± 5 nm divided by an extension length of 0.36 nm/residue yields unfolding of 161±14 residues. This corresponds well to complete unfolding of the predicted 177-residue A2 domain.
Figure 2. Unfolding of the A2 domain.
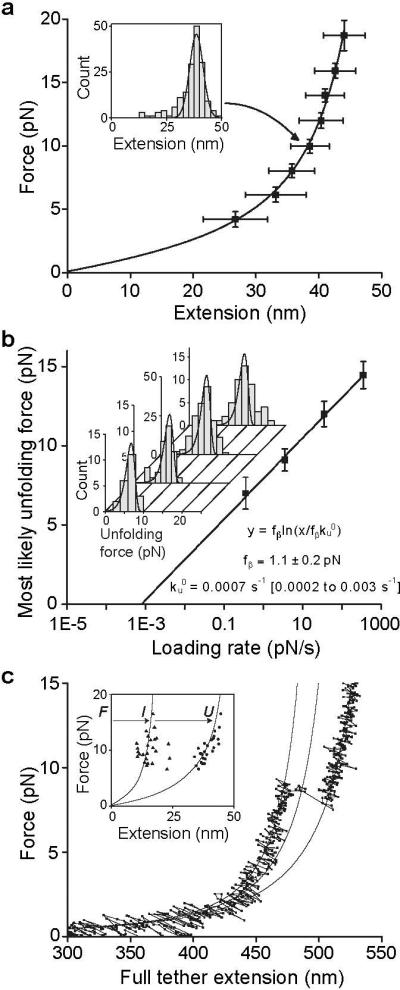
a. A2 domain force-extension data with an error-weighted least squares fit to the worm-like chain model (line) (14). Extension distances were sorted by unfolding force into 2 pN bins. A histogram of extensions for each bin (inset) was fit to a Gaussian (inset, solid line) to find peak extension, and force averaged for that bin. Uncertainty in extension is shown as the half width of the Gaussian fit and uncertainty in force is shown as one standard deviation. b. Unfolding force as a function of loading rate. Unfolding forces were binned by loading rate and plotted as histograms (inset). The peak of each histogram was plotted against the loading rate; uncertainty in y is shown as half of the bin width. A linear fit to the data (line) predicts the distributions of unfolding force (inset, lines), which agree well the histograms (inset). c. Representative force-extension trace for a tether pausing at an intermediate state, with three
WLC curves (solid lines) representing DNA + folded A2, DNA + partially unfolded A2, and DNA + fully unfolded A2. Inset: Extensions of A2 to intermediate (I) and unfolded (U) lengths, fit to the WLC model (lines).
Over a range of force loading rates, unfolding force was determined and plotted against the logarithm of the loading rate (Fig. 2b). The fit to a single-barrier kinetic model (16) yields an unfolding rate in the absence of force, , of 0.0007 s−1 (confidence band of 0.0002 s−1 to 0.003 s−1), and a force scale, fβ, which exponentially increases the unfolding rate , of 1.1 ± 0.2 pN.
A subset of about 20% of unfolding events included a discernable pause (defined by 4 or more data points at a short-lived (Fig. S3), partially unfolded intermediate state, which was directly observed in force-extension curves (Fig. 2c). Fit to the worm-like chain model of the A2 extension distances (Fig. 2c, inset) shows that the intermediate state usually lies 40% of the distance between the fully folded and unfolded states.
During the pause at a clamped force between each cycle of force decrease and increase, the A2 domain had the opportunity to refold (Fig. 1c). Subsequent unfolding revealed folding during the pause (Fig. 1c,d, cycle ii), while a lack of unfolding suggested an absence of refolding (Fig. 1c,d, cycle iii). The binary state of the domain was further confirmed with force extension curves, which have distinct branches for the unfolded and folded states (Fig. 1d). The force dependence of refolding (Fig. 3) was fit using maximum likelihood to an f2 model, which takes into account the soft compliance of the unfolded state (16-19): (see also Fig. S3). We found a refolding rate in absence of force , and compliance κ = 0.18 +/− 0.04 pN/nm.
Figure 3. A2 domain refolding kinetics.

Binary refolding events were binned by clamp force and time. Standard errors (bars) were calculated as (p*(1-p)/n)0.5, where p is fraction refolded and n is number of events. Overlaid on the data are the exponential curves predicted by maximum likelihood estimation (i.e. on the data without binning) using the τ∼ exp(f2) model.
Using the folding and unfolding rates in the absence of force, we can estimate the free energy difference between the two states: . This is close to the ΔG of 5.9±0.8 kBT ( 3.5±0.5 kcal/mol) estimated from urea-induced unfolding of an E. coli A2 fragment (20).
To test the hypothesis that A2 unfolding is required for cleavage by ADAMTS13, A2 was mechanically unfolded in the absence or presence of ADAMTS13, and relaxed to a clamped force of 5 pN (Fig. 4a). At this force, the lifetime of the unfolded state is >140 s, making refolding unlikely during the incubation with ADAMTS13. Cleavage by ADAMTS13 was detected as a drop in force on the tether to 0 pN (Fig. 4a, panel 1). Spontaneous rupture at 5 pN, i.e. the background with no enzyme (Fig. 4a, panel 2), was rare (Fig. 4b inset). In experiments with a lower force ramp, unfolding sometimes did not occur, as shown by lack of the characteristic force-extension signature. No cleavage of folded A2 at 5 pN with 100 nM or 1 μM enzyme was observed.
Figure 4. Mechanoenzymatic cleavage of A2 by ADAMTS13.

a. Representative traces showing cleavage in presence of enzyme ( panel 1) and no cleavage in absence of enzyme (panel 2) b. Enzyme kinetics. The hyperbolic dependence of catalytic rate on enzyme concentration was fit with the single-molecule Michaelis-Menten equation (21) (solid line). Data points and standard error were determined from single parameter exponential fits to the survival fraction as a function of time (inset).
With unfolded A2 in presence of enzyme, the fraction of surviving tethers decreased exponentially with time, demonstrating first-order reaction kinetics, and yielding the time constant τ for cleavage at three different enzyme concentrations (Fig. 4b inset). The observed enzymatic rate, i.e. reciprocal of τ, was fit with the single-molecule Michaelis-Menten equation (21), 1/τ = kcat[ADAMTS13]/([ADAMTS13]+ KM) (Fig. 4b).
As the largest known soluble protein, VWF has more force exerted on it than any other free protein in the vasculature. Hydrodynamics and the overall shape and orientation of VWF multimers in flow are relevant to understanding the tensile force exerted on A2 domains within ULVWF, and trimming by ADAMTS13 (5). In shear flow, the rate of fluid flow increases from the wall toward the center (Fig. 5a,b). The product of shear rate and viscosity, shear stress, in units of force per area, imparts force to particles in shear flow that is related to their surface area. Compared to VWF free in flow, the hydrodynamic force at a given shear is much higher on VWF immobilized on a vessel wall or bridging two platelets free in flow, and at intermediate levels for VWF bound to a single platelet free in flow (9, 22). Because of weak attractive interactions between domains within each multimer, VWF multimers have an overall compact, yarn ball-like shape in stasis (2, 23-25). Above a critical shear stress of 50 dyn/cm2 (see supplement), the attractive forces are overcome by hydrodynamic drag, and VWF free in flow periodically elongates and contracts (2, 25) (Fig. 5c). Shear flow can be conceptualized as the superposition of rotational flow and elongational flow (Fig. 5b). The rotational flow causes particles to tumble (Fig. 5c). Tumbling is very evident for polymers such as DNA (26); however, the attractive forces between VWF monomers appear to keep it largely zipped up during tumbling, but with alternating cycles of elongation and compaction (Fig. 5c) (2, 25).
Figure 5. Model for mechanoenzymatic cleavage of ULVWF in the circulation.

a. Shear flow in a vessel and elongational flow at a site of bleeding. b. Shear flow may be represented as elongational flow superimposed on rotational flow (modified from (26)). c. Cartoon of VWF elongating, compressing, and tumbling in shear flow. d. Peak force as function of monomer position in a VWF multimer chain of 200, 100, or 50 monomers at 100 dyn/cm2. Dashed line shows the most likely unfolding force for the A2 domain at a loading rate of 25 pN/s. e. Schematic of VWF, with N-end as triangle, A2 as spring, and C-end as circle. Elongation results in unfolding of some A2 domains, some of which are cleaved (arrows). The resulting fragments are shown.
Several concepts from the field of polymer dynamics have not previously been introduced in the VWF literature. Importantly, for an extended VWF multimer with N monomers, the force on a monomer increases with distance from the nearest end of the multimer (Fig. 5d), and force at the middle of the multimer is proportional to N2 (Fig. 5d) (27)(Supplement). Force increases with the square of length because both multimer size, and the difference in velocity between shear lamina in which the two ends of the multimer find themselves, increase with length ((9, 27) and Supplement). This previously unremarked second power dependence has important implications not only for unfolding of the A2 domain and cleavage by ADAMTS13 (Fig. 5d), but also for shear-induced aggregation of platelets through the A1 domain of VWF, since it explains the much greater potency of longer than shorter VWF multimers both in hemostasis and thrombosis (1, 5).
Could the tensile force on VWF free in the circulation reach levels in vivo sufficient to explain unfolding of the A2 domain and cleavage by ADAMTS13? The tensile force is estimated (Supplement) to reach 10 pN on the middle of a VWF 200-mer at the maximal shear stress of 100 dyn/cm2 (shear rate of 5,000 s−1) found in healthy vessels in vivo (5, 28) (Fig. 5d). Using a loading rate of 25 pN/s estimated from the VWF tumbling rate in shear (Supplement), the A2 domain typically unfolds at about 11 pN (peak of the unfolding force distribution, Fig. 2b). The upper size limit of VWF in the circulation is variously estimated to correspond to a 100-mer (1) or a 200-mer (2, 23, 24) (Supplement). Thus, our single molecule data on the A2 domain successfully predicts the observed upper size limit of VWF multimers in vivo as ∼200 monomers (Fig. 5d). Caveats include uncertainty in the angle of maximally extended VWF with respect to flow direction, which could influence the magnitude of the peak force estimate by several fold (Supplement), simplifying assumptions made in the calculations, and a possible contribution of platelets to VWF trimming (22, 29). The dynamics of VWF in shear flow is an important area of future investigation for understanding susceptibility to ADAMTS13 as well as activation in hemostasis.
The existence of a clear threshold for the lengths of VWF multimers has been shown in vivo; a bolus of ULVWF released from endothelium into the circulation is trimmed to the pre-existing equilibrium length distribution of circulating VWF multimers within 2 h by ADAMTS13 (4). Our analysis illustrates the principles that dictate the maximum length of circulating VWF multimers in vivo, and the previously unappreciated concept that the force on VWF free in the circulation is sufficient to induce unfolding of the A2 domain and cleavage by ADAMTS13.
Another important concept known in polymer dynamics (26) not previously introduced in the VWF literature is elongational flow (Fig. 5a,b). In contrast to shear flow, which has both rotational and elongational components (Fig. 5a left), close to a site of hemorrhage flow will transition to elongational (Fig. 5a right). Although the actual flow pattern would be complex, the overall picture is that tumbling and alternating cycles of compression will tend to cease, and VWF will only experience elongation. Alignment of extended VWF with the flow direction could increase peak tensile force approximately 10-fold over that experienced in shear flow (Supplement).
We have definitively established that unfolding is required for cleavage of the A2 domain by ADAMTS13. Intriguingly, in a proportion of unfolding events, a transient intermediate state was observed. In VWF A2, the N-terminal β1-strand is central in the fold, whereas the C-terminal α6-helix is peripheral. Therefore, unfolding induced by elongational force will begin at the C-terminus, as confirmed by simulations (15). Unfolding of 40% of the contour length in the intermediate state would thus correspond to the unfolding of about 70 C-terminal residues, up to and including the β4-strand, which contains the scissile Tyr1605-Met1606 peptide bond. Studies with peptide fragments show that C-terminal, but not N-terminal segments distal from the cleavage site are recognized by ADAMTS13 (30). Thus it is possible that ADAMTS13 could recognize and cleave the intermediate unfolded state.
Our single-molecule kcat for the ADAMTS13 enzyme of 0.14 s−1 is in the range of 0.14 to 1.3 s−1 determined in bulk phase with unfolded peptide substrates corresponding to the C-terminal 70 residues of A2 (11, 31). However, our KM of 0.16 μM is lower than previous estimates of 1.7 and 1.6 μM (11, 31). The lower KM value determined here may reflect a more physiologic state of the substrate. Notably, different domains within ADAMTS13 recognize different portions of the unfolded peptide substrate that are far apart in sequence (30, 31). Whereas peptide substrates have essentially random configurations, tension applied to the unfolded A2 domain partially orders it in one dimension, and this more linear configuration may be more optimal for recognition by different domains within ADAMTS13.
VWF will only be exposed to peak shear intermittently during each tumbling cycle, and only to high shear during transit through arterioles and capillaries. The lifetime of about 2 s of the unfolded state in the absence of force is longer than the time period of peak force exposure (9) (Supplement), and provides a window of opportunity after unfolding for cleavage of circulating VWF by ADAMTS13. Refolding to the correct low energy state after tension is released is another property of the A2 domain that is important to its function in vivo. Aberrant refolding could permit cleavage by ADAMTS13, as is observed with some A2 domain preparations from E. coli (32).
Our single molecule enzyme assays suggest that the rate of VWF cleavage is limited by ADAMTS13 concentration in vivo, which at 6 nM (33), is substantially below the KM of 160 nM, and yields a timescale for cleavage in vivo of ∼200 s. Although the numbers may be altered for cleavage of unfolded A2 within intact VWF, these rough estimates are relevant to understanding events in vivo. Thus, over the short time periods of < 1 s important in hemostasis, binding of VWF through the A1 domain to gpIb on platelets and through the A3 domain to collagen on the subendothelium should win out over cleavage of the A2 domain by ADAMTS13. These kinetic differences enable the same biophysical principles, i.e. the relation of force to the second power of multimer length, and the transition from shear to elongational flow at sites of hemorrhage, to activate both hemostasis by VWF, and cleavage by ADAMTS13.
A further wrinkle is added by a cis-Pro recently discovered in the A2 structure (15) consistent with a small number of A2 tethers that suddenly stopped refolding, and after a long delay, resumed refolding (Supplement). VWF, bound to platelets at sites of hemorrhage, would be exposed to forces sufficient to accelerate cis-to-trans peptide isomerization (34) in unfolded A2. A trans-Pro would be a long-lasting (100 to 1000 s) impediment to refolding that would enhance cleavage by ADAMTS13 during wound repair.
Its unique lack of protection by disulfide bonds within VWF (Fig. 1a), and low resistance to unfolding, suggest that the A2 domain has evolved to be the shear bolt domain of VWF. A shear bolt breaks above a designed force threshold, to protect other parts of a machine from accidental damage. Similarly, the A2 domain unfolds when present in VWF multimers that experience high tensile force and is cleaved by ADAMTS13, resulting in down regulation of the hemostatic activity.
Supplementary Material
Acknowledgements
Supported by NIH HL-48675 (TAS), the Rowland Junior Fellows (WPW), and AHA 525918T (XZ). The authors are indebted to Drs. Carlos Bustamante, Susan Marqusee, and Ciro Cecconi for protocols for DNA-protein coupling. We thank Drs. J. Evan Sadler, Diane Schaak, Jongseong Kim, Joonil Seog, Chafen Lu, and Alfredo Alexander-Katz for reagents and insightful discussions, and Graham Dempsey for work on the early stages of this project.
References
- 1.Sadler JE. Annu. Rev. Biochem. 1998;67:395. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- 2.Schneider SW, et al. Proc. Natl. Acad. Sci. U. S .A. 2007;104:7899. doi: 10.1073/pnas.0608422104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sadler JE. Annu. Rev. Med. 2005;56:173. doi: 10.1146/annurev.med.56.082103.104713. [DOI] [PubMed] [Google Scholar]
- 4.Batlle J, et al. Blood. 1987;70:173. [PubMed] [Google Scholar]
- 5.Tsai HM. Semin. Thromb. Haemost. 2003;29:479. doi: 10.1055/s-2003-44556. [DOI] [PubMed] [Google Scholar]
- 6.Tsai HM. Blood. 1996;87:4235. [PubMed] [Google Scholar]
- 7.Furlan M, Robles R, Lamie B. Blood. 1996;87:4223. [PubMed] [Google Scholar]
- 8.Dong JF, et al. Blood. 2002;100:4033. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 9.Shankaran H, Neelamegham S. Biophys. J. 2004;86:576. doi: 10.1016/S0006-3495(04)74136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutherland JJ, O'Brien LA, Lillicrap D, Weaver DF. J. Mol. Model. 2004;10:259. doi: 10.1007/s00894-004-0194-9. [DOI] [PubMed] [Google Scholar]
- 11.Zanardelli S, et al. J. Biol. Chem. 2006;281:1555. doi: 10.1074/jbc.M508316200. [DOI] [PubMed] [Google Scholar]
- 12.Moffitt JR, Chemla YR, Smith SB, Bustamante C. Annu. Rev. Biochem. 2008;77:205. doi: 10.1146/annurev.biochem.77.043007.090225. [DOI] [PubMed] [Google Scholar]
- 13.Greenleaf WJ, Woodside MT, Block SM. Annu. Rev. Biophys. Biomol. 2007;36:171. doi: 10.1146/annurev.biophys.36.101106.101451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bustamante C, Marko JF, Siggia ED, Smith S. Science (New York, N.Y .) 1994;265:1599. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Q, Zhou Y-F, Zhang C.-z., Springer TA. Proc. Natl. Acad. Sci. U. S .A. 2009 in press. [Google Scholar]
- 16.Evans E, Ritchie K. Biophys. J. 1997;72:1541. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanggi P, Talkner P, Borkovec M. Rev. Mod. Phys. 1990;62:251. [Google Scholar]
- 18.Evans E, Halvorsen K, Kinoshita K, Wong WP. In: Handbook of Single Molecule Biophysics. Hinterdorfer P, editor. Springer; 2009. [Google Scholar]
- 19.Wong WP. Harvard University; 2006. [Google Scholar]
- 20.Auton M, Cruz MA, Moake J. J. Mol. Biol. 2007;366:986. doi: 10.1016/j.jmb.2006.10.067. [DOI] [PubMed] [Google Scholar]
- 21.Kou SC, Cherayil BJ, Min W, English BP, Xie XS. J. Phys. Chem. B. 2005;109:19068. doi: 10.1021/jp051490q. [DOI] [PubMed] [Google Scholar]
- 22.Shim K, Anderson PJ, Tuley EA, Wiswall E, Sadler JE. Blood. 2008;111:651. doi: 10.1182/blood-2007-05-093021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fowler WE, Fretto LJ, Hamilton KK, Erickson HP, McKee PA. J. Clin. Invest. 1985;76:1491. doi: 10.1172/JCI112129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slayter H, Loscalzo J, Bockenstedt P, Handin RI. J. Biol. Chem. 1985;260:8559. [PubMed] [Google Scholar]
- 25.Alexander-Katz A, Schneider MF, Schneider SW, Wixforth A, Netz RR. Phys. Rev. Lett. 2006;97:138101. doi: 10.1103/PhysRevLett.97.138101. [DOI] [PubMed] [Google Scholar]
- 26.Smith DE, Babcock HP, Chu S. Science (New York, N.Y .) 1999;283:1724. doi: 10.1126/science.283.5408.1724. [DOI] [PubMed] [Google Scholar]
- 27.Odell JA, Keller A. J. Chem. Phys. 1988;88:4022. [Google Scholar]
- 28.Ruggeri ZM, Mendolicchio GL. Circ. Res. 2007;100:1673. doi: 10.1161/01.RES.0000267878.97021.ab. [DOI] [PubMed] [Google Scholar]
- 29.Genderen v., Budde U, JJ M, R v. S., HH v. V. Br. J. Haematol. 1996;93:962. doi: 10.1046/j.1365-2141.1996.d01-1729.x. [DOI] [PubMed] [Google Scholar]
- 30.Gao W, Anderson PJ, Sadler JE. Blood. 2008;112:1713. doi: 10.1182/blood-2008-04-148759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao W, Anderson PJ, Majerus EM, Tuley EA, Sadler JE. Proc. Natl. Acad. Sci. U. S .A. 2006;103:19099. doi: 10.1073/pnas.0607264104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitelock JL, et al. J. Thromb. Haemost. 2004;2:485. doi: 10.1111/j.1538-7836.2004.00601.x. [DOI] [PubMed] [Google Scholar]
- 33.Feyes HB, et al. J. Thromb. Haemost. 2005;4:955. [Google Scholar]
- 34.Valiaev A, Lim DW, Oas TG, Chilkoti A, Zauscher S. J. Am. Chem. Soc. 2007;129:6491. doi: 10.1021/ja070147r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.