

Abstract
Chlorophyllin (CHL) is a water-soluble derivative of chlorophyll that exhibits cancer chemopreventive properties, but which also has been studied for its possible cancer therapeutic effects. We report here that human colon cancer cells treated with CHL accumulate in S-phase of the cell cycle, and this is associated with reduced expression levels of p53, p21, and other G1/S checkpoint controls. At the same time, E2F1 and E2F4 transcription factors become elevated and exhibit increased DNA binding activity. In CHL-treated colon cancer cells, bromodeoxyuridine pulse-chase experiments provided evidence for the inhibition of DNA synthesis. Ribonucleotide reductase (RR), a pivotal enzyme for DNA synthesis and repair, was reduced at the mRNA and protein level after CHL treatment, and the enzymatic activity was inhibited in a concentration-dependent manner both in vitro and in vivo. Immunoblotting revealed that expression levels of RR subunits R1, R2, and p53R2 were reduced by CHL treatment in HCT116 (p53+/+) and HCT116 (p53−/−) cells, supporting a p53-independent mechanism. Prior studies have shown that reduced levels of RR small subunits can increase the sensitivity of colon cancer cells to clinically-used DNA-damaging agents and RR inhibitors. We conclude that by inhibiting R1, R2, and p53R2, CHL has the potential to be effective in the clinical setting, when used alone or in combination with currently available cancer therapeutic agents.
Keywords: Colorectal cancer, chlorophyllin, ribonucleotide reductase, Rb/E2F pathway
Introduction
Human ribonucleotide reductase (RR) catalyzes the conversion of ribonucleoside diphosphates (rNDPs) to deoxyribonucleoside diphosphates (dNDPs). To form an active enzyme, two heterodimers consisting of two large (R1) and two small (R2) subunits are required.1 The R1 subunit contains the enzyme active site, whereas the R2 subunit harbors a catalytically essential tyrosyl free radical and an oxygen-linked diferric iron center. Both subunits are crucial for enzyme activity.1,2 Multiple levels of RR activity control have been reported. In addition to allosteric regulation,3–5 transcription of both R1 and R2 genes is controlled by the cell cycle, leading to optimal levels of expression during S phase.6 The protein level of R1 remains constant and in excess throughout the cell cycle, whereas R2 reaches the highest level during S phase and undergoes proteasome-mediated degradation when the cell enters mitosis.7–10 An interesting link between RR and cancer has emerged since the discovery of p53R2 (p53-inducible), an analog of R2 with 80% homology in mammalian cells,11,12 which plays a significant role in mitochondrial DNA synthesis.13,14 Among several different features between the two small subunits, p53R2 is a transcriptional target of p53 in response to genotoxic stress, supplying deoxyribonucleotides for DNA damage repair. On the other hand, R2 is regulated by cell cycle-associated factors NF-Y and E2F,8,15,16 and is responsible for DNA synthesis in proliferating cells. Interestingly, whereas R2 was reported to be associated with cellular transformation, tumorigenesis, and malignancy,17,18 p53R2 was negatively related to metastasis of colon adenocarcinoma.19 Because of its pivotal role in DNA synthesis and repair, RR has been recognized as a promising target for several anti-cancer drugs, including subunit-specific RR inhibitors.20–22
E2F transcription factors regulate genes that encode proteins with pivotal roles in cell cycle progression and DNA synthesis, such as Cyclins E and A, cdc2 (cdk1), thymidine kinase, DNA polymerase-α, proliferating cell nuclear antigen (PCNA), and RR.23,24 E2F factors form heterodimers with DP-family proteins and promote progression through G1 and into S-phase. Association of E2F-DP with the retinoblastoma protein (Rb) or pocket proteins p107 and p130 can convert E2F factors from transcriptional activators to transcriptional repressors.23 This is regulated by the interplay among various proteins, including Cyclins D and E, cyclin-dependent kinases and their inhibitors, and protein phosphatases.25,26 In addition to binding of the pocket proteins, Cyclin A, Sp1, p53, MDM2, and the ubiquitin-proteasome pathway regulate E2F activity.27–29 Deregulation of the Rb/E2F pathway and cell cycle control has been associated with cancers of the liver, pancreas, lung, prostate, and colon.30–35 Thus, the Rb/E2F pathway may be an attractive target for chemopreventive and chemotherapeutic agents.
Chlorophyllin (CHL) is a water-soluble derivative of chlorophyll that possesses anticarcinogenic and antimutagenic properties.36–45 In human colon cancer cells, CHL induced apoptosis via caspase-8 activation, release of apoptosis-inducing factor from mitochondria, and cleavage of nuclear lamins.36 Unlike chemopreventive agents such as butyrate, salicylate, vitamin D, and curcumin,46–49 apoptosis induced by CHL was cytochrome c-independent. Based on these observations, we sought to examine the cell cycle checkpoint controls that might be altered by CHL treatment in human colon cancer cells. The results presented here indicate that CHL-treated cells accumulate in S-phase, with inhibition of DNA synthesis and marked changes in the expression of key cell cycle regulators, and this was associated with the marked loss of RR activity.
Materials and methods
Cell culture and treatment with chlorophyllin
Human colon cancer cell lines were obtained from American Type Culture Collection (Manassas, VA), namely HCT116 (CCL-247) carcinoma, SW480 (CCL-228) adenoma, SW48 (CCL-231) adenocarcinoma, and HT29 (HTB-38) adenocarcinoma cells. Cells were grown in modified (+L-glutamine) McCoy's 5A medium (Invitrogen, Carlsbad, CA) supplemented with 10% (v/v) fetal bovine serum (Invitrogen), penicillin (100 units/ml), and streptomycin (0.1 mg/ml) (Sigma Chemical Co, St. Louis, MO), and maintained at 37°C in a 5% CO2 atmosphere. Cells typically were seeded at 1 × 106 cells/T25 flask and treated with CHL (Sigma) 3 d later, as reported before.36 In some experiments, HCT116 (p53+/+) and HCT116 (p53−/−) cells (gift of Dr. B. Vogelstein) were seeded at 1.5 × 106 cells on 100 mm × 20 mm dishes and treated at 70% confluency. Cells were harvested 24 h after CHL treatment, unless stated otherwise. Experiments were conducted under subdued lighting, and CHL stock solution (20 mM) was prepared fresh in double-distilled water immediately before use.
Cell cycle analyses
Cell cycle analyses were performed as reported by Díaz et al.36 In brief, adherent cells were detached, fixed in 70% ethanol, and stored at −20°C overnight. Cells were pelleted, resuspended in PBS containing 20 μg/ml propidium iodide (Sigma), 0.15 μg/ml RNase (Boehringer Mannheim, Indianapolis, IN), and incubated at 37°C for 30 min in the dark. Cells were left overnight at 4°C in the dark before analysis on a fluorescence-activated cell sorter (FACS). Cell cycle distribution was determined using MultiCycle software (Phoenix Flow Systems, San Diego, CA). In some experiments cells were treated with CHL and pulse-labeled with 40 μM bromodeoxyuridine (BrdU) for 10–15 min before bivariate FACS analysis. The protocol used anti-BrdU-FITC antibody (Becton-Dickinson, Franklin Lakes, NJ) and propidium iodide, as reported elsewhere.50–52
Quantitative real-time RT-PCR
Total RNA was isolated using an RNeasy kit (Qiagen, Valencia, CA), as described elsewhere.53 cDNAs were amplified from 4 μg total RNA using an Omniscript Reverse Transcriptase Kit (Qiagen). Primers were as follows: R1, 5'-G G C A C C C C G T A T A T G C T C T A-3' (forward) and 5'-C C A G G G A A G C C A A A T T A C A A- 3' (reverse); R2, 5'-A C A G A A G C C C G C T G T T T C T A-3' (forward) and 5'-C C C A G T C T G C C T T C T T C T T G-3' (reverse); glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5'-G A A G G T G A A G G T C G G A G T C-3' (forward) and 5'-G A A G A T G G T G A T G G G A T T T C-3' (reverse). Amplifications were carried out in an Opticon Monitor 2 system (Finnzymes, Finland), after initial denaturation at 95°C for 10 min, with 42 cycles of 94°C for 10s; 58°C for 20s, and 72°C for 20s. The levels of R1 and R2 were normalized to GAPDH.
Immunoblotting
Fractionated or total lysates of untreated and CHL-treated cells were resolved on NuPAGE 4–12% Bis-Tris gel (Invitrogen), and proteins were transferred to nitrocellulose membrane (0.45 μm pore size; Invitrogen). Equal protein loading was confirmed with Amido Black staining and β-actin expression. The membrane was blocked with 2% BSA in PBS for 1 h at room temperature, followed by overnight incubation with primary antibody at 4°C. The membrane was washed with PBS-T and incubated for 1 h at room temperature with the corresponding secondary antibodies coupled to horseradish peroxidase (goat anti-mouse IgG-HRP and goat anti-rabbit IgG-HRP from Bio-Rad, Hercules, CA); donkey anti-goat IgG-HRP from Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and revealed with Western Lightning Chemiluminescence Reagents Plus ECL (Perkin Elmer Life Science, Boston, MA). Primary antibodies were as follows: mouse anti-p53 monoclonal (DO-1; Santa Cruz Biotechnology, Inc.), 1:200 dilution; rabbit anti-p21 polyclonal (H-164; Santa Cruz Biotechnology, Inc.), 1:200 dilution; mouse anti-Cyclin D1 monoclonal (DCS-6; Biosource International, Camarillo, CA), 1:250 dilution; mouse anti-Rb monoclonal (1F8; Biosource International), 2 μg/ml; mouse anti-MDM2 monoclonal (SMP14; Santa Cruz Biotechnology, Inc.), 1:100 dilution; mouse anti-E2F1 monoclonal (SQ71; Biosource International), 1:200 dilution; mouse anti-E2F4 monoclonal (RK-13; Santa Cruz Biotechnology, Inc.), 1:100 dilution; mouse anti-DP-1 monoclonal (TFD10; MBL International, Woburn MA), 2.5 μg/ml; mouse anti-p107 monoclonal (SD9; Santa Cruz Biotechnology, Inc.), 1:100 dilution; mouse anti-Cyclin E monoclonal (HE12; Biosource International), 1 μg/ml; mouse anti-Cyclin A monoclonal (E72; Biosource International), 1:100 dilution; mouse anti-cdk2 monoclonal (8D4; Biosource International), 1 μg/ml; mouse anti-Cyclin B1 monoclonal (GNS1; Santa Cruz Biotechnology, Inc.), 1:100 dilution; mouse anti-cdc2 p34 monoclonal (sc-54; Santa Cruz Biotechnology, Inc.), 1:100 dilution; mouse anti-PCNA monoclonal clone PC10 (Biosource International), 1:400 dilution; goat anti-ribonucleotide reductase R1 polyclonal (T-16; Santa Cruz Biotechnology, Inc.), 1:200 dilution; rabbit anti-ribonucleotide reductase R2 polyclonal (165–174; Calbiochem), 2.5 μg/ml; polyclonal anti-p53R2 (N-16, Santa Cruz Biotechnology) 1:200 dilution; and β-actin (Sigma).
Electrophoretic mobility-shift assays
These experiments were performed as reported by Li et al.54 In brief, nuclear extracts from CHL-treated cells were incubated with a 32P end-labeled DNA fragment containing the specific E2F recognition sequence.55 For competition experiments, excess unlabeled oligonucleotide (oligo) was included in the incubation reaction, containing either the wild type or mutant sequence.55 Supershift bands were generated using antibodies to E2F1, DP-1, Rb, E2F4, p107, or Cyclin A (listed above). Raji nuclear extract (Active Motif, Carlsbad, CA) served as a reference control.
Ribonucleotide reductase activity
Cells were treated with CHL and 24 h later they were harvested at 4°C, washed three times with ice-cold PBS, and centrifuged at 1,500 rpm for 5 min. Cell pellets were resuspended in 0.5 ml ice-cold 0.1 M HEPES buffer, pH 8 before sonication at 5 W three times for 15 sec, each with a 30-sec pause in between (on ice at all times). After lysis, cell debris was removed by centrifugation at 14,000 rpm (17,000 × g) at 4°C. Protein concentration was determined by the Bradford assay.
Ribonucleotide reductase assay was performed as previously described, with slight modification.56 Cell extracts were added to a reaction mixture containing 100 mM HEPES buffer, pH 8, 10 mM DTT, 4 mM AMP-PNP, 20 μM FeCl3, 2 mM magnesium acetate, 50 μM CDP, and 3H-CDP (100 cpm/pmol CDP). Reactions (40 μl) were incubated at 37°C for 30 min. The tubes were transferred to an ice-bath before adding 4 μl 10 M perchloric acid. After 15 min on ice the precipitate was removed by centrifugation at 14,000 rpm for 2 min. The supernatant was transferred to new tubes, after which boiling was carried out for 20 min. After cooling, 4 μl of a marker solution containing 60 mM CMP, 60 mM dCMP, and 60 mM dUMP plus 12 μl 5 M KOH was added to the supernatant (pH > 8), followed by incubation on ice for 15 min and centrifugation at 14,000 rpm for 5 min. Nucleotides were resolved on a thin-layer chromatography (TLC) plate with fluorescent indicator (Cellulose 300, F-254). Solvents used were composed of 95% ethanol, saturated sodium tetraborate, 5 M ammonium acetate, pH 9.8 and 0.25 M EDTA, pH 8 at a ratio of 220:80:20:1. After 12–15 h the TLC plate was dried, and dCMP-dUMP spots were cut and placed in scintillation vials. To each vial was added 0.5 M HCl (1 ml) to elute nucleotides for 15 min before counting in 5 ml EcoLite scintillation fluid.
Results
CHL induces cell cycle arrest in human colon cancer cells
Four widely-used colon cancer cell lines were treated with increasing concentrations of CHL. After 24 h there was an increase in the percentage of floating cells and a decrease in the attached cell yield (Fig. 1A). In general, HT29 cells were more resistant to CHL treatment than SW48, SW480, and HCT116 cells. At the highest concentration of 250 μM CHL, 12% of HCT116 cells were floating and had apoptotic morphology, such as nuclear condensation and membrane blebbing (data not presented). Cell cycle analyses of the adherent cell population revealed a sub-G1 peak, indicative of apoptosis, in both HCT116 and HT29 cells 24 h after treatment with CHL (Fig. 1B). Treatment with 5–25 μM CHL produced a slight increase in the percentage of HCT116 cells occupying G1 of the cell cycle, and there was a concomitant loss of cells in S-phase (Fig. 1C). However, concentrations >50 μM CHL increased markedly the proportion of HCT116 cells in S-phase. At the highest concentration of 250 μM CHL, 40% of HCT116 cells were in G1 and 7–8% occupied G2/M, but >50% were present in S-phase.
Figure 1.
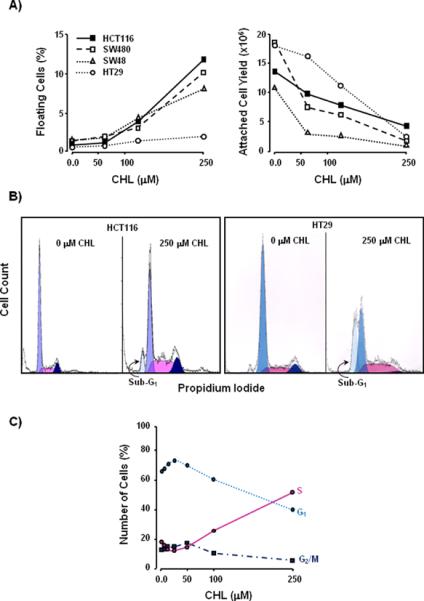
Growth inhibition and cell cycle arrest in human colon cancer cells treated with CHL. (A) HCT116, SW480, SW48, and HT29 cells were treated with CHL for 24 h, and the floating cells (%) and attached cell yield were determined. (B) FACS analyses of CHL-treated cells, showing a sub-G1 peak in the adherent cell population. (C) Changes in the proportion of HCT116 cells occupying G1, S, and G2/M phases of the cell cycle, across a broad range of CHL concentrations. Data shown in the figures are representative findings from at least three independent experiments.
CHL increases the expression of E2F1 and E2F4 transcription factors
To investigate the mechanism of the CHL-induced S-phase arrest in HCT116 cells, immunoblot analyses were conducted on various cell cycle regulators (Fig. 2). Expression levels of p53, p21, Cyclin D1, Rb, and MDM2 were decreased markedly by CHL treatment (Fig. 2A). In contrast, the transcription factor E2F1 was increased at all concentrations of CHL (Fig. 1B). The E2F1 binding partner DP-1 exhibited two immunoreactive bands; the lower band, which is associated with E2F DNA binding activity,27 was reduced following CHL treatment (Fig. 2B). We also examined other members of the E2F family and found E2F4 to be increased relative to the 0 μM CHL controls (Fig. 2B). The pocket protein p107 was diminished with increasing concentrations of CHL (Fig. 2B), whereas p130 was unchanged (data not presented).
Figure 2.

Altered expression levels of cell cycle checkpoint controls in CHL-treated colon cancer cells. HCT116 cells were treated with CHL and 24 h later whole cell lysates were subjected to immunoblotting. (A) Loss of p53, p21, Cyclin D1, Rb, and MDM2 with dose of CHL. Rb and pRb indicates hypo- and hyperphosphorylated forms, respectively. (B) Increased expression of E2F1 and E2F4 transcription factors, and diminished expression of DP-1 (DNA binding form, lower band) and p107. (C) Expression levels of Cyclins E, A, B1, Cdk2, cdc2, and PCNA. Equal loading was confirmed by Amido Black staining and β-actin (not shown). Data are representative findings from three or more independent experiments.
Cyclin E, Cyclin A, and Cyclin B1 levels were essentially unchanged up to 250 μM CHL, and only at the highest concentration of 500 μM CHL was there a noticeable loss in expression of these cyclins (Fig. 2C). Cdk2 and cdc2 were unchanged at the lowest concentration of 62.5 μM CHL, but both proteins were reduced slightly at higher CHL concentrations. Finally, PCNA levels remained constant across all concentrations of CHL (Fig. 2C). Collectively, these studies identified several important G1/S checkpoint controls as being reduced by CHL treatment, along with increases in the expression of E2F1 and E2F4 transcription factors.
CHL enhances the DNA binding activity of E2F1 and E2F4
Nuclear extracts from CHL-treated HCT116 cells were subjected to mobility-shift assays, using the methodology reported before.54 In the presence of radiolabeled E2F-specific oligo a band-shift was detected with nuclear extracts from CHL-treated HCT116 cells (Fig. 3A, lanes 1–3). The band of interest was competed away by excess wild type unlabeled “cold” oligo (lanes 4–6), but not by excess mutant oligo (lanes 7–9). A supershift was detected upon addition of E2F1-specific antibody (lanes 10–12), as seen in nuclear extract from Raji cells (lanes 13–14).
Figure 3.
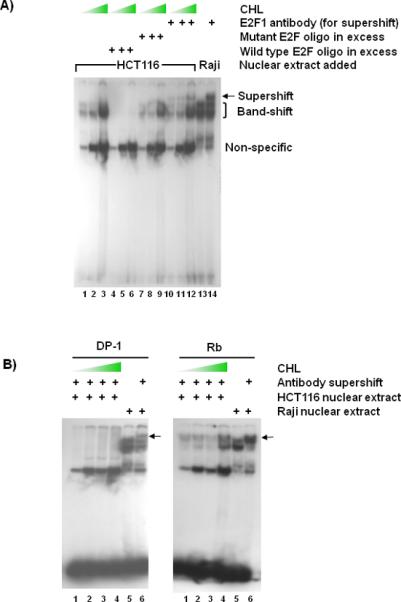
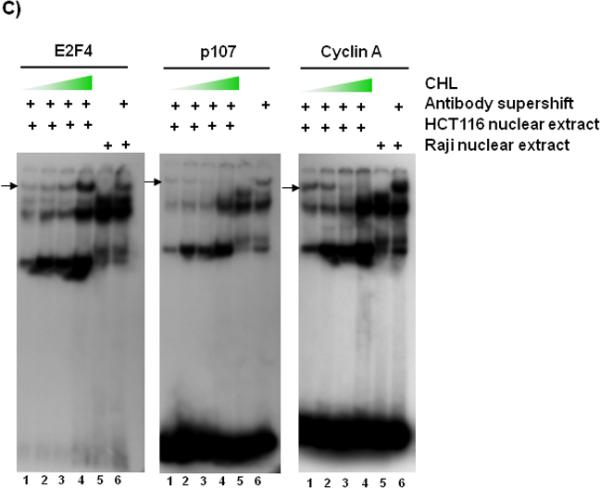
Electrophoretic mobility-shift assays with nuclear extracts from CHL-treated colon cancer cells. (A) HCT116 cells were treated with increasing CHL concentrations and 24 h later the nuclear extracts were used in mobility-shift assays, according to a published methodology.54 Double-stranded oligo containing an E2F binding site was end-labeled with 32P and incubated with 10 μg nuclear extract from CHL-treated HCT116 cells (lanes 1–12) or with Raji nuclear extract as a reference control (lanes 13 and 14). To determine specificity of binding, competition assays were performed with 100-fold excess unlabeled wild type or mutant oligo.55 Monoclonal antibody against human E2F1 was used to create a supershift. Positions of the specific band-shift (bracket), supershift (arrow), and non-specific band are indicated adjacent to lane 14. Wedge symbol = 0, 62.5, 500 μM CHL. Supershift assays also were performed using monoclonal antibodies against (B) DP-1 and Rb or (C) E2F4, p107, and Cyclin A. In (B) and (C), the supershift band is indicated by an arrow and the wedge symbol = 0, 62.5, 125, and 250 μM CHL. Data are representative findings from two or more independent experiments.
The supershift experiment was repeated with antibodies specific for DP-1, Rb, E2F4, p107, and Cyclin A. Under the current assay conditions, DP-1 antibody completely competed away the binding activity in HCT116 nuclear extracts, whereas Rb antibody generated a supershift band at all CHL concentrations (Fig. 3B). Following incubation with anti-E2F4 antibody (Fig. 3C), a distinct supershift band was detected that increased with CHL concentration, and under the same conditions p107 and Cyclin A supershift bands were progressively diminished.
Inhibition of DNA synthesis and ribonucleotide reductase activity by CHL
HCT116 cells were pretreated with CHL and pulse-labeled with BrdU before undergoing bivariate FACS analysis (Fig. 4A). In the absence of CHL treatment, most of the BrdU labeling (96%) was associated with the S-phase population, but this declined to 16% and 0.75% in cells treated with 62.5 and 250 μM CHL, respectively. These findings resemble the effects reported for antineoplastic drugs that block DNA synthesis and induce S-phase arrest by inhibiting RR activity.51,57,58
Figure 4.
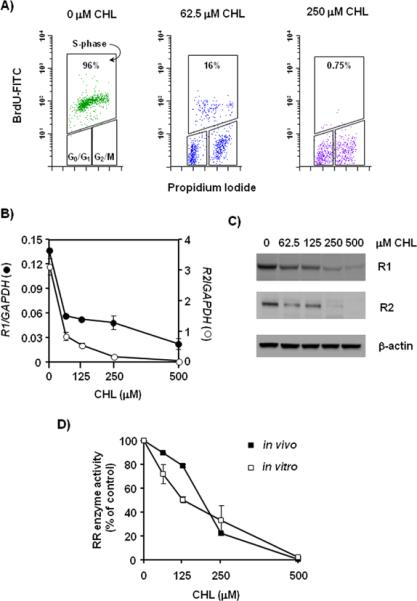
Inhibition of DNA synthesis and ribonucleotide reductase activity in CHL-treated colon cancer cells. (A) HCT116 cells were pretreated with 0, 62.5, or 250 μM CHL for 24 h and pulse-labeled with BrdU for 10–15 min prior to bivariate FACS analysis with anti-BrdU-FITC antibody/propidium iodide, as reported.50–52 The percentage of S-phase cells staining positive for BrdU incorporation is as indicated in the figure. (B) mRNA expression levels of the large (R1; solid circles) and small (R2; open circles) subunits of RR were measured by qRT-PCR and expressed relative to GAPDH. (C) Protein expression levels of R1 and R2 detected by immunoblotting; β-actin, loading control. (D) RR enzyme activity was measured by the CDP reduction assay (see Materials and methods). Assays were performed on total lysates prepared from HCT116 cells treated with 0, 62.5, 125, 250, and 500 μM CHL for 24 h (in vivo). In a separate set of experiments, CHL was added directly to the reaction mixture containing whole cell extract of untreated HCT116 cells (in vitro). Data were normalized to the corresponding 0 μM CHL treatment and expressed as percent of control. In (B) and (D), error bars indicate the variance from duplicates, and data are representative of results from three independent experiments.
In qRT-PCR analyses of RR mRNA expression, transcription of the genes encoding for both R1 and R2 subunits was inhibited markedly by CHL in HCT116 cells (Fig. 4B). There was a decrease in the corresponding protein expression levels (Fig. 4C), as well as inhibition of the activity of the holoenzyme in HCT116 cells (Fig. 4D, closed symbols). Interestingly, when CHL was added directly to a reaction mixture containing an extract of untreated HCT116 cells, RR enzyme activity also was inhibited in a concentration-dependent fashion (Fig. 4D, open symbols). The activities assayed both in vivo and in vitro were inhibited 70–80% at 250 μM CHL, relative to the untreated control, and they were undetectable at 500 μM CHL.
CHL-mediated inhibition of RR is p53-independent
In addition to R1 and R2, there is growing interest in the p53-inducible small subunit, p53R2, as a target for cancer therapy.22 In both HCT116 (p53+/+) and HCT116 (p53−/−) cells, the protein expression levels of R1, R2, and p53R2 were reduced markedly 48 h after CHL treatment (Fig. 5A). In total cell lysates, RR activity was inhibited by CHL in a concentration-dependent manner, and the extent of inhibition was similar in HCT116 (p53+/+) and HCT116 (p53−/−) cells (Fig. 5B). Thus, these data supported a p53-independent mechanism of RR inhibition in CHL-treated colon cancer cells.
Figure 5.

Inhibition of RR activity by CHL is p53-independent. (A) HCT116 (p53+/+) and HCT116 (p53−/−) cells were treated with 0 or 125 μM CHL and immunoblotting was performed on whole cell lysates with antibodies specific for R1, R2, and p53R2. β-Actin served as the loading control. (B) RR enzyme activity assays were performed as described in Materials and methods, on total lysates from HCT116 (p53+/+) and HCT116 (p53−/−) cells treated with the indicated CHL concentrations for 24 h. Error bars indicate the variance from duplicates, and data are representative of results from three independent experiments.
Discussion
We reported previously on the induction of apoptosis by CHL in HCT116 cells.36 In the present study, CHL affected the growth and survival of other colon cancer cell lines, causing an increase in the population of floating cells and a decrease in the attached cell yield. HCT116 cells (p53-wild type) were sensitive to CHL treatment whereas HT29 cells (p53-mutant) were more resistant. However, following treatment with 250 μM CHL, both cell lines exhibited a sub-G1 peak in the adherent cell population at 24 h, indicative of apoptosis. As a critical regulator of the cell cycle and apoptosis, p53 is often mutated in human cancers, although some p53 mutations can be gain-of-function.59 Thus, we became interested in the p53 status and how it influences sensitivity to CHL treatment.
In HCT116 cells, low doses of CHL induced a slight increase in the population of cells occupying G1 with loss of cells in S phase, whereas at higher CHL concentrations a striking S-phase arrest was detected. The differential response at low versus high CHL dose is worthy of further investigation, but we elected to focus here on the S-phase arrest. Immunoblot analyses confirmed the loss of several checkpoint controls that regulate G1/S transition, including p53, p21, and Cyclin D1. Cyclin D1 depletion also has been observed in CHL-treated MCF-7 breast cancer cells.60 Two other important cell cycle regulators, Rb and MDM2, were decreased at the protein level following CHL treatment. Despite the loss of Cyclin D1, hyperphosphorylated Rb (pRb) was detected even at the highest CHL concentrations. It is possible that pRb was generated by active Cyclin E/Cdk2 or Cyclin A/Cdk2 complexes.61
We were particularly interested in determining how CHL might affect the interplay between E2F family members and their associated binding partners. The levels of E2F2, E2F3, and E2F5 were not altered in whole cell lysates (data not presented), but E2F1 and E2F4 proteins were elevated markedly by CHL treatment. When bound to the DNA-binding partner DP-1, E2F1 becomes a transcriptional activator and E2F4 a transcriptional repressor.55,62 Interestingly, Cyclin A-Cdk2 interacts with an amino-terminal domain on E2F1 and inhibits E2F1/DP-1 transcriptional activity, but E2F4, which lacks this domain, associates indirectly with Cyclin A-Cdk2 through one of the pocket proteins, p130 or p107.62 We saw no changes in p130 levels after CHL treatment (data not presented), but p107 protein expression was decreased in a dose-dependent manner. This suggested a scenario in which the loss of p107 might limit the interactions of Cyclin A-Cdk2 with E2F4, freeing E2F4 to act as a transcriptional repressor. Support for this idea came from the mobility-shift assays, with the increased DNA binding activity of E2F4 in nuclear extracts from CHL-treated cells, and the loss of p107 and Cyclin A. Prior studies have shown that Rb is a binding partner for both E2F1 and E2F4, and that once cells enter S-phase there is a pool of free E2F1 and E2F4 in equal mixture.55 Mobility-shift assays provided evidence for increased E2F1 and Rb in nuclear extracts of CHL-treated cells, but the levels of binding typically were much weaker than observed for E2F4. This might be because E2F4 competed with E2F1 for a limited supply of nuclear DP-1, but further studies are needed to clarify this question.
The experiments to this point implicated E2F4 as a transcriptional repressor during CHL-induced S-phase arrest in HCT116 cells. Subsequent work using BrdU/PI covariate cell cycle analysis demonstrated that CHL inhibited DNA synthesis, and we turned our attention to RR. The promoter region of the mouse R2 gene has been reported to bind E2F4 as a negative regulator,8 and we considered the possibility that increased E2F4 levels in CHL-treated cells might transcriptionally downregulate R2 expression. Interestingly, both R1 and R2 subunits were decreased in a concentration-dependent fashion in HCT116 cells, R2 being particularly sensitive to CHL treatment.
When R2 is unavailable, the p53-inducible subunit p53R2 can associate with R1 to form an active RR enzyme.22 However, there was a marked loss of p53R2 expression in both HCT116 (p53+/+) and HCT116 (p53−/−) cells following CHL treatment, in addition to reduced R1 and R2 levels (Fig. 5). It is interesting to consider these findings in the context of prior work that used knockdown strategies to target either R2 or p53R2. Lin et al.63 observed that R2 levels critically determine the sensitivity of HCT116 (p53−/−) cells to RR inhibitors, and that ectopic expression of p53R2 does not compensate for the decreased levels of R2 brought about by R2-siRNA. Yoshida et al.64 knocked down either p53 or p53R2 in a panel of cell lines and demonstrated a reduction in the DNA repair response induced by short-chain fatty acids. Interestingly, HCT116 cells have a mutant form of p53R2; as a consequence, the DNA repair response is attenuated and p53 or p53R2 siRNAs had little or no effect on DNA repair activity.64 An important conclusion from the knockdown studies is that the level of R2 protein reflects the ability of cells to participate in DNA repair, and is a critical determinant of the sensitivity to DNA damaging agents.63,64 Given that CHL markedly inhibited R2 expression in HCT116 (p53+/+) and HCT116 (p53−/−) cells (Fig. 5), we conclude that CHL might be effective in sensitizing cancer cells to DNA-damaging agents, regardless of the p53 status.
It is noteworthy that, in addition to the transcriptional downregulation of RR subunits, CHL had a direct inhibitory effect on RR enzymatic activity. This was demonstrated by taking HCT116 cells that had not been treated with CHL, and using the cell extracts in the RR enzyme assay, where CHL produced dose-dependent inhibition. These findings resemble the reported effects of inhibitors such hydroxyurea, which quench the tyrosyl free radical in the active site of R2.58 In this context, it is interesting to consider briefly the evidence for CHL as a free radical-scavenging agent.
CHL has been shown to react with hydroxyl radical as well as deoxyribose peroxyl radical generated by pulse radiolysis in vitro.65 The stable 1,1-diphenyl-2-picrylhydrazyl radical also was scavenged by CHL. An electron paramagnetic resonance study provided evidence that CHL inhibited formation of 5,5-dimethyl-1-pyrroline-N-oxide-hydroxyl radical and 2,2,6,6-tetramethyl-piperidine oxide radical, generated by γ-radiation or by photosensitization of methylene blue with visible light.66 CHL also inhibited oxidation of low-density lipoprotein, and scavenged tyrosyl radicals that were generated by myeloperoxidase in the presence of H2O2 and tyrosine.67 Porphyrins in general are strong protein binders, and CHL itself interacts with human serum albumin with an affinity constant of 7.0 × 103 M−1.68 In the case of RR, CHL binding might displace rNDP substrates and/or allosteric modifiers from their corresponding binding sites by interacting with one or both subunits of the enzyme. Alternatively, CHL might cause conformational changes that no longer favor binding of substrates and/or allosteric effectors to R1. Further studies are needed to clarify the precise mechanism(s) by which CHL inhibits RR activity, and the extent to which this sensitizes cancer cells to DNA-damaging agents or anticancer therapies specifically targeting RR.20
In summary, we conducted a detailed investigation of cell cycle checkpoint controls that are altered during CHL-induced S-phase arrest in colon cancer cells. With loss of p53, p21, and other key regulators of the G1/S transition there was increased expression of E2F1 and E2F4 transcription factors. The role of E2F4 as a reported transcriptional repressor of RR, coupled with the inhibition of DNA synthesis by CHL, led to studies of R1, R2, and p53R2. CHL reduced markedly the expression levels of all three subunits, with R2 being particularly sensitive, and there was loss of RR enzyme activity. We conclude that CHL might provide a useful new avenue for cancer treatment in the clinical setting, helping to sensitize cancer cells to the actions of anticancer agents.69 In vivo studies to corroborate this possibility are now in progress.
Acknowledgements
Dr. B. Vogelstein (Johns Hopkins University, Baltimore, MD) kindly supplied HCT116 (p53+/+) and HCT116 (p53−/−) cells. This work was supported in part by NIH grants CA090890, CA65525, and CA122959. Cell cycle analyses were performed in the Cell Imaging & Analysis Core of the Environmental Health Sciences Center, funded by NIEHS center grant P30 ES00210.
Abbreviations
- RR
ribonucleotide reductase
- rNDPs
ribonucleoside diphosphates
- dNDPs
deoxyribonucleoside diphosphates
- PCNA
proliferating cell nuclear antigen
- Rb
retinoblastoma tumor suppressor protein
- CHL
chlorophyllin
- FACS
fluorescence-activated cell sorter
- BrdU
bromodeoxyuridine
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- TLC
thin-layer chromatography
- oligo
oligonucleotide
References
- 1.Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 2.Larsson KM, Jordan A, Eliasson R, Reichard P, Logan DT, Nordlund P. Structural mechanism of allosteric substrate specificity regulation in a ribonucleotide reductase. Nat Struct Mol Biol. 2004;11:1142–9. doi: 10.1038/nsmb838. [DOI] [PubMed] [Google Scholar]
- 3.Hendricks SP, Mathews CK. Allosteric regulation of vaccinia virus ribonucleotide reductase, analyzed by simultaneous monitoring of its four activities. J Biol Chem. 1998;273:29512–8. doi: 10.1074/jbc.273.45.29512. [DOI] [PubMed] [Google Scholar]
- 4.Chimploy K, Mathews CK. Mouse ribonucleotide reductase control: influence of substrate binding upon interactions with allosteric effectors. J Biol Chem. 2001;276:7093–7100. doi: 10.1074/jbc.M006232200. [DOI] [PubMed] [Google Scholar]
- 5.Chimploy K, Tassotto ML, Mathews CK. Ribonucleotide reductase, a possible agent in deoxyribonucleotide pool asymmetries induced by hypoxia. J Biol Chem. 2000;275:39267–71. doi: 10.1074/jbc.M006233200. [DOI] [PubMed] [Google Scholar]
- 6.Björklund S, Skog S, Tribukait B, Thelander L. S-phase-specific expression of mammalian ribonucleotide reductase R1 and R2 subunit mRNAs. Biochemistry. 1990;29:5452–8. doi: 10.1021/bi00475a007. [DOI] [PubMed] [Google Scholar]
- 7.Engström Y, Eriksson S, Jildevik I, Skog S, Thelander L, Tribukait B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J Biol Chem. 1985;260:9114–6. [PubMed] [Google Scholar]
- 8.Chabes AL, Björklund S, Thelander L. S phase-specific transcription of the mouse ribonucleotide reductase R2 gene requires both a proximal repressive E2F-binding site and an upstream promoter activating region. J Biol Chem. 2004;279:10796–807. doi: 10.1074/jbc.M312482200. [DOI] [PubMed] [Google Scholar]
- 9.Chabes AL, Pfleger CM, Kirschner MW, Thelander L. Mouse ribonucleotide reductase R2 protein: a new target for anaphase-promoting complex-Cdh1-mediated proteolysis. Proc Natl Acad Sci USA. 2003;100:3925–3929. doi: 10.1073/pnas.0330774100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chabes AL, Thelander L. Controlled protein degradation regulates ribonucleotide reductase activity in proliferating mammalian cells during the normal cell cycle and in response to DNA damage and replication blocks. J Biol Chem. 2000;275:17747–53. doi: 10.1074/jbc.M000799200. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, Takei Y, Nakamura Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–9. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 12.Nakano K, Bálint E, Ashcroft M, Vousden KH. Ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene. 2000;19:4283–9. doi: 10.1038/sj.onc.1203774. [DOI] [PubMed] [Google Scholar]
- 13.Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chrétien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, Nakamura Y, Munnich A, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–80. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 14.Thelander L. Ribonucleotide reductase and mitochondrial DNA synthesis. Nat Genet. 2007;39:703–4. doi: 10.1038/ng0607-703. [DOI] [PubMed] [Google Scholar]
- 15.Filatov D, Thelander L. Role of a proximal NF-Y binding promoter element in S phase-specific expression of mouse ribonucleotide reductase R2 gene. J Biol Chem. 1995;270:25239–43. doi: 10.1074/jbc.270.42.25239. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Zhou B, Xue L, Qiu W, Shih J, Zheng S, Yen Y. Nuclear factor Y regulation and promoter transactivation of human ribonucleotide reductase subunit M2 gene in a Gemcitabine resistant KB clone. Biochem Pharmacol. 2004;67:1499–1511. doi: 10.1016/j.bcp.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 17.Fan H, Villegas C, Wright JA. Ribonucleotide reductase R2 component is a novel malignancy determinant that cooperates with activated oncogenes to determine transformation and malignant potential. Proc Natl Acad Sci USA. 1996;93:14036–40. doi: 10.1073/pnas.93.24.14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou BS, Tsai P, Ker R, Tsai J, Ho R, Yu J, Shih J, Yen Y. Overexpression of transfected human ribonucleotide reductase M2 subunit in human cancer cells enhances their invasive potential. Clin Exp Metastasis. 1998;16:43–9. doi: 10.1023/a:1006559901771. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Zhou B, Xue L, Shih J, Tye K, Lin W, Qi C, Chu P, Un F, Wen W, Yen Y. Metastasis-suppressing potential of ribonucleotide reductase small subunit p53R2 in human cancer cells. Clin Cancer Res. 2006;12:6337–44. doi: 10.1158/1078-0432.CCR-06-0799. [DOI] [PubMed] [Google Scholar]
- 20.Heidel JD, Yu Z, Liu JY, Rele SM, Liang Y, Zeidan RK, Kornbrust DJ, Davis ME. Administration in non-human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA. Proc Natl Acad Sci USA. 2007;104:5715–21. doi: 10.1073/pnas.0701458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao J, Zhou B, Chu B, Yen Y. Ribonucleotide reductase inhibitors and future drug design. Curr Cancer Drug Targets. 2006;6:409–31. doi: 10.2174/156800906777723949. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Zhenchuk A, Wiman KG, Albertioni F. Regulation of p53R2 and its role as potential target for cancer therapy. Cancer Lett. 2009;276:1–7. doi: 10.1016/j.canlet.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 23.Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annu Rev Pharmacol Toxicol. 1999;39:295–312. doi: 10.1146/annurev.pharmtox.39.1.295. [DOI] [PubMed] [Google Scholar]
- 24.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 25.Genovese C, Trani D, Caputi M, Claudio PP. Cell cycle control and beyond: emerging roles for the retinoblastoma gene family. Oncogene. 2006;25:5201–9. doi: 10.1038/sj.onc.1209652. [DOI] [PubMed] [Google Scholar]
- 26.Johnson DG, Schneider-Broussard R. Role of E2F in cell cycle control and cancer. Front Biosci. 1998;3:d447–58. doi: 10.2741/a291. [DOI] [PubMed] [Google Scholar]
- 27.Martin K, Trouche D, Hagemeier C, Sørensen TS, La Thangue NB, Kouzarides T. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature. 1995;375:691–4. doi: 10.1038/375691a0. [DOI] [PubMed] [Google Scholar]
- 28.O'Connor DJ, Lam EW, Griffin S, Zhong S, Leighton LC, Burbidge SA, Lu X. Physical and functional interactions between p53 and cell cycle co-operating transcription factors, E2F1 and DP1. EMBO J. 1995;14:6184–92. doi: 10.1002/j.1460-2075.1995.tb00309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sørensen TS, Girling R, Lee CW, Gannon J, Bandara LR, La Thangue NB. Functional interaction between DP-1 and p53. Mol Cell Biol. 1996;16:5888–95. doi: 10.1128/mcb.16.10.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayhew CN, Carter SL, Fox SR, Sexton CR, Reed CA, Srinivasan SV, Liu X, Wikenheiser-Brokamp K, Boivin GP, Lee JS, Aronow BJ, Thorgeirsson SS, et al. RB loss abrogates cell cycle control and genome integrity to promote liver tumorigenesis. Gastroenterology. 2007;133:976–84. doi: 10.1053/j.gastro.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 31.Nacusi LP, Sheaff RJ. Deregulation of cell cycle machinery in pancreatic cancer. Pancreatology. 2007;7:373–7. doi: 10.1159/000107398. [DOI] [PubMed] [Google Scholar]
- 32.Xing J, Spitz MR, Lu C, Zhao H, Yang H, Wang W, Stewart DJ, Wu X. Deficient G2-M and S checkpoints are associated with increased lung cancer risk: a case-control analysis. Cancer Epidemiol Biomarkers Prev. 2007;16:1517–22. doi: 10.1158/1055-9965.EPI-07-0111. [DOI] [PubMed] [Google Scholar]
- 33.Wiman KG. The retinoblastoma gene: role in cell cycle control and cell differentiation. FASEB J. 1993;7:841–5. doi: 10.1096/fasebj.7.10.8393817. [DOI] [PubMed] [Google Scholar]
- 34.Wang HL, Wang J, Xiao SY, Haydon R, Stoiber D, He TC, Bissonnette M, Hart J. Elevated protein expression of cyclin D1 and Fra-1 but decreased expression of c-Myc in human colorectal adenocarcinomas overexpressing beta-catenin. Int J Cancer. 2002;101:301–10. doi: 10.1002/ijc.10630. [DOI] [PubMed] [Google Scholar]
- 35.Kaiser S, Park YK, Franklin JL, Halberg RB, Yu M, Jessen WJ, Freudenberg J, Chen X, Haigis K, Jegga AG, Kong S, Sakthivel B, et al. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol. 2007;8:R131. doi: 10.1186/gb-2007-8-7-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Díaz GD, Li Q, Dashwood RH. Caspase-8 and apoptosis-inducing factor mediate a cytochrome c-independent pathway of apoptosis in human colon cancer cells induced by the dietary phytochemical chlorophyllin. Cancer Res. 2003;63:1254–61. [PubMed] [Google Scholar]
- 37.Arimoto S, Fukuoka S, Itome C, Nakano H, Rai H, Hayatsu H. Binding of polycyclic planar mutagens to chlorophyllin resulting in inhibition of the mutagenic activity. Mutat Res. 1993;287:293–305. doi: 10.1016/0027-5107(93)90022-8. [DOI] [PubMed] [Google Scholar]
- 38.Breinholt V, Schimerlik M, Dashwood R, Bailey G. Mechanisms of chlorophyllin anticarcinogenesis against aflatoxin B1: complex formation with the carcinogen. Chem Res Toxicol. 1995;8:506–514. doi: 10.1021/tx00046a004. [DOI] [PubMed] [Google Scholar]
- 39.Dashwood RH. Protection by chlorophyllin against the covalent binding of 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) to rat liver DNA. Carcinogenesis. 1992;13:113–8. doi: 10.1093/carcin/13.1.113. [DOI] [PubMed] [Google Scholar]
- 40.Dashwood RH, Breinholt V, Bailey GS. Chemopreventive properties of chlorophyllin: inhibition of aflatoxin B1 (AFB1)-DNA binding in vivo and anti-mutagenic activity against AFB1 and two heterocyclic amines in the Salmonella mutagenicity assay. Carcinogenesis. 1991;12:939–42. doi: 10.1093/carcin/12.5.939. [DOI] [PubMed] [Google Scholar]
- 41.Egner PA, Wang JB, Zhu YR, Zhang BC, Wu Y, Zhang QN, Qian GS, Kuang SY, Gange SJ, Jacobson LP, Helzlsouer KJ, Bailey GS, et al. Chlorophyllin intervention reduces aflatoxin-DNA adducts in individuals at high risk for liver cancer. Proc Natl Acad Sci USA. 2001;98:14601–6. doi: 10.1073/pnas.251536898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo D, Horio DT, Grove JS, Dashwood RH. Inhibition by chlorophyllin of 2-amino-3-methylimidazo[4,5-f]quinoline-induced tumorigenesis in the male F344 rat. Cancer Lett. 1995;95:161–5. doi: 10.1016/0304-3835(95)03882-w. [DOI] [PubMed] [Google Scholar]
- 43.Guo D, Schut HA, Davis CD, Snyderwine EG, Bailey GS, Dashwood RH. Protection by chlorophyllin and indole-3-carbinol against 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced DNA adducts and colonic aberrant crypts in the F344 rat. Carcinogenesis. 1995;16:2931–7. doi: 10.1093/carcin/16.12.2931. [DOI] [PubMed] [Google Scholar]
- 44.Reddy AP, Harttig U, Barth MC, Baird WM, Schimerlik M, Hendricks JD, Bailey GS. Inhibition of dibenzo[a,l]pyrene-induced multi-organ carcinogenesis by dietary chlorophyllin in rainbow trout. Carcinogenesis. 1999;20:1919–26. doi: 10.1093/carcin/20.10.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tachino N, Guo D, Dashwood WM, Yamane S, Larsen R, Dashwood R. Mechanisms of the in vitro antimutagenic action of chlorophyllin against benzo[a]pyrene: studies of enzyme inhibition, molecular complex formation and degradation of the ultimate carcinogen. Mutat Res. 1994;308:191–203. doi: 10.1016/0027-5107(94)90154-6. [DOI] [PubMed] [Google Scholar]
- 46.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2004;101:18030–5. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung YM, Bae YS, Lee SY. Molecular ordering of ROS production, mitochondrial changes, and caspase activation during sodium salicylate-induced apoptosis. Free Radic Biol Med. 2003;34:434–42. doi: 10.1016/s0891-5849(02)01301-1. [DOI] [PubMed] [Google Scholar]
- 48.Narvaez CJ, Welsh J. Role of mitochondria and caspases in vitamin D-mediated apoptosis of MCF-7 breast cancer cells. J Biol Chem. 2001;276:9101–7. doi: 10.1074/jbc.M006876200. [DOI] [PubMed] [Google Scholar]
- 49.Shankar S, Srivastava RK. Involvement of Bcl-2 family members, phosphatidylinositol 3'-kinase/AKT and mitochondrial p53 in curcumin (diferulolylmethane)-induced apoptosis in prostate cancer. Int J Oncol. 2007;30:905–918. [PubMed] [Google Scholar]
- 50.Rancourt RC, Keng PC, Helt CE, O'Reilly MA. The role of p21CIP1/WAF1 in growth of epithelial cells exposed to hyperoxia. Am J Lung Cell Mol Physiol. 2001;280:L617–26. doi: 10.1152/ajplung.2001.280.4.L617. [DOI] [PubMed] [Google Scholar]
- 51.Ababou M, Dumaire V, Lécluse Y, Amor-Guéret M. Bloom's syndrome protein response to ultraviolet-c radiation and hydroxyurea-mediated DNA synthesis inhibition. Oncogene. 2002;21:2079–88. doi: 10.1038/sj.onc.1205246. [DOI] [PubMed] [Google Scholar]
- 52.Joerges C, Kuntze I, Herzinger T. Induction of a caffeine-sensitive S-phase cell cycle checkpoint by psoralen plus ultraviolet A radiation. Oncogene. 2003;22:6119–28. doi: 10.1038/sj.onc.1206613. [DOI] [PubMed] [Google Scholar]
- 53.Li Q, Dashwood WM, Zhong X, Nakagama H, Dashwood RH. Bcl-2 overexpression in PhIP-induced colon tumors: cloning of the rat Bcl-2 promoter and characterization of a pathway involving beta-catenin, c-Myc and E2F1. Oncogene. 2007;26:6194–6202. doi: 10.1038/sj.onc.1210438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Q, Dashwood WM, Zhong X, Al-Fageeh M, Dashwood RH. Cloning of the rat β-catenin (Ctnnb1) gene promoter and its functional analysis compared with the Catnb and CTNNB1 promoters. Genomics. 2004;83:231–242. doi: 10.1016/j.ygeno.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 55.Moberg K, Starz MA, Lees JA. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol. 1996;16:1436–49. doi: 10.1128/mcb.16.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slabaugh MB, Johnson TL, Mathews CK. Vaccinia virus induces ribonucleotide reductase in primate cells. J Virology. 1984;52:507–14. doi: 10.1128/jvi.52.2.507-514.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsimberidou AM, Alvarado Y, Giles FJ. Evolving role of ribonucleotide reductase inhibitors in hematologic malignancies. Expert Rev Anticancer Ther. 2002;2:437–48. doi: 10.1586/14737140.2.4.437. [DOI] [PubMed] [Google Scholar]
- 58.Yarbro JW. Mechanism of action of hydroxyurea. Semin Oncol. 1992;19:1–10. [PubMed] [Google Scholar]
- 59.van Oijen MG, Slootweg PJ. Gain-of-function mutations in the tumor suppressor gene p53. Clin Cancer Res. 2000;6:2138–45. [PubMed] [Google Scholar]
- 60.Chiu LC, Kong CK, Ooi VE. The chlorophyllin-induced cell cycle arrest and apoptosis in human breast cancer MCF-7 cells is associated with ERK deactivation and Cyclin D1 depletion. Int J Mol Med. 2005;16:735–40. [PubMed] [Google Scholar]
- 61.Yamazaki L. Role of the RB tumor suppressor in cancer. Cancer Treat Res. 2003;115:209–30. doi: 10.1007/0-306-48158-8_9. [DOI] [PubMed] [Google Scholar]
- 62.Woo MS, Sánchez I, Dynlacht BD. p130 and p107 use a conserved domain to inhibit cellular cyclin-dependent kinase activity. Mol Cell Biol. 1997;17:3566–79. doi: 10.1128/mcb.17.7.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin ZP, Belcourt MF, Cory JG, Sartorelli AC. Stable suppression of the R2 subunit of ribonucleotide reductase by R2-targeted short interference RNA sensitizes p53(−/−) HCT-116 colon cancer cells to DNA-damaging agents and ribonucleotide reductase inhibitors. J Biol Chem. 2004;279:27030–8. doi: 10.1074/jbc.M402056200. [DOI] [PubMed] [Google Scholar]
- 64.Yoshida T, Haga S, Numata Y, Yamashita K, Mikami T, Ogawa T, Ohkusa T, Okayasu I. Disruption of the p53-p53R2 DNA repair system in ulcerative colitis contributes to colon tumorigenesis. Int J Cancer. 2006;118:1395–1403. doi: 10.1002/ijc.21538. [DOI] [PubMed] [Google Scholar]
- 65.Kumar SS, Chaubey RC, Devasagayam TP, Priyadarsini KI, Chauhan PS. Inhibition of radiation-induced DNA damage in plasmid pBR322 by chlorophyllin and possible mechanism(s) of action. Mutat Res. 1999;425:71–9. doi: 10.1016/s0027-5107(98)00250-4. [DOI] [PubMed] [Google Scholar]
- 66.Kumar SS, Devasagayam TP, Bhushan B, Verma NC. Scavenging of reactive oxygen species by chlorophyllin: an ESR study. Free Radic Res. 2001;35:563–74. doi: 10.1080/10715760100301571. [DOI] [PubMed] [Google Scholar]
- 67.Kapiotis S, Hermann M, Exner M, Laggner H, Gmeiner BM. Copper- and magnesium protoporphyrin complexes inhibit oxidative modification of LDL induced by hemin, transition metal ions and tyrosyl radicals. Free Radic Res. 2005;39:1193–1202. doi: 10.1080/10715760500138981. [DOI] [PubMed] [Google Scholar]
- 68.Ouameur AA, Marty R, Tajmir-Riahi HA. Human serum albumin complexes with chlorophyll and chlorophyllin. Biopolymers. 2005;77:129–36. doi: 10.1002/bip.20173. [DOI] [PubMed] [Google Scholar]
- 69.Te C, Gentile JM, Baguley BC, Pearson AE, Gregory T, Ferguson LR. In vivo effects of chlorophyllin on the antitumor agent cyclophosphamide. Int J Cancer. 1997;70:84–9. doi: 10.1002/(sici)1097-0215(19970106)70:1<84::aid-ijc13>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]