

Abstract
This invited paper reviews the study of protein glycosylation, commonly known as glycoproteomics, beginning with the origins of the subject area in the early 1970s shortly after mass spectrometry was first applied to protein sequencing. We go on to describe current analytical approaches to glycoproteomic analyses, with exemplar projects presented in the form of the complex story of human glycodelin and the characterisation of blood group H eptitopes on the O-glycans of gp273 from Unio elongatulus. Finally, we present an update on the latest progress in the field of automated and semi-automated interpretation and annotation of these data in the form of GlycoWorkBench, a powerful informatics tool that provides valuable assistance in unravelling the complexities of glycoproteomic studies.
Keywords: Carbohydrate sequencing, glycodelin, glycoproteomics, GlycoWorkBench, gp273, mass spectrometry, peptide sequencing
Introduction and Historical Perspective
Glycoproteomics, as distinct from proteomics or glycomics, is the study of the glycosylation of proteins, a covalent modification which confers altered physico-chemical properties and functional activity on the nascent protein chain. There are two broad classes of protein glycosylation in nature, those ‘O-linked’ to Serine or Threonine residues in the protein backbone, and those ‘N-linked’ to Asparagine residues. Mass Spectrometry (MS) has played a key and irreplaceable role in defining the structures of glycoproteins over the past 30 years [1,2] using methods developed from the earlier studies on Antarctic fish blood “antifreeze” and prothrombin glycoproteins [3,4] together with the general ‘mass mapping’ strategy [5] of determining and screening the masses of peptides/glycopeptides produced from specific proteolytic or chemical digests, which itself evolved from earlier ‘mixture analysis’ approaches to protein and glycoprotein sequencing [6,7]. The concept of mapping (sometimes called fingerprinting) derives from the realisation in the late 1970s that the data set comprised of peptide molecular ions M+. or quasimolecular ions [M+H]+ produced by digesting any given protein is likely to be unique (especially if more than one digest is used), and therefore it provides a reasonable diagnostic for characterisation or identification of the protein, distinguishing it from others, importantly without the need for sequencing. From 1981, early research applications of mass mapping ranged from the screening of recombinant proteins and glycoproteins for the Biotech industry, detecting errors of translation or confirming mass matching and thus identity [8] using a software mass-search aid ProtMap, through to assisting in the structural characterisation of new peptide hormones [9] and the detection and characterisation of glycosylation in human Interleukin 2 [10]. With the later advent of comprehensive computerised protein databases, the peptide maps could then be used to interrogate those databases for matches to, and thus identification of unknown proteins [11], which in turn has stimulated the general development of the field of proteomics.
Two unique strengths of mass mapping were, and still are, the ability to map (and therefore visualise) the N-terminal and C-terminal domains of a protein with equal probability, and most importantly, the ability to discover post-translational modifications (PTMs) including glycosylation by detecting mass shifts in component peptides in the mass map or by locking on to sugar mass differences in the map, created by facile glycosidic bond cleavage. Once detection is achieved in this way, a whole battery of techniques including MS/MS can then be applied to determine even the most complex structures, and this laboratory has reported many such novel glycosylation studies over the past twenty years including defining the glycosylation of tissue plasminogen activator [12], of pro-opiomelanocortin (POMC) [13] of glycodelins A [14] and S [15], cytoplasmic glycosylation of Skp 1 [16,17], multiple ‘O-linked’ glycosylation of CD8 [18] and an unexpected novel ‘N-linked’ glycosylation in C.jejuni glycoproteins [19].
Despite those advances, the field of glycoproteomics remains a difficult one to enter for the new researcher, largely due to the sheer complexity and variability in the protein glycosylation we observe in most areas of biological research. In this paper, we attempt to demonstrate the further refinement of the strategies outlined above, with the aim of defining a generic approach to glycoproteomics, illustrated with advanced studies in which the interactive informatic tools which we are currently using to assist in detailed interpretation of MS and MS/MS data are described.
Glycoproteomic strategies
A number of reviews have been published which document the historical perspectives, principles and practice of glycoproteomic analysis [1, 2, 20-25]. The aims of this section are to highlight general issues and to suggest where efforts need to be focused to enable glycoproteomics research to be carried out more effectively. Firstly, of course, a key basic requirement is a well-found laboratory, for example at Imperial this includes 3 electrospray (ES) Q-TOF type instruments (including a Q-Star), 2 matrix-assisted laser desorption ionisation (MALDI) 4800 TOF-TOFs plus a range of ancillary equipment, such as gas chromatography (GC)-MS for composition and linkage analysis and nano-liquid chromatography(LC) for sample presentation both to the Q-TOFs and TOF-TOFs, in the latter case via a Probot auto-spotter. Broadly speaking, the majority of laboratories engaged in glycoproteomic analyses employ all or part of what has become a generic workflow as illustrated in Figure 1, with specific methodologies being dictated by available infrastructure, instrumentation and expertise. The type of sample being analysed will also influence the choice of methodology. For example, although not always applicable to large and/or highly heterogeneous glycoproteins, molecular weight profiling of intact glycoproteins (purple arrows in Figure 1) can sometimes provide very useful information on the type and extent of glycosylation. Such “top-down” methods have proven especially powerful, in bacterial glycoproteomics where novel glycans are frequently observed [26-28], and in studies on intact antibodies for the Biopharmaceutical industry, where M-Scan routinely screens intact masses at around 150 kilodaltons by both MALDI-TOF and ES-Q-TOF to give confirmatory total mass analysis when reconstructing the detailed protein and carbohydrate profiles from mass mapping studies.
Figure 1.

A simplified glycoproteomic experimental workflow is shown, illustrating common approaches to glycoproteomic analysis. Samples take the form of slices or spots excised from single or multi-dimensional polyacrylamide gels, or batches of cells, fluids, immunoprecipitates or tissue extracts. Analytical approaches can broadly be categorised as “top down” or “bottom up”. The former, illustrated by a purple arrow, begins with work on purified samples of glycoproteins in an attempt to identify the intact molecular weight profile by direct MALDI-TOF MS or by ES-MS. By subtracting the known or inferred mass of the protein component, the type and extent of glycosylation may then be deduced. In “bottom up” approaches, which incidentally are essential for describing the detailed glycosylation profile of any protein, the glycoprotein is digested enzymatically and/or chemically, ideally with high-specificity procedures, and the resulting peptide/glycopeptide mixture is mapped mass spectrometrically either by on-line (red arrows) LC-ES-MS followed by MS/MS analysis of signals of interest, or by off-line (blue arrows) strategies involving ES- or MALDI-MS and MS/MS approaches. Prior to these separation and mapping procedures, various strategies for enrichment of glycopeptides may be introduced, including lectin binding (see text). Parallel glycomic analyses (illustrated by the green arrow) are an invaluable feature of the “bottom up” approach involving enzymatic or chemical release of the glycans followed by MALDI-TOF MS or ES-MS mapping of the glycan populations, usually as permethyl derivatives (see text), providing information about specific glycans and their relative amounts, which can then be compared and matched with data at the glycopeptide and overall glycoprotein levels.
Central to all general glycoproteomic strategies is the mass spectrometric analysis of glycopeptides, usually after chromatographic separation, either on-line (red arrows) or off-line (blue arrows), to simplify the maps produced. Glycopeptides are normally obtained by specific proteolytic or chemical digestion of glycoproteins present in gel bands, immunoprecipitates or tissue extracts.
“Bottom-up” analysis, as the name suggests, begins with the analysis of individual glycans, building back through an analytical tree towards the intact glycoprotein, gathering information such as glycan structure, glycan repertoire, heterogeneity and sites of attachment. In the off-line approach, where chromatographic fractions are collected for individual analysis, this is of course generally more time-consuming and less suitable to automation, but it benefits from the fact that one can then apply a wider array of mass spectrometric equipment and methods, with far fewer issues than on-line analysis, where the compatibility of the eluents with mass spectrometric analysis and other limitations arising from the coupling of the chromatographic output to the ionisation source may restrict the applications somewhat.
Parallel glycomic analyses (illustrated by the green arrow) are also invaluable to glycoproteomic studies, providing information concerning the specific glycans present and the relative levels of the individual structures. Typically, specific glycan populations are released by enzymatic and chemical digestion of the peptide/glycopeptide mixture, derivatised by permethylation in order to enhance the separation, fragmentation, detection and stability of the constituent structures and then subjected to mass spectrometric analysis. A variety of enzymatic and chemical digestions of the released glycans can be employed prior to analysis if more specific structural information is desired.
Information gathered from these analytical approaches, the quantity of which can be vast, can then be used to search for and identify putative glycopeptides via characteristic fragment ions, with compositions being assigned through the use of both biosynthetic information and MS and MS/MS data.
Individual glycopeptide glycoforms are often very minor constituents compared with the peptides derived from the proteolytic digestion. Hence enrichment of glycoproteins and/or glycopeptides may be essential prior to analysis to ensure that glycoproteomic information is not obscured by vast quantities of proteomic data. Lectins [29] and hydrophilic affinity gels [30] are useful tools for glycoprotein/glycopeptide enrichment. Lectins with relatively broad specificity such as concanavalin A (conA), which recognises high mannose, hybrid and biantennary complex-type N-glycans, are showing considerable promise for enriching serum glycoproteins [31], whilst more specific lectins such as as Vicia villosa lectin (VVL), which preferentially binds to alpha- or beta-linked terminal GalNAc, are valuable tools for purifying glycoproteins that carry the cognate structure. For example bovine pregnancy associated glycoproteins which are rich in glycans carrying the Sda epitope (NeuAcα2-3(GalNAcβ1-4)Gal-) can be efficiently purified from placental tissue using VVL affinity columns [32]. Despite many examples of successful glycopeptide enrichment, the development of such methodologies that are applicable to a wide spectrum of samples is still a major challenge for glycoproteomics.
An even greater challenge occurs at the end of the workflow due to the paucity of informatic tools that are available to aid data interpretation (see below). To illustrate the data handling issues that need to be addressed in glycoproteomic experiments, exemplar data from on-line nanoLC-ES-MS analysis of a tryptic digest of a sample of human Glycodelin [33] are shown in Figure 2. The upper panel shows the total ion chromatogram for the complete LC-MS run whilst the middle panel shows a summation of the mass spectra corresponding to the part of the chromatogram that is highlighted in yellow. This region of the chromatogram has been chosen for scrutiny because the presence of characteristic sugar fragment ions (m/z 204, 366, 407 and 512; see annotations on Figure 2B) indicate that glycopeptides are present. Putative glycopeptide molecular ions are observed at low abundance throughout the green shaded region which is magnified in the bottom panel (Figure 2C) for clarity. Parts of the spectrum are expanded in the inserts to illustrate the richness of the data and to give insights into the assignment process (see legend). Interpretation of such complex data is greatly facilitated by prior knowledge of the compositions of the glycans in the glycoprotein sample being studied [33]. For this reason it is often advisable for the glycoproteomic workflow to include glycomics experiments which define the total glycan repertoire of the samples under study (see Figure 1, green arrows) [21,34,35]. A recent study reporting on the glycoproteome of mouse uterine luminal fluid [36] is a useful example of how to optimise the complementarity of the pathways shown in blue, red and green in the Figure 1 workflow.
Figure 2.

Nano-LC-ES-MS of a tryptic digest of a human glycodelin sample. The sample was reduced, carboxymethylated and dialysed prior to digestion with trypsin. A few micrograms of the digest products were subjected to on-line reverse-phase nanoLC coupled to a Q-STAR mass spectrometer.
Panel A shows the total ion chromatogram (TIC) for a 90-min experiment. Extracted ion chromatograms of glycan fragment ions were used to determine when glycopeptides elute and Panel B shows the summation of all spectra acquired between 7.5 and 11 min which were identified as corresponding to a glycopeptide elution time window. The diagnostic glycan fragment ions are annotated in the low-mass region of the spectrum shown in Panel B. Panels C, D and E detail portions of the spectrum with special emphasis at middle mass (Panel D) and high mass (Panel E) which have been expanded to show typical patterns for mixtures of multiply charged glycopeptides. For simplicity, the majority of the signals in the complete spectrum are not labelled but all multiply charged signals can be attributed to glycoforms of two major peptides containing the same N-glycosylation site at Asn-63. Sugar symbols used throughout this chapter are those employed by the Consortium for Functional Glycomics. Circles represent hexoses (yellow: Galactose, green: Mannose), squares represent N-acetylhexosamines (yellow: N-acetylgalactosamine, blue: N-acetylglucosamine), red triangle: Fucose, purple diamond: N-acetylneuraminic acid.
Glycoinformatic tools
Automated identification of proteins from MS and MS/MS spectra is now almost routine via the usage of informatic tools such as Mascot (http://www.matrixscience.com/). A major factor restricting progress in the glycomics/glycoproteomics field is the lack of rapid, accurate and flexible automated tools capable of retrieving structural information from MS data. The complexity of the glycan structures and the variety of techniques that are used for their study, pose additional obstacles to the development of a single automated tool that could have the same impact on glycoproteomics as tools such as Mascot have had for proteomics. Library-based sequencing tools for MS data interpretation, similar to the methods now commonly used for proteins, are limited by the lack of availability of comprehensive and well-curated collections of glycan sequences. De-novo sequencing tools and composition analysis tools are not restricted to previously characterized structures, but expert knowledge is fundamental to restrict the number of solutions matching experimental data and to obtain reasonable results. Probably the most successful of these tools is Cartoonist which has been designed to incorporate the same assumptions used by human expert annotators. Information about biosynthetic pathways is encoded in Cartoonist as a library of several hundred archetype glycans and a set of rules to modify these structures. Additional constraints are enforced to further limit the amount of possible structures. The peaks are then annotated by searching for all the structures that can be generated from the archetypes that match the given mass. A software calibration is performed for each spectrum to match observed and predicted masses. Calibration results together with isotope envelope shapes are then used to assign confidence scores to peak annotations. Finally, a graphical output is created by superimposition of the assigned structures on the actual spectra [37]. The Cartoonist algorithms continue to be developed with the objective of reducing the amount of prior expertise required [38]. An additional development has been to expand the tool’s use to glycoproteomics. Peptoonist now automatically identifies the glycans present at each N-glycosylation site of a glycoprotein [39]. Similar objectives have been achieved by combining generated glycopeptide MS/MS data with in silico workflows. These consist of comparing the original spectra to a N-glycopeptide library to assign the peptide sequence and predicting the N-glycan composition. Both processes are statistically validated to obtain best fit glycopeptides [40].
One of the major advances in our efficiency has come from the development of GlycoWorkbench. The aim of this tool is to provide complete support to the routine interpretation of glycomic mass spectrometric data and to form the basis for the development of fully automated assignment software. GlycoWorkbench comprises several features designed to help the user annotate their MS data. For example, the visual editor of glycan structures, the GlycanBuilder [41] enables a rapid assembly of graphical representations of structural models. Indeed with GlycanBuilder, a glycan can be rapidly specified starting from the reducing end by sequentially adding monosaccharides, modifications, or reducing-end markers (for example 2-aminopyridine, 2-aminobenzamide and 2-aminobenzoic acid) to the already drawn structure, simultaneously computing the corresponding theoretical m/z value. Different possible chemical derivatisations of the glycan structures, such as permethylation and acetylation, can also be integrated into calculations of theoretical m/z calculations.
When the structure is too complex and encompasses too many possible arrangements to be easily drawn, GlycoWorkbench offers the possibility to the user of entering the m/z value of the unknown species and then defining a set of parameters such as the presence of chemical modification(s), the nature of the reducing end or the set of possible monosaccharides that could be biosynthetically utilised by the organism studied. These parameters limit the number of possible arrangements. GlycoWorkbench will then compute the theoretical m/z values of the various possible compositions, given the restrictions defined by the user, match them with the experimental m/z value and create a report in which compositions are listed together with the m/z accuracy.
GlycoWorkbench also offers the possibility to assist annotations of MS/MS data in an interactive manner. Various ways of predicting fragments, from the drawn structures or from given m/z values are offered to the user. Spectra can also be downloaded and a list of peaks manually selected. If two possible structures are predicted to be present, then both can be matched against the list of observed fragment ions. The insilico fragmentation engine computes a complete list of theoretical fragments (multiple glycosidic cleavages and all the possible ring fragments). The annotation engine automatically matches the theoretical list of fragment masses with the manually defined experimental peak-list. The proposed annotations are presented using comprehensive and easily understandable reports that allow the comparison of the different annotations from the structure candidates.
The user can create and maintain multiple sets of candidate structures, peak-lists, mass spectra and annotated peak-lists in a single workspace so that all the information generated in an experiment can be organized and stored in a file. The software is publicly available for download from the EUROCarbDB Web site [42].
The capabilities of this tool are exemplified by our current glycoproteomic studies of a glycoprotein called gp273 which is found in the extracellular coat of the freshwater mollusc bivalve Unio elongatulus, and is believed to be the ligand for sperm-egg interaction during fertilization [43]. This glycoprotein has attracted the attention of the mammalian developmental biology community because it has been shown to bind to human sperm and to induce the acrosomal reaction [44]. Fucosylated epitopes on the O-glycans of gp273 appear to play a role in this binding. Moreover, it has been suggested that an anti-gp273 IgG antibody recognizes a Lewisa (Galβ1-3(Fucα1-4)GlcNAc)-like epitope on gp273. The binding of human sperm to gp273 was found to be reversed by solubilised human zona pellucidae [45] suggesting that gp273 might have structural features in common with the genuine sperm receptor ligands on human eggs whose identities remain a mystery. Characterization of gp273 glycosylation could, therefore, provide the first clues as to their likely structures. Previous structural studies of gp273 have been confined to defining its approximate molecular weight by MALDI-MS (273 kDa) [43] and showing by MALDI-MS and nuclear magnetic resonance (NMR) that its major N-glycans are Glc1Man9GlcNAc2 and Man9GlcNAc2 [46]. Nothing has so far been documented concerning its O-glycosylation other than the aforementioned possibility that Lewisa-like structures are present. Information on the nature of the O-glycome is now beginning to emerge from our glycomic analyses exploiting high sensitivity MALDI-TOF/TOF instrumentation and the GlycoWorkBench informatics tool. This research is revealing that gp273 carries a diverse repertoire of O-glycans, many of which are rich in fucose. This is illustrated by the MS and MS/MS data in Figures 3 and 4, respectively. As shown in the exemplar annotations in Figure 3, the GlycoWorkBench tool facilitates the assignment of possible sugar compositions to each of the molecular ions in the MALDI-MS profile. Putative compositions can then be ruled in or out by information provided by MS/MS experiments. For example, the GlycoWorkBench-annotated MS/MS data in Figure 4 reveal that m/z 912 and 1680 have the compositions Hex2HexNAc1dHex1 and Hex2HexNAc2dHex4 respectively, and not the other compositions shown in Figure 3. Note that the structures shown in the Figure 4 annotations take into account information from linkage analysis experiments (data not shown) which defined the types of monosaccharides in the gp273 glycans. Significantly the MS/MS data showed unequivocally that the fucose residues are mostly found in the context of the blood group H-antigen. Work is in progress to establish whether any Lewisa-like structures are actually present in gp273.
Figure 3.

MALDI-TOF MS profiling of Unio elongatulus glycoprotein gp273 O-glycans. Around 250 micrograms of gp273 were reduced, carboxymethylated and digested with trypsin. N-glycans were removed using PNGase A then separated from the pool of peptides and O-glycopeptides. The latter were subjected to reductive elimination and reduced O-glycans were purified prior to being permethylated and analysed using MALDI-TOF instrumentation. The Unio elongatulus genome is unknown so far. Therefore information about possible glycan structures cannot be readily deduced from biosynthetic knowledge. The use of a semi-automated tool such as GlycoWorkbench proved to be essential for an unbiased and comprehensive annotation of this MS spectrum. The putative compositions shown in boxes on this figure have been obtained using the GlycoPeakFinder feature of GlycoWorkbench (see Text). The type of monosaccharides considered were restricted to Hexose (10 maximum, symbol: blank circle), N-acetylhexosamine (10 maximum, symbol: blank square), Pentose (1 maximum, symbol: blank star), deoxy-Hexose (10 maximum, symbol: blank triangle), Hexuronic Acid (3 maximum, symbol: blank 2-halved-diamond) and Neuraminic acid (1 Maximum, symbol: blank diamond).
Figure 4.

GlycoWorkbench annotated MALDI-TOF/TOF MS/MS spectrum of the Unio elongatulus O-glycan species at m/z 912 (panel A) and 1680 (panel B) obtained using MALDI TOF/TOF instrumentation. According to the semi-automated annotation performed using GycoPeakFinder the species at m/z 912 and 1680 could correspond to 2 and 6 compositions, respectively. For each of the two MS/MS spectra, a list of fragment ions was manually selected. This list was then compared with the lists of predicted ions calculated for each of the possible structures corresponding to the computed compositions. A report showing the scores for each proposed arrangement was produced and the highest score glycan structures are shown on the top right corner of the spectra. Once the structures are selected, GlycoWorkbench can generate annotated spectra as shown here. For monosaccharide keys, see Figure 2. The reduced reducing end is represented by the  symbol. Keys related to fragmentation are the following:
symbol. Keys related to fragmentation are the following: 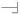 represents a fragmentation on the reducing end side of the glycosidic bond (also known as B or Z ions) and
represents a fragmentation on the reducing end side of the glycosidic bond (also known as B or Z ions) and  represents a fragmentation on the non-reducing end side of the glycosidic bond (also known as C or Y ions).
represents a fragmentation on the non-reducing end side of the glycosidic bond (also known as C or Y ions).
Conclusion and Future Prospects
The advances in glycoproteomics over the past four decades have been very significant, in no small part due to key advances in mass spectrometric instrumentation both with regard to ionisation techniques and instrument geometries, but also to the development of strategies, both chemical and biochemical, for dealing with complex glycoproteomic problems.
These advances, building upon the early concepts of biomolecular mass spectrometric strategy and tactics, have been illustrated in the present paper which outlines current glycoproteomic research projects aimed at gaining a better understanding of the first stages of life itself, including sperm-egg binding. We have stressed the important role of new glycoinformatic tools in this work, and the further development of Cartoonist, GlycoWorkbench and other similar software, together with the associated databases, will in future provide a goldmine of expert interpretive knowledge available to all. In summary, we can conclude that in perhaps only one or two more decades at most, glycoproteomic analysis will be carried out as effectively and efficiently and in as automated a fashion as is currently achievable in the related field of proteomics!
Acknowledgements
This research was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) grant numbers BBF0083091 and B19088 and the Analytical Glycotechnology Core of the Consortium for Functional Glycomics (GM62116). A.C. was supported by the sixth European Union Research Framework Programme (EUROCarbDB RIDS Contract Number 011952). AD was a BBSRC Professorial Research Fellow.
Abbreviations
- conA
Concanavalin A
- dHex
Deoxyhexose
- ES
Electrospray
- Fuc
Fucose
- Gal
Galactose
- GalNAc
N-acetylgalactosamine
- GC
Gas chromatography
- Glc
Glucose
- GlcNAc
N-acetylglucosamine
- Hex
Hexose
- HexNAc
N-acetylhexosamine
- LC
Liquid chromatography
- Man
Mannose
- MALDI-
Matrix assisted laser desorption ionisation
- NeuAc
N-acetylneuraminic acid
- MS
Mass spectrometry
- MS/MS
Tandem mass spectrometry
- NMR
Nuclear magnetic resonance
- POMC
Pro-opiomelanocortin
- PTM
Post-translational modification
- Q-TOF
Quadrupole orthogonal acceleration time of flight
- TOF
Time of flight
- TIC
Total ion chromatogram
- VVL
Vicia villosa
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Dell A, Morris HR. Glycoprotein structure determination by mass spectrometry. Science. 2001;291:2351–6. doi: 10.1126/science.1058890. [DOI] [PubMed] [Google Scholar]
- [2].Morris HR, Chalabi S, Panico M, Sutton-Smith M, Clark GF, Goldberg D, Dell A. Glycoproteomics: Past, present and future. International Journal of Mass Spectrometry. 2007;259:16–31. [Google Scholar]
- [3].Morris HR, Thompson MR, Osuga DT, Ahmed AI, Chan SM, Vandenheede JR, Feeney RE. Antifreeze glycoproteins from the blood of an antarctic fish. The structure of the proline-containing glycopeptides. J Biol Chem. 1978;253:5155–62. [PubMed] [Google Scholar]
- [4].Taylor GW, Morris HR, Peterson TE, Magnusson S. Advances in Mass Spectrometry. 1980;18:1090. [Google Scholar]
- [5].Morris HR, Panico M, Taylor GW. FAB-mapping of recombinant-DNA protein products. Biochem Biophys Res Commun. 1983;117:299–305. doi: 10.1016/0006-291x(83)91575-9. [DOI] [PubMed] [Google Scholar]
- [6].Morris HR, Geddes AJ, Graham GN. Some problems associated with the amino acid-sequence analysis of proteins by mass spectrometry. Biochem J. 1969;111:38P. doi: 10.1042/bj1110038p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Morris HR, Williams DH, Ambler RP. Determination of the sequences of protein-derived peptides and peptide mixtures by mass spectrometry. Biochem J. 1971;125:189–201. doi: 10.1042/bj1250189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Morris HR, Greer FM. Mass-spectrometry of natural and recombinant proteins and glycoproteins. Trends in Biotechnology. 1988;6:140–147. [Google Scholar]
- [9].Morris HR, Panico M, Etienne T, Tippins J, Girgis SI, MacIntyre I. Isolation and characterization of human calcitonin gene-related peptide. Nature. 1984;308:746–8. doi: 10.1038/308746a0. [DOI] [PubMed] [Google Scholar]
- [10].Robb RJ, Kutny RM, Panico M, Morris HR, Chowdhry V. Amino acid sequence and post-translational modification of human interleukin 2. Proc Natl Acad Sci U S A. 1984;81:6486–90. doi: 10.1073/pnas.81.20.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pappin DJ, Hojrup P, Bleasby AJ. Rapid identification of proteins by peptide-mass fingerprinting. Curr Biol. 1993;3:327–32. doi: 10.1016/0960-9822(93)90195-t. [DOI] [PubMed] [Google Scholar]
- [12].Chan AL, Morris HR, Panico M, Etienne AT, Rogers ME, Gaffney P, Creighton-Kempsford L, Dell A. A novel sialylated N-acetylgalactosamine-containing oligosaccharide is the major complex-type structure present in Bowes melanoma tissue plasminogen activator. Glycobiology. 1991;1:173–85. doi: 10.1093/glycob/1.2.173. [DOI] [PubMed] [Google Scholar]
- [13].Siciliano RA, Morris HR, McDowell RA, Azadi P, Rogers ME, Bennett HP, Dell A. The Lewis x epitope is a major non-reducing structure in the sulphated N-glycans attached to Asn-65 of bovine pro-opiomelanocortin. Glycobiology. 1993;3:225–39. doi: 10.1093/glycob/3.3.225. [DOI] [PubMed] [Google Scholar]
- [14].Dell A, et al. Structural analysis of the oligosaccharides derived from glycodelin, a human glycoprotein with potent immunosuppressive and contraceptive activities. J Biol Chem. 1995;270:24116–26. doi: 10.1074/jbc.270.41.24116. [DOI] [PubMed] [Google Scholar]
- [15].Morris HR, et al. Gender-specific glycosylation of human glycodelin affects its contraceptive activity. J Biol Chem. 1996;271:32159–67. doi: 10.1074/jbc.271.50.32159. [DOI] [PubMed] [Google Scholar]
- [16].Teng-umnuay P, Morris HR, Dell A, Panico M, Paxton T, West CM. The cytoplasmic F-box binding protein SKP1 contains a novel pentasaccharide linked to hydroxyproline in Dictyostelium. J Biol Chem. 1998;273:18242–9. doi: 10.1074/jbc.273.29.18242. [DOI] [PubMed] [Google Scholar]
- [17].van Der Wel H, Morris HR, Panico M, Paxton T, North SJ, Dell A, Thomson JM, West CM. A non-Golgi alpha 1,2-fucosyltransferase that modifies Skp1 in the cytoplasm of Dictyostelium. J Biol Chem. 2001;276:33952–63. doi: 10.1074/jbc.M102555200. [DOI] [PubMed] [Google Scholar]
- [18].Moody AM, et al. Sialic acid capping of CD8beta core 1-O-glycans controls thymocyte-major histocompatibility complex class I interaction. J Biol Chem. 2003;278:7240–6. doi: 10.1074/jbc.M210468200. [DOI] [PubMed] [Google Scholar]
- [19].Wacker M, et al. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science. 2002;298:1790–3. doi: 10.1126/science.298.5599.1790. [DOI] [PubMed] [Google Scholar]
- [20].Geyer H, Geyer R. Strategies for analysis of glycoprotein glycosylation. Biochim Biophys Acta. 2006;1764:1853–69. doi: 10.1016/j.bbapap.2006.10.007. [DOI] [PubMed] [Google Scholar]
- [21].Haslam SM, North SJ, Dell A. Mass spectrometric analysis of N- and O-glycosylation of tissues and cells. Curr Opin Struct Biol. 2006;16:584–91. doi: 10.1016/j.sbi.2006.08.006. [DOI] [PubMed] [Google Scholar]
- [22].Wuhrer M, Catalina MI, Deelder AM, Hokke CH. Glycoproteomics based on tandem mass spectrometry of glycopeptides. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;849:115–28. doi: 10.1016/j.jchromb.2006.09.041. [DOI] [PubMed] [Google Scholar]
- [23].Zaia J. Mass spectrometry and the emerging field of glycomics. Chem Biol. 2008;15:881–92. doi: 10.1016/j.chembiol.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Amon S, Zamfir AD, Rizzi A. Glycosylation analysis of glycoproteins and proteoglycans using capillary electrophoresis-mass spectrometry strategies. Electrophoresis. 2008;29:2485–507. doi: 10.1002/elps.200800105. [DOI] [PubMed] [Google Scholar]
- [25].Bindila L, Peter-Katalinic J. Chip-mass spectrometry for glycomic studies. Mass Spectrom Rev. 2009;28:223–53. doi: 10.1002/mas.20197. [DOI] [PubMed] [Google Scholar]
- [26].Hitchen PG, Dell A. Bacterial glycoproteomics. Microbiology. 2006;152:1575–80. doi: 10.1099/mic.0.28859-0. [DOI] [PubMed] [Google Scholar]
- [27].Aas FE, Egge-Jacobsen W, Winther-Larsen HC, Lovold C, Hitchen PG, Dell A, Koomey M. Neisseria gonorrhoeae type IV pili undergo multisite, hierarchical modifications with phosphoethanolamine and phosphocholine requiring an enzyme structurally related to lipopolysaccharide phosphoethanolamine transferases. J Biol Chem. 2006;281:27712–23. doi: 10.1074/jbc.M604324200. [DOI] [PubMed] [Google Scholar]
- [28].Twine SM, et al. Flagellar glycosylation in Clostridium botulinum. FEBS J. 2008;275:4428–44. doi: 10.1111/j.1742-4658.2008.06589.x. [DOI] [PubMed] [Google Scholar]
- [29].Sharon N. Lectins: past, present and future. Biochem Soc Trans. 2008;36:1457–60. doi: 10.1042/BST0361457. [DOI] [PubMed] [Google Scholar]
- [30].Tajiri M, Yoshida S, Wada Y. Differential analysis of site-specific glycans on plasma and cellular fibronectins: application of a hydrophilic affinity method for glycopeptide enrichment. Glycobiology. 2005;15:1332–40. doi: 10.1093/glycob/cwj019. [DOI] [PubMed] [Google Scholar]
- [31].Madera M, Mann B, Mechref Y, Novotny MV. Efficacy of glycoprotein enrichment by microscale lectin affinity chromatography. J Sep Sci. 2008;31:2722–32. doi: 10.1002/jssc.200800094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Klisch K, Jeanrond E, Pang PC, Pich A, Schuler G, Dantzer V, Kowalewski MP, Dell A. A tetraantennary glycan with bisecting N-acetylglucosamine and the Sd(a) antigen is the predominant N-glycan on bovine pregnancy-associated glycoproteins. Glycobiology. 2008;18:42–52. doi: 10.1093/glycob/cwm113. [DOI] [PubMed] [Google Scholar]
- [33].Pang PC, et al. Effects of differential glycosylation of glycodelins on lymphocyte survival. J Biol Chem. 2009 doi: 10.1074/jbc.M807960200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jang-Lee J, North SJ, Sutton-Smith M, Goldberg D, Panico M, Morris H, Haslam S, Dell A. Glycomic profiling of cells and tissues by mass spectrometry: fingerprinting and sequencing methodologies. Methods Enzymol. 2006;415:59–86. doi: 10.1016/S0076-6879(06)15005-3. [DOI] [PubMed] [Google Scholar]
- [35].Wada Y, et al. Comparison of the methods for profiling glycoprotein glycans--HUPO Human Disease Glycomics/Proteome Initiative multi-institutional study. Glycobiology. 2007;17:411–22. doi: 10.1093/glycob/cwl086. [DOI] [PubMed] [Google Scholar]
- [36].Kuo CW, Chen CM, Lee YC, Chu ST, Khoo KH. Glycomics and proteomics analyses of mouse uterine luminal fluid revealed a predominance of Lewis Y and X epitopes on specific protein carriers. Mol Cell Proteomics. 2009;8:325–42. doi: 10.1074/mcp.M800320-MCP200. [DOI] [PubMed] [Google Scholar]
- [37].Goldberg D, Sutton-Smith M, Paulson J, Dell A. Automatic annotation of matrix-assisted laser desorption/ionization N-glycan spectra. Proteomics. 2005;5:865–75. doi: 10.1002/pmic.200401071. [DOI] [PubMed] [Google Scholar]
- [38].Goldberg D, Bern M, North SJ, Haslam SM, Dell A. Glycan family analysis for deducing N-glycan topology from single MS. Bioinformatics. 2009;25:365–71. doi: 10.1093/bioinformatics/btn636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Goldberg D, Bern M, Parry S, Sutton-Smith M, Panico M, Morris HR, Dell A. Automated N-glycopeptide identification using a combination of single- and tandem-MS. J Proteome Res. 2007;6:3995–4005. doi: 10.1021/pr070239f. [DOI] [PubMed] [Google Scholar]
- [40].Joenvaara S, Ritamo I, Peltoniemi H, Renkonen R. N-glycoproteomics - an automated workflow approach. Glycobiology. 2008;18:339–49. doi: 10.1093/glycob/cwn013. [DOI] [PubMed] [Google Scholar]
- [41].Ceroni A, Dell A, Haslam SM. The GlycanBuilder: a fast, intuitive and flexible software tool for building and displaying glycan structures. Source Code Biol Med. 2007;2:3. doi: 10.1186/1751-0473-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ceroni A, Maass K, Geyer H, Geyer R, Dell A, Haslam SM. GlycoWorkbench: a tool for the computer-assisted annotation of mass spectra of glycans. J Proteome Res. 2008;7:1650–9. doi: 10.1021/pr7008252. [DOI] [PubMed] [Google Scholar]
- [43].Focarelli R, Lupetti P, Seraglia R, Rosati F. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and electron microscopy for the characterization of the vitelline coat glycoproteins of the polarized egg of Unio elongatulus. Mol Reprod Dev. 1997;48:511–7. doi: 10.1002/(SICI)1098-2795(199712)48:4<511::AID-MRD12>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- [44].Monache S. Delle, et al. Gp273, the ligand molecule for sperm-egg interaction in the bivalve mollusk, Unio elongatulus, binds to and induces acrosome reaction in human spermatozoa through a protein kinase C-dependent pathway. Biol Reprod. 2003;69:1779–84. doi: 10.1095/biolreprod.103.019976. [DOI] [PubMed] [Google Scholar]
- [45].Focarelli R, Capone A, Ermini L, Del Buono F, La Sala G. Battista, Balasini M, Rosati F. Immunoglobulins against gp273, the ligand for sperm-egg interaction in the mollusc bivalve Unio elongatulus, are directed against charged O-linked oligosaccharide chains bearing a Lewis-like structure and interact with epitopes of the human zona pellucida. Mol Reprod Dev. 2003;64:226–34. doi: 10.1002/mrd.10241. [DOI] [PubMed] [Google Scholar]
- [46].Di Patrizi L, Capone A, Focarelli R, Rosati F, Gallego RG, Gerwig GJ, Vliegenthart JF. Structural characterization of the N-glycans of gp273, the ligand for sperm-egg interaction in the mollusc bivalve Unio elongatulus. Glycoconj J. 2001;18:511–8. doi: 10.1023/a:1019617728660. [DOI] [PubMed] [Google Scholar]