

Abstract
Schizophrenia represents a pervasive deficit in brain function, leading to hallucinations and delusions, social withdrawal and a decline in cognitive performance. As the underlying genetic and neuronal abnormalities in schizophrenia are largely unknown, it is challenging to measure the severity of its symptoms objectively, or to design and evaluate psychotherapeutic interventions. Recent advances in neurophysiological techniques provide new opportunities to measure abnormal brain functions in patients with schizophrenia and to compare these with drug-induced alterations. Moreover, many of these neurophysiological processes are phylogenetically conserved and can be modelled in preclinical studies, offering unique opportunities for use as translational biomarkers in schizophrenia drug discovery.
Schizophrenia is a major mental disorder characterized by positive and negative symptoms, as well as persistent neurocognitive deficits. All currently approved medications for schizophrenia function by blocking dopamine D2 receptors, and have proved effective primarily against positive symptoms. By contrast, negative and cognitive symptoms frequently persist, and lead to persistent disability and poor long-term outcome. Brain processes underlying persistent cognitive deficits in schizophrenia can be probed using various techniques, including structural and functional imaging, positron emission tomography and single photon emission computed tomography receptor assays, and neurophysiological measures such as electroencephalography (EEG) and event-related potentials (ERPs).
Neurophysiological measures as a group have several characteristics that make them well suited for use as biomarkers in drug development studies. First, many can be recorded in passive paradigms in which no attention, task engagement or behavioural readout is required. These measures are therefore ideal for use in populations that may be difficult to engage in behavioural studies. Second, because of their high temporal resolution, neurophysiological biomarkers can uniquely be used to trace the flow of information in the brain from sensory through to association brain regions, and hence can be used to determine the earliest stages at which information processing is impaired. Last, because they index underlying neuronal activity, neurophysiological biomarkers may be seen as objective indices of cognitive dysfunction — a prominent feature of patients with schizophrenia. Here, we review the best available biomarkers to date, and the strengths and limitations of their use in the development of drugs for schizophrenia.
Neurophysiological signals
Neurophysiological measures are generally recorded using standard EEG techniques in which an array of up to 256 electrodes are affixed to specific scalp locations and the pattern of electrical activity is monitored while subjects participate in specific experimental paradigms. Scalp recorded electrical activity primarily represents spatial and temporal summation of synchronous current flow through postsynaptic dendritic membranes of cortical pyramidal neurons that, because of their parallel alignment in the cortex and asymmetric morphology, serve as ‘open field’ generators in the brain and give rise to local field potentials.
The basic physiological data underlying most bio-markers that are presently available are similar to that used in qualitative or quantitative EEG assessment. A critical difference is the use of signal averaging. Brain responses to individual sensory, cognitive or motor events are small relative to the background EEG, whereas signal averaging permits such events to stand out. Neurophysiological biomarkers are typically time-locked to sensory events (for example, auditory or visual stimuli). Nevertheless, other types of events can also be defined. For example, in response tasks, electrical responses can be back-averaged from motor responses and can be used to analyse preceding activity. In smooth pursuit eye movement (SPEM) tasks, neurophysiological activity related to eye movements can be back-averaged from the onset of a saccade to detail underlying brain processes. However, the absence of electrical activity cannot be taken as evidence of the lack of response of a given region as the majority of electrical activity generated in the brain is invisible to surface recordings. Nevertheless, over recent years, neurophysiological measures that probe brain activity in regions that are of relevance to schizophrenia have been developed.
Time versus frequency domain analyses
Once neurophysiological data have been recorded (BOX 1), two complementary analytic approaches are used for data analysis. In the first approach, brain activations are viewed simply as a series of amplitude deflections that vary in time and space over the surface of the scalp. Analysis of response amplitude over time has traditionally been referred to as a time-domain analysis or, more accurately, as time-amplitude domain analysis. The sequence of deflections triggered by a given event is referred to as an ERP.
The second approach views brain activity as a sum of superimposed oscillations maintained within and between brain regions giving rise to event-related evoked or induced oscillations1. This approach, also known as time-frequency analysis, captures more information on underlying brain activity than the traditional ERP approach, but is computationally more complex and less standardized across subjects.
Source localization
For both time and frequency domain analyses, additional data concerning underlying mechanisms can be obtained using source analysis approaches that evaluate the electrical sources of scalp-derived activity within the brain. Source localization approaches make specific assumptions regarding the propagation of electrical activity through the brain and scalp, which may lead to localization imprecision. In practice, however, good spatial agreement is typically obtained between functional magnetic resonance imaging (fMRI) and ERP (for example, REF. 2) or oscillation-based (for example, REF. 3) measures of convergent phenomena.
Validation of a neurophysiological measure in the spatial domain with imaging methods that are based on blood flow offers critical advantages: the signal-to-noise ratio improves tenfold (when activity is generated in the cortex) and time-frequency information, which is currently not amenable to other functional imaging tools, becomes available. By extension, neurophysiological biomarkers may be more sensitive to drug-induced changes compared with other functional imaging modalities.
Magnetoencephalography
Although most research with biomarkers has been performed using electrically based techniques, magneto-encephalography (MEG) can be used instead to detect the magnetic fields that accompany electrical current flow through the brain. As magnetic fields are less susceptible to distortion by volume conduction of currents than electrical fields, reconstruction of MEG sources is possible with higher spatial accuracy than those of EEG. Furthermore, MEG offers the advantage of avoiding cumbersome electrode fixation to the scalp. As with EEG-based measures, MEG recordings can be analysed in either the time or the frequency domain. As a result of the electromagnetic field distribution, however, MEG is limited by its insensitivity to sources radial to the scalp surface (that is, pointed perpendicular, rather than parallel, to the skull surface) and by its cost: state-of-the-art MEG equipment is still more expensive than EEG-equipment and thus its use is currently limited to a few research centres. In contrast to the term ERP, which is used to refer to time-locked electrical activity, the term event-related field (ERF) is commonly applied to MEG data.
Neurophysiological measures as biomarkers
Because of the widespread availability of neurophysiological equipment and expertise, neurophysiological measures have been widely applied in schizophrenia research. Some well-validated measures, such as prepulse inhibition (PPI) or P50 gating, reflect the ‘gating out’ of particular sensory information4. Such measures, however, reflect only a portion of the psychopathology of schizophrenia, and are not closely linked to neurocognitive dysfunction4,5. Over recent years, newer neurophysiological measures have become available that index information processing abnormalities and correlate well with negative symptoms and cognitive deficits, as well as global functional outcome. Furthermore, these biomarkers reflect the integrity of differential brain regions and cognitive pathways (FIG. 1), and so can be used to probe specific underlying regionalized and neurochemical hypotheses of schizophrenia.
Figure 1. This figure reflects the multiple brain regions implicated in schizophrenia, and source of likely generators for specific biomarkers used in the study of schizophrenia.

AX-CPT, AX-type visual continuous performance task; MMN, mismatch negativity; Ncl, closure negativity; SPEM, smooth pursuit eye movements.
Well-studied measures include auditory P300 and mismatch negativity (MMN), both of which are commonly elicited using an auditory oddball paradigm (that is, when a sequence of repetitive standard stimuli is interrupted infrequently and unexpectedly by a physically deviant oddball stimulus), as well as measures of impaired visual processing and SPEMs. In addition, significant advances have been made in recent years in analysing these deficits not only in terms of response amplitude within the time domain, but also in terms of the spectral content of underlying neural oscillations within the frequency domain. These different biomarkers have differential strengths and weaknesses with regard to sensitivity, specificity and timing relative to disease onset, and thus may be viewed as complementary approaches to neurophysiological characterizations of schizophrenia and of treatment responses (TABLE 1).
Table 1.
Characteristics of selected biomarkers in schizophrenia
| Marker | Effect in unaffected family members |
Effect in prodromes |
Effect at first-episode |
Effect in SPD |
EFfect in other disorders |
Response to clozapine |
Human challenge/ linkage studies |
Animal models |
|---|---|---|---|---|---|---|---|---|
| Gating measures | ||||||||
| Prepulse inhibition | Abnormal192 | ND | Abnormal193 | Normal192 | Abnormal in AD194; BPD195 | Yes130,196 | DA197,198; NMDA129,131,199 | Rodent128; primate129,196 |
| P50 gating | Abnormal200 | Abnormal201 | Abnormal202 | Abnormal203 | Abnornal in AD204; BPD205; cocaine abuse92,206 | Yes93 | DA92,206,207; nACh7 (RefS 151, 206,208); (REF. 183); 5-HT3 NMDA130 | Rodent81,198,209 |
| Information-processing measures | ||||||||
| Auditory P300 (P3) | Abnormal18,210 | Abnormal/normal211 | Abnormal212,213 | Abnormal214 | Abnormal in AD215; BPD64; ADHD216 | Abnormal/normal 15,217,218 | DA19,96,98,219; ACh110; NMDA220 | Mouse87; rat86 |
| Mismatch negativity | Normal31,221; abnormal32 | Abnormal/normal33,211,222 | Normal14,27,29 | ND | Normal in AD223; BPD24 | No15,224 | NMDA133–135; 5-HT2A (REF. 137) | Primate133,225; rodent23 |
| Auditory N1 | Abnormal215,226 | ND | Abnormal37,38 | Normal227 | Abnornal in cocaine abuse228; normal in BPD229 | ND | ND | Rodent83,85; primate46 |
| Visual P1 | Abnormal41 | ND | ND | ND | ND | ND | NMDA43 | Primate230 |
| Neural-synchrony measures | ||||||||
| Gamma: auditory steady-state response | Abnormal63 | ND | Abnormal62 | Normal227 | Abnormal in autism231; BPD62, 64; cannabis abuse182 | ND | ND | Rodent232 |
| Gamma: Evoked/induced oscillations | ND | ND | Abnormal233 | ND | ND | ND | mACh220; DA234,235; NMDA236 | ND |
| Prefrontal slow-wave synchrony (‘noise’) | Abnormal18,65 | ND | Abnormal60 | Abnormal/normal60 | Normal in AD60 | ND | DA19,101 | Rodent237 |
| Other neurophysiological measures | ||||||||
| Smooth pursuit eye movements | Abnormal74,210,238 | Abnormal239 | Abnormal240,241 | ND | ND | Worsens242 | nACh164,243,249; NMDA138,139 | ND |
5-HT, 5-hydroxytryptamine (serotonin); Ach, acetylcholine; AD, Alzheimer’s disease; ADHD, attention-deficit/hyperactive disorder; BPD, bipolar disorder; DA, dopamine; mACh, muscarinic acetylcholine; nACh, nicotinic acetylcholine; ND, not determined; NMDA, N-methyl-D-aspartate; SPD, schizotypal personality disorder.
Auditory P300
The earliest and best studied cognitive ERP abnormality in schizophrenia is the amplitude reduction of the auditory P300 component6. P300 is elicited most commonly in the context of the auditory oddball paradigm in which subjects are instructed to attend to the sequence of tones, and either count the deviants or press a button in response to the deviants. Under such conditions, P300 occurs approximately 300 ms following deviant tone presentation. P300 is typically divided into two subcomponents, an early P3-like potential (P3a) that is elicited by novel stimuli, even in the absence of a detection task, and is located relatively frontally, and a later, more parietal component, P3b, that is elicited only during target detection.
Subjects who are chronically ill with schizophrenia show a reduction of P300 amplitude of approximately one standard deviation relative to healthy subjects7,8, and include P3a as well as P3b components9,10. Furthermore, deficits in P3b track the severity of negative symptoms both longitudinally and cross-sectionally9,10. A limitation of P300 is its relative nonspecificity with regard to illness; schizophrenia-like P300 reductions are also observed in alcohol-dependent subjects11, bipolar disorder12 and Alzheimer’s disease13 among other diagnoses. P300 is typically preceded by an N200 component that localizes to secondary auditory regions. Like P300, generation of N200 is typically reduced in schizophrenia14. Deficits in P300 generation are not affected by either typical or atypical antipsychotics, although they may be partially ameliorated by clozapine15,16.
Box 1 | Neurophysiological data analysis
Electroencephalography (EEG) data are recorded from an electrode array sampling the electric field across the scalp (a). In the classical event-related potential (ERP) approach, single-trial recording epochs (b) are averaged together (c). The average ERP consists of voltage deflections that have been characterized as particular components, which are typically defined by latency, polarity (P for positive, N for negative), scalp topography and variation with experimental manipulations. For example, the P1 and N170 are occipito-temporal positive and negative components occurring early in the stream of visual processing, approximately 100 ms and 170 ms following stimulation, and represent discrete stages of sensory/perceptual processing (c). The amplitude of the P1 is reduced in schizophrenia42, and this deficit represents one biomarker for schizophrenia. Since the advent of signal averaging more than 40 years ago tens of thousands of ERP studies have been published, providing a catalogue of components that are well-suited for probing the functioning of specific brain regions and cognitive processes. In contrast to the classical ERP approach, which discards most of the oscillatory information in the EEG through signal averaging, contemporary time-frequency approaches (d–g) represent changes in oscillatory activity as a function of time3, typically using Fourier or wavelet transforms1.
Interest in time-frequency analysis has been motivated by the hypothesis that synchronous neural activity, mediated by oscillations in different frequency bands (h), enables information coded by individual neurons to be linked together (for example, REF. 187). High-frequency oscillations (for example, gamma; 30–70 Hz) may be predominantly relevant for coupling local assemblies of neurons55,188,189, whereas low-frequency oscillations (for example, theta; 4–-8 Hz) may coordinate activity across widespread cortical regions. Very high frequency oscillations (70–300 Hz) might reflect axonal action potentials189.
Oscillatory responses are typically characterized by phase synchronization locking (g) and average power (μV2) (e–f) across trials. Evoked oscillations are strictly phase-locked to the stimulus (e), whereas induced oscillations occur with variable phase (f)1.

Heritability of P300-amplitude is high (70–80%)17, suggesting that P300 may serve as a risk or trait marker of the genetic risk for schizophrenia18. In fact, several recent studies have reported associations of P300 abnormalities with genetic variations that are thought to contribute to this genetic risk19,20. In contrast to the parietal P300 amplitude, which is reduced in individuals at risk for schizophrenia, recent work suggests that early prefrontal P300 amplitude may even be increased18. Despite the reduction of P300 in family members with schizophrenia as a whole, negative results with P300 have been reported among offspring of schizophrenia probands21.
MMN and additional sensory components
Compared with P300, MMN is elicited by unattended, as well as attended, deviants within an auditory oddball task, and thus appears to reflect a pre-attentive stage of auditory information processing22 (FIG. 2). For MMN generation, oddball stimuli may differ from standards in any of a number of physical dimensions, including sensory modality, frequency, duration, intensity or location. In studies in which both MMN and P300 are measured simultaneously, the latencies of the two components increase in parallel, suggesting that two components are sequentially related.
Figure 2. Schematic diagram of mismatch negativity (MMN) generators in schizophrenia.

a. MMN is elicited in an auditory oddball paradigm in which a sequence of repetitive standard stimuli (blue boxes) are interrupted by stimuli that differ in a physical stimulus dimension such as pitch or duration (green boxes). The deviant probability equals the number of deviants divided by the total number of stimuli. MMN reflects N-methyl-D-aspartate (NMDA)-dependent processing of stimulus deviance within the auditory sensory cortex. b. Schematic diagram of MMN generators within the auditory cortex (located in the superior temporal lobe, shown in red). Because of the orientation of MMN generators, the MMN reverses in polarity between the frontal midline electrode (Fz) and the left (LM) and right (RM) mastoids. Because pitch deviance can be detected at stimulus onset, but duration deviance can only be detected at the time of standard stimulus offset, duration MMN (pale blue line) is delayed relative to pitch (frequency) MMN (pink line). The dashed arrow indicates the orientation of the electrical field originating from the auditory cortex. Activity from auditory cortex characteristically inverts between the central midline electrode (Fz) and the mastoids (RM, LM), relative to a nose reference (not shown). c. Characteristic waveforms at Fz from patients with recent onset or chronic schizophrenia versus controls. Peak MMN responses (arrows) are significantly reduced in patients with schizophrenia relative to controls, for both pitch (top line) and duration (bottom line). Dashed lines illustrate the latency shift in response to pitch versus duration to deviant stimuli25. Δf, pitch difference between standard and deviant; ERP, event-related potential; ISI, interstimulus interval. FIGS 2a and 2c are modified with permission from REF. 23 © (2005) and REF. 14 © (2006) Elsevier Science, respectively.
Like P300, deficits in MMN generation have been extensively replicated in schizophrenia, with mean effect size of approximately one standard deviation across studies23. Compared with P300, however, MMN deficits are relatively selective for schizophrenia over other neuropsychiatric disorders24. Deficits in MMN generation persist following treatment with both typical and atypical antipsychotics, including clozapine15,16. Furthermore, in chronically ill patients the severity of MMN deficits correlates with severity of negative symptoms25 and impaired global outcome26, suggesting that it indexes a core deficit of the disorder.
Compared with P300, which appears to be reduced even at illness onset, three independent studies have observed relatively normal MMN generation in individuals at first presentation of schizophrenia and well-established MMN deficits in patients that are 18 months or more into their illness14,27–29, suggesting that deficits may become established during the initial course of their illness. The heritability of MMN is also high, suggesting a strong genetic contribution17,30. Nevertheless, of the two family studies that have been conducted with MMN to date, one study found a reduction in MMN generation in unaffected family members of patients with schizophrenia31, whereas the second study did not32. In both studies, MMN was decreased in patients with established schizophrenia. In one study that evaluated MMN in patients showing prodromal symptoms of schizophrenia, no significant reduction was observed, although mean MMN amplitude among prodromal patients was intermediate between that of patients and controls. Furthermore, intersubject variance was high, consistent with the concept that MMN deficits might be present in only a subset of patients before the onset of illness33.
Primary generators for MMN have been localized to the primary auditory cortex using multiple techniques including EEG and MEG dipole localization, fMRI and direct intracranial recordings from the auditory cortex of both humans and monkeys34. Deficits in auditory cortical activation have been demonstrated in an fMRI adaptation of the MMN paradigm35, supporting the idea that impaired MMN generation in schizophrenia reflects dysfunction at the level of the primary auditory cortex.
Although MMN is the best-studied sensory measure in schizophrenia, other measures that index early auditory and visual processing may also be affected. For example, deficits are also reported in the generation of auditory N10036–38, an auditory potential that reflects response to repetitive standard stimuli, and to visual P1, a measure that reflects responses within the dorsal visual stream39–41 (FIG. 3). Steady-state visual evoked potential (ssVEP) responses to magnocellularly biased stimuli are observed as well42,43. In all cases, ERP components can be measured even in the absence of a behavioural task, rendering these components well suited for both clinical and translational biomarker studies.
Figure 3. Visual P1 deficit in schizophrenia using local autoregressive average (LAURA) model.
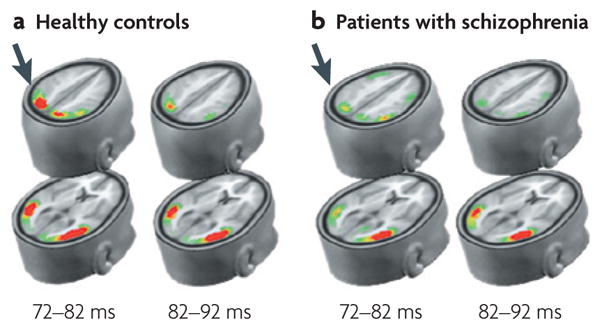
LAURA depicts the degree of brain electrical activity (current source density) within derived source regions190. P1 reflects early stimulus-elicited activity within the dorsal (top rows) and ventral (bottom rows) visual streams, occurring within the first 100 ms following stimulation. In schizophrenia, activation is normal within the ventral stream, but is markedly reduced within the dorsal stream (arrows), reflecting impaired activity within the magnocellular system. This image is modified with permission from REF. 190 © (2005) Oxford University Press.
Visual P300
Although P300-like activity has been best studied in the auditory system, deficits have also been observed in the visual system. In the visual system, deficits in simple visual discrimination (for example, oddball44 or enumeration45 tasks) are not typically observed in patients with stabilized schizophrenia, and if a deficit is found it is typically smaller than that found for auditory P300 (REF. 8). By contrast, more robust deficits in visual P300 have been found in tasks with greater cognitive demands46,47.
Neurophysiological deficits have been demonstrated particularly in continuous performance tasks (CPTs), such as the AX-CPT in which subjects must press a button whenever they see the letter A followed by the letter X, but withhold response to other letter sequences. Patients with schizophrenia have behavioural deficits in the ability to withhold response following invalid cues (that is, letters other than A), suggesting a preferential deficit in the ability to utilize context48. Such deficits are accompanied by reduced amplitude of the P300 responses to the invalid cues46.
Behavioural deficits are observed in both chronic and first-episode treatment-naive patients, and are relatively selective to schizophrenia versus non-schizophrenic psychosis49,50. Generators of the visual P3 response in the AX-CPT have also been localized to prefrontal and cingulate cortex using fMRI50,51 and ERP52 approaches, as well as intracranial recordings in monkeys53.
In the AX-CPT53 and similar visual tasks, a slow frontal negativity, termed the contingent negative variation is seen following cue presentation; this indexes response preparation, and so may serve as an additional target in schizophrenia. AX-CPT deficits can be ameliorated in schizophrenia by modifying the stimuli used, suggesting that deficits in sensory processing might be, at least in part, responsible for failures in subsequent stages of processing54.
Neural synchrony measures
Traditional ERP measures analyse brain electrical activity as a function of amplitude over time, ignoring precise dynamics of underlying neural oscillations. However, with increases in computing power, it is now possible to extract information on neural synchrony from ongoing EEG activity using approaches such as Fourier transform or wavelet analysis. These oscillations reflect synchronous brain activity within and between brain regions, and serve to link clinical recordings with burgeoning preclinical literature regarding oscillatory brain activity during cognitive operations.
The neural synchrony approach examines naturally arising brain rhythms that occur while the brain performs specific sensory, cognitive or motor operations. Oscillatory measures are typically divided into multiple frequency ranges, with faster rhythms (gamma and beta for example) being phase-locked to slower rhythms (theta and delta for example)55 (BOX 1). Gamma measures have been of particular interest in schizophrenia as they are linked with the concept of perceptual feature binding and interbrain-area synchronization (FIG. 4). Deficits in gamma generation have been observed in both auditory56 and visual57,58 discrimination tasks, although contrary results have also been reported59. Furthermore, deficits do not appear to be unique to gamma-band responses, as deficits are also observed in the beta range59 and slower frequencies (for example, 0.5–12 Hz)60. Reductions in neural synchrony at slower frequencies have been associated with attentional performance in both patients with schizophrenia and their unaffected relatives (FIG. 5). This finding, which is interpreted as reflecting increased noise (that is, brain activity in the absence of stimulus) in the low-frequency range in schizophrenia, is consistent with long-standing findings of increased trial-by-trial variability of ERP responses in patients with schizophrenia60.
Figure 4. Time/frequency domain electroencephalography measures from a control subject and a patient with chronic schizophrenia.

The average event-related potential (ERP) (n = 88 trials) reveals that both P1 and N1 components are markedly reduced in the patient with schizophrenia compared with the control subject. Reduced activity across the frequency spectrum is apparent in the maps of evoked power, induced power and phase-locking synchrony (colour scales same as in BOX 1).
Figure 5. Cortical response variability is increased in patients with schizophrenia.

a. Frequency domain analyses showing increased prefrontal noise, that is, increased variability of slow-wave oscillations, in patients with schizophrenia and their clinically unaffected siblings60. Increased variability of slow-wave oscillations results from impaired phase-locking of these oscillations in schizophrenia65. b. An analogous increase in variability of blood-oxygen-level dependent response is observed in patients with schizophrenia3: group contrast analysis compared with controls. Prefrontal noise is modulated by synaptic dopamine signalling (catechol-O-methyltransferase (COMT)-genotype) as measured by electrophysiology and functional magnetic resonance imaging3,60,101. This image is reproduced with permission from REF. 60 © (2004) and REF. 191 © (2006) American Psychiatric Association.
A related, but distinct, approach to assess neural synchrony is the evaluation of auditory steady-state responses (ASSRs) or visual ssVEPs. In ssVEPs, sensory systems are driven at the frequency of interest and amplitude of the cortical response is assessed as a function of the stimulation rate. Thus, activity is not generated endogenously but is driven exogenously. The auditory system shows a characteristic resonance at 40 Hz (that is, within the gamma range), whereas the visual system shows maximal response at 8–16 Hz. Deficits in 40 Hz auditory responses have been observed in some61,62, but not all63 studies in schizophrenia. Reductions of the 40 Hz ASSR have also been found in patients with bipolar disorder62,64 and in unaffected family members of schizophrenia probands63. Preliminary data suggest that the reduction of the 40 Hz ASSR and the increased low-frequency noise found in chronic patients is also present at first hospitalization62,65.
Patients with schizophrenia also show deficits in visual steady-state responses. Reduced amplitudes have been observed consistently at high (beta/gamma-range) driving frequencies66 and at lower frequencies (theta/alpha) in some42,67, but not all66, studies. Patients also show increased noise particularly at low frequencies, resulting in reduced signal-to-noise ratios across frequency bands66. Timing of the response may also be altered, with patients taking more time both to reach maximal response and to decay following cessation of stimulation68. Deficits may also be stimulus dependent, with patients showing greater deficits to stimuli biased towards the magnocellular system versus the parvocellular system43, and for even versus odd harmonics of ‘windmill dartboard’-type stimuli69. So, as with ASSR, ssVEP may index basic neurophysiological deficits in schizophrenia, although the ideal paradigms for implementation in biomarker studies remain to be determined.
SPEMs
The SPEM system appeared phylogenetically late — developing fully only in primates. The SPEM functions to keep the image of a slowly moving object on to the fovea (FIG. 6). SPEM does not occur in the absence of motion information, and normally the slippage of the image of the object of interest (target) away from the fovea stimulates the initiation of SPEM with one of the shortest latencies in the human motor system (~135 ms). The short latency is partly due to the automatic nature of the response. Anticipation of target motion, particularly based on previous target behaviour, has a crucial role in further reducing the pursuit latency to near zero with repeated presentations of target motion in the laboratory70. The initiation response is accurate and fast, and within 300–500 ms the eye matches the target velocity. Paradoxically, this removes the retinal velocity error (target image velocity minus eye velocity) that stimulated the pursuit response in the first place. Under these circumstances subjects elicit a predictive pursuit response based on the internal representations of the target image velocity71. These representations are derived from the efference copy of the motor command, as well as the memory trace of the earlier motion percept. Thus, SPEMs are maintained by the combination of predictive response based on the internal representation of the target motion and adjustments based on the visual feedback of velocity and/or position errors72.
Figure 6. Components of the smooth pursuit eye movements: the underlying neural circuit.

The smooth pursuit eye movement system functions to maintain the image of a moving object on the fovea by minimizing error between the target velocity and the eye velocity. Broadly, the system is divided into two components: the response based on the internal representation of the target motion (that is, the extraretinal motion information), and the corrections based on the visual feedback of discrepancies between the eye and target velocities, the so-called retinal error. The pursuit maintenance in a control subject (red graph) and a patient with schizophrenia (blue graph) is shown (a). The healthy individual is able to accurately match the target velocity, as this response is mostly driven by the extraretinal motion signals during the pursuit maintenance, whereas the patient with schizophrenia shows lower eye velocity, because of inadequate extraretinal motion signals that create a retinal error. In response to the visual feedback of the retinal error, the subject makes corrective responses in the form of a saccade or an increase in eye velocity (marked by Z in panel a). However, this increase in velocity is transient as the retinal error signal becomes weak as the eye velocity matches the target velocity. This interaction between the responses to retinal and extraretinal motion signals is modelled by the regression equation72: maintenance of eye velocity = be × retinal error + ber × extraretinal error (where be and ber are coefficients associated with retinal error and extraretinal error, respectively). Data suggest that compared with control subjects, individuals with schizophrenia depend more on retinal error and less on extraretinal motion signals to maintain pursuit72. When schizophrenia and control subjects were matched on how well they maintained pursuit, schizophrenia subjects showed more activation of the medio-temporal cortex (MT) (b), a region known to process motion, than the healthy control subjects. However, these patients showed less activation of the medio-superior temporal cortex (MST), the posterior-parietal cortex (PPC) and the frontal eye-field regions (FEF), areas that are known to process extraretinal motion signals (c); see REF. 74 for more details. ACC, anterior cingulate cortex; SEF, supplementary eye fields. FIG. 6c is modified with permission from REF. 80 © (2005) Elsevier Science.
SPEM is examined in the laboratory by asking subjects to follow a small moving target, and the eye position is recorded using electro-oculography, magnetic search coil, corneal reflection tracker or high sampling rate video cameras. Although initiation and even later responses are semi-automatic, the subject’s cooperation is a much larger issue than for ERP measures. SPEM abnormalities in schizophrenia were first observed at the beginning of the 1900s73. Although the SPEM abnormality is observed in relatives with no overt psychosis, it tends to co-aggregate with subtle traits and cognitive deficits in these individuals that are defined by schizophrenia spectrum personality disorders. A recent sibling-pair study estimated a heritability of around 91% for the predictive pursuit abnormality in families with a history of schizophrenia74.
There are major generator regions of SPEM: one related to the initiation and the other is associated with predictive pursuit response. The motion of the target image is processed by the medio-temporal (MT) region, whereas the smooth pursuit initiation response is mediated by neural circuitry that involves the MT region, the cerebellum and the brain-stem oculomotor pathways75,76 (FIG. 6). The subsequent predictive smooth pursuit response is based on the internal representation of the previous target image motion mediated by the posterior parietal neurons, and the medio-superior temporal cortex that encodes the efference copy of the motor command77,78. Frontal eye fields integrate the extraretinal motion information into a predictive pursuit response79. The deficit in schizophrenia is thought to be due to deficits in this extraretinal circuitry80, with patients showing decreased activation in multiple brain regions including the medio-superior temporal cortex, the frontal eye fields and the secondary eye fields (FIG. 6). In addition, patients with schizophrenia who have enduring negative symptoms (and their relatives) show deficits in smooth pursuit initiation, which may be mediated by the MT cortex and the cerebellum vermis.
Preclinical translation
In addition to serving as robust indices of brain dysfunction in schizophrenia, neurophysiological biomarkers have a distinct advantage of being translatable into animal models for drug discovery. For many of the measures, either homologous or analogous processes can be studied in rodent or primate models. Furthermore, neurophysiological measures can be obtained in awake, unrestrained animals, as well as restrained, anaesthetized or ex vivo preparations.
For example, auditory measures resembling those in humans have been demonstrated in both rodent and primate models. In rodents, auditory-gating phenomena can be studied using EEG/field potential recordings from the cortex and hippocampus CA3 region, or single-unit recordings from the amygdala, thalamic reticular nucleus and brain-stem reticular formation in response to acoustic stimulation paradigms that are equivalent to those used clinically81. Although absolute latencies of cortical and subcortical auditory evoked potentials in rodents (that is, 13–80 ms) are typically much shorter than those in humans (that is, 50–300 ms), the processes appear to be closely similar in response to parametric manipulation and response to pharmacological agents81.
MMN-like23,82 and N1-like83,84 activity can be recorded in awake rodents, supporting its role in early stage drug development. Furthermore, in rodents, generation of MMN-like and N1-like activity is inhibited by NMDA (N-methyl-D-aspartate) antagonists such as ketamine, which is similar to findings in humans85. Putative rodent models of the P300 have also been developed86,87. Overall, substantial data demonstrate that abnormal information processing of schizophrenia could be reproduced in pre-clinical animal models using pharmacological, genetic or environmental manipulations. Ideally, these disrupting factors should reflect genetic alterations in schizophrenia or replicate the dysfunctional neuronal activity or neurotransmission that is observed in patients with the disease.
Auditory and visual processes similar to those observed in humans may also be reproduced in primate models. Thus, both MMN34,46 and auditory P300 (REFS 88,89) can be recorded in monkeys. Primates also show visual sensory90 and AX-CPT53 responses that are highly similar to those in humans when similar stimulation and recording procedures are used. Although less amenable to genetic manipulation than rodents, primate models offer the advantage of extremely close homology between laboratory and clinical measures.
Underlying pathophysiological mechanisms
In addition to indexing the function of specific brain regions, biomarkers for schizophrenia may be differentially sensitive to the involvement of specific underlying neurochemical systems. Schizophrenia has traditionally been linked to abnormal dopaminergic function, although more recent theories emphasize additional disturbances in glutamatergic, GABA (γ-aminobutyric acid)-ergic, serotonergic and cholinergic mechanisms. The contribution of these measures to biomarker deficits has been evaluated based on challenge studies, animal models and informative genetic linkages.
Dopamine
Dopaminergic models of schizophrenia are supported by the ability of dopaminergic agents, such as amphetamine, to stimulate schizophrenia-like psychosis, as well as the ability of dopamine D2 antagonists to ameliorate such symptoms. However, acute amphetamine administration does not cause either negative symptoms or cognitive deficits like those observed in schizophrenia, leaving the role of dopamine in neurocognitive dysfunction unresolved.
Dopaminergic hyperfunction is most relevant to older biomarkers, such as PPI or P50 gating deficits, which may be reproduced in humans by amphetamine or other dopamine agonists91,92. Amphetamine and cocaine also disrupt auditory gating in the rat hippocampus, reticular thalamus and cortex in a D2-dependent manner81. However, deficits in P50 gating91,93 and PPI5 can be reversed in schizophrenia by atypical (especially clozapine) but not typical antipsychotics, suggesting involvement of additional neurotransmitter systems.
Genetic studies have provided further insight into the connection between dopamine neurotransmission and information processing94,95. Preliminary evidence associates the late temporoparietal P300 component with specific genetic variations in the dopamine D3 and D2 receptor genes96–99, whereas variations in the gene that codes for the dopamine catabolizing enzyme catechol-O-methyltransferase (COMT) have been associated with variations in prefrontal activity (noise)19,100,101 and SPEM102. However, other studies have not found significant associations between dopamine-related genes and P300-amplitude103–105. In line with these findings, it has been argued that reduced synaptic dopamine levels may result in a diminished signal-to-noise ratio of pre-frontal neurons during attention-requiring and working memory tasks60,101,106.
GABA
GABA disturbances have been linked to schizophrenia on the basis of post-mortem findings of a reduction in the number of parvalbumin-expressing interneurons in the hippocampus and frontal brain regions107,108, as well as specific abnormalities in the modulation of pyramidal cell spiking by parvalbumin-expressing, fast-spiking interneurons. GABAergic neurotransmission has been suggested to be a major determinant of the frequency, power and synchrony of gamma oscillations109.
Because GABAergic processes have a prominent role in regulating excitatory neurotransmission in the cortex, it is not surprising that GABAergic links can be found to most biomarkers. GABAergic drugs have been shown to alter P300 parameters, with amplitudes being reduced and P300 latencies delayed after the application of GABAergic drugs110. Similarly, the generation of MMN is thought to depend on intracortical inhibition, reflecting GABAergic inhibition of local GABAergic interneurons in the auditory cortex34. Similarly, local GABAergic interneurons in the MT cortical region modulate motion perception111,112, which contributes to the control of SPEM.
Among neural oscillatory measures, beta frequencies, even in background EEG, may be related to GABAergic feedback. Benzodiazepines, such as triazolam, produce a well-replicated increase in beta amplitude that indexes effects of these compounds on psychomotor performance113. By contrast, GABA inverse agonists decrease beta amplitude114. Associations between beta amplitude and GABAA receptor polymorphisms have also been described in healthy volunteers115 and familial alcoholics116, although the significance of these findings to schizophrenia remains to be determined.
Glutamate
NMDA receptors are one of several receptor types for the excitatory neurotransmitter glutamate. NMDA receptor antagonists such as ketamine or phencyclidine (PCP) induce symptoms and neuropsychological deficits that closely resemble those of schizophrenia, implicating NMDA receptors in the pathogenesis of schizophrenia117,118. Such models have been supported recently by demonstration of reduced markers of glutamatergic neuronal integrity119, as well as reduced concentrations of amino acids such as glycine120,121 or D-serine122 in the plasma and/or cerebrospinal fluid of patients with schizophrenia. Several schizophrenia susceptibility genes, such as neuregulin, ERBB4, dysbindin or D-amino acid oxidase (DAOA; also known as G72) affect glutamate neurotransmission and NMDA receptor function123. Similarly, several environmental alterations, such as reduced glutathione levels124, increased kynurenic acid125 or increased homocysteine levels121 also converge on NMDA receptors, suggesting a central role of NMDA dysfunction in the disorder. Treatment with NMDA antagonists such as ketamine or PCP leads to schizophrenia-like dopaminergic dysregulation in human126 and rodent127 models, suggesting that disturbances in dopaminergic function in schizophrenia might be secondary to underlying glutamatergic dysfunction, consistent with reciprocal interaction between systems94.
NMDA antagonists, such as ketamine, disrupt PPI in rodents128 and primates129. However, in humans, ketamine does not induce P50 gating deficits130 and may enhance, rather than inhibit, PPI131. Furthermore, in humans, PPI deficits are associated with increased, rather than decreased, plasma levels of the amino-acid glycine, which serves as an NMDA co-agonist in vivo132. Thus, relationships between phenomenology and NMDA dysfunction appear to differ across species. In animals, NMDA antagonist-induced PPI deficits are reversed by clozapine, but not by most typical or atypical antipsychotics128,129.
Of the considered neurophysiological measures, MMN has been most extensively investigated with regard to glutamatergic mechanisms, particularly involving NMDA receptors. Deficits in MMN generation may be induced in both monkeys133 and humans134,135 following administration of NMDA antagonists. Furthermore, in healthy volunteers, MMN amplitude predicts the degree of psychosis induced by ketamine across subjects136. In contrast to effects of ketamine, MMN was not affected by psilocybin, a psychotomimetic agent that functions via stimulation of 136. Deficits in 5-hydroxytryptamine 2A (5-HT2A) receptors MMN generation persist despite treatment with available antipsychotic agents, including 5-HT2A-selective compounds such as clozapine, olanzapine and risperidone, suggesting that dopaminergic and serotonergic systems are not closely related to MMN generation.
Abnormal glutamate transmission has been suggested in other impaired neurophysiological measures in schizophrenia. For example, schizophrenia-like deficits in AX-CPT processing are induced by ketamine, supporting a role of NMDA dysfunction134. However, such deficits are reproduced by psilocybin as well, suggesting that they are less specific for NMDA dysfunction than MMN137. Alteration in visual P1-evoked potentials, which indicate abnormal early stage visual processing in schizophrenia, has also been linked to glutamate hypofunction and/or reduced NMDA receptor activity43. Furthermore, ketamine administration mimics several features of SPEM deficits in healthy subjects, such as the initiation response138,139.
NMDA receptors may also have a role in modulation of oscillatory activity. Infusion of NMDA agonists into the hippocampus or septum directly induces theta activity, whereas NMDA antagonists suppress spontaneous theta activity and activity that is induced by behavioural manipulation, such as tail pinch, while having non-significant effects on gamma activity140,141. Similarly, NMDA antagonists reduce beta, but not gamma, activity in the hippocampal slice142. In humans, ketamine attenuates the increases in cortical delta and beta activity that are observed during opiate withdrawal143. Thus, NMDA dysfunction in schizophrenia would be anticipated to cause deficits in neural synchrony in schizophrenia, particularly in slower frequency ranges.
Acetylcholine (ACh)
Finally, disturbances in cholinergic function have been associated with schizophrenia based on linkages to the α7 nicotinic receptor, as well as increased smoking rates among patients with schizophrenia144. Nicotinic mechanisms may be particularly relevant to the phenomenon of P50 gating, in that nicotine improves, although transiently, P50 auditory-gating deficit in patients and their relatives145. In preclinical animal models, pharmacologically, genetically and environmentally induced gating deficits are normalized by nicotinic receptor agonists and modulators146–150, thus confirming a role of nicotinic mechanisms in auditory gating. Since it was shown that a partial nicotinic α7 agonists improves the gating deficit in patients with schizophrenia151, it appears that this neurophysiological measure can be used as translational biomarker, at least for nicotinic targets.
Involvement of cholinergic mechanisms in auditory-evoked P300 has also been observed. ACh and cholinergic drugs appear to strongly increase P300 amplitude. The opposite observation (a decrease of P300 amplitude) was made for anticholinergic substances. Both effects are likely to be mediated by muscarinic and perhaps also nicotinergic receptors152–161. In line with these observations in humans, septal cholinergic lesions abolish auditory P300 in cats162. As abnormal cholinergic transmission appears to be involved in aspects of schizophrenia pathophysiology and, in particular, cognitive deficits including memory and attentional deficits163, P300 may be considered a useful biomarker to track pharmacological interventions with cholinergic drugs. The immediate response to a novel presentation of target motion, which is dependent on early motion processing, is also improved by the administration of nicotine in patients with schizophrenia164, suggesting that this marker could be used as well.
Additionally, cholinergic mechanisms have been implicated in neuronal network oscillations. Visually evoked gamma-band oscillation patterns and neuronal synchronization are greatly reduced after intracortical administration of the muscarinic ACh receptor antagonist scopolamine165. Furthermore, α7 nicotinic ACh receptors appear to modulate theta and gamma-band oscillations in both in vitro and in vivo experiments in animals, in part through modulation of GABA activity146,166. Thus, even if deficits in cholinergic activity are not causative in schizophrenia, nicotinic receptors, particularly α7, may serve as a target for intervention.
In summary, several neurotransmitter systems have been linked to schizophrenia, either through the analysis of the action of drugs of abuse such as PCP or amphetamine, of through the analysis of genetic polymorphisms. Several of the proposed biomarkers show particular sensitivity to deficits in specific neurochemical pathways, and thus might contribute to targeted drug development. The strongest associations are found between P50 gating and nicotinic mechanisms, dopamine and prefrontal noise measurements, and NMDA receptors and sensory disturbances in both the auditory and visual modalities. In general, later components, such as P300, have a more complex pharmacology than earlier potentials and are more likely to reflect an interplay between multiple neurotransmitter systems.
Future directions
At present, several described biomarkers, such as auditory MMN or P300, visual P1 or SPEM disturbances, appear sufficiently established so that they may serve as appropriate translational measures for drug discovery projects. In addition, the use of neurophysiological measures has been greatly extended over recent years by the evaluation of neural synchrony measures along with simple response amplitude. Across regions, activity appears to be noisy, reflecting the inability to synchronize brain oscillations appropriately across a wide frequency range including, but apparently not limited to, the gamma frequency band.
Although only a few biomarkers have progressed to the point in which they can be reliably applied in drug development studies, neurophysiological measures are undergoing continuous development, suggesting that the type and variety of measures will increase in the coming years. Moreover, targets for drug treatment of schizophrenia are broadening continually (for reviews see REFS 167,168). These targets include specific subtypes of monoamine receptors, such as the 5-HT2c (REFS 169,170), 5-HT3 (REF. 171), 5-HT6 (REF. 172) and histamine (H3)173 receptors, various ligands and modulators of glutamate receptors, such as the metabotropic (mGluR2/3) glutamate receptors174, and other diverse targets of intracellular processes and/or second messenger systems, such as the phosphodiesterases175. Some of these novel targets have a strong association with genes considered to be candidate susceptibility factors for schizophrenia, such as phosphodiesterase 4B (PDE4B) interacting with the disrupted in schizophrenia 1 (DISC1) gene176. Other targets have emerged from epidemiological studies, such as the cannabinoid receptors (for example, CB1) or endocannabinoids177.
Moreover, it is known that many experimental compounds acting on these targets profoundly affect neuronal network oscillations178–185 or other electro-physiological measures of information processing (such as auditory gating). Examples include ligands of 5-HT3, CB1 and H3 receptors, as well as inhibitors of PDE4 and PDE10 enzymes173,179,183–185. Once these drug candidates enter clinical development, neurophysiological biomarkers discussed here will provide a better understanding of the underlying neurophysiological abnormalities in schizophrenia, and their relation to the various positive, negative and cognitive symptoms of the disease. These novel compounds designed to treat a range of symptoms of schizophrenia are currently being evaluated in various preclinical models of neurophysiological biomarkers. These findings, combined with additional clinical neurophysiological biomarker studies, will provide an opportunity to validate not only novel targets, but also validate the therapeutic predictive value of these neurophysiological animal models.
Importantly, different neurophysiological measures probe different aspects of brain function. Thus, biomarkers are probably best used in concert with each other, rather than in isolation. For example, although measures such as P50 gating, MMN and P300 all show high heritability and reproducibility on their own, they show relatively low cross-correlation17, suggesting relatively little genetic and mechanistic overlap. Individual measures may also differ in terms of sensitivity and specificity for schizophrenia, as well as underlying neurochemical mechanisms. The use of combined, rather than individual measures, has recently been demonstrated for diagnostic purposes186. A similar situation is likely to hold for drug development. Finally, multimodal imaging, in which neurophysiological biomarkers are combined with structure-based neuroimaging approaches (for example, fMRI), may provide more information than either modality alone.
Although brain activity is impaired in schizophrenia across a range of anatomical regions and physiological processes, not all medications will affect all underlying neurochemical deficits equally. Thus, the choice of biomarkers in drug development depends not only on the pathophysiology of the underlying disorder, but also on the expected mechanism of action of the drug to be investigated. The availability of analogous measures in animal research represents an outstanding strength of neurophysiological biomarkers, therefore permitting their use as translational measures during early drug development, and as proof-of-concept or proof-of-mechanism measures in both healthy subjects and individuals with schizophrenia.
Over the coming years, additional refinements in both behavioural paradigms and analytical approaches will undoubtedly lead to further development of biomarker approaches for schizophrenia.
Acknowledgments
This work was supported in part by K02 MH01439 and R01 MH49,334 (D.C.J.), R03 MH76,760 and a NARSAD Young Investigator Award (K.M.S.), and RO1 MH-49826 and RO1 MH-67,014 (G.K.T.). The authors wish to thank E. Saccente for her technical assistance.
- Local field potential
The summed postsynaptic potentials recorded from neurons neighbouring a microelectrode
- Prepulse inhibition
(PPI). Prepulse inhibition is a measure of sensory gating in which a weak prestimulus (prepulse) reduces the startle response elicited by a subsequent intense auditory stimulus
- P50
An early auditory potential reflecting initial sensory activation. P50 gating refers to the decreased P50 amplitude to the second stimulus in a paired click compared to the first
- Prodromal symptoms
Symptoms that arise before the onset of fully diagnosed schizophrenia
- Perceptual feature binding
The process(es) by which elemental sensory information is combined into the representation of a perception, for example, a visual object
- Magnocellular system
In the primate visual system, the magnocellular system is specialized for rapid conduction of low resolution visual representations and motion information
- Parvocellular system
In the primate visual system, the parvocellular system is for slow conduction of high resolution visual representations and colour
Footnotes
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
COMT | DISC1 | DOAO | ERBB4 | PDE4B
OMIM: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIMSchizophrenia
FURTHER INFORMATION
National Institute of Mental Health — Schizophrenia: http://www.nimh.nih.gov/health/publications/schizophrenia/complete-publication.shtml
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Tallon-Baudry C, Bertrand O. Oscillatory gamma activity in humans and its role in object representation. Trends Cogn Sci. 1999;3:151–162. doi: 10.1016/s1364-6613(99)01299-1. Reviews evidence for gamma-band synchrony in humans and its possible roles, as well as methods for non-invasive measurement of neural synchrony. [DOI] [PubMed] [Google Scholar]
- 2.Sehatpour P, Molholm S, Javitt DC, Foxe JJ. Spatiotemporal dynamics of human object recognition processing: an integrated high-density electrical mapping and functional imaging study of “closure” processes. Neuroimage. 2006;29:605–618. doi: 10.1016/j.neuroimage.2005.07.049. [DOI] [PubMed] [Google Scholar]
- 3.Winterer G, et al. Complex relationship between BOLD signal and synchronization/desynchronization of human brain MEG oscillations. Hum Brain Mapp. 2007;28:805–816. doi: 10.1002/hbm.20322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Potter D, Summerfelt A, Gold J, Buchanan RW. Review of clinical correlates of P50 sensory gating abnormalities in patients with schizophrenia. Schizophr Bull. 2006;32:692–700. doi: 10.1093/schbul/sbj050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swerdlow NR, et al. Startle gating deficits in a large cohort of patients with schizophrenia: relationship to medications, symptoms, neurocognition, and level of function. Arch Gen Psychiatry. 2006;63:1325–1335. doi: 10.1001/archpsyc.63.12.1325. [DOI] [PubMed] [Google Scholar]
- 6.Roth WT, Cannon EH. Some features of the auditory evoked response in schizophrenics. Arch Gen Psychiatry. 1972;27:466–471. doi: 10.1001/archpsyc.1972.01750280034007. [DOI] [PubMed] [Google Scholar]
- 7.Ford JM. Schizophrenia: the broken P300 and beyond. Psychophysiology. 1999;36:667–682. [PubMed] [Google Scholar]
- 8.Jeon YW, Polich J. Meta-analysis of P300 and schizophrenia: patients, paradigms, and practical implications. Psychophysiology. 2003;40:684–701. doi: 10.1111/1469-8986.00070. [DOI] [PubMed] [Google Scholar]
- 9.Frodl-Bauch T, Gallinat J, Meisenzahl EM, Moller HJ, Hegerl U. P300 subcomponents reflect different aspects of psychopathology in schizophrenia. Biol Psychiatry. 1999;45:116–126. doi: 10.1016/s0006-3223(98)00108-5. [DOI] [PubMed] [Google Scholar]
- 10.Mathalon DH, Ford JM, Pfefferbaum A. Trait and state aspects of P300 amplitude reduction in schizophrenia: a retrospective longitudinal study. Biol Psychiatry. 2000;47:434–449. doi: 10.1016/s0006-3223(99)00277-2. [DOI] [PubMed] [Google Scholar]
- 11.Polich J, Bloom FE. P300 and alcohol consumption in normals and individuals at risk for alcoholism. A preliminary report. Prog Neuropsychopharmacol Biol Psychiatry. 1986;10:201–210. doi: 10.1016/0278-5846(86)90074-6. [DOI] [PubMed] [Google Scholar]
- 12.Hall MH, et al. Genetic overlap between bipolar illness and event-related potentials. Psychol Med. 2007;37:667–678. doi: 10.1017/S003329170600972X. [DOI] [PubMed] [Google Scholar]
- 13.Turetsky BI, et al. Neurophysiological endophenotypes of schizophrenia: the viability of selected candidate measures. Schizophr Bull. 2007;33:69–94. doi: 10.1093/schbul/sbl060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Umbricht DS, Bates JA, Lieberman JA, Kane JM, Javitt DC. Electrophysiological indices of automatic and controlled auditory information processing in first-episode, recent-onset and chronic schizophrenia. Biol Psychiatry. 2006;59:762–772. doi: 10.1016/j.biopsych.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 15.Umbricht D, et al. Effects of clozapine on auditory event-related potentials in schizophrenia. Biol Psychiatry. 1998;44:716–725. doi: 10.1016/s0006-3223(97)00524-6. [DOI] [PubMed] [Google Scholar]
- 16.Umbricht D, et al. Effects of risperidone on auditory event-related potentials in schizophrenia. Int J Neuropsychopharmcol. 1999;2:299–304. doi: 10.1017/S1461145799001595. [DOI] [PubMed] [Google Scholar]
- 17.Hall MH, et al. Genetic overlap between P300, P50, and duration mismatch negativity. Am J Med Genet B Neuropsychiatr Genet. 2006;141:336–343. doi: 10.1002/ajmg.b.30318. [DOI] [PubMed] [Google Scholar]
- 18.Winterer G, et al. P300 and genetic risk for schizophrenia. Arch Gen Psychiatry. 2003;60:1158–1167. doi: 10.1001/archpsyc.60.11.1158. [DOI] [PubMed] [Google Scholar]
- 19.Gallinat J, et al. Association of the G1947A COMT (Val(108/158)Met) gene polymorphism with prefrontal P300 during information processing. Biol Psychiatry. 2003;54:40–48. doi: 10.1016/s0006-3223(02)01973-x. [DOI] [PubMed] [Google Scholar]
- 20.Blackwood DH, Muir WJ. Clinical phenotypes associated with DISC1, a candidate gene for schizophrenia. Neurotox Res. 2004;6:35–41. doi: 10.1007/BF03033294. [DOI] [PubMed] [Google Scholar]
- 21.Friedman D, Cornblatt B, Vaughan H, Jr, Erlenmeyer-Kimling L. Auditory event-related potentials in children at risk for schizophrenia: the complete initial sample. Psychiatry Res. 1988;26:203–221. doi: 10.1016/0165-1781(88)90075-3. [DOI] [PubMed] [Google Scholar]
- 22.Naatanen R. Mismatch negativity: clinical research and possible applications. Int J Psychophysiol. 2003;48:179–188. doi: 10.1016/s0167-8760(03)00053-9. [DOI] [PubMed] [Google Scholar]
- 23.Umbricht D, Krljes S. Mismatch negativity in schizophrenia: a meta-analysis. Schizophr Res. 2005;76:1–23. doi: 10.1016/j.schres.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 24.Umbricht D, et al. How specific are deficits in mismatch negativity generation to schizophrenia? Biol Psychiatry. 2003;53:1120–1131. doi: 10.1016/s0006-3223(02)01642-6. [DOI] [PubMed] [Google Scholar]
- 25.Javitt DC, Doneshka P, Grochowski S, Ritter W. Impaired mismatch negativity generation reflects widespread dysfunction of working memory in schizophrenia. Arch Gen Psychiatry. 1995;52:550–558. doi: 10.1001/archpsyc.1995.03950190032005. [DOI] [PubMed] [Google Scholar]
- 26.Light GA, Braff DL. Mismatch negativity deficits are associated with poor functioning in schizophrenia patients. Arch Gen Psychiatry. 2005;62:127–136. doi: 10.1001/archpsyc.62.2.127. [DOI] [PubMed] [Google Scholar]
- 27.Salisbury DF, Shenton ME, Griggs CB, Bonner-Jackson A, McCarley RW. Mismatch negativity in chronic schizophrenia and first-episode schizophrenia. Arch Gen Psychiatry. 2002;59:686–694. doi: 10.1001/archpsyc.59.8.686. [DOI] [PubMed] [Google Scholar]
- 28.Javitt DC, Shelley AM, Silipo G, Lieberman JA. Deficits in auditory and visual context-dependent processing in schizophrenia: defining the pattern. Arch Gen Psychiatry. 2000;57:1131–1137. doi: 10.1001/archpsyc.57.12.1131. Documents disturbances in both auditory and visual processing in schizophrenia as biomarkers for cognitive dysfunction. [DOI] [PubMed] [Google Scholar]
- 29.Salisbury DF, Kuroki N, Kasai K, Shenton ME, McCarley RW. Progressive and interrelated functional and structural evidence of post-onset brain reduction in schizophrenia. Arch Gen Psychiatry. 2007;64:521–529. doi: 10.1001/archpsyc.64.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall MH, et al. Heritability and reliability of P300, P50 and duration mismatch negativity. Behav Genet. 2006;36:845–857. doi: 10.1007/s10519-006-9091-6. [DOI] [PubMed] [Google Scholar]
- 31.Michie PT, Innes-Brown H, Todd J, Jablensky AV. Duration mismatch negativity in biological relatives of patients with schizophrenia spectrum disorders. Biol Psychiatry. 2002;52:749–758. doi: 10.1016/s0006-3223(02)01379-3. [DOI] [PubMed] [Google Scholar]
- 32.Bramon E, et al. Mismatch negativity in schizophrenia: a family study. Schizophr Res. 2004;67:1–10. doi: 10.1016/s0920-9964(03)00132-4. [DOI] [PubMed] [Google Scholar]
- 33.Brockhaus-Dumke A, et al. Impaired mismatch negativity generation in prodromal subjects and patients with schizophrenia. Schizophr Res. 2005;73:297–310. doi: 10.1016/j.schres.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 34.Javitt DC. Intracortical mechanisms of mismatch negativity dysfunction in schizophrenia. Audiol Neurootol. 2000;5:207–215. doi: 10.1159/000013882. [DOI] [PubMed] [Google Scholar]
- 35.Wible CG, et al. A functional magnetic resonance imaging study of auditory mismatch in schizophrenia. Am J Psychiatry. 2001;158:938–943. doi: 10.1176/appi.ajp.158.6.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shelley AM, Silipo G, Javitt DC. Diminished responsiveness of ERPs in schizophrenic subjects to changes in auditory stimulation parameters: implications for theories of cortical dysfunction. Schizophr Res. 1999;37:65–79. doi: 10.1016/s0920-9964(98)00138-8. [DOI] [PubMed] [Google Scholar]
- 37.Gallinat J, et al. Frontal and temporal dysfunction of auditory stimulus processing in schizophrenia. Neuroimage. 2002;17:110–127. doi: 10.1006/nimg.2002.1213. [DOI] [PubMed] [Google Scholar]
- 38.Mulert C, et al. Reduced event-related current density in the anterior cingulate cortex in schizophrenia. Neuroimage. 2001;13:589–600. doi: 10.1006/nimg.2000.0727. [DOI] [PubMed] [Google Scholar]
- 39.Foxe JJ, Doniger GM, Javitt DC. Early visual processing deficits in schizophrenia: impaired P1 generation revealed by high-density electrical mapping. Neuroreport. 2001;12:3815–3820. doi: 10.1097/00001756-200112040-00043. [DOI] [PubMed] [Google Scholar]
- 40.Schechter I, et al. Impairments in generation of early-stage transient visual evoked potentials to magno- and parvocellular-selective stimuli in schizophrenia. Clin Neurophysiol. 2005;116:2204–2215. doi: 10.1016/j.clinph.2005.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeap S, et al. Early visual sensory deficits as endophenotypes for schizophrenia: high-density electrical mapping in clinically unaffected first-degree relatives. Arch Gen Psychiatry. 2006;63:1180–1188. doi: 10.1001/archpsyc.63.11.1180. [DOI] [PubMed] [Google Scholar]
- 42.Butler PD, et al. Dysfunction of early stage visual processing in schizophrenia. Am J Psychiatry. 2001;158:1126–1133. doi: 10.1176/appi.ajp.158.7.1126. [DOI] [PubMed] [Google Scholar]
- 43.Butler PD, et al. Early-stage visual processing and cortical amplification deficits in schizophrenia. Arch Gen Psychiatry. 2005;62:495–504. doi: 10.1001/archpsyc.62.5.495. Demonstrates the three-way relationship between magnocellular deficits in schizophrenia, impaired NMDA activity and structural changes in optic radiations as demonstrated by diffusion tensor imaging. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duncan CC. Event-related brain potentials: a window on information processing in schizophrenia. Schizophr Bull. 1988;14:199–203. doi: 10.1093/schbul/14.2.199. [DOI] [PubMed] [Google Scholar]
- 45.Bruder G, et al. The time course of visuospatial processing deficits in schizophrenia: an event-related brain potential study. J Abnorm Psychol. 1998;107:399–411. doi: 10.1037//0021-843x.107.3.399. [DOI] [PubMed] [Google Scholar]
- 46.Javitt DC, Jayachandra M, Lindsley RW, Specht CM, Schroeder CE. Schizophrenia-like deficits in auditory P1 and N1 refractoriness induced by the psychomimetic agent phencyclidine (PCP) Clin Neurophysiol. 2000;111:833–836. doi: 10.1016/s1388-2457(99)00313-2. [DOI] [PubMed] [Google Scholar]
- 47.Kayser J, et al. ERP/CSD indices of impaired verbal working memory subprocesses in schizophrenia. Psychophysiology. 2006;43:237–252. doi: 10.1111/j.1469-8986.2006.00398.x. [DOI] [PubMed] [Google Scholar]
- 48.Servan-Schreiber D, Cohen JD, Steingard S. Schizophrenic deficits in the processing of context. A test of a theoretical model. Arch Gen Psychiatry. 1996;53:1105–1112. doi: 10.1001/archpsyc.1996.01830120037008. [DOI] [PubMed] [Google Scholar]
- 49.Barch DM, Carter CS, MacDonald AW, 3rd, Braver TS, Cohen JD. Context-processing deficits in schizophrenia: diagnostic specificity, 4-week course, and relationships to clinical symptoms . J Abnorm Psychol. 2003;112:132–143. [PubMed] [Google Scholar]
- 50.MacDonald AW, 3rd, et al. Specificity of prefrontal dysfunction and context processing deficits to schizophrenia in never-medicated patients with first-episode psychosis. Am J Psychiatry. 2005;162:475–484. doi: 10.1176/appi.ajp.162.3.475. [DOI] [PubMed] [Google Scholar]
- 51.Barch DM, et al. Selective deficits in prefrontal cortex function in medication-naive patients with schizophrenia. Arch Gen Psychiatry. 2001;58:280–288. doi: 10.1001/archpsyc.58.3.280. [DOI] [PubMed] [Google Scholar]
- 52.Dias EC, Foxe JJ, Javitt DC. Changing plans: a high density electrical mapping study of cortical control. Cereb Cortex. 2003;13:701–715. doi: 10.1093/cercor/13.7.701. [DOI] [PubMed] [Google Scholar]
- 53.Dias EC, et al. Changing plans: neural correlates of executive control in monkey and human frontal cortex. Exp Brain Res. 2006;174:279–291. doi: 10.1007/s00221-006-0444-4. [DOI] [PubMed] [Google Scholar]
- 54.Javitt DC, Rabinowicz E, Silipo G, Dias EC. Encoding vs. retention: differential effects of cue manipulation on working memory performance in schizophrenia. Schizophr Res. 2007;91:159–168. doi: 10.1016/j.schres.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lakatos P, et al. An oscillatory hierarchy controlling neuronal excitability and stimulus processing in the auditory cortex. J Neurophysiol. 2005;94:1904–1911. doi: 10.1152/jn.00263.2005. [DOI] [PubMed] [Google Scholar]
- 56.Haig AR, et al. Gamma activity in schizophrenia: evidence of impaired network binding? Clin Neurophysiol. 2000;111:1461–1468. doi: 10.1016/s1388-2457(00)00347-3. [DOI] [PubMed] [Google Scholar]
- 57.Spencer KM, et al. Abnormal neural synchrony in schizophrenia. J Neurosci. 2003;23:7407–7411. doi: 10.1523/JNEUROSCI.23-19-07407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spencer KM, et al. Neural synchrony indexes disordered perception and cognition in schizophrenia. Proc Natl Acad Sci USA. 2004;101:17288–17293. doi: 10.1073/pnas.0406074101. References 57 and 58 present evidence for disturbances in stimulus-evoked and perception-related gamma oscillations in schizophrenia, and suggest relationships to particular symptoms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uhlhaas PJ, et al. Dysfunctional long-range coordination of neural activity during Gestalt perception in schizophrenia. J Neurosci. 2006;26:8168–8175. doi: 10.1523/JNEUROSCI.2002-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Winterer G, et al. Prefrontal broadband noise, working memory, and genetic risk for schizophrenia. Am J Psychiatry. 2004;161:490–500. doi: 10.1176/appi.ajp.161.3.490. Is the first report on abnormal electrophysiological ‘noise’ as being a genetically determined risk factor for schizophrenia and cognitive deficits. [DOI] [PubMed] [Google Scholar]
- 61.Kwon JS, et al. Gamma frequency-range abnormalities to auditory stimulation in schizophrenia. Arch Gen Psychiatry. 1999;56:1001–1005. doi: 10.1001/archpsyc.56.11.1001. The first direct evidence for impaired gamma-band oscillations in schizophrenia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spencer KM, Salisbury DF, Shenton ME, McCarley RW. Gamma-band steady-state responses are impaired in first episode psychosis. Soc Neurosci Abstr. 2006;36:122.2. doi: 10.1016/j.biopsych.2008.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hong LE, et al. Evoked gamma band synchronization and the liability for schizophrenia. Schizophr Res. 2004;70:293–302. doi: 10.1016/j.schres.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 64.O’Donnell BF, et al. Neural synchronization deficits to auditory stimulation in bipolar disorder. Neuroreport. 2004;15:1369–1372. doi: 10.1097/01.wnr.0000127348.64681.b2. [DOI] [PubMed] [Google Scholar]
- 65.Winterer G, et al. Schizophrenia: reduced signal-to-noise ratio and impaired phase-locking during information processing. Clin Neurophysiol. 2000;111:837–849. doi: 10.1016/s1388-2457(99)00322-3. Is the first report on abnormal electrophysiological phase-synchrony (noise) being increased in schizophrenia and that it predicts cortical activation abnormalities in schizophrenia with high diagnostic specificity. [DOI] [PubMed] [Google Scholar]
- 66.Krishnan GP, et al. Steady state visual evoked potential abnormalities in schizophrenia. Clin Neurophysiol. 2005;116:614–624. doi: 10.1016/j.clinph.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 67.Jin Y, Castellanos A, Solis ER, Potkin SG. EEG resonant responses in schizophrenia: a photic driving study with improved harmonic resolution. Schizophr Res. 2000;44:213–220. doi: 10.1016/s0920-9964(99)00211-x. [DOI] [PubMed] [Google Scholar]
- 68.Clementz BA, Keil A, Kissler J. Aberrant brain dynamics in schizophrenia: delayed buildup and prolonged decay of the visual steady-state response. Brain Res Cogn Brain Res. 2004;18:121–129. doi: 10.1016/j.cogbrainres.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 69.Kim D, Zemon V, Saperstein A, Butler PD, Javitt DC. Dysfunction of early-stage visual processing in schizophrenia: harmonic analysis. Schizophr Res. 2005;76:55–65. doi: 10.1016/j.schres.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 70.Avila MT, Hong LE, Moates A, Turano KA, Thaker GK. Role of anticipation in schizophrenia-related pursuit initiation deficits. J Neurophysiol. 2006;95:593–601. doi: 10.1152/jn.00369.2005. [DOI] [PubMed] [Google Scholar]
- 71.Barnes GR, Barnes DM, Chakraborti SR. Ocular pursuit responses to repeated, single-cycle sinusoids reveal behavior compatible with predictive pursuit. J Neurophysiol. 2000;84:2340–2355. doi: 10.1152/jn.2000.84.5.2340. [DOI] [PubMed] [Google Scholar]
- 72.Thaker GK, et al. A model of smooth pursuit eye movement deficit associated with the schizophrenia phenotype. Psychophysiology. 2003;40:277–284. doi: 10.1111/1469-8986.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Diefendor AR, Dodge R. An experimental study of the ocular reactions of the insane from photographic records. Brain. 1908;31:451–489. [Google Scholar]
- 74.Hong LE, et al. Familial aggregation of eye-tracking endophenotypes in families of schizophrenic patients. Arch Gen Psychiatry. 2006;63:259–264. doi: 10.1001/archpsyc.63.3.259. Provides heritability estimates of the SPEM abnormality in families of schizophrenia probands. [DOI] [PubMed] [Google Scholar]
- 75.Tusa RJ, Ungerleider LG. Fiber pathways of cortical areas mediating smooth pursuit eye movements in monkeys. Ann Neurol. 1988;23:174–183. doi: 10.1002/ana.410230211. [DOI] [PubMed] [Google Scholar]
- 76.Takagi M, Zee DS, Tamargo RJ. Effects of lesions of the oculomotor cerebellar vermis on eye movements in primate: smooth pursuit. J Neurophysiol. 2000;83:2047–2062. doi: 10.1152/jn.2000.83.4.2047. [DOI] [PubMed] [Google Scholar]
- 77.Newsome WT, Wurtz RH, Komatsu H. Relation of cortical areas MT and MST to pursuit eye movements. II. Differentiation of retinal from extraretinal inputs. J Neurophysiol. 1988;60:604–620. doi: 10.1152/jn.1988.60.2.604. This with its accompanying paper dissects the SPEM function to highlight the critical role of the predictive mechanism in maintaining the image of a moving target on the fovea. [DOI] [PubMed] [Google Scholar]
- 78.Assad JA, Maunsell JH. Neuronal correlates of inferred motion in primate posterior parietal cortex. Nature. 1995;373:518–521. doi: 10.1038/373518a0. [DOI] [PubMed] [Google Scholar]
- 79.Tanaka M, Fukushima K. Neuronal responses related to smooth pursuit eye movements in the periarcuate cortical area of monkeys. J Neurophysiol. 1998;80:28–47. doi: 10.1152/jn.1998.80.1.28. [DOI] [PubMed] [Google Scholar]
- 80.Hong LE, et al. Specific motion processing pathway deficit during eye tracking in schizophrenia: a performance-matched functional magnetic resonance imaging study. Biol Psychiatry. 2005;57:726–732. doi: 10.1016/j.biopsych.2004.12.015. Describes neuronal circuitry that mediates the predictive smooth pursuit and provides evidence of reduced activity in this circuit in schizophrenia. [DOI] [PubMed] [Google Scholar]
- 81.Hajos M. Targeting information-processing deficit in schizophrenia: a novel approach to psychotherapeutic drug discovery. Trends Pharmacol Sci. 2006;27:391–398. doi: 10.1016/j.tips.2006.05.005. Reviews the neurophysiological abnormalities in patients with schizophrenia and their potential use for preclinical research, with a particular emphasis on auditory gating. [DOI] [PubMed] [Google Scholar]
- 82.Eriksson J, Villa AE. Event-related potentials in an auditory oddball situation in the rat. Biosystems. 2005;79:207–212. doi: 10.1016/j.biosystems.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 83.Maxwell CR, et al. Effects of chronic olanzapine and haloperidol differ on the mouse N1 auditory evoked potential. Neuropsychopharmacology. 2004;29:739–746. doi: 10.1038/sj.npp.1300376. [DOI] [PubMed] [Google Scholar]
- 84.Umbricht D, et al. Midlatency auditory event-related potentials in mice: comparison to midlatency auditory ERPs in humans. Brain Res. 2004;1019:189–200. doi: 10.1016/j.brainres.2004.05.097. [DOI] [PubMed] [Google Scholar]
- 85.Maxwell CR, et al. Ketamine produces lasting disruptions in encoding of sensory stimuli. J Pharmacol Exp Ther. 2006;316:315–324. doi: 10.1124/jpet.105.091199. [DOI] [PubMed] [Google Scholar]
- 86.Slawecki CJ, Thomas JD, Riley EP, Ehlers CL. Neonatal nicotine exposure alters hippocampal EEG and event-related potentials (ERPs) in rats. Pharmacol Biochem Behav. 2000;65:711–718. doi: 10.1016/s0091-3057(99)00258-0. [DOI] [PubMed] [Google Scholar]
- 87.Ehlers CL, Somes C. Long latency event-related potentials in mice: effects of stimulus characteristics and strain. Brain Res. 2002;957:117–128. doi: 10.1016/s0006-8993(02)03612-0. [DOI] [PubMed] [Google Scholar]
- 88.Pineda JA, Foote SL, Neville HJ, Holmes TC. Endogenous event-related potentials in monkey: the role of task relevance, stimulus probability, and behavioral response. Electroencephalogr Clin Neurophysiol. 1988;70:155–171. doi: 10.1016/0013-4694(88)90115-0. [DOI] [PubMed] [Google Scholar]
- 89.Pineda JA, Foote SL, Neville HJ. Effects of locus coeruleus lesions on auditory, long-latency, event-related potentials in monkey. J Neurosci. 1989;9:81–93. doi: 10.1523/JNEUROSCI.09-01-00081.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen CM, et al. Functional anatomy and interaction of fast and slow visual pathways in macaque monkeys. Cereb Cortex. 2007;17:1561–1569. doi: 10.1093/cercor/bhl067. [DOI] [PubMed] [Google Scholar]
- 91.Light GA, Geyer MA, Clementz BA, Cadenhead KS, Braff DL. Normal P50 suppression in schizophrenia patients treated with atypical antipsychotic medications. Am J Psychiatry. 2000;157:767–771. doi: 10.1176/appi.ajp.157.5.767. [DOI] [PubMed] [Google Scholar]
- 92.Boutros NN, Gooding D, Sundaresan K, Burroughs S, Johanson CE. Cocaine-dependence and cocaine-induced paranoia and mid-latency auditory evoked responses and sensory gating. Psychiatry Res. 2006;145:147–154. doi: 10.1016/j.psychres.2006.02.005. Demonstrates reduced auditory-evoked potentials and impaired auditory gating (P50, N100, P200) in chronic cocaine abusers. [DOI] [PubMed] [Google Scholar]
- 93.Adler LE, et al. Varied effects of atypical neuroleptics on P50 auditory gating in schizophrenia patients. Am J Psychiatry. 2004;161:1822–1828. doi: 10.1176/ajp.161.10.1822. [DOI] [PubMed] [Google Scholar]
- 94.Winterer G, Weinberger DR. Genes, dopamine and cortical signal-to-noise ratio in schizophrenia. Trends Neurosci. 2004;27:683–690. doi: 10.1016/j.tins.2004.08.002. Is a comprehensive overview on the noise concept in schizophrenia and how different molecular mechanisms (dopamine, GABA, glutamate) contribute to cortical microcircuit stability by changing neural synchrony (noise). [DOI] [PubMed] [Google Scholar]
- 95.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 96.Hill SY, et al. Genetic association between reduced P300 amplitude and the DRD2 dopamine receptor A1 allele in children at high risk for alcoholism. Biol Psychiatry. 1998;43:40–51. doi: 10.1016/s0006-3223(97)00203-5. [DOI] [PubMed] [Google Scholar]
- 97.Anokhin AP, Todorov AA, Madden PA, Grant JD, Heath AC. Brain event-related potentials, dopamine D2 receptor gene polymorphism, and smoking. Genet Epidemiol. 1999;17 (Suppl 1):37–42. doi: 10.1002/gepi.1370170707. [DOI] [PubMed] [Google Scholar]
- 98.Mulert C, et al. A Ser9Gly polymorphism in the dopamine D3 receptor gene (DRD3) and event-related P300 potentials. Neuropsychopharmacology. 2006;31:1335–1344. doi: 10.1038/sj.npp.1300984. [DOI] [PubMed] [Google Scholar]
- 99.Berman SM, et al. P300 development during adolescence: effects of DRD2 genotype. Clin Neurophysiol. 2006;117:649–659. doi: 10.1016/j.clinph.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 100.Ehlis AC, Reif A, Herrmann MJ, Lesch KP, Fallgatter AJ. Impact of catechol-O-methyltransferase on prefrontal brain functioning in schizophrenia spectrum disorders. Neuropsychopharmacology. 2007;32:162–170. doi: 10.1038/sj.npp.1301151. [DOI] [PubMed] [Google Scholar]
- 101.Winterer G, et al. COMT genotype predicts BOLD signal and noise characteristics in prefrontal circuits. Neuroimage. 2006;32:1722–1732. doi: 10.1016/j.neuroimage.2006.05.058. Demonstrates that dopamine reduces cortical noise as measured with fMRI. [DOI] [PubMed] [Google Scholar]
- 102.Thaker GK, Wonodi I, Avila MT, Hong LE, Stine OC. Catechol O-methyltransferase polymorphism and eye tracking in schizophrenia: a preliminary report. Am J Psychiatry. 2004;161:2320–2322. doi: 10.1176/appi.ajp.161.12.2320. [DOI] [PubMed] [Google Scholar]
- 103.Lin CH, Yu YW, Chen TJ, Tsa SJ, Hong CJ. Association analysis for dopamine D2 receptor Taq1 polymorphism with P300 event-related potential for normal young females. Psychiatr Genet. 2001;11:165–168. doi: 10.1097/00041444-200109000-00010. [DOI] [PubMed] [Google Scholar]
- 104.Tsai SJ, Yu YW, Chen TJ, Chen MC, Hong CJ. Association analysis for dopamine D3receptor, dopamine D4 receptor and dopamine transporter genetic polymorphisms and P300 event-related potentials for normal young females. Psychiatr Genet. 2003;13:51–53. doi: 10.1097/00041444-200303000-00009. [DOI] [PubMed] [Google Scholar]
- 105.Bramon E, et al. Is there an association between the COMT gene and P300 endophenotypes? Eur Psychiatry. 2006;21:70–73. doi: 10.1016/j.eurpsy.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 106.Winterer G, et al. Prefrontal electrophysiologic “noise” and catechol-O-methyltransferase genotype in schizophrenia. Biol Psychiatry. 2006;60:578–584. doi: 10.1016/j.biopsych.2006.03.023. Demonstrates that dopamine reduces electrophysiological noise. [DOI] [PubMed] [Google Scholar]
- 107.Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 108.Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nature Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. Reviews the evidence for abnormalities in inhibitory interneurons in schizophrenia. [DOI] [PubMed] [Google Scholar]
- 109.Whittington MA, Traub RD. Interneuron diversity series: inhibitory interneurons and network oscillations in vitro. Trends Neurosci. 2003;26:676–682. doi: 10.1016/j.tins.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 110.Frodl-Bauch T, Bottlender R, Hegerl U. Neurochemical substrates and neuroanatomical generators of the event-related P300. Neuropsychobiology. 1999;40:86–94. doi: 10.1159/000026603. [DOI] [PubMed] [Google Scholar]
- 111.Crook JM, Kisvarday ZF, Eysel UT. GABA-induced inactivation of functionally characterized sites in cat striate cortex: effects on orientation tuning and direction selectivity. Vis Neurosci. 1997;14:141–158. doi: 10.1017/s095252380000883x. [DOI] [PubMed] [Google Scholar]
- 112.Tadin D, Lappin JS, Blake R. Fine temporal properties of center-surround interactions in motion revealed by reverse correlation. J Neurosci. 2006;26:2614–2622. doi: 10.1523/JNEUROSCI.4253-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Greenblatt DJ, Gan L, Harmatz JS, Shader RI. Pharmocokinetics and pharmacodynamics of single-dose triazolam: electroencephalography compared with the Digit-Symbol Substitution Test. Br J Clin Pharmacol. 2005;60:244–248. doi: 10.1111/j.1365-2125.2005.02409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Roschke J, et al. Electrophysiological evidence for an inverse benzodiazepine receptor agonist in panic disorder. J Psychiatr Res. 1999;33:1–5. doi: 10.1016/s0022-3956(98)00050-8. [DOI] [PubMed] [Google Scholar]
- 115.Porjesz B, et al. Linkage disequilibrium between the beta frequency of the human EEG and a GABAA receptor gene locus. Proc Natl Acad Sci USA. 2002;99:3729–3733. doi: 10.1073/pnas.052716399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Edenberg HJ, et al. Variations in GABRA2, encoding the alpha 2 subunit of the GABA(A) receptor, are associated with alcohol dependence and with brain oscillations. Am J Hum Genet. 2004;74:705–714. doi: 10.1086/383283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. Reviews the role of NMDA dysfunction in the pathophysiology of schizophrenia. [DOI] [PubMed] [Google Scholar]
- 118.Coyle JT, Tsai G. NMDA receptor function, neuroplasticity, and the pathophysiology of schizophrenia. Int Rev Neurobiol. 2004;59:491–515. doi: 10.1016/S0074-7742(04)59019-0. [DOI] [PubMed] [Google Scholar]
- 119.Tsai G, et al. Abnormal excitatory neurotransmitter metabolism in schizophrenic brains. Arch Gen Psychiatry. 1995;52:829–836. doi: 10.1001/archpsyc.1995.03950220039008. [DOI] [PubMed] [Google Scholar]
- 120.Sumiyoshi T, Jin D, Jayathilake K, Lee M, Meltzer HY. Prediction of the ability of clozapine to treat negative symptoms from plasma glycine and serine levels in schizophrenia. Int J Neuropsychopharmacol. 2005;8:451–455. doi: 10.1017/S1461145705005237. [DOI] [PubMed] [Google Scholar]
- 121.Neeman G, et al. Relation of plasma glycine, serine, and homocysteine levels to schizophrenia symptoms and medication type. Am J Psychiatry. 2005;162:1738–1740. doi: 10.1176/appi.ajp.162.9.1738. [DOI] [PubMed] [Google Scholar]
- 122.Hashimoto K, et al. Reduced D-serine to total serine ratio in the cerebrospinal fluid of drug naive schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:767–769. doi: 10.1016/j.pnpbp.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 123.Harrison PJ, West VA. Six degrees of separation: on the prior probability that schizophrenia susceptibility genes converge on synapses, glutamate and NMDA receptors. Mol Psychiatry. 2006;11:981–983. doi: 10.1038/sj.mp.4001886. [DOI] [PubMed] [Google Scholar]
- 124.Steullet P, Neijt HC, Cuenod M, Do KQ. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: relevance to schizophrenia. Neuroscience. 2006;137:807–819. doi: 10.1016/j.neuroscience.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 125.Nilsson LK, et al. Elevated levels of kynurenic acid in the cerebrospinal fluid of male patients with schizophrenia. Schizophr Res. 2005;80:315–322. doi: 10.1016/j.schres.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 126.Kegeles LS, et al. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biol Psychiatry. 2000;48:627–640. doi: 10.1016/s0006-3223(00)00976-8. [DOI] [PubMed] [Google Scholar]
- 127.Javitt DC, et al. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-D-aspartate receptor/glycine-site agonists. Neuropsychopharmacology. 2004;29:300–307. doi: 10.1038/sj.npp.1300313. [DOI] [PubMed] [Google Scholar]
- 128.Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology. 2001;156:117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- 129.Linn GS, Javitt DC. Phencyclidine (PCP)-induced deficits of prepulse inhibition in monkeys. Neuroreport. 2001;12:117–120. doi: 10.1097/00001756-200101220-00031. [DOI] [PubMed] [Google Scholar]
- 130.Oranje B, Gispen-de Wied CC, Verbaten MN, Kahn RS. Modulating sensory gating in healthy volunteers: the effects of ketamine and haloperidol. Biol Psychiatry. 2002;52:887–895. doi: 10.1016/s0006-3223(02)01377-x. [DOI] [PubMed] [Google Scholar]
- 131.Abel KM, Allin MP, Hemsley DR, Geyer MA. Low dose ketamine increases prepulse inhibition in healthy men. Neuropharmacology. 2003;44:729–737. doi: 10.1016/s0028-3908(03)00073-x. [DOI] [PubMed] [Google Scholar]
- 132.Heresco-Levy U, et al. High glycine levels are associated with prepulse inhibition deficits in chronic schizophrenia patients. Schizophr Res. 2007;91:14–21. doi: 10.1016/j.schres.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 133.Javitt DC, Steinschneider M, Schroeder CE, Arezzo JC. Role of cortical N-methyl-D-aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc Natl Acad Sci USA. 1996;93:11962–11967. doi: 10.1073/pnas.93.21.11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Umbricht D, et al. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry. 2000;57:1139–1147. doi: 10.1001/archpsyc.57.12.1139. References 133 and 134 demonstrate the ability of NMDA antagonists to reproduce neurophysiological markers of schizophrenia in animal and human models, respectively. [DOI] [PubMed] [Google Scholar]
- 135.Kreitschmann-Andermahr I, et al. Effect of ketamine on the neuromagnetic mismatch field in healthy humans. Brain Res Cogn Brain Res. 2001;12:109–116. doi: 10.1016/s0926-6410(01)00043-x. [DOI] [PubMed] [Google Scholar]
- 136.Umbricht D, Koller R, Vollenweider FX, Schmid L. Mismatch negativity predicts psychotic experiences induced by NMDA receptor antagonist in healthy volunteers. Biol Psychiatry. 2002;51:400–406. doi: 10.1016/s0006-3223(01)01242-2. [DOI] [PubMed] [Google Scholar]
- 137.Umbricht D, et al. Effects of the 5-HT2A agonist psilocybin on mismatch negativity generation and AX-continuous performance task: implications for the neuropharmacology of cognitive deficits in schizophrenia. Neuropsychopharmacology. 2003;28:170–181. doi: 10.1038/sj.npp.1300005. [DOI] [PubMed] [Google Scholar]
- 138.Avila MT, Weiler MA, Lahti AC, Tamminga CA, Thaker GK. Effects of ketamine on leading saccades during smooth-pursuit eye movements may implicate cerebellar dysfunction in schizophrenia. Am J Psychiatry. 2002;159:1490–1496. doi: 10.1176/appi.ajp.159.9.1490. [DOI] [PubMed] [Google Scholar]
- 139.Weiler MA, Thaker GK, Lahti AC, Tamminga CA. Ketamine effects on eye movements. Neuropsychopharmacology. 2000;23:645–653. doi: 10.1016/S0893-133X(00)00156-1. [DOI] [PubMed] [Google Scholar]
- 140.Bland BH, Declerck S, Jackson J, Glasgow S, Oddie S. Septohippocampal properties of N-methyl-D-aspartate-induced theta-band oscillation and synchrony. Synapse. 2007;61:185–197. doi: 10.1002/syn.20357. [DOI] [PubMed] [Google Scholar]
- 141.Leung LS, Shen B. Glutamatergic synaptic transmission participates in generating the hippocampal EEG. Hippocampus. 2004;14:510–525. doi: 10.1002/hipo.10199. [DOI] [PubMed] [Google Scholar]
- 142.Faulkner HJ, Traub RD, Whittington MA. Anaesthetic/amnesic agents disrupt beta frequency oscillations associated with potentiation of excitatory synaptic potentials in the rat hippocampal slice. Br J Pharmacol. 1999;128:1813–1825. doi: 10.1038/sj.bjp.0702948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Freye E, Latasch L, Levy JVS. (+)-Ketamine attenuates increase in electroencephalograph activity and amplitude height of sensory-evoked potentials during rapid opioid detoxification. Anesth Analg. 2006;102:1439–1444. doi: 10.1213/01.ane.0000202382.82847.64. [DOI] [PubMed] [Google Scholar]
- 144.Freedman R, et al. The genetics of sensory gating deficits in schizophrenia. Curr Psychiatry Rep. 2003;5:155–161. doi: 10.1007/s11920-003-0032-2. [DOI] [PubMed] [Google Scholar]
- 145.Adler LE, et al. Schizophrenia, sensory gating, and nicotinic receptors. Schizophr Bull. 1998;24:189–202. doi: 10.1093/oxfordjournals.schbul.a033320. [DOI] [PubMed] [Google Scholar]
- 146.Hajos M, et al. The selective α7 nicotinic acetylcholine receptor agonist PNU-282987 [N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4- chlorobenzamide hydrochloride] enhances GABAergic synaptic activity in brain slices and restores auditory gating deficits in anesthetized rats. J Pharmacol Exp Ther. 2005;312:1213–1222. doi: 10.1124/jpet.104.076968. Describes the effects of a selective α7 nicotinic receptor agonist on auditory gating and hippocampal oscillations in rats. [DOI] [PubMed] [Google Scholar]
- 147.O’Neill HC, Rieger K, Kem WR, Stevens KE. DMXB, an α7 nicotinic agonist, normalizes auditory gating in isolation-reared rats. Psychopharmacology. 2003;169:332–339. doi: 10.1007/s00213-003-1482-2. [DOI] [PubMed] [Google Scholar]
- 148.Stevens KE, Kem WR, Freedman R. Selective α 7 nicotinic receptor stimulation normalizes chronic cocaine-induced loss of hippocampal sensory inhibition in C3H mice. Biol Psychiatry. 1999;46:1443–1450. doi: 10.1016/s0006-3223(99)00200-0. [DOI] [PubMed] [Google Scholar]
- 149.Stevens KE, Kem WR, Mahnir VM, Freedman R. Selective α7-nicotinic agonists normalize inhibition of auditory response in DBA mice. Psychopharmacology. 1998;136:320–327. doi: 10.1007/s002130050573. [DOI] [PubMed] [Google Scholar]
- 150.Hurst RS, et al. A novel positive allosteric modulator of the α7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J Neurosci. 2005;25:4396–4405. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Olincy A, et al. Proof-of-concept trial of an α7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry. 2006;63:630–638. doi: 10.1001/archpsyc.63.6.630. Shows that a partial agonist on α7 nicotinic receptors improve P50 and neurocognitive function in patients with schizophrenia. [DOI] [PubMed] [Google Scholar]
- 152.Hollander E, et al. RS 86 in the treatment of Alzheimer’s disease: cognitive and biological effects. Biol Psychiatry. 1987;22:1067–1078. doi: 10.1016/0006-3223(87)90049-7. [DOI] [PubMed] [Google Scholar]
- 153.Callaway E, Halliday R, Naylor H, Schechter G. Effects of oral scopolamine on human stimulus evaluation. Psychopharmacology. 1985;85:133–138. doi: 10.1007/BF00428401. [DOI] [PubMed] [Google Scholar]
- 154.Meador KJ. Cholinergic, serotonergic, and GABAergic effects on the ERP. Electroencephalogr Clin Neurophysiol Suppl. 1995;44:151–155. [PubMed] [Google Scholar]
- 155.Dierks T, Frolich L, Ihl R, Maurer K. Event-related potentials and psychopharmacology. Cholinergic modulation of P300. Pharmacopsychiatry. 1994;27:72–74. doi: 10.1055/s-2007-1014282. [DOI] [PubMed] [Google Scholar]
- 156.Anokhin AP, et al. The P300 brain potential is reduced in smokers. Psychopharmacology. 2000;149:409–413. doi: 10.1007/s002130000387. [DOI] [PubMed] [Google Scholar]
- 157.Thomas A, Iacono D, Bonanni L, D’Andreamatteo G, Onofrj M. Donepezil, rivastigmine, and vitamin E in Alzheimer disease: a combined P300 event-related potentials/neuropsychologic evaluation over 6 months. Clin Neuropharmacol. 2001;24:31–42. doi: 10.1097/00002826-200101000-00007. [DOI] [PubMed] [Google Scholar]
- 158.Knott V, Mohr E, Mahoney C, Engeland C, Ilivitsky V. Effects of acute nicotine administration on cognitive event-related potentials in tacrine-treated and non-treated patients with Alzheimer’s disease. Neuropsychobiology. 2002;45:156–160. doi: 10.1159/000054957. [DOI] [PubMed] [Google Scholar]
- 159.Knott VJ, et al. Acute nicotine fails to alter event-related potential or behavioral performance indices of auditory distraction in cigarette smokers. Nicotine Tob Res. 2006;8:263–273. doi: 10.1080/14622200600576669. [DOI] [PubMed] [Google Scholar]
- 160.Neuhaus A, et al. Persistent dysfunctional frontal lobe activation in former smokers. Psychopharmacology. 2006;186:191–200. doi: 10.1007/s00213-006-0366-7. [DOI] [PubMed] [Google Scholar]
- 161.Werber EA, Gandelman-Marton R, Klein C, Rabey JM. The clinical use of P300 event related potentials for the evaluation of cholinesterase inhibitors treatment in demented patients. J Neural Transm. 2003;110:659–669. doi: 10.1007/s00702-003-0817-9. [DOI] [PubMed] [Google Scholar]
- 162.Harrison JB, Buchwald JS, Kaga K, Woolf NJ, Butcher LL. ‘Cat P300’ disappears after septal lesions. Electroencephalogr Clin Neurophysiol. 1988;69:55–64. doi: 10.1016/0013-4694(88)90035-1. [DOI] [PubMed] [Google Scholar]
- 163.Tamminga CA. The neurobiology of cognition in schizophrenia. J Clin Psychiatry. 2006;67 (Suppl 9):9–13. [PubMed] [Google Scholar]
- 164.Sherr JD, et al. The effects of nicotine on specific eye tracking measures in schizophrenia. Biol Psychiatry. 2002;52:721–728. doi: 10.1016/s0006-3223(02)01342-2. [DOI] [PubMed] [Google Scholar]
- 165.Rodriguez R, Kallenbach U, Singer W, Munk MH. Short- and long-term effects of cholinergic modulation on gamma oscillations and response synchronization in the visual cortex. J Neurosci. 2004;24:10369–10378. doi: 10.1523/JNEUROSCI.1839-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Siok CJ, Rogers JA, Kocsis B, Hajos M. Activation of α7 acetylcholine receptors augments stimulation-induced hippocampal theta oscillation. Eur J Neurosci. 2006;23:570–574. doi: 10.1111/j.1460-9568.2005.04560.x. [DOI] [PubMed] [Google Scholar]
- 167.Buchanan RW, Freedman R, Javitt DC, Abi-Dargham A, Lieberman JA. Recent advances in the development of novel pharmacological agents for the treatment of cognitive impairments in schizophrenia. Schizophr Bull. 2007;33:1120–1130. doi: 10.1093/schbul/sbm083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gray JA, Roth BL. The pipeline and future of drug development in schizophrenia. Mol Psychiatry. 2007;12:904–922. doi: 10.1038/sj.mp.4002062. [DOI] [PubMed] [Google Scholar]
- 169.Dunlop J, et al. Pharmacological profile of the 5-HT(2C) receptor agonist WAY-163909; therapeutic potential in multiple indications. CNS Drug Rev. 2006;12:167–177. doi: 10.1111/j.1527-3458.2006.00167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Siuciak JA, et al. CP-809,101, a selective 5-HT2C agonist, shows activity in animal models of antipsychotic activity. Neuropharmacology. 2007;52:279–290. doi: 10.1016/j.neuropharm.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 171.Zhang ZJ, et al. Beneficial effects of ondansetron as an adjunct to haloperidol for chronic, treatment-resistant schizophrenia: a double-blind, randomized, placebo-controlled study. Schizophr Res. 2006;88:102–110. doi: 10.1016/j.schres.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 172.Mitchell ES, Neumaier JF. 5-HT6 receptors: a novel target for cognitive enhancement. Pharmacol Ther. 2005;108:320–333. doi: 10.1016/j.pharmthera.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 173.Fox GB, et al. Pharmacological properties of ABT-239[4-(2-{2-[(2R)-2-Methylpyrrolidinyl]ethyl}-benzofuran-5-yl)benzonitrile]: II. Neurophysiological characterization and broad preclinical efficacy in cognition and schizophrenia of a potent and selective histamine H3 receptor antagonist. J Pharmacol Exp Ther. 2005;313:176–190. doi: 10.1124/jpet.104.078402. [DOI] [PubMed] [Google Scholar]
- 174.Patil ST, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nature Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- 175.Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nature Rev Drug Discov. 2006;5:660–670. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 176.Porteous DJ, Thomson P, Brandon NJ, Millar JK. The genetics and biology of DISC1 — an emerging role in psychosis and cognition. Biol Psychiatry. 2006;60:123–131. doi: 10.1016/j.biopsych.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 177.D’Souza DC. Cannabinoids and psychosis. Int Rev Neurobiol. 2007;78:289–326. doi: 10.1016/S0074-7742(06)78010-2. [DOI] [PubMed] [Google Scholar]
- 178.Gill CH, Soffin EM, Hagan JJ, Davies CH. 5-HT7 receptors modulate synchronized network activity in rat hippocampus. Neuropharmacology. 2002;42:82–92. doi: 10.1016/s0028-3908(01)00149-6. [DOI] [PubMed] [Google Scholar]
- 179.Hoffmann WE, Siok CJ, Kocsis B, Hajos M. Society of Neuroscience web site, Program No. 498.1. 2007 Neuroscience Meeting Planner. San Diego, CA : 2007. Activation of cannabinoid-1 receptors disrupts sensory gating and neuronal oscillation: relevance to schizophrenia. [online], < http://www.sfn.org/am2007/index.cfm?pagename=call_for_abstracts>. [Google Scholar]
- 180.Staubli U, Xu FB. Effects of 5-HT3 receptor antagonism on hippocampal theta rhythm, memory, and LTP induction in the freely moving rat. J Neurosci. 1995;15:2445–2452. doi: 10.1523/JNEUROSCI.15-03-02445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Robbe D, et al. Cannabinoids reveal importance of spike timing coordination in hippocampal function. Nature Neurosci. 2006;9:1526–1533. doi: 10.1038/nn1801. [DOI] [PubMed] [Google Scholar]
- 182.Skosnik PD, Krishnan GP, Aydt EE, Kuhlenshmidt HA, O’Donnell BF. Psychophysiological evidence of altered neural synchronization in cannabis use: relationship to schizotypy. Am J Psychiatry. 2006;163:1798–1805. doi: 10.1176/ajp.2006.163.10.1798. [DOI] [PubMed] [Google Scholar]
- 183.Adler LE, et al. Improved p50 auditory gating with ondansetron in medicated schizophrenia patients. Am J Psychiatry. 2005;162:386–388. doi: 10.1176/appi.ajp.162.2.386. [DOI] [PubMed] [Google Scholar]
- 184.Maxwell CR, Kanes SJ, Abel T, Siegel SJ. Phosphodiesterase inhibitors: a novel mechanism for receptor-independent antipsychotic medications. Neuroscience. 2004;129:101–107. doi: 10.1016/j.neuroscience.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 185.Schmidt CJ, et al. Preclinical characterization of selective PDE10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J Pharmacol Exp Ther. 2007 doi: 10.1124/jpet.107.132910. (in the press) [DOI] [PubMed] [Google Scholar]
- 186.Price GW, et al. A multivariate electrophysiological endophenotype, from a unitary cohort, shows greater research utility than any single feature in the Western Australian family study of schizophrenia. Biol Psychiatry. 2006;60:1–10. doi: 10.1016/j.biopsych.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 187.Singer W. Neuronal synchrony: a versatile code for the definition of relations? . Neuron. 1999;24:49–65. 111–125. doi: 10.1016/s0896-6273(00)80821-1. [DOI] [PubMed] [Google Scholar]
- 188.Pfurtscheller G, Lopes da Silva FH. Event-related EEG/MEG synchronization and desynchronization: basic principles. Clin Neurophysiol. 1999;110:1842–1857. doi: 10.1016/s1388-2457(99)00141-8. [DOI] [PubMed] [Google Scholar]
- 189.Canolty RT, et al. High gamma power is phase-locked to theta oscillations in human neocortex. Science. 2006;313:1626–1628. doi: 10.1126/science.1128115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Foxe JJ, Murray MM, Javitt DC. Filling-in in schizophrenia: a high-density electrical mapping and source-analysis investigation of illusory contour processing. Cereb Cortex. 2005;15:1914–1927. doi: 10.1093/cercor/bhi069. [DOI] [PubMed] [Google Scholar]
- 191.Winterer G, et al. Instability of prefrontal signal processing in schizophrenia. Am J Psychiatry. 2006;163:1960–1968. doi: 10.1176/ajp.2006.163.11.1960. Is the first report on abnormal ‘noise’ in schizophrenia as measured with fMRI. [DOI] [PubMed] [Google Scholar]
- 192.Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology. 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- 193.Mackeprang T, Kristiansen KT, Glenthoj BY. Effects of antipsychotics on prepulse inhibition of the startle response in drug-naive schizophrenic patients. Biol Psychiatry. 2002;52:863–873. doi: 10.1016/s0006-3223(02)01409-9. [DOI] [PubMed] [Google Scholar]
- 194.Ueki A, Goto K, Sato N, Iso H, Morita Y. Prepulse inhibition of acoustic startle response in mild cognitive impairment and mild dementia of Alzheimer type. Psychiatry Clin Neurosci. 2006;60:55–62. doi: 10.1111/j.1440-1819.2006.01460.x. [DOI] [PubMed] [Google Scholar]
- 195.Perry W, Minassian A, Feifel D, Braff DL. Sensorimotor gating deficits in bipolar disorder patients with acute psychotic mania. Biol Psychiatry. 2001;50:418–424. doi: 10.1016/s0006-3223(01)01184-2. [DOI] [PubMed] [Google Scholar]
- 196.Linn GS, Negi SS, Gerum SV, Javitt DC. Reversal of phencyclidine-induced prepulse inhibition deficits by clozapine in monkeys. Psychopharmacology. 2003;169:234–239. doi: 10.1007/s00213-003-1533-8. [DOI] [PubMed] [Google Scholar]
- 197.Swerdlow NR, et al. Dopamine agonist effects on startle and sensorimotor gating in normal male subjects: time course studies. Psychopharmacology. 2002;161:189–201. doi: 10.1007/s00213-002-1040-3. [DOI] [PubMed] [Google Scholar]
- 198.Swerdlow NR, et al. Convergence and divergence in the neurochemical regulation of prepulse inhibition of startle and N40 suppression in rats. Neuropsychopharmacology. 2006;31:506–515. doi: 10.1038/sj.npp.1300841. Compares neurochemical/pharmacological attributes of prepulse inhibition and auditory gating in rats. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Duncan EJ, et al. Clinical and sensorimotor gating effects of ketamine in normals. Neuropsychopharmacology. 2001;25:72–83. doi: 10.1016/S0893-133X(00)00240-2. [DOI] [PubMed] [Google Scholar]
- 200.Clementz BA, Geyer MA, Braff DL. Poor P50 suppression among schizophrenia patients and their first-degree biological relatives. Am J Psychiatry. 1998;155:1691–1694. doi: 10.1176/ajp.155.12.1691. [DOI] [PubMed] [Google Scholar]
- 201.Myles-Worsley M, Ord L, Blailes F, Ngiralmau H, Freedman R. P50 sensory gating in adolescents from a pacific island isolate with elevated risk for schizophrenia. Biol Psychiatry. 2004;55:663–667. doi: 10.1016/j.biopsych.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 202.Yee CM, Nuechterlein KH, Morris SE, White PM. P50 suppression in recent-onset schizophrenia: clinical correlates and risperidone effects. J Abnorm Psychol. 1998;107:691–698. doi: 10.1037//0021-843x.107.4.691. [DOI] [PubMed] [Google Scholar]
- 203.Cadenhead KS, Light GA, Geyer MA, Braff DL. Sensory gating deficits assessed by the P50 event-related potential in subjects with schizotypal personality disorder. Am J Psychiatry. 2000;157:55–59. doi: 10.1176/ajp.157.1.55. [DOI] [PubMed] [Google Scholar]
- 204.Cancelli I, et al. Sensory gating deficit assessed by P50/Pb middle latency event related potential in Alzheimer’s disease. J Clin Neurophysiol. 2006;23:421–425. doi: 10.1097/01.wnp.0000218991.99714.ee. [DOI] [PubMed] [Google Scholar]
- 205.Schulze KK, et al. P50 Auditory evoked potential suppression in bipolar disorder patients with psychotic features and their unaffected relatives. Biol Psychiatry. 2007;62:121–128. doi: 10.1016/j.biopsych.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 206.Adler LE, et al. Reversal of diminished inhibitory sensory gating in cocaine addicts by a nicotinic cholinergic mechanism. Neuropsychopharmacology. 2001;24:671–679. doi: 10.1016/S0893-133X(00)00242-6. [DOI] [PubMed] [Google Scholar]
- 207.Light GA, et al. Amphetamine disrupts P50 suppression in normal subjects. Biol Psychiatry. 1999;46:990–996. doi: 10.1016/s0006-3223(99)00034-7. [DOI] [PubMed] [Google Scholar]
- 208.Freedman R, et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci USA. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.de Bruin NM, et al. Sensory gating of auditory evoked potentials in rats: effects of repetitive stimulation and the interstimulus interval. Biol Psychol. 2001;55:195–213. doi: 10.1016/s0301-0511(00)00084-3. [DOI] [PubMed] [Google Scholar]
- 210.Blackwood DH, St Clair DM, Muir WJ, Duffy JC. Auditory P300 and eye tracking dysfunction in schizophrenic pedigrees. Arch Gen Psychiatry. 1991;48:899–909. doi: 10.1001/archpsyc.1991.01810340031004. [DOI] [PubMed] [Google Scholar]
- 211.Schreiber H, Stolz-Born G, Kornhuber HH, Born J. Event-related potential correlates of impaired selective attention in children at high risk for schizophrenia. Biol Psychiatry. 1992;32:634–651. doi: 10.1016/0006-3223(92)90294-a. [DOI] [PubMed] [Google Scholar]
- 212.Salisbury DF, et al. First-episode schizophrenic psychosis differs from first-episode affective psychosis and controls in P300 amplitude over left temporal lobe. Arch Gen Psychiatry. 1998;55:173–180. doi: 10.1001/archpsyc.55.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Hirayasu Y, et al. Abnormalities of auditory event-related potentials in schizophrenia prior to treatment. Biol Psychiatry. 1998;43:244–253. doi: 10.1016/S0006-3223(97)00275-8. [DOI] [PubMed] [Google Scholar]
- 214.Niznikiewicz MA, et al. Lateralized P3 deficit in schizotypal personality disorder. Biol Psychiatry. 2000;48:702–705. doi: 10.1016/s0006-3223(00)00938-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Polich J, Corey-Bloom J. Alzheimer’s disease and P300: review and evaluation of task and modality. Curr Alzheimer Res. 2005;2:515–525. doi: 10.2174/156720505774932214. [DOI] [PubMed] [Google Scholar]
- 216.Lopez V, et al. Attention-deficit hyperactivity disorder involves differential cortical processing in a visual spatial attention paradigm. Clin Neurophysiol. 2006;117:2540–2548. doi: 10.1016/j.clinph.2006.07.313. [DOI] [PubMed] [Google Scholar]
- 217.Galletly CA, Clark CR, McFarlane AC. Clozapine improves working memory updating in schizophrenia. Eur Neuropsychopharmacol. 2005;15:601–608. doi: 10.1016/j.euroneuro.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 218.Gallinat J, et al. P300 and symptom improvement in schizophrenia. Psychopharmacology. 2001;158:55–65. doi: 10.1007/s002130100835. [DOI] [PubMed] [Google Scholar]
- 219.Blum K, Braverman ER, Dinardo MJ, Wood RC, Sheridan PJ. Prolonged P300 latency in a neuropsychiatric population with the D2 dopamine receptor A1 allele. Pharmacogenetics. 1994;4:313–322. doi: 10.1097/00008571-199412000-00004. [DOI] [PubMed] [Google Scholar]
- 220.Ahveninen J, et al. Scopolamine augments transient auditory 40-Hz magnetic response in humans. Neurosci Lett. 1999;277:115–118. doi: 10.1016/s0304-3940(99)00854-x. [DOI] [PubMed] [Google Scholar]
- 221.Ahveninen J, et al. Inherited auditory-cortical dysfunction in twin pairs discordant for schizophrenia. Biol Psychiatry. 2006;60:612–620. doi: 10.1016/j.biopsych.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 222.Bar-Haim Y, Marshall PJ, Fox NA, Schorr EA, Gordon-Salant S. Mismatch negativity in socially withdrawn children. Biol Psychiatry. 2003;54:17–24. doi: 10.1016/s0006-3223(03)00175-6. [DOI] [PubMed] [Google Scholar]
- 223.Pekkonen E, Jousmaki V, Kononen M, Reinikainen K, Partanen J. Auditory sensory memory impairment in Alzheimer’s disease: an event-related potential study. Neuroreport. 1994;5:2537–2540. doi: 10.1097/00001756-199412000-00033. [DOI] [PubMed] [Google Scholar]
- 224.Schall U, Catts SV, Karayanidis F, Ward PB. Auditory event-related potential indices of fronto-temporal information processing in schizophrenia syndromes: valid outcome prediction of clozapine therapy in a three-year follow-up. Int J Neuropsychopharmcol. 1999;2:83–93. doi: 10.1017/S1461145799001418. [DOI] [PubMed] [Google Scholar]
- 225.Javitt DC, Schroeder CE, Steinschneider M, Arezzo JC, Vaughan HG., Jr Demonstration of mismatch negativity in the monkey. Electroencephalogr Clin Neurophysiol. 1992;83:87–90. doi: 10.1016/0013-4694(92)90137-7. [DOI] [PubMed] [Google Scholar]
- 226.Frangou S, et al. The Maudsley Family Study, II: Endogenous event-related potentials in familial schizophrenia. Schizophr Res. 1997;23:45–53. doi: 10.1016/S0920-9964(96)00089-8. [DOI] [PubMed] [Google Scholar]
- 227.Brenner CA, Sporns O, Lysaker PH, O’Donnell BF. EEG synchronization to modulated auditory tones in schizophrenia, schizoaffective disorder, and schizotypal personality disorder. Am J Psychiatry. 2003;160:2238–2240. doi: 10.1176/appi.ajp.160.12.2238. [DOI] [PubMed] [Google Scholar]
- 228.Boutros N, et al. Cocaine use and the mid-latency auditory evoked responses. Psychiatry Res. 2000;96:117–126. doi: 10.1016/s0165-1781(00)00207-9. [DOI] [PubMed] [Google Scholar]
- 229.O’Donnell BF, Vohs JL, Hetrick WP, Carroll CA, Shekhar A. Auditory event-related potential abnormalities in bipolar disorder and schizophrenia. Int J Psychophysiol. 2004;53:45–55. doi: 10.1016/j.ijpsycho.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 230.Schroeder CE, et al. N-methyl-D-aspartate enhancement of phasic responses in primate neocortex. Exp Brain Res. 1997;114:271–278. doi: 10.1007/pl00005635. [DOI] [PubMed] [Google Scholar]
- 231.Wilson TW, Rojas DC, Reite ML, Teale PD, Rogers SJ. Children and adolescents with autism exhibit reduced MEG steady-state gamma responses. Biol Psychiatry. 2007;62:192–197. doi: 10.1016/j.biopsych.2006.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Santarelli R, et al. Effects of isoflurane on auditory middle latency (MLRs) and steady-state (SSRs) responses recorded from the temporal cortex of the rat. Brain Res. 2003;973:240–251. doi: 10.1016/s0006-8993(03)02520-4. [DOI] [PubMed] [Google Scholar]
- 233.Gallinat J, Winterer G, Herrmann CS, Senkowski D. Reduced oscillatory gamma-band responses in unmedicated schizophrenic patients indicate impaired frontal network processing. Clin Neurophysiol. 2004;115:1863–1874. doi: 10.1016/j.clinph.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 234.Ahveninen J, et al. Suppression of transient 40-Hz auditory response by haloperidol suggests modulation of human selective attention by dopamine D2 receptors. Neurosci Lett. 2000;292:29–32. doi: 10.1016/s0304-3940(00)01429-4. [DOI] [PubMed] [Google Scholar]
- 235.Demiralp T, et al. DRD4 and DAT1 polymorphisms modulate human gamma band responses. Cereb Cortex. 2007;17:1007–1019. doi: 10.1093/cercor/bhl011. [DOI] [PubMed] [Google Scholar]
- 236.Cunningham MO, et al. Region-specific reduction in entorhinal gamma oscillations and parvalbumin-immunoreactive neurons in animal models of psychiatric illness. J Neurosci. 2006;26:2767–2776. doi: 10.1523/JNEUROSCI.5054-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Holsheimer J. Generation of theta activity (RSA) in the cingulate cortex of the rat. Exp Brain Res. 1982;47:309–312. doi: 10.1007/BF00239391. [DOI] [PubMed] [Google Scholar]
- 238.Thaker GK, et al. Smooth pursuit eye movements to extraretinal motion signals: deficits in relatives of patients with schizophrenia. Arch Gen Psychiatry. 1998;55:830–836. doi: 10.1001/archpsyc.55.9.830. [DOI] [PubMed] [Google Scholar]
- 239.Rosenberg DR, et al. Eye-tracking dysfunction in offspring from the New York High-Risk Project: diagnostic specificity and the role of attention. Psychiatry Res. 1997;66:121–130. doi: 10.1016/s0165-1781(96)02975-7. [DOI] [PubMed] [Google Scholar]
- 240.Sweeney JA, et al. Eye tracking abnormalities in schizophrenia: evidence for dysfunction in the frontal eye fields. Biol Psychiatry. 1998;44:698–708. doi: 10.1016/s0006-3223(98)00035-3. [DOI] [PubMed] [Google Scholar]
- 241.Ross RG. Early expression of a pathophysiological feature of schizophrenia: saccadic intrusions into smooth-pursuit eye movements in school-age children vulnerable to schizophrenia. J Am Acad Child Adolesc Psychiatry. 2003;42:468–476. doi: 10.1097/01.CHI.0000046818.95464.42. [DOI] [PubMed] [Google Scholar]
- 242.Litman RE, Hommer DW, Radant A, Clem T, Pickar D. Quantitative effects of typical and atypical neuroleptics on smooth pursuit eye tracking in schizophrenia. Schizophr Res. 1994;12:107–120. doi: 10.1016/0920-9964(94)90068-x. [DOI] [PubMed] [Google Scholar]
- 243.Avila MT, Sherr JD, Hong E, Myers CS, Thaker GK. Effects of nicotine on leading saccades during smooth pursuit eye movements in smokers and nonsmokers with schizophrenia. Neuropsychopharmacology. 2003;28:2184–2191. doi: 10.1038/sj.npp.1300265. [DOI] [PubMed] [Google Scholar]
- 244.Olincy A, Johnson LL, Ross RG. Differential effects of cigarette smoking on performance of a smooth pursuit and a saccadic eye movement task in schizophrenia. Psychiatry Res. 2003;117:223–236. doi: 10.1016/s0165-1781(03)00022-2. [DOI] [PubMed] [Google Scholar]