

Abstract
Raman crystallography is the application of Raman spectroscopy to single crystals. This technique has been applied to a variety of protein molecules where it has provided unique information about biopolymer folding, substrate binding, and catalysis. Here, we describe the application of Raman crystallography to functional RNA molecules. RNA represents unique opportunities and challenges for Raman crystallography. One issue that confounds studies of RNA is its tendency to adopt multiple non-functional folds. Raman crystallography has the advantage that it isolates a single state of the RNA within the crystal and can evaluate its fold, metal ion binding properties (ligand identity, stoichiometry, and affinity), proton binding properties (identity, stoichiometry, and affinity), and catalytic potential. In particular, base-specific stretches can be identified and then associated with the binding of metal ions and protons. Because measurements are carried out in the hanging drop at ambient, rather than cryo, conditions and because RNA crystals tend to be approximately 70% solvent, RNA dynamics and conformational changes become experimentally accessible. This review focuses on experimental setup and procedures, acquisition and interpretation of Raman data, and determination of physicochemical properties of the RNA. Raman crystallographic and solution biochemical experiments on the HDV RNA enzyme are summarized and found to be in excellent agreement. Remarkably, characterization of the crystalline state has proven to help rather than hinder functional characterization of functional RNA, most likely because the tendency of RNA to fold heterogeneously is limited in a crystalline environment. Future applications of Raman crystallography to RNA are briefly discussed.
Keywords: Raman spectroscopy, RNA crystallography, metal ion binding, pKa determination, RNA folding, RNA dynamics, conformational changes, HDV ribozyme
1. Introduction
RNA is involved in myriad biological function and controls gene expression at almost all levels. The leading models for the origin of life posit that life began with a so-called ‘RNA World’, in which RNA served genetic, structural, and functional roles [1–3]. Such biological complexity necessitates molecular complexity. Indeed, it was demonstrated more than thirty years ago that tRNA can adopt complex folds with extensive non-Watson-Crick base pairing, intricate helical stacking, and diverse metal ion binding interactions [4,5].
In the ensuing years, functional RNAs have been discovered that catalyze chemical reactions (ribozymes) [6,7] and bind small molecules and alter gene expression (riboswitches) [8–10]. Over the past 15 years in particular, atomic-level experimental structures of ribozymes, riboswitches, and the ribosome have been determined and reveal intricate structures with buried active sites [11]. Most of these structures have been solved by X-ray crystallography at −170°C, with some contributions from solution NMR spectroscopy [12]. In addition, NMR spectroscopy and molecular dynamics (MD) are revealing how dynamics contribute to RNA function [13,14]. An important goal in the field of RNA biology is to relate three-dimensional structures and dynamics to biological function.
A variety of solution studies, both in bulk solution and at the single molecule level, have revealed that a given RNA sequence can adopt multiple, stable structures and only a fraction of these can be functional (e.g. can bind substrates and catalyze chemical reactions) while others are non-functional [15,16]. For example, rate-pH profiles of the HDV ribozyme showed that certain constructs have multi-exponential kinetics, which reflect 2 or more populations of the ribozyme only one of which has direct catalytic potential [17–20]. In addition, single molecule studies on the hairpin ribozyme revealed four distinct species, which have ‘memory effects’ and do not interconvert [21]. The tendency of RNA to misfold also applies to larger RNAs [22]. These properties indicate that structural and functional studies of RNA must be reconciled.
Ribozymes provide an ideal vehicle for assessing RNA folding because their native states have one or more specific, testable functional properties, including metal ion binding, proton binding, and bond cleavage. Because RNA tends to crystallize in a single conformation, crystallography offers the potential to isolate and describe single ribozyme states [23]. However, determining whether these states are functionally relevant has proven challenging, in part because crystals are typically studied under cryo-conditions near 100K, which limits dynamics and the ability to perform biophysical experiments, and also because modifications are often needed to keep RNAs from reacting in the crystal, which can be perturbing. In an effort to characterize the functional properties of RNA crystals, we have applied Raman spectroscopy to single crystals of a small ribozyme under ambient conditions in the hanging drop [24–26]. This article will focus entirely on RNA and primarily on one crystal form of a small ribozyme from the authors’ laboratories and on a study on tRNA. Although these are the only studies available in the literature, the methodology is also applicable to proteins, with many examples from the Carey lab [27]. The generality of Raman crystallography of RNA will be determined as additional RNA systems are examined in the future.
2. Advantages and disadvantages of Raman crystallography for characterizing RNA
In this section we consider some of the advantages and disadvantages of Raman spectroscopy versus other spectroscopies for characterizing RNA, followed by some of the advantages and disadvantages of working with crystals.
2.1. Advantages and disadvantages of Raman spectroscopy
Raman spectroscopy provides a vibrational spectrum of the ground electronic state of the RNA and is derived specifically from those vibrational motions that cause a change in the polarizability of the RNA. Because electrons promoted into an excited virtual state immediately relax to a vibrational level of the ground state, transitions in Raman are very fast (less than 10−14 s), providing essentially an instantaneous snapshot of the molecule [27]. As such, there is no spectral broadening from chemical exchange, slower relaxation processes, or other RNA dynamics.
Inelastic scattering of incident photons with vibrational modes in RNA crystals leads to loss or gain in energy of the incident radiation (i.e. the Raman shift), which give rise to the Raman photons that produce the Stokes and anti-Stokes spectrum, respectively. Because the shifts in energy giving rise to the Raman peaks are small, it is important that the incident light be monochromatic, and, therefore, a laser is typically used. The Raman shift by its nature is independent of the wavelength of the incident light; in an effort to mininize photodamage to the RNA, we therefore choose a wavelength in the visible region to excite the sample. The Carey lab has found that red excitation (647 nm) proves to be a good choice for efficient Raman scattering, which is inversely proportional to the 4th power of the wavelength (i.e. shorter wavelengths give more scattering), detector efficiency, and minimization of fluorescent background [27]. The Stokes lines that result are much more intense than the anti-Stokes lines, especially at larger wavenumbers and ambient temperatures, and therefore Stokes lines are measured in Raman spectroscopy.
Many sugar, phosphate, and base-specific vibrations of RNA are Raman active, well characterized, and spectrally disperse (see Section 6.5), making Raman spectroscopy an excellent method for characterization RNA folding, and especially powerful for assessing the conformational and environmental changes that occur upon substrate or cofactor binding and catalysis. As described in Section 6.4, much of the data are processed as difference spectra, in which Mg2+ or pH is changed, in order to allow the user to focus on bound ligands and to identify conformational changes of the RNA that occur during the experiment. Other advantages of Raman spectroscopy is that the RNA does not have to be labeled, saving time, expense, and avoiding potentially perturbing changes to the RNA, and that the experiment is conducted at ambient temperatures, which allows potentially important dynamics to occur.
This article will be concerned exclusively with ‘normal’ (i.e. non-resonance) Raman spectroscopy. Resonance Raman spectroscopy, which uses an excitation wavelength tuned to an intense absorption transition, can be performed with dilute solutions or chromophoric crystals (e.g. from heme proteins), where it takes advantage of the intense Raman signal from the chromophoric group [28]. However, resonance Raman has a number of limitations, especially for RNA. First, there are no unique chromophores in RNA: the four bases, which provide most of the absorption, overlap extensively in their spectra. Second, resonance Raman reduces the number of bands in the spectrum, which severely limits useful molecular-level information about RNA folding. Third, because the electronic transitions needed for resonance Raman are typically of higher energy than the input for nonresonance Raman, luminescence background from solution contaminants or from the RNA itself can become problematic. Finally, because of the higher energy of the incident radiation in resonance Raman, often in the UV, photodegradation can occur, which could result in covalent crosslinking.
Raman provides several unique advantages when compared to other spectroscopies, including infrared (IR), fluorescence, and NMR spectroscopy [29]. Infrared spectroscopy, like Raman, provides valuable and complementary information about bond vibrations in a molecule; however, water absorbs strongly in IR but not Raman spectroscopy. Because RNA crystals typically contain approximately 70% solvent and because the crystals are kept in a largely aqueous mother liquor, as described below, IR is of limited applicability. Fluorescence spectroscopy provides much less detail than Raman does about the fold of the RNA and requires labeling of the RNA (the nucleobases themselves have very poor quantum yields), which is laborious, costly, and potentially perturbing. However, fluorescence has a much slower timescale (10−6 to 10−8 s) than Raman, which allows it to be used to follow RNA dynamics on this time scale. NMR spectroscopy can provide structural level information, but is highly insensitive, requiring ~600 L of millimolar solutions, and is subject to line broadening by chemical and conformational exchange events on the millisecond to microsecond timescale. Moreover, NMR is limited to relatively smaller RNAs (less than ~100 nt). In addition, water has a large background signal in NMR studies of exchangeable protons, although this can be largely suppressed with specialized pulse sequences. X-ray crystallography shares many similarities with Raman crystallography, however there are several limitations of X-ray crystallography. At ambient temperature where dynamics may be observed, X-rays damage the RNA. This damage is reduced at cryogenic temperatures; however, dynamics are not observable under these conditions. Since non-resonance Raman spectroscopy is carried out with visible light, photodegradation at ambient temperature is obviated. Finally, X-ray crystallography is limited by the need to obtain crystals that diffract to high resolution.
Non-resonance Raman spectroscopy is not without its disadvantages. One of the major drawbacks is that it too is insensitive. At most 0.001% of the source intensity is transferred to the Raman lines [29]. The issue of poor signal-to-noise is exacerbated in difference spectra, where 99% or more of the signal is often subtracted out [30] and which typically involves at least three subtractions, which further increases noise (see Section 6.4). Since Raman intensities are typically proportional to sample concentration, the main way to increase the Raman signal-to-noise ratio is to increase the concentration of the RNA. Lasers are used to provide power to improve signal-to-noise, although too much power can damage the crystal (see Section 5). In principle, decreasing the wavelength of the laser can also increase the intensity of Raman scattering, as discussed above, but this approach is seldom used due to the risk of high-energy photons causing degradation of the sample.
Another problem with any form of Raman spectroscopy is that it is prone to interference from luminescence due to extrinsic impurities or intrinsic chromophores. As described in the next subsection, working with RNA crystals and a Raman microscope allows both the issues of sensitivity and fluorescence background to be addressed. Lastly, interpretation of Raman data is less software driven as compared to NMR and X-ray, and therefore involves more human intervention, which is time consuming and can be challenging.
2.2. Advantages and disadvantages of conducting experiments in RNA crystals
Raman spectra of RNA crystals can be obtained using a Raman microscope (Fig. 1) (described fully in Section 5). One of the major advantages of working with RNA crystals is that the RNA is very concentrated in the crystal, with typical concentrations on the order of 20 to 30 mM, see footnote (38) in [24]. As mentioned in Section 2.1, increasing the concentration of the RNA is the simplest and best way to increase the signal-to-noise ratio. RNA concentrations in the crystal are typically 10 to 100-fold greater than those that can be achieved in solution; moreover, high concentrations of RNA in solution tend to induce aggregation, which is less likely to occur in the crystal [27].
Fig. 1.
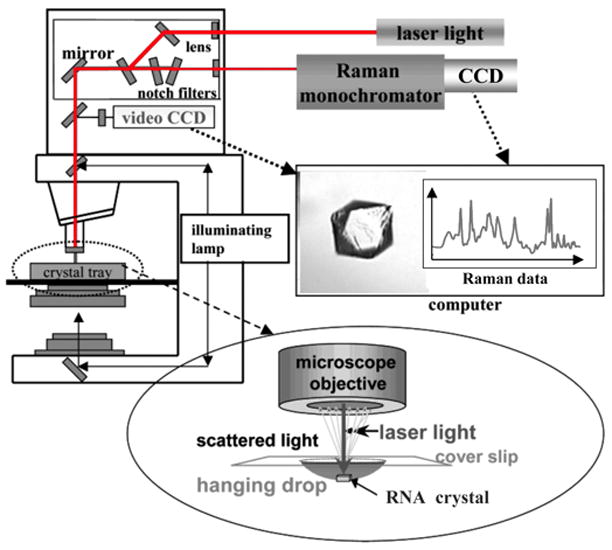
Diagram of the Raman microscope setup. Red laser light at 647 nm from a krypton ion laser (Coherent Innova 70C) is used to excite the RNA (follow red lines in diagram). The laser travels through a fiber optic cable to the top of a HoloLab Raman microscope. From there it moves through a series of lenses and mirrors and then impinges on an RNA crystal in a 5 L hanging drop in a crystal tray (see blow-up at bottom). The Raman photons that are backscattered from the crystal are collected through a microscope objective set to 50× and directed to a high-throughput Raman spectrograph with a CCD detector, which records the Raman data (see spectrum in red). To remove the intense Rayleigh scattering, this output travels through a series of notch filters prior to arriving at the monochromator. An illuminating lamp (follow black lines in diagram) allows the crystal to be visualized through a video CCD (see crystal of HDV ribozyme). This feature facilitates positioning of the crystal in the center of the laser beam by use of a microscope stage. Adapted with permission from [73]. (Copyright, American Chemical Society.)
Another advantage of working with RNA crystals is that they tend to have much lower luminescent background than solutions of RNA. This probably results because a crystallization step stringently purifies the RNA. Thus, working with crystals of RNA addresses the two main concerns for Raman spectroscopy mentioned in Section 2.1. In addition, we recently discovered that working with the Raman microscope helps in cases where Raman spectra of RNA solutions are needed. By pipetting ~5 L of ~2 mM RNA on a siliconized glass cover slip and inverting it over a crystal tray in the Raman microscope, good quality Raman data can be obtained (Chen, Carey, and Dayie, unpublished data); this volume is ~1/100th that of similarly concentrated samples needed for NMR.
Another advantage of working with crystals is that RNA folding is likely more homogeneous in the crystal. As mentioned in Section 1, RNA molecules in solution often adopt multiple folds, which confounds interpretation of biophysical experiments. Because single crystals tend to cause RNA to adopt a single conformation, biophysical studies of RNA are simplified. Moreover, the bandwidth of spectral features tends to be narrowed due to the damping of RNA motions and excursions into non-functional conformations in crystals, which results in sharp and well-resolved spectra (Fig. 2). Another concern with working with crystals is that the RNA may be precluded from large-scale conformational changes, although small motions required for ion binding have been shown possible as mentioned above. Indeed, despite being in a crystal, studies to date indicate that the HDV ribozyme is still able to bind magnesium ions and protons in a native fashion, as determined by comparison to biochemical studies; moreover, these ions can be ‘soaked in’ or ‘soaked out’ of the crystal with similar results (see Section 6.3). This dynamic nature of crystalline RNA is likely possible because the studies are carried out at ambient temperature and because RNA crystals are ~70% solvent—a value similar to the water content of the cell [31]. A survey of solvent content of 20 RNA crystals by Ferré-D’Amaré and Doudna gave a range of solvent content from 50% to 81%, with an average of 67% [32]. Whether RNA crystals with less solvent prove to be less dynamic will await further experimentation. A final advantage of carrying out Raman measurement in crystals is that the crystals do not have to diffract to high resolution. We recently obtained excellent Raman data in crystals that diffract to only ~5 Å (Fig. 2) [26]. Moreover, successful experiments can be conducted in crystals even in the absence of a structure.
Fig. 2.
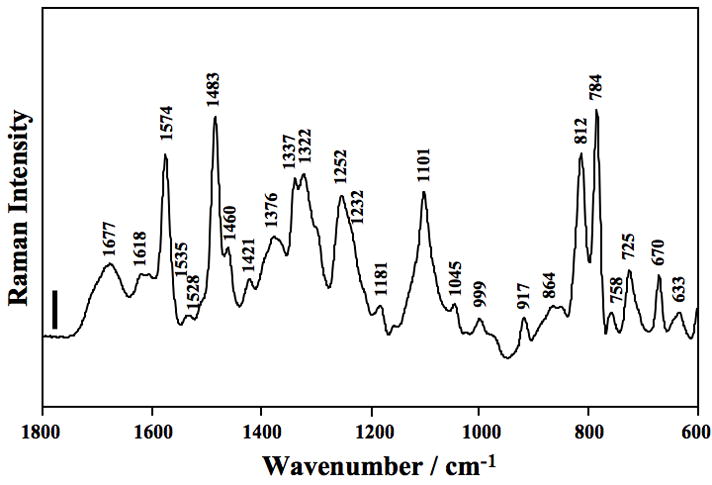
Raman spectrum of HDV ribozyme crystals. The spectrum of the wild-type ribozyme (using 2′-OCH3 substitution at the U-1 position of the inhibitor strand) in the region of 1800–600 cm−1. The crystal was surrounded by cacodylate buffer containing 20 mM Mg2+ at pH 7.0 at room temperature (22 °C). The frequencies of major Raman bands are marked in the spectrum and assignments of these bands are shown in Table 3. The vertical bar represents a 5000 photon event, and the y-axis scales linearly with the number of photons. Very high signal-to–noise ratios are obtained, ≈ 300:1. Reproduced with permission from [24]. (Copyright, American Chemical Society.)
Some of the possible disadvantages of working with crystals is that first and foremost, crystals of the RNA have to be grown, which has proven challenging for some RNAs and for certain mutants, although methods are constantly improving [23,33,34]. Also, crystal contacts can disrupt native interactions, which can be especially problematic for smaller, single-helix RNAs; see, for example, studies on the lead-dependent ribozyme [35–37]. As with all crystallographic studies, comparison between Raman studies in the crystal and Raman and biochemical studies in solution are critical (see Section 3.2). In fact, Raman is one of the few spectroscopic methods that can directly compare molecular properties in solution and crystal phases.
Another limitation to Raman crystallography is that it can only follow slow time changes (>10 s) since data collection takes that long. A second temporal factor is the diffusion of ions or substrate into the crystal. Changes in the crystal can be brought about by changing conditions in mother liquor surrounding crystal (see Section 6.3); for example, metal ion diffusing into the crystal can set up a new ‘state’. It usually takes tens of seconds before the equilibrium is set up throughout the crystal and Raman data can be collected. In addition, the outer layers of crystal reach equilibrium first, creating an ‘onion effect’ [27].
A final potential concern with Raman crystallography is that the intensity of Raman scattering depends on the relative orientation of the electric vector of the laser beam and the molecule scattering the photons [38]. Thus, in some cases, intensity can depend on the orientation of the crystal. Orientation dependence is especially sensitive when each target molecule in the crystal is oriented in the same direction [39]. If there is more than one target molecule orientation within the crystal and if the laser excitation is not polarized, little or no crystal orientation dependence is observed. We have found little orientation dependence with cubic space group crystals of the HDV ribozyme [24–26], which provide essentially isotropic conditions. Orientation dependence does not make Raman crystallographic studies intractable, but extra care has to be exercised to try to orient the crystal in the same fashion for each measurement (see Section 6.2). Moreover, if the crystals diffract poorly, complementary structural studies of that RNA may not be possible in that state. In principle, polarized Raman spectra can provide group orientations in ordered samples, and Thomas, Tsuboi and co-workers have demonstrated this for fibers of double-stranded RNA [40]. However, for single crystals the experiments are challenging, requiring a method for controlling crystal orientation and a simple target group that has a single orientation with respect to the crystal axes.
3. Chemical information obtainable by Raman crystallography of RNA
One of the main advantages of Raman spectroscopy is that it allows unique and extensive chemical information to be obtained about RNA (summarized in Table 1). This is best illustrated through consideration of literature on Raman crystallography of RNA. In the first subsection, we consider the early, albeit limited, Raman studies of RNA crystals and the molecular insight into RNA that was gained. In the second subsection, we describe some of our recent Raman studies of RNA crystals and physicochemical measurements made. Comparison between biochemical and Raman crystallographic studies, where available, reveals excellent agreement.
Table 1.
Summary of chemical information obtainable by Raman crystallography of RNA
| Chemical Information | Example |
|---|---|
| Proton-binding: identification of RNA base and determination of pKa | pKa of C75 in HDV ribozyme crystal near 6.5 |
| Metal-binding interactions: identification of metal | Bound innersphere Mg2+ itself has peak at ~322 cm−1. |
| Metal-binding: quantification of number of metals and affinity | Agreement between metals from Raman and from crystal structure. |
| Metal-binding interactions: identification of RNA ligands | Innersphere Mg2+ -PO2− interaction has a differential near 1100 cm−1. Innersphere Mg2+ N7 of guanine interaction has a differential near 1490 cm−1. |
| Isotope effects on metal binding | Determination of H218O and 2H2O effects on bound innersphere Mg2+ ion. |
| RNA conformational changes | ribose, phosphate backbone |
| Compare crystal behavior to solution | Similar pKa, metal binding found for HDV ribozyme. |
| Chemical reaction in crystal; identify reaction intermediates and determine optimal conditions for trapping target species for X-ray analysis | Not demonstrated for RNA yet, but shown for proteins |
3.1. Early studies of Raman crystallography of RNA
Since the late 1960s, Raman spectroscopy has been widely used to study nucleic acids and their biological complexes. However, nucleic acid crystals, especially RNA crystals, were rarely used as Raman samples, mainly due to difficulties of obtaining pure RNA or DNA suitable for crystallization, and to technical limitations of conventional Raman instrumentation (e.g. it is very difficult to position and focus laser light in crystals in a controlled manner). Pioneering work on Raman spectroscopic application in RNA crystals was done in 1975, in which Chen and coworkers recorded Raman spectra of orthorhombic and hexagonal yeast phenylalanine tRNA as well as tRNA in solution [41]. They compared these spectra and drew conclusions that the structure of tRNA is identical in both crystal forms and also appears to be the same in the crystal and in solution. This was a crucial result because it had yet to be proven that the L-shaped structure observed in the tRNA crystals was relevant to its biological function.
In the early 1990s, G.J. Thomas’s group performed structural studies of bean pod mottle virus (BPMV) capsid and RNA in crystalline and solution states by Raman spectroscopy. They observed minor structural differences of RNA in the crystal from that in the solution, which is indicated by 8% difference of phosphodiester conformation markers of RNAs in crystalline and solution states [42]. They also found that Raman spectral difference between crystal and solution of BPMV is mainly due to different electrostatic environments of the packaged RNA molecules in the two states [43]. With technical innovations, a Raman microscope has been integrated into the conventional Raman system, which has allowed us to directly acquire the Raman spectrum of a single protein or nucleic acid crystal with very high signal-to-noise ratio. This approach is named ‘Raman crystallography’ and is described in the next subsection.
3.2. Recent studies of Raman crystallography of RNA
In the last several years, we have employed Raman crystallography to determine information about metal ion binding, proton binding, and conformational changes in RNA, and have used these to make comparisons to solution [24–26] (see Table 1). RNA is a polyanion and therefore its folding and function often depends on interaction with divalent cations, such as magnesium [44]. Magnesium ions can interact with RNA in multiple ways, including diffuse, outersphere (interacting with RNA though water ligands), and innersphere (interacting directly with RNA ligands) modes [45,46]. Given this complexity, defining the mode and site of Mg2+ binding to RNA is challenging. NMR [47,48] and phosphorothioate rescue experiments [49,50] have made valuable contributions, although the former is limited in its ability to distinguish metal ion binding modes, while the latter requires chemical modification of the RNA. We have applied difference Raman spectroscopy to metal soaking experiments (Section 6.3) along with solvent isotope effects to identify bound metal ions as innersphere, quantify the approximate number of bound metal ions, define ligands of metal ions such as PO2− and N7 of guanine, and identify accompanying conformational changes in the RNA [25,26]. The number of ions detected by Raman crystallography is in good agreement with values from crystallography. Moreover, binding affinities for these ions from Raman crystallography agrees well with biochemical values (manuscript in preparation).
The nucleobases in small ribozymes can participate in general acid-base chemistry, which can be enhanced by having pKa’s shifted towards neutrality [51]. We have applied Raman spectroscopy to determine a pKa shift to 7 in the precleaved HDV ribozyme, quantify the number of bases ionizing as 1, and assign the pKa to a specific residue (C75) [24]. This study represented the first measurement of a pKa of neutrality by a physical method, and was in excellent agreement with solution studies [52,53]. In addition, Raman crystallography revealed that the pKa couples anticooperatively with Mg2+, consistent with biochemical studies. It is noteworthy that similar studies of the pKa of this residue by NMR did not lead to a measurable pKa shift on the precleaved RNA, possibly because of conformational heterogeneity or non-ideal dynamics [54]. Thus, crystallography appears to help rather than hinder functional characterization of functional RNAs, at least in some instances, perhaps because heterogeneity is limited in the crystal and because the timescale for Raman is essentially instantaneous. Lastly, we have observed RNA conformational changes associated with metal and proton binding, including changes in ribose pucker and phosphate backbone conformation [26,55].
4. Sample preparation
This section is concerned with the design, synthesis, purification, and crystallization of RNA samples for Raman crystallography. Each of these topics has been extensively reviewed in the literature and so is summarized only briefly here, with issues relevant to Raman crystallography emphasized.
4.1. Design of constructs for Raman crystallography
The first consideration in sample preparation is design of appropriate constructs. As mentioned in Section 1, RNA samples are prone to adopting multiple stable folds, which can complicate interpretation of data and crystallization. Mapping of the secondary structures of such misfolds for the HDV ribozyme, along with characterization of appropriate single and higher-order mutants, has led to the rational design of sequences that react with single exponential and fast kinetics [17–20] that approach the intrinsic ‘speed-limit’ for small ribozymes [56], with half lives in the millisecond regime. Such optimized constructs have proven amenable to crystallization [24,57], suggesting that their kinetic properties reflect a single, well-folded structure.
4.2. Synthesis and purification of RNA for Raman crystallography
Synthesis of large (milligram) quantities of RNA is necessary for Raman studies and can be accomplished by either chemical or enzymatic approaches. Solid-phase synthesis is the primary chemical approach for making RNA, and several companies offer chemically synthesized RNA, including Dharmacon Inc. and Integrated DNA Technologies, Inc., and there are national facilities such as the Keck Facility at Yale that will make RNA from commercially available phosphoramidites from suppliers such as Glen Research and Chemgenes. Solid-phase synthesis is used routinely for preparing RNA oligonucleotides up to about 50 nt. Advantages of solid-phase synthesis are that modifications including isotopic substitutions can be readily introduced into the RNA and time is saved. Disadvantages are cost, although prices for RNA have dropped in recent years, and the inability to obtain longer synthetic RNAs.
Large scale in vitro transcription with T7 RNA polymerase is the primary enzymatic approach to making RNA [23,58–60]. Transcription can be performed using either commercially available kits from companies such as Ambion Inc. and Epicentre Biotechnologies, or T7 polymerase and transcription reagents prepared in the laboratory, which saves cost and allows fine tuning of conditions to optimize yield. The DNA template for transcription can be prepared from a hemi-duplex, PCR product, or plasmid. An advantage of cloning the template into a plasmid is that large amounts of DNA can be easily obtained using the plasmid purification kits from Qiagen Inc.; moreover, the sequence, once established, can be readily mutagenized using commercially available kits such as QuikChange (Stratagene). On the other hand, transcription from a PCR product or hemi-duplex has the advantage that a large number of sequences can be rapidly screened. Advantages of enzymatic synthesis over chemical synthesis are that large RNAs can be prepared and the approach is relatively inexpensive. Disadvantages are that chimeric RNAs containing site-specific modifications or isotopic substitutions cannot be prepared.
Lastly, the chemical and enzymatic approaches can be combined in so-called semi-synthetic approaches. We have done so in the preparation of HDV RNA constructs for Raman crystallography, using Watson-Crick base pairing to bind a synthetic oligonucleotide with an inactivating modification at a critical 2′OH to an enzymatic transcript [24,57]. Similar approaches have been used to crystallize other small ribozymes, including the hairpin ribozyme [61]. Another way to combine chemical and enzymatic RNAs is to covalently attach them using T4 DNA ligase and a DNA splint [62], although this approach generally leads to significant sample loss, which could be problematic for crystallization and Raman studies.
We purify RNA for Raman crystallography by standard methods. Chemically synthesized shorter RNAs (<10 nt) generally have negligible amounts of shorter ‘failure’ sequences and so do not need to be size purified. This can be tested by fractionating a small quantity of the RNA on a high percentage denaturing polyacrylamide gel. If purity is not sufficient, high percentage polyacrylamide gels or TLC can be used to purify the full-length RNA [26]. Short RNAs are desalted using a C18 reversed phase Sep-Pak column (Waters); samples are eluted with acetonitrile and dried in a speed-vac to remove the acetonitrile. Longer RNAs need to be size-purified by denaturing polyacrylamide gel electrophoresis using standard detection, elution, and ethanol precipitation methods [23]. These RNAs are then suitable for use in crystallography, although they should be dialyzed (Float-A-Lyzer, Spectrum Laboratories) to remove small molecules and fluorescent impurities, especially if solution studies in the hanging drop are to be performed (Section 2.2).
4.3. Crystallization of RNA for Raman crystallography
The final step in sample preparation is crystallization of the RNA (summarized in Table 2, left-hand column). Concentrations of RNA needed for crystallization are approximately 1–5 mM for small RNAs and 3–10 mg/mL for larger RNAs, with several hundred L of RNA solution typically needed for screening [23]. In order to encourage native folding, the RNA is usually renatured prior to preparing crystallization trays. Such renaturations often include a step of heating in the absence of Mg2+ to a temperature near 90 °C to break base pairing, cooling to restore base pairing, and reheating in the presence of Mg2+ to an intermediate temperature near 50 °C to encourage native tertiary folding. Effects of different renaturation procedures on RNA function can be tested using ribozyme assays [60], where the goal is to try to find a renaturation that leads to single exponential and fast kinetics.
Table 2.
Experimental parameters for a RNA Raman crystallography experiment
| RNA crystal | Raman microscope system | ||
|---|---|---|---|
| Size | Minimum is 30 μm edge; typical is 100–300 μm | Excitation source | 647.1 nm Kr+ laser |
| Quality | Stable to changes of experimental conditions (e.g. pH, ions, …) | Laser power | 50–100 mW |
| Reservoir solution | Stabilization buffer: 20 mM MgCl2, 2 mM spermine, 50% MPD, 50 mM buffer | Microscope objective | 50× (or 20×) long- focal-length |
| Hanging drop | ~5 μL stabilization buffer | Operation mode | Nonconfocal mode |
| Symmetry | High symmetry is less orientation-dependent (e.g. cubic) | Data collection time | 100 s (=10 accumulations for 10 sec each) |
| Resolution | Low resolution (e.g. 5 Å) crystals work well | Operation temperature | Room temperature |
| Temperature | Crystal is in hanging drop at ambient temperature (i.e. no cryo- conditions used) | Detector | Charge-coupled device at −70 °C |
Screening for RNA crystals is typically done using a sparse matrix approach, and several screens for RNA have been published, some of which are commercially available [23]. Components for crystallization include a buffer, a precipitant such as 2-methyl-2,4-pentanediol (MPD), a polyamine, and monovalent and divalent salts. RNA crystals are usually grown by vapor diffusion, using hanging drop or sitting drop plates, and can take several days to months to grow; details on growing crystals of the HDV ribozyme for Raman crystallography are provided elsewhere [24]. Siliconized glass cover slips (Hampton Research) are used both for growing RNA crystals and for acquiring data with the Raman microscope. Crystallization is a routine method for purifying organic molecules and it appears to purify RNA as well, as we have found RNA crystals to be relatively free of luminescent impurities as compared to RNA solutions.
For Raman crystallography, crystals larger than 100 m in the minimum dimension are easier to work with and control, and crystals of this size are generally attainable for RNA; however, crystals as small as 30 m are also tractable. Diffraction quality crystals are not needed, and, as mentioned above, we have obtained excellent Raman data with crystals that diffract to only 5 Å [24–26]. As mentioned in Section 2.2, crystals in which each RNA molecule is oriented in the same direction are highly orientation dependent, which is less desirable. Thus, it is advantageous to test several different crystal types that may arise from the sparse matrix, as some may be more ideal for Raman crystallography, and/or have different physicochemical properties in soaking experiments. Another prime experimental requirement of the crystals is that they be stable to soaking conditions required for difference spectroscopy (see Section 6.3), which must be determined empirically.
5. The Raman microscope
In the 1980s and 1990s, the method of Raman microscopy was further developed wherein an optical microscope is coupled to a Raman spectrometer to collect Raman scattering from micron-sized samples [27]. The Raman crystallography setup used in our laboratory is depicted in Figure 1 and summarized in the right-hand column of Table 2. A krypton ion laser (Coherent Innova 70C; Coherent, Inc.) produces a 647.1 nm red light (see first horizontal red line in Fig. 1), which scatters from the RNA. The laser is operated at ~0.55 W and is focused onto a fiber optic cable that directs the beam to a series of lenses and mirrors that send it along the optic axis of the Raman microscope (see vertical red line in Fig. 1). As discussed in Section 2.1, laser light in the red region of the spectrum is ideal because it produces photons of relatively low energy, which reduces the possibility of photodegradation of the RNA but still gives acceptable Raman scattering.
Raman microscopes are available from several commercial vendors, and we use a HoloLab Series 5000 Raman microscope (Kaiser Optical Systems), which is operated in the nonconfocal mode [27]. One important consideration for the microscope is a long focal length objective (ca. 1 cm). In order to measure the actual power impinging on the sample, a small handheld power meter is placed under the objective at the point in space where the cover slip will be positioned (see ‘crystal tray’ in Fig. 1). A typical power reading at this spot is 50–100 mW, or approximately 1/10 that of the input laser power. Because Raman scattering by its nature is weak, it is important to have a high photon flux, but too much power (in excess of ~100 mW at 647.1 nm) can cause crystal damage and lead to excessive heating (see below). The handheld power meter is also used to check for a clean laser focal point: by raising and lowering the meter, the position of focus can be determined both by visual inspection of the red laser spot and by determination of maximal readout.
Raman photons that are backscattered (through 180°) from the crystal are collected through the microscope objective (20× or 50×) (see blow-up at bottom of Fig. 1), and directed to a high-throughput Raman spectrograph via a fiber optic system (see second horizontal red line in Fig. 1). A back-illuminated red-light sensitive charge-coupled device (CCD) detector, operating at −70 °C to reduce dark current, is used to detect the Raman photons and generate a Raman spectrum on the computer monitor using the HoloGRAMS software bundled with the HoloLab Raman microscope (see spectrum in Fig. 1). Prior to detection, the scattered light is passed through a series of band-stop notch filters, which function to remove (‘notch out’) the undesired and much more intense elastically scattered Rayleigh photons.
Prior to collecting the Raman spectrum, a microscope stage is used to precisely position the plastic crystal tray having the crystal in a 5 L hanging drop (see blow-up at bottom of Fig. 1). An illuminating lamp, whose output is detected with a separate CCD (see horizontal black line in Fig. 1), is used to view the crystal in the hanging drop (see photograph of crystal in Fig. 1). The illumination feature of the microscope allows the beam to be focused cleanly inside of the stationary crystal.
6. Conducting Raman experiments
Obtaining adequate Raman data on RNA requires attention to a number of experimental details. In this section, we consider the major steps involved in conducting Raman experiments including instrument calibration; crystal preparation; soaking of ligands; acquisition, processing and interpretation of Raman data; and measurement of physicochemical properties of RNA. Typical experimental parameters both for the Raman microscope system and the RNA crystal are presented in Table 2. These parameters are fine-tuned from experiment to experiment.
6.1. Calibration of the microscope
The first step in conducting an experiment is to calibrate the microscope and photon detection. A neon source accessory is placed under the 10× objective and used as a standard to calibrate the wavenumber to ± 1 cm−1. The source is then placed under the 50× (or 20×) objective and used to calibrate the intensity response of the detector. For the Hololab Raman microscope, this is done with assistance of the HoloGRAMS software. To avoid stray light reaching the detector, this procedure is done in total darkness. Generally, calibration needs to be done only once per day.
6.2. Preparation of the crystal in the microscope
Crystals are suspended in the ~5 L hanging drop (sometimes a sitting drop) on a siliconized glass cover slip covering a well in the crystal tray. Data can be collected in the same drop in which the crystal was grown; however, it is common to transfer the crystal to a new hanging drop in a stabilization buffer (artificial mother liquor), especially for the soaking in and out experiments (see Section 6.3). For pKa measurements on the HDV ribozyme, we grew crystals in 35% MPD, 10 mM MgCl2, 0.5 mM spermine, and 50 mM K-cacodylate (pH 6.5), and transferred crystals to a stabilization buffer typically containing 50% MPD, 20 mM MgCl2, 2 mM spermine, and acetate or cacodylate buffer (50 mM) at a variety of pH values [24].
In addition, we have developed a protocol for transferring RNA crystals between labs using express shipping. Crystals are placed in the low pH (5.0–5.4) acetate stabilization buffer for 30 min and then 5 L of crystal-containing solution is pipetted into Nalge Nunc 870 PFA tubing (Nalgene, Inc.) with 1/4 in ID and 3/8 in OD, where the material forms a drop inside the tubing. An extra drop of stabilization buffer is added to each end of the tubing, and the tubing is sealed with wax (Hampton Research). The tubing is surrounded by ice bags to keep the sample cool and sent by overnight express shipping.
A 24-well plastic tray used for growing crystals is placed on the microscope stage and the beam is focused on the sample using the bright field illumination lamp. Detection is by the video CCD and readout on the computer screen (Fig. 1). It is helpful to have the drop near the center of the cover slip. In this process, the tray is first positioned manually by hand and then precisely by fine adjustments to the stage. After the first acquisition, it is important to rotate the crystal in the stage and reacquire data to check for orientation dependence, as discussed in Section 2.2.
6.3. Soaking of ligands into and out of the crystal
Measurement of RNA physicochemical properties generally requires that a change be made to the crystal conditions. We accomplish this by soaking the crystal in artificial mother liquors in which a variable of interest (e.g. pH or Mg2+ concentration) is altered (Figs. 3 and 4). For example, to study Mg2+-HDV interactions, we usually transfer HDV crystals in one artificial mother liquor (containing Mg2+) to a new artificial mother liquor containing a different Mg2+ concentration [25]. In some instances, the crystal is soaked in EDTA to remove bound Mg2+ from the crystals, using a background of 150 mM Na+ to help maintain folding [25]; this process was shown to be reversible, as ‘resoaking’ the crystals with Mg2+ allowed spectra indistinguishable from the original Mg2+-containing spectra to be obtained [55]. Spectra were acquired as a function of time to assure equilibration had occurred. As mentioned in Section 4.3, it is important that crystals be stable to these changes in experimental conditions, which might be problematic under conditions that disfavor RNA folding or stable crystal packing, such as extremes of pH or no Mg2+. The crystals are soaked in each artificial mother liquor for 15–20 min. Over the course of this time, the new artificial mother liquor is replenished several times (at least 3 soaks) to assure that the equilibrium is achieved. Replenishing is important given the high concentration of RNA in the crystal.
Fig. 3.
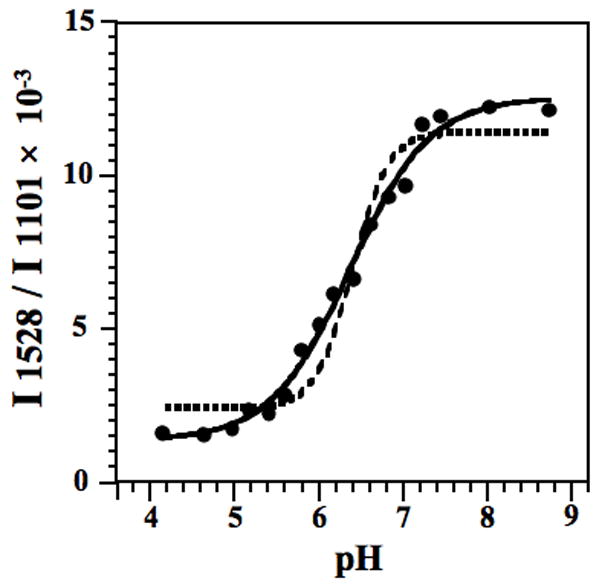
pH titration of HDV ribozyme crystals. The pKa of cytosine in an HDV ribozyme crystal (using the 2′-OCH3 substitution) containing 2 mM Mg2+ was fit using Eq. (2), as shown by the solid line. For comparison, the best fit using Eq. (2) with a Hill constant of 2 is shown with a dashed line. The model with a Hill constant of 1 provides the best description of the observed data. The y-axis is the relative intensity ratio of 1528 cm−1 band (neutral cytosine) to the intensity of the band at 1101 cm−1 (PO2−, internal standard). A pKa of 6.40 ± 0.05 at 2 mM Mg2+ was obtained. Reproduced with permission from [24]. (Copyright, American Chemical Society.)
Fig. 4.
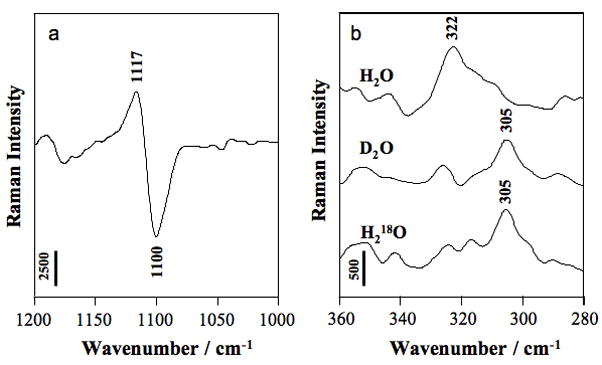
Raman difference spectra of HDV crystals [HDV + 20 mM Mg2+] minus [HDV no Mg2+] pH 6.0. Vertical bar represents photon events. (a) Partial Raman spectrum showing PO2− symmetric stretch of phosphate groups bound innersphere to Mg2+ at 1117 cm−1; the negative differential at 1100 cm−1 is due to phosphate symmetric stretch of metal-free groups. (b) Raman signatures of Mg hydrate (pentahydrate and tetrahydrate are both possible) bound innersphere to PO2− oxygen. Adapted with permission from [25]. (Copyright, American Chemical Society.)
6.4. Acquisition and processing of Raman data
Before collecting data, the room is darkened to avoid stray late reaching the CCD. Typically, ten 10 s accumulations are recorded, for a total acquisition time of 100 s. With a power of 100 mW and a mother liquor of 5 L bathing the crystal, this leads to heating of ~15 °C above ambient, or temperatures near 37 °C [27], which is often an experimentally desirable temperature. Temperature increases can be modulated to some extent by increasing the volume of the mother liquor; waiting between accumulations; or attenuating the laser beam, albeit with less signal.
Raman data analysis is performed using GRAMS/32 software. In general, data are viewed as difference spectra, which allows the user to focus on the occupancy of bound ligands and the portions of the RNA that change during the experiment. Features of the RNA that are gained during soaking appear as positive peaks in the difference spectrum, while features that are lost appear as negative peaks (see Fig. 4A). The following is a general procedure used to process the data and obtain difference spectra, adapted from [63]. Prior to subtraction of Raman spectra, a buffer spectrum is subtracted from each RNA crystal spectrum. This is accomplished by acquiring a spectrum of the RNA crystal under condition 1 (abbreviated with ‘(1)’), followed by a spectrum of its mother liquor, obtained by translating the hanging drop ~50 m so that the laser focal point is shifted away from the crystal. These spectra are subtracted, [RNA crystal (1) + buffer (1)] − [buffer (1)], to give a Raman difference spectrum of RNA crystal, referred to as [RNA crystal (1)]. This processing step removes Raman bands due to buffer and crystallization media present in the crystal channels. In the subtraction procedures, a scaling factor is used to reduce the contribution of buffer peaks to zero. The same crystal is then soaked into a new buffer to give condition 2, and a similar spectral subtraction is performed to give [RNA crystal (2)]. Finally, a Raman spectrum of the ligand bound in the RNA crystal plus any accompanying conformational changes of the RNA is obtained by subtracting the two subtractions:
| (1) |
Lastly, we note that having to perform three subtractions in Eq. (1) increases the noise in the data. Callender and Deng [30] have provided a detailed analysis of the sources of error in Raman difference spectroscopy. However, the prime factor leading to high quality difference data is the overlap achieved between the spectra for the first two terms in Eq. (1). When good overlap occurs the experienced operator knows immediately that high quality difference data will result. The two considerations that lead to this situation are high signal-to-noise in the parent spectra with the Raman peaks sitting on a low, flat optical background.
6.5. Interpretation of Raman data
There are two major approaches for interpreting Raman spectral features. One is based on decades of research in IR and Raman spectroscopy on nucleic acids and related systems. Specific Raman bands (position and intensity) have been associated with molecular groupings and tabulated (Tables 3 and 4 provide assignments for the HDV ribozyme) [24,26,38,64,65]. These spectral features are then used as marker bands to follow the fate of the corresponding groupings during chemical or conformational changes. Tables 3 and 4 summarize assignments from experiments we have published on the HDV ribozyme. Features assignable to sugar, phosphate, and bases are found. Importantly, there are regions of the spectrum unique to each base, including regions unique to the protonated bases, which allow molecular states to be assigned to specific bases and atoms within the RNA (see Section 6.6). Although usually semi-quantitative, this method of interpreting Raman data can yield powerful biochemical insights.
Table 3.
Assignment of major Raman bands observed in HDV ribozyme crystals
| Wavenumber (cm−1) | Assignment |
|---|---|
| 1677 (m) | base C=O |
| 1574 (s) | A, G |
| 1535 (w) | C |
| 1528 (w) | C |
| 1483 (vs) | G, A |
| 1460 (w) | C-H (ring) |
| 1421 (w) | A, G |
| 1376 (m) | A, G |
| 1337 (s) | A |
| 1322 (s) | G |
| 1252 (s) | C |
| 1232 (sh) | U |
| 1181 (w) | G |
| 1101 (s) | PO2− |
| 1045 (w) | ribose |
| 999 (w) | ribose |
| 917 (w) | ribose |
| 812 (s) | O-P-O |
| 784 (vs) | C, U |
| 725 (m) | A |
| 670 (m) | G |
| 633 (w) | G |
Vs, very strong; s, strong; m, medium; w, weak; sh, shoulder. The features assigned to the bases are due to ring modes. Reproduced with permission from [24]. (Copyright, American Chemical Society.)
Table 4.
Positions (cm−1) and assignment of major Raman difference bands observed in Raman difference spectra of [HDV pH 7.5] minus [HDV pH 5.0] and [CMP pH 6.0] minus [CMP pH 3.0]
| HDV | CMP | ||
|---|---|---|---|
| Wavenumber (cm−1) | Assignment | Wavenumber (cm−1) | Assignment |
| 1547 | C+ | ||
| 1528 | C | 1529 | C |
| 1489/1473 | G | ||
| 1341 | A | ||
| 1298 | C | 1298 | C |
| 1258 | C+ | 1258 | C+ |
| 983 | PO32− | ||
| 929 | ribose | ||
| 916/896 | ribose | ||
| 892 | ribose | ||
| 815 | -O-P-O- | ||
| 790 | C+ | 790 | C+ |
| 780 | C | 778 | C |
| 728 | A | ||
| 599 | C | 599 | C |
| 323 | Mg(H2O)x2+ (≤5) | ||
C, neutral cytosine; C+, protonated cytosine; Mg(H2O)x2+ (x≤5), inner-sphere coordinated Mg-hydrate. The features assigned to the bases are due to ring modes. Reproduced with permission from [26]. (Copyright, American Chemical Society.)
The second, more recent approach of analyzing Raman spectra uses computer software. Using commercially available quantum programs such as Gaussian [66], it is possible to calculate the vibrational spectrum (IR and Raman) of a molecule or molecular fragment of up to 30–40 atoms. This can be accomplished on a modern personal computer in a matter of hours. This innovation provides deep insight into molecular properties and their changes via analysis of the Raman data. In brief, full geometry optimization of the entire molecule is performed using Gaussian software, which is used for the subsequent estimation of harmonic force constants at the density functional theory (DFT) level of B3LYP (Becke’s three-parameter hybrid method using the correlation functional of Lee, Yang, and Parr ) [67,68]. A Gaussian basis set of 6–31+G(d) was used for our previous DFT calculations [24,25]. The calculated Raman data at the DFT/B3LYP level were converted by a single scaling factor of 0.9692 through a least-square approach on the basis of the work of Scott and Radom [69].
We used Gaussian to analyze the vibrational modes of neutral and N3-protonated cytosine, using a methyl group in place of a ribose at the N1 site (1-MeC) [24]. Briefly, we were able to identify exocyclic bending and N3-C4 endocyclic stretching modes in neutral C, as well as N3-H stretches in protonated C. Through analysis of these calculations, we then identified spectral features that correlate with the occupancy of neutral C, which was used in pKa determinations.
More recently, we performed quantum mechanical calculations with Gaussian to analyze metal ion binding modes [25]. Ab initio quantum mechanical calculations were performed to predict the Raman spectra of [Mg•(H2O)6]2+, [Mg•(H2O)5]2+−(H2PO4), and [Mg•(H2O)4]2+−2(H2PO4). These calculations gave values for 2H-substituted and 18O-substituted molecules that had trends with isotopic substitution that helped assign experimental data to innersphere complexes. Recently, similar calculations were carried out for guanine and guanine complexed to [Mg•(H2O)5]2+ through its N7, which helped assign binding of a magnesium ion as an innersphere complex to the N7 of G1 [26].
6.6. Measurement of physicochemical properties of RNA
There are a variety of physical and chemical properties of RNA that can potentially be measured by Raman spectroscopy. To date, we have pursued primarily proton and metal ion binding as summarized in Table 1. Other potential applications of Raman are mentioned in Section 7. As discussed in Section 3.2, the nucleobases in small ribozymes have been implicated in general acid-base chemistry [51]. In particular, we have focused on C75 in the genomic HDV ribozyme, which has been shown to participate in general acid-base chemistry [70], with some data implicating it as the general acid for bond cleavage and some as the general base. The proton transfer function of C75 would be enhanced by a pKa shifted from the typical value near 4 to a value near biological pH [71]. As mentioned above, we were able to use Gaussian calculations to identify a spectral feature that tracked with loss of protonation of cytosine (centered at ~1528 cm−1) [24]. This feature overlapped with another peak and required fitting to a mixed Gaussian/Lorentzian peak shape to extract its value. The 1528 cm−1 feature was divided by a PO2− peak centered at 1101 cm−1 whose intensity did not change throughout the titration and so provided an internal standard. It is noteworthy that this analysis was done without taking difference spectra, which increases signal-to-noise.
A few features are worth mentioning about these titrations. First, the identity of the titrating species as a cytosine was readily made because of its unique spectral characteristics assignable to a cytosine, as determined from tabulated base features (Tables 3 and 4), Gaussian calculations, and experiments conducted on a cytosine 5′-monophosphate (CMP) standard [24]. Second, the pKa and number of ionizing bases was determined by curve fitting to an equation derived from a standard two-state model, divided through by a reference peak
| (2) |
where Iobs is the observed intensity at the 1528 cm−1 feature; IIS is the intensity of the internal standard feature at 1101 cm−1;IRH+ and IR are the intensities of the fully protonated and deprotonated states, respectively; and nHill is the Hill coefficient, which provides a lower limit on the number of protons binding. Of these constants, only Iobs is a function of pH. Fits to 2 or more ionizations (where nHill was fixed at 2) did not accurately model the observed data. The data are best modeled as a single ionization event with a pKa of 6.40 ± 0.05 (Fig. 3). The number of ionizing cytosines was independently determined to be unity by comparing the integrated intensity near 1528 cm−1 from an RNA crystal to a CMP calibration curve and correlating to the concentration of the RNA in the crystal of 26 mM. Third, the ionizing base, once identified as a single cytosine, was assigned to C75 by mutating it to a U, crystallizing C75U, and showing that the titration was lost; future studies aim to employ isotopic substitutions to help with identifying participating residues and to eliminate the need to make crystallizable mutants (see Section 7). Lastly, it is worth noting that C75, with its pKa near 6.5, was probably an ideal case for Raman crystallography. Good baselines demand about 2 pH units in either direction of the pKa. Because the pKa was near 6.5, we could titrate between pH 4.5 and 8.5 (Fig. 3) and thereby avoid acid- or alkaline-denaturing the sample.
The other measurements made on RNA crystals in our labs to date has been studying metal ion binding [25,26]. As summarized in Table 1, Raman crystallography has been used to identify bound metal ions as innersphere, quantify the approximate number of metal ions bound to the RNA, and to identify some of the ligands to the ion. Innersphere metal ions are identified by an unusual magnesium hydrate feature that arises at ~322 cm−1 (Fig. 4B), as determined from early studies on electrolyte solutions [72] and Gaussian calculations (see Section 6.5), as well as studies on model compound dimethyl phosphate plus magnesium hydrate [25].
The number of bound metal ions was estimated by dividing the area of the attenuated PO2− Raman peak in the presence of Mg2+ (the area of the negative limb in the differential at 1100 cm−1 in the difference spectrum in Fig. 4A) to the area of the parent spectrum (i.e. the unsubtracted spectrum) in the absence of Mg2+. This ratio was ~0.07 in 20 mM Mg2+, which multiplied by the number of phosphates in HDV, provided ~5 ions bound on average to HDV, which was in good agreement with consideration of crystallographic studies [25]. In another study, the number of bound ions that change upon lowering the pH from 7.5 and 5.0 was estimated by taking the ratio of the innersphere feature of 323 cm−1 in the pH difference spectrum to the same feature in the parent spectrum at pH 7.5 [26]. This ratio was found to be about ~1/6th the total number of 5 ions, giving 1 ion ejected upon lowering the pH. This behavior is consistent with anticooperative interaction between proton binding and Mg2+ ion binding observed in biochemical studies [52]. Ejection of a single Mg2+ ion was further supported by complete loss of the 323 cm−1 feature in the pH 7.5 and 5.0 difference spectrum by modification of a single atom in the ribozyme, the N7 at position 1[26].
Ligands to innersphere-bound Mg2+ ions have been identified by detecting changes in the intensity of specific Raman bands upon changing either the solution or mutating specific atoms. Direct coordination of phosphate oxygens to metal ions can be discerned by the decrease in the negative differential peak near 1100 cm−1 in the difference spectra. This signal tracks with an increase in the resonance 323 cm−1, corresponding to [Mg(H2O)x]2+ where x ≤ 5. Both signals increase as the metal ion concentration is increased [25].
Interaction of a magnesium ion and an N7 of a guanine within the HDV ribozyme could be discerned by a differential feature at the guanine N7 resonance (~1489 cm−1) observed in pH 7.5–pH 5.0 difference spectrum [26] (Fig. 5B). Notably, this spectrum is remarkably similar to that of a pH 6.0–pH 3.0 difference spectrum of CMP (compare Figs. 5A and 5B; Table 4), which supported deprotonation of a cytosine in the HDV ribozyme (identified as C75, as per ref [24] and above); the HDV ribozyme difference spectrum also contains an additional feature (at 323 cm−1) for uptake of an innersphere Mg2+ ion, the above mentioned differential feature (at ~1489 cm−1) for a guanine N7 coordination, and several features associated with changes in the conformation of the sugar-phosphate backbone. Assignment of this guanosine was accomplished by growing crystals in which a 7-deazaguanosine modification was introduced at G1. In spectra of the 7-deazaguanosine mutant, both the innersphere feature at 323 cm−1 and the differential feature at ~1489 cm−1 were lost [26]. This experiment provided positive data supporting coordination of a [Mg(H2O)x]2+ ion to the N7 of G. One other feature was striking about the pH-difference spectrum: no evidence was found for innersphere coordination of the active site metal ion to PO2−, providing evidence against phosphate coordination in this instance. In summary, Raman can be used to identify metal ions as innersphere; calculate stoichiometries of metal ions, as well as subpopulations that change only with pH; and identify innersphere PO2− and N7 ligands, which can be mutually exclusive in at least some instances. Quantification of metal ion binding affinities can also be performed (manuscript in preparation).
Fig. 5.
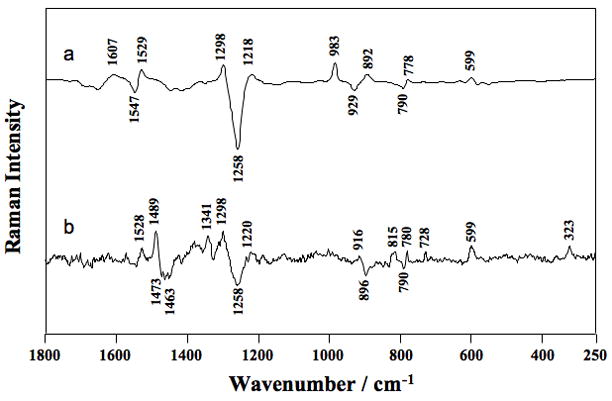
Raman difference spectra of (a) [CMP pH 6.0] minus [CMP pH 3.0] and (b) [HDV pH 7.5] minus [HDV pH 5.0]. The HDV ribozyme crystal was surrounded by cacodylate buffer (pH7.5) or acetate buffer (pH5.0) containing 20 mM Mg2+. Peak assignments are provided in Table 4. Adapted with permission from [26]. (Copyright, American Chemical Society.)
7. Concluding remarks
Raman crystallography has provided unique insights into RNA physicochemical properties, including pKas near neutrality; metal ion binding mode, quantification, affinity, and ligands; and conformational changes and affinity. Where comparisons are possible, agreement between biochemical and Raman crystallography has been superb. Native folding of the HDV ribozyme in solution and in the crystal, with its high solvent content, likely gives rise to this agreement. Based on this, we are encouraged that advanced approaches into RNA function by Raman crystallography will yield even deeper insight. In the future, Raman crystallography should also prove applicable to other functional RNAs, including riboswitches and larger ribozymes.
Selective isotopic labeling (isotope editing) of specific functional groups in the RNA should allow for assignment of ionizing bases and ligands without the need for deletion or substitution of functional groups. This is because the isotope-labeled atoms typically shift to a new region of the spectrum where they can be observed in isolation. In addition, isotope editing of scissile bonds may allow ribozyme reactions to be followed in the crystal and any reaction intermediates or tautomers of the bases, which typically have unusual vibrations, to be observed. An additional advantage of following the reaction in the crystal is that conditions that populate reaction intermediates can be determined and then used to freeze crystals for structural characterization [27]. As mentioned in Section 2.2, the rate of diffusion of material throughout the crystal is tens of seconds, and if the reaction is too fast, its rate can be slowed by changing the pH, temperature, or metal ions. Raman crystallography should continue to give new and exciting insights into the structure and function of RNA.
Acknowledgments
NSF Grant MCB-0527102 and the Pennsylvania State University for sabbatical support. BLG: Purdue University Department of Biochemistry, and the Purdue University Cancer Center. PRC: NIH Grant GM-54072
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Joyce GF, Orgel LE. In: RNA World. 3. Gesteland RF, Cech TR, Atkins JF, editors. Cold Spring Harbor Press; Cold Spring Harbor, New York: 2006. p. 768. [Google Scholar]
- 2.Turner DH, Bevilacqua PC. In: The RNA World. Gesteland RF, Atkins JF, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1993. pp. 447–464. [Google Scholar]
- 3.Benner SA, Carrigan MA, Ricardo A, Frye F. In: RNA World. 3. Gesteland RF, Cech TR, Atkins JF, editors. Cold Spring Harbor Press; Cold Spring Harbor, New York: 2006. p. 768. [Google Scholar]
- 4.Robertus JD, Ladner JE, Finch JT, Rhodes D, Brown RS, Clark BF, Klug A. Nature. 1974;250:546–551. doi: 10.1038/250546a0. [DOI] [PubMed] [Google Scholar]
- 5.Kim SH, Suddath FL, Quigley GJ, McPherson A, Sussman JL, Wang AH, Seeman NC, Rich A. Science. 1974;185:435–440. doi: 10.1126/science.185.4149.435. [DOI] [PubMed] [Google Scholar]
- 6.Fedor MJ, Williamson JR. Nat Rev Mol Cell Biol. 2005;6:399–412. doi: 10.1038/nrm1647. [DOI] [PubMed] [Google Scholar]
- 7.Doudna JA, Lorsch JR. Nat Struct Mol Biol. 2005;12:395–402. doi: 10.1038/nsmb932. [DOI] [PubMed] [Google Scholar]
- 8.Breaker RR. Science. 2008;319:1795–1797. doi: 10.1126/science.1152621. [DOI] [PubMed] [Google Scholar]
- 9.Henkin TM. Genes Dev. 2008;22:3383–3390. doi: 10.1101/gad.1747308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nudler E, Mironov AS. Trends Biochem Sci. 2004;29:11–17. doi: 10.1016/j.tibs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 11.Holbrook SR. Curr Opin Struct Biol. 2005;15:302–308. doi: 10.1016/j.sbi.2005.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott LG, Hennig M. Methods Mol Biol. 2008;452:29–61. doi: 10.1007/978-1-60327-159-2_2. [DOI] [PubMed] [Google Scholar]
- 13.Krasovska MV, Sefcikova J, Reblova K, Schneider B, Walter NG, Sponer J. Biophys J. 2006;91:626–638. doi: 10.1529/biophysj.105.079368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaiana AC, Westhof E, Auffinger P. Biochimie. 2006;88:1061–1073. doi: 10.1016/j.biochi.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Treiber DK, Williamson JR. Curr Opin Struct Biol. 1999;9:339–345. doi: 10.1016/S0959-440X(99)80045-1. [DOI] [PubMed] [Google Scholar]
- 16.Bevilacqua PC, Cerrone-Szakal AL, Siegfried NA. Q Rev Biophys. 2007;40:55–85. doi: 10.1017/S0033583507004593. [DOI] [PubMed] [Google Scholar]
- 17.Chadalavada DM, Knudsen SM, Nakano S, Bevilacqua PC. J Mol Biol. 2000;301:349–367. doi: 10.1006/jmbi.2000.3953. [DOI] [PubMed] [Google Scholar]
- 18.Chadalavada DM, Senchak SE, Bevilacqua PC. J Mol Biol. 2002;317:559–575. doi: 10.1006/jmbi.2002.5434. [DOI] [PubMed] [Google Scholar]
- 19.Brown TS, Chadalavada DM, Bevilacqua PC. J Mol Biol. 2004;341:695–712. doi: 10.1016/j.jmb.2004.05.071. [DOI] [PubMed] [Google Scholar]
- 20.Chadalavada DM, Cerrone-Szakal AL, Bevilacqua PC. RNA. 2007;13:2189–2201. doi: 10.1261/rna.778107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhuang X, Kim H, Pereira MJ, Babcock HP, Walter NG, Chu S. Science. 2002;296:1473–1476. doi: 10.1126/science.1069013. [DOI] [PubMed] [Google Scholar]
- 22.Russell R. Front Biosci. 2008;13:1–20. doi: 10.2741/2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golden BL. Methods Mol Biol. 2007;363:239–257. doi: 10.1007/978-1-59745-209-0_12. [DOI] [PubMed] [Google Scholar]
- 24.Gong B, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR. J Am Chem Soc. 2007;129:13335–13342. doi: 10.1021/ja0743893. [DOI] [PubMed] [Google Scholar]
- 25.Gong B, Chen Y, Christian EL, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR. J Am Chem Soc. 2008;130:9670–9672. doi: 10.1021/ja801861s. [DOI] [PubMed] [Google Scholar]
- 26.Chen JH, Gong B, Bevilacqua PC, Golden BL, Carey PR. Biochemistry. 2009 in press. [Google Scholar]
- 27.Carey PR. Annu Rev Phys Chem. 2006;57:527–554. doi: 10.1146/annurev.physchem.57.032905.104521. [DOI] [PubMed] [Google Scholar]
- 28.Spiro TG, Stein P. Annu Rev Phys Chem. 1977;28:501–521. [Google Scholar]
- 29.Skoog DA. Principles of Instrumental Analysis. CBS College Publishing; New York: 1985. [Google Scholar]
- 30.Callender R, Deng H. Annu Rev Biophys Biomol Struct. 1994;23:215–245. doi: 10.1146/annurev.bb.23.060194.001243. [DOI] [PubMed] [Google Scholar]
- 31.Zimmerman SB, Minton AP. Annu Rev Biophys Biomol Struct. 1993;22:27–65. doi: 10.1146/annurev.bb.22.060193.000331. [DOI] [PubMed] [Google Scholar]
- 32.Ferre-D’Amare AR, Doudna JA. In: Current Protocols in Nucleic Acid Chemistry. Beaucage SL, Bergstrom DE, Glick GD, Jones RA, editors. John Wiley & Sons, Inc; New York: 2000. pp. 7.6.1–7.6.13. [Google Scholar]
- 33.Wedekind JE, McKay DB. Methods Enzymol. 2000;317:149–168. doi: 10.1016/s0076-6879(00)17013-2. [DOI] [PubMed] [Google Scholar]
- 34.Ferre-D’Amare AR, Doudna JA. Curr Protoc Nucleic Acid Chem Chapter. 2001;7 doi: 10.1002/0471142700.nc0706s00. Unit 7 6. [DOI] [PubMed] [Google Scholar]
- 35.Wedekind JE, McKay DB. Nat Struct Biol. 1999;6:261–268. doi: 10.1038/6700. [DOI] [PubMed] [Google Scholar]
- 36.Wedekind JE, McKay DB. Biochemistry. 2003;42:9554–9563. doi: 10.1021/bi0300783. [DOI] [PubMed] [Google Scholar]
- 37.Yajima R, Proctor DJ, Kierzek R, Kierzek E, Bevilacqua PC. Chem Biol. 2007;14:23–30. doi: 10.1016/j.chembiol.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 38.Carey PR. Biochemical application of Raman and resonance Raman spectroscopies. Academic Press; New York: 1982. [Google Scholar]
- 39.Altose MD, Zheng Y, Dong J, Palfey BA, Carey PR. Proc Natl Acad Sci U S A. 2001;98:3006–3011. doi: 10.1073/pnas.061029598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benevides JM, Tsuboi M, Bamford JK, Thomas GJ., Jr Biophys J. 1997;72:2748–2762. doi: 10.1016/S0006-3495(97)78917-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen MC, Giege R, Lord RC, Rich A. Biochemistry. 1975;14:4385–4391. doi: 10.1021/bi00691a007. [DOI] [PubMed] [Google Scholar]
- 42.Li TS, Chen ZG, Johnson JE, Thomas GJ., Jr Biochemistry. 1990;29:5018–5026. doi: 10.1021/bi00473a004. [DOI] [PubMed] [Google Scholar]
- 43.Li T, Chen Z, Johnson JE, Thomas GJ., Jr Biochemistry. 1992;31:6673–6682. doi: 10.1021/bi00144a006. [DOI] [PubMed] [Google Scholar]
- 44.Draper DE. Biophys J. 2008;95:5489–5495. doi: 10.1529/biophysj.108.131813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Misra VK, Draper DE. Biopolymers. 1998;48:113–135. doi: 10.1002/(SICI)1097-0282(1998)48:2<113::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 46.Draper DE. RNA. 2004;10:335–343. doi: 10.1261/rna.5205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kieft JS, Tinoco I., Jr Structure. 1997;5:713–721. doi: 10.1016/s0969-2126(97)00225-6. [DOI] [PubMed] [Google Scholar]
- 48.Maderia M, Horton TE, DeRose VJ. Biochemistry. 2000;39:8193–8200. doi: 10.1021/bi000140l. [DOI] [PubMed] [Google Scholar]
- 49.Hougland JL, Kravchuk AV, Herschlag D, Piccirilli JA. PLoS Biol. 2005;3:1536–1548. doi: 10.1371/journal.pbio.0030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maderia M, Hunsicker LM, DeRose VJ. Biochemistry. 2000;39:12113–12120. doi: 10.1021/bi001249w. [DOI] [PubMed] [Google Scholar]
- 51.Bevilacqua PC, Brown TS, Nakano S, Yajima R. Biopolymers. 2004;73:90–109. doi: 10.1002/bip.10519. [DOI] [PubMed] [Google Scholar]
- 52.Nakano S, Chadalavada DM, Bevilacqua PC. Science. 2000;287:1493–1497. doi: 10.1126/science.287.5457.1493. [DOI] [PubMed] [Google Scholar]
- 53.Cerrone-Szakal AL, Siegfried NA, Bevilacqua PC. J Am Chem Soc. 2008;130:14504–14520. doi: 10.1021/ja801816k. [DOI] [PubMed] [Google Scholar]
- 54.Luptak A, Ferre-D’Amare AR, Zhou K, Zilm KW, Doudna JA. J Am Chem Soc. 2001;123:8447–8452. doi: 10.1021/ja016091x. [DOI] [PubMed] [Google Scholar]
- 55.Gong B. Thesis: Raman spectroscopic studies of the hepatitis delta virus. Case Western Reserve University; Cleveland, OH: 2008. [Google Scholar]
- 56.Emilsson GM, Nakamura S, Roth A, Breaker RR. RNA. 2003;9:907–918. doi: 10.1261/rna.5680603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yajima R. Thesis: Insight into ribozyme structure and function using conformationally-restricted RNA: Applications to X-ray crystallography. The Pennsylavnia State University; University Park: 2006. [Google Scholar]
- 58.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milligan JF, Uhlenbeck OC. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 60.Bevilacqua PC, Brown TS, Chadalavada D, Parente AD, Yajima R. Kinetic Analysis of Ribozyme Cleavage. Oxford University Press; Oxford: 2003. [Google Scholar]
- 61.Rupert PB, Ferre-D’Amare AR. Nature. 2001;410:780–786. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- 62.Moore MJ, Sharp PA. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- 63.Dong J, Swift K, Matayoshi E, Nienaber VL, Weitzberg M, Rockway T, Carey PR. Biochemistry. 2001;40:9751–9757. doi: 10.1021/bi010955+. [DOI] [PubMed] [Google Scholar]
- 64.Thomas GJ, Jr, Wang AH-J. Nucleic Acids Mol Biol. 1988;2:1–30. [Google Scholar]
- 65.Thomas GJ, Jr, Tsuboi M. Adv Biophys Chem. 1993;3:1–70. [Google Scholar]
- 66.Frisch MJ, et al. Gaussian, Inc; Pittsburgh, PA: 1998. [Google Scholar]
- 67.Lee CT, Yang WT, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 68.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 69.Scott AP, Radom L. J Phys Chem. 1996;100:16502–16513. [Google Scholar]
- 70.Perrotta AT, Shih I, Been MD. Science. 1999;286:123–126. doi: 10.1126/science.286.5437.123. [DOI] [PubMed] [Google Scholar]
- 71.Bevilacqua PC. Biochemistry. 2003;42:2259–2265. doi: 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- 72.Peleg M. J Phys Chem. 1972;76:1019–1025. [Google Scholar]
- 73.Carey PR, Dong J. Biochemistry. 2004;43:8885–8893. doi: 10.1021/bi049138a. [DOI] [PubMed] [Google Scholar]