

Abstract
Defects in DNA damage responses may underlie genetic instability and malignant progression in melanoma. Cultures of normal human melanocytes (NHMs) and melanoma lines were analyzed to determine whether global patterns of gene expression could predict the efficacy of DNA damage cell cycle checkpoints that arrest growth and suppress genetic instability. NHMs displayed effective G1 and G2 checkpoint responses to ionizing radiation-induced DNA damage. A majority of melanoma cell lines (11/16) displayed significant quantitative defects in one or both checkpoints. Melanomas with B-RAF mutations as a class displayed a significant defect in DNA damage G2 checkpoint function. In contrast the epithelial-like subtype of melanomas with wild-type N-RAS and B-RAF alleles displayed an effective G2 checkpoint but a significant defect in G1 checkpoint function. RNA expression profiling revealed that melanoma lines with defects in the DNA damage G1 checkpoint displayed reduced expression of p53 transcriptional targets, such as CDKN1A and DDB2, and enhanced expression of proliferation-associated genes, such as CDC7 and GEMININ. A Bayesian analysis tool was more accurate than significance analysis of microarrays for predicting checkpoint function using a leave-one-out method. The results suggest that defects in DNA damage checkpoints may be recognized in melanomas through analysis of gene expression.
INTRODUCTION
During the period 1973-1999, the incidence rate for melanoma among Caucasian American men >65 years of age rose over four-fold from 20 to 90 per 100,000 and the death rate doubled (Geller et al., 2002). Sunlight, or solar radiation, is an important etiologic factor, with epidemiologic studies indicating that severe childhood sunburns contribute the greatest risk (Gilchrest et al., 1999). There is also genetic susceptibility to melanoma as demonstrated by several familial cancer syndromes, xeroderma pigmentosum (Kraemer et al., 1989), familial atypical mole and malignant melanoma syndrome (Gibbs et al., 2002), retinoblastoma (Fletcher et al., 2004), and Li-Fraumeni syndrome (Cohen et al., 2005). Melanomagenesis is associated with defects in nucleotide excision repair of solar radiation-induced DNA damage, as in xeroderma pigmentosum, and cell cycle checkpoints that arrest growth after DNA damage and oncogene activation, as in familial atypical mole and malignant melanoma syndrome, retinoblastoma, and Li-Fraumeni syndrome. Lastly, MC1R polymorphisms (associated with red hair and freckling) are also associated with melanoma risk (Pho et al., 2006).
Somatic mutations in B-RAF or N-RAS are key genetic alterations that contribute to development of melanoma (Eskandarpour et al., 2003; Smalley and Herlyn, 2004). Activating mutations in these genes are comparatively common in melanoma, with about 50-75% of melanomas having mutations in B-RAF and 10-15% having mutations in N-RAS (Daniotti et al., 2004; Thomas et al., 2004). A recent molecular epidemiologic study of melanoma in North Carolina identified a significant association of these oncogene mutations with age (Thomas et al., 2007). Although clinically similar in terms of progression and vertical growth, melanomas with B-RAF mutations appeared 15 years earlier than melanomas with N-RAS mutations. Mutations in these genes also occur very early in melanomagenesis, as similar frequencies of melanocytic nevi and melanomas express these mutant oncogenes (Kumar et al., 2004).
Mutations in N-RAS and B-RAF may stimulate growth, in part, through activation of ERK1/2 (Satyamoorthy et al., 2003; Shields et al., 2007) and enhance genetic instability by attenuation of cell cycle checkpoint function. DNA double-strand breaks (DSBs) are known to activate a cell cycle checkpoint response that blocks progression of G2 cells into mitosis (G2 checkpoint) (Paules et al., 1995). Mitotic entry normally requires activation of cyclin B1/Cdk1 kinase activity (mitosis-promoting factor) within the interphase nucleus. In response to DNA DSB, ataxia telangiectasia-mutated (ATM), and ATM and rad3-related (ATR)-dependent activation of Chk1 leads to inhibition of mitosis-promoting factor and inhibition of Plk1 prevents the accumulation of mitosis-promoting factor within the nucleus (reviewed in Kaufmann et al., 2002). Ras oncogenes have been reported to enhance expression of cyclin B1 and cyclin-dependent kinase 1 (Santana et al., 2002), and to attenuate G2 checkpoint function (Agapova et al., 2004; Knauf et al., 2006). Thus, expression of oncogenic N-RAS and B-RAF may attenuate G2 checkpoint function in cells with defects in p16 and p53, thereby enhancing genetic instability in cells with DNA damage.
Although p53 mutations are comparatively rare in sporadic melanoma, the demonstration of UV signature mutations in p53 in melanomas from xeroderma pigmentosum patients indicates that melanocyte p53 is a target of UV and suggests that p53 signaling suppresses melanomagenesis (Giglia-Mari and Sarasin, 2003). A recent report showed that inactivation of p53 cooperates with induction of telomerase and oncogenic N-RAS to induce melanomas in human xenobiotic skin grafts (Chudnovsky et al., 2005). Alterations in the p53 signaling pathway could contribute to melanoma genesis through attenuation of DNA repair, cell cycle checkpoint function, or apoptosis (Kastan and Bartek, 2004), leading to enhanced mutation and reduced cell death. p53 function is regulated by ARF, a second product of the CDKN2A/INK4A locus that is commonly deleted in melanoma (Grafstrom et al., 2005).
Recent studies demonstrate that melanomas and melanoma cell lines display large numbers of nuclear foci that are positive for phospho-histone H2AX as a marker of replication stress and DNA DSBs (Gorgoulis et al., 2005; Warters et al., 2005). Melanomas also display severe chromosomal instability, with many amplifications and deletions, as determined by array comparative genomic hybridization (Bauer and Bastian, 2006). As the p53-dependent G1 checkpoint should arrest the growth of cells with DNA DSBs, these results suggest that inactivation of G1 checkpoint function may be common in melanoma, permitting melanoma cells to proliferate with broken and rearranged chromosomes (Gorgoulis et al., 2005). How the DNA damage signal is ignored in melanomas with wild-type p53 is unknown.
Analysis of global gene expression through microarray technology can discover patterns of co-regulated genes that reveal unanticipated biological relationships. The basal cell subtype of breast cancer was discovered in this way (Alizadeh et al., 2005). Several studies have examined the global patterns of gene expression in melanomas (Seykora et al., 2003; Hoek et al., 2004; Haqq et al., 2005; Nambiar et al., 2005; Shields et al., 2007). These studies showed an association of TWIST expression with epithelial-mesenchymal transition and worsened outcome (Hoek et al., 2004), identified a large suite of genes regulated by the extracellular signal-regulated kinase ERK (Shields et al., 2007), and established the occurrence of an epithelial-like subtype of melanoma with wild-type N-RAS and B-RAF proto-oncogenes (Bloethner et al., 2005; Shields et al., 2007). In this study, the patterns of gene expression in melanoma cell lines were analyzed and correlated with quantitative variation in DNA damage checkpoint function. We were interested in whether patterns of gene expression could be used to identify melanomas with defects in DNA damage checkpoint function. The results show that melanoma cell lines with defective G1 and G2 checkpoint functions can often be recognized by their patterns of gene expression in the basal, undamaged state.
RESULTS
DNA damage cell cycle checkpoint function was quantified in secondary cultures of normal human melanocytes (NHMs) and in the melanoma cell lines detailed in Table 1. Because DNA DSBs appear to be a feature of early and late stages of melanomagenesis (Bartkova et al., 2005; Warters et al., 2005), we considered it appropriate to use ionizing radiation (IR) to induce DNA DSBs in melanocytes and melanoma cell lines. DNA damage checkpoints acting in G1 and G2 cells were analyzed for functional activity using flow cytometric assays. By monitoring efficacy using quantitative assays of cell cycle arrest in response to DNA DSBs, melanoma lines with defective checkpoint function were identified and metrics that estimated the degree of dysfunction were generated.
Table 1.
Sources of melanoma cell lines and their N-RAS/B-RAF mutation status
| Melanoma line | N-RAS | B-RAF | Source |
|---|---|---|---|
| PMWK | WT | WT | J Arbiser (Emory) |
| Mel505 | WT | WT | J Hansson (Karolinska) |
| SK-Mel-187 | WT | WT | A Houghton (Sloan Kettering) |
| RPMI8322 | WT | WT | J Hansson (Karolinska) |
| SK-Mel-173 | Q61K | WT | A Houghton (Sloan Kettering) |
| VMM39 | Q61K | WT | C Slingluff (UVA) |
| Mel224 | Q61R | WT | J Hansson (Karolinska) |
| SK-Mel-103 | Q61R | WT | A Houghton (Sloan Kettering) |
| SK-Mel-147 | Q61R | WT | A Houghton (Sloan Kettering) |
| SK-Mel-119 | Q61R | WT | A Houghton (Sloan Kettering) |
| UACC257 | WT | V600E | M Soengas (U Michigan) |
| A375 | WT | V600E | J Arbiser (Emory) |
| WM2664 | WT | V600D | J Arbiser (Emory) |
| A2058 | WT | V600E | J Arbiser (Emory) |
| SK-Mel-24 | WT | V600E | American Type Culture Collection |
| SK-Mel-28 | WT | V600E | American Type Culture Collection |
WT, wild type.
NHMs express a functional DNA damage G1 checkpoint
DNA damage G1 checkpoint function was quantified by treating cells with 1.5 Gy IR (or sham treatment for control) and then labeling cells with BrdU for 2 hours beginning 6 hours post-treatment. S phase nuclei that incorporated BrdU into DNA were stained with a fluorescein-labeled anti-BrdU antibody and counterstained with propidium iodide. Two-channel flow cytometry was performed to quantify the fraction of cells in the first half of the S phase; such cells had 2-3N DNA content and positive labeling with BrdU. The IR-induced reduction in the percentage of cells in the first half of S provides a quantitative index of G1 checkpoint function (Doherty et al., 2003). Foreskin fibroblasts with effective G1 checkpoint function (NHF1-hTERT) displayed >90% reduction in the fraction of cells in early S phase, 6-8 hours after irradiation, as irradiated G1 cells delayed entry into S phase (Figure 1a). NHMs also displayed a significant reduction in the fraction of cells in the first half of S phase, after treatment with IR (Figure 1a). The degree of inhibition in NHMs averaged 57%, in the range previously reported for cultures of human bladder epithelial cells (Doherty et al., 2003). Expression of a dominant-negative p53 allele (p53H179Q) or HPV16E6 in the NHM-2 melanocytes by retroviral gene transfer ablated the IR-induced G1 arrest (NHM-2-179, NHM-2-E6; Figure 1b). These results indicated that NHMs express an effective p53-dependent G1 checkpoint response to IR-induced DNA damage.
Figure 1. G1 checkpoint function in NHMs and melanoma cell lines.

(a). Flow cytometric analysis of BrdU incorporation versus DNA content. Normal human fibroblasts (F1-hTERT) and melanocytes (NHM-2) were incubated with BrdU at 6-8 hours after 1.5 Gy IR or sham treatment, and then processed for analysis of the fraction of cells in the first half of S phase (BrdU-labeled and 2-3N DNA content, enclosed by boxes). The IR-induced reduction in this fraction was a quantitative measure of DNA damage G1 checkpoint function. (b) Quantification of G1 checkpoint function in fibroblasts, melanocytes, and melanoma lines. Cells were treated with 1.5 Gy IR. The fraction of IR-treated cells in the first half of S phase, 6-8 hours after treatment, was expressed as a percentage of the sham-treated control as a quantitative index of G1 checkpoint function. Results represent the mean percent of control; error bars enclose one standard deviation above the mean (n = 2-5). *P<0.05 versus NHMs, by Dunnett's t-test, with adjustment for multiple comparisons; #P< 0.05 versus NHM-2 (Student's t-test).
Western immunoblot was used to monitor checkpoint signaling in response to DNA damage in the NHM-4 melanocyte culture. Cells were harvested 2, 6, and 24 hours after 1.5 Gy, or 6 hours after sham treatment as the control. After quantification of protein concentrations, equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis. The NHM-4 preparation expressed the checkpoint kinases, ATM, ATR and the effector of G1 checkpoint function, p53 (Figure 2). There was little change in p53 protein after IR treatment although there was a clear induction of phosphorylation at ser-15 in p53, which appeared to peak at 2 hours and diminish by 24 hours after IR. This phosphorylation of p53 is ATM-dependent in IR-treated normal human fibroblasts (Kaufmann et al., 2003). NHM-4 also expressed the checkpoint-transducing kinases Chk1 and Chk2. Treatment with IR produced little phosphorylation of Chk1 at ser-317, but strong phosphorylation of Chk2 at ser-68. This IR-induced phosphorylation of Chk2 is also ATM dependent (Brown et al., 1999). The NHM-4 cells expressed p21Waf1 and p16; p21Waf1 was induced 2, 6, and 24 hours after IR while p16 appeared to be induced by IR modestly at 24 hours. These results indicated that ATM signaling to p53 and Chk2, as well as induction of p21Waf1, was effective in the IR-treated NHM-4 melanocyte strain.
Figure 2. Western immunoblot analysis of checkpoint protein expression in NHMs.
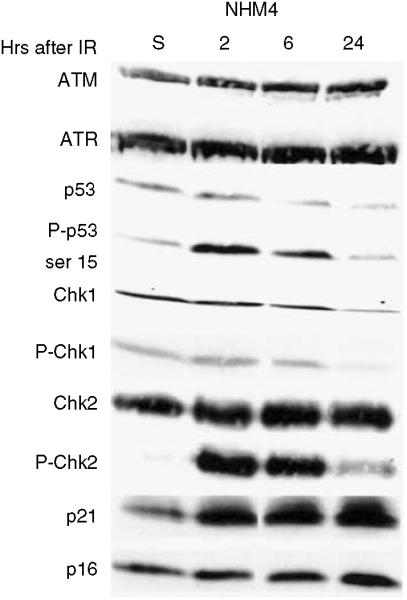
NHM-4 cells were treated with 1.5 Gy IR then harvested 2, 6, and 24 hours later. Controls (S) were harvested 6 hours after sham treatment. Protein concentration in cell lysates was determined, so that equal protein was loaded for separation by SDS-polyacrylamide gel electrophoresis. Proteins were transferred to nitrocellulose membranes and probed for expression of various checkpoint proteins.
Defective G1 checkpoint function in melanoma cell lines
Melanoma cell lines displayed a continuous range of responses to IR, with two lines delaying S phase entry by 75% and five lines displaying less than 10% inhibition of S phase entry (Figure 1b). Statistical evaluation of the data set indicated that 10 of the 16 melanoma lines displayed a defective G1 checkpoint response to IR-induced DNA damage in comparison to NHMs (Table 2).
Table 2.
DNA damage checkpoint function in melanoma cell lines
| Melanoma line | Mutation status | G2 checkpoint | G2 checkpoint |
|---|---|---|---|
| PMWK | WT | Defective | Effective |
| Mel505 | WT | Defective | Effective |
| SK-Mel-187 | WT | Defective | Effective |
| RPMI8322 | WT | Defective | Effective |
| Mel224 | N-RAS | Defective | Effective |
| SK-Mel-119 | N-RAS | Defective | Effective |
| SK-Mel-103 | N-RAS | Defective | Effective |
| SK-Mel-147 | N-RAS | Effective | (Effective)1 |
| SK-Mel-173 | N-RAS | Effective | Defective |
| VMM39 | N-RAS | Defective | (Effective) |
| SK-Mel-24 | B-RAF | Effective | (Effective) |
| A375 | B-RAF | Effective | Effective |
| WM2664 | B-RAF | Effective | (Effective) |
| UACC257 | B-RAF | Effective | (Effective) |
| SK-Mel-28 | B-RAF | Defective | Defective |
| A2058 | B-RAF | Defective | Defective |
WT, wild type.
The qualification of checkpoint status as (Effective) denotes melanoma cell lines that were significantly different (P<0.05) than NHMs when adjustments for multiple comparisons were not made.
Western immunoblot analysis was performed on six melanoma lines that were compared to normal human fibroblasts (F1-hTERT) for their response to IR-induced DNA damage. This analysis included two melanoma lines with wild-type N-RAS and B-RAF alleles (RPMI8322 and SK-Mel-187), two lines with mutant B-RAF (SK-Mel-24 and A2058), and two with mutant N-RAS (SK-Mel-173 and -103). Protein was quantified in cell lysates and equal amounts were loaded for electrophoretic separation on polyacrylamide gels. Levels of expression of proteins and specific post-translational modifications were quantified by densitometry and the relative pixel intensities were normalized to the F1-hTERT sham (included on every blot). This analysis revealed substantial variation among melanoma lines in the levels of expression of checkpoint-associated proteins and their post-translational modifications.
Protein expression within the G1 checkpoint signaling pathway (Figure 3) was well correlated with functional capacity (Figure 1b). Two G1 checkpoint-defective melanomas (RPMI8322 and SK-Mel-187) displayed enhanced expression of p53, suggestive of inactivating mutations that increase protein half-life, with little or no induction of p21Waf1 post-IR. The G1 checkpoint-defective A2058 line expressed nearly normal levels of p53, which was phosphorylated at ser-15 after treatment with IR. However, this line overexpressed p16 substantially and did not express p21Waf1. The line may grow in the presence of high levels of p16, because RB function is defective (Noonan et al., 2005). The defects in both RB and p21Waf1 can explain the defect in G1 checkpoint function in the A2058 line. Two lines had effective G1 checkpoint function (SK-Mel-24 and -173); these lines activated p53 through phosphorylation of ser-15 and induced p21Waf1 after treatment with IR. SK-Mel-103 displayed a moderate but significant attenuation of G1 checkpoint function. ATM expression appeared to be reduced in this line, and although p53 was activated by IR, induction of p21Waf1 by IR was delayed. There was little induction at 2 and 6 hours, and only a modest two-fold induction at 24 hours after irradiation. Among the six melanomas tested, four expressed no detectable p16 and one expressed very high levels. These results are consistent with the common inactivation of the p16/RB pathway in melanoma. The two G1 checkpoint-effective melanomas (SKMel-173 and -24) displayed normal signaling through p53 to p21Waf1. As these lines did not express p16, the results confirm that p16 is not required for G1 checkpoint response to IR-induced DNA damage.
Figure 3. Immunoblot analysis of G1 checkpoint protein expression in melanoma lines.

Details of cell treatment and analysis of protein expression are as in the legend to Figure 2. NHF1-hTERT cells were included as a normalization control in all analyses. This figure represents a composite of results from three different membranes. Below each immunoblot image are depicted pixel intensity values relative to the sham-treated NHF1-hTERT control that was included on the same membrane. Only a representative image of the fibroblast controls is shown. Mutation status at the B-RAF and N-RAS loci and G1 checkpoint function are indicated for each cell line below and above the composite image, respectively.
NHMs express a functional DNA damage G2 checkpoint
DNA damage G2 checkpoint function was scored by monitoring a mitosis-specific form of phospho-histone H3, using a fluoresceinated antibody (Figure 4a). Mitotic cells with 4N DNA content stained positively with this antibody, allowing their quantification by flow cytometry. Normal human fibroblasts (F1-hTert) responded to 1.5 Gy IR, with >95% inhibition of mitosis, as cells in G2 delayed entry into mitosis, and mitotic cells completed cell division and entered G1. Similarly, irradiation of the NHM-2 melanocyte strain produced >90% inhibition of mitosis (Figure 4a and b). Secondary cultures of NHMs responded to IR, with a mean 90% inhibition of mitosis, indicative of effective DNA damage G2 checkpoint function (Figure 4b).
Figure 4. DNA damage G2 checkpoint function in NHMs and melanoma lines.

(a) Flow cytometric quantification of the percentage of mitotic cells with 4N DNA content and expression of phospho-histone H3 (enclosed by boxes). Normal human fibroblasts (NHF1-hTERT) and melanocytes (NHM-2) were treated with 1.5 Gy IR or sham treated, then harvested 2 hours later for quantification of mitotic cells. The IR-induced reduction in the fraction of mitotic cells was used as a quantitative measure of DNA damage G2 checkpoint function. (b) Quantification of DNA damage G2 checkpoint function in fibroblasts, melanocytes, and melanoma lines. The fraction of IR-treated cells in mitosis 2 hours after treatment was expressed as a percentage of the sham-treated control as a quantitative index of G2 checkpoint function. Results represent the mean percent of control; error bars enclose one standard deviation above the mean (n = 2-7). *P<0.05 versus NHMs, by Dunnett's t-test, with adjustment for multiple comparisons.
Defective G2 checkpoint function in melanoma lines with mutant B-RAF
As was seen for the G1 checkpoint response, melanoma cell lines displayed a continuous range of G2 checkpoint response to IR, with some lines inhibiting mitosis by >95% and one line inhibiting mitosis by <40% (Figure 4b). In comparison to the average response in NHMs, three of 16 melanoma lines displayed a statistically significant defect in DNA damage G2 checkpoint function (P<0.05 after correction for multiple comparisons). Five other lines (SK-Mel-147, SK-Mel-24, UACC257, VMM39, and WM2664) displayed a significant (P<0.05) defect in G2 checkpoint function in comparison to NHMs, when the analysis was not corrected for multiple comparisons (Table 2). Analysis of ATM, ATR, Chk1, NBS1, and Myt1 protein expression in the melanoma lines depicted in Figure 3 did not reveal clear differences that could explain the variation in G2 checkpoint function among the lines.
Association between N-RAS and B-RAF mutations and DNA damage checkpoint function
Activated mutant Ras oncogenes have been shown to attenuate DNA damage G2 checkpoint function (Agapova et al., 2004), and melanomas commonly harbor mutations in N-RAS or its downstream effector B-RAF. To determine whether the variation in checkpoint function among melanomas was related to mutations in these components of the mitogen-activated protein kinase signaling pathway, N-RAS and B-RAF alleles were sequenced at the codons that are typically mutated in melanomas (Thomas et al., 2007). This analysis identified two highly significant associations between checkpoint function and oncogene mutation status (Figure 5). As a class, melanoma lines with wild-type N-RAS and B-RAF alleles displayed effective G2 checkpoint response to IR, with <5% of G2 cells evading the checkpoint. This same class of melanomas displayed a defective G1 checkpoint response to IR, with >90% of G1 cells evading the checkpoint. As a class, melanomas with a mutant B-RAF allele displayed an effective G1 checkpoint response to IR. However, these lines displayed significantly attenuated G2 checkpoint function, with an average of 38% of G2 cells evading the checkpoint. The class of melanomas with a mutant N-RAS allele displayed an average of 21% of G2 cells evading the checkpoint, a moderate but insignificant attenuation in comparison to NHMs. This class of melanomas also displayed variable G1 checkpoint response that was not significantly different from NHMs.
Figure 5. Defective DNA damage checkpoint function in melanoma subtypes based on oncogene mutation status.

DNA damage G1 (a) and G2 (b) checkpoint functions were quantified in normal human fibroblasts (NHF, n = 3), ataxia telangiectasia fibroblasts (AT, n = 3), normal human melanocytes (NHM, n = 5), melanoma lines with wild-type N-RAS and B-RAF (WT, n = 4), melanoma lines with mutant N-RAS (NRAS, n = 6), and melanoma lines with mutant B-RAF (BRAF, n = 6). After log-transformation of data, class averages were compared by Student's t-test.
Defects in cell cycle checkpoint function were comparatively common in melanoma cell lines, supporting the hypothesis that genetic instability in melanoma may be due in part to defects in the system of response to DNA damage (Table 2). Defects in checkpoint function did not appear to sort randomly among the melanoma lines. For example, the four melanoma lines with wild-type N-RAS and B-RAF alleles and effective DNA damage G2 checkpoint function all displayed defective DNA damage G1 checkpoint function. These results suggest that defects in G1 and G2 checkpoints arise in melanomas from separate genetic alterations. Thus, there are at least two ways that DNA damage checkpoint function can be disturbed during the development of melanoma, inactivation of G1 checkpoint independent of B-RAF and N-RAS mutations, and attenuation of G2 checkpoint associated with B-RAF mutations.
Signatures of defective DNA damage checkpoint function
Having established the occurrence of functional defects in DNA damage cell cycle checkpoints in melanoma lines, global gene expression was evaluated to search for signatures that were associated with the defects. The 44K Agilent human oligonucleotide microarray was used for expression profiling and each cell line was analyzed once. We have previously reported that the melanoma lines with wild-type N-RAS and B-RAF alleles displayed a unique signature of gene expression in comparison to the lines with mutations in N-RAS and B-RAF (Shields et al., 2007). The oncogene mutations were associated with high levels of phospho-ERK1/2 and enhanced expression of >100 ERK1/2-responsive mRNAs. Statistical evaluation of the microarray data sets also identified gene sets that were associated with the functional activity of DNA damage checkpoints.
Two methods of analysis tested for genes that varied among the NHM strains and melanoma lines according to the efficacy of checkpoint function. Significance analysis of microarrays (SAM) (Tusher et al., 2001) has been commonly applied to microarray data to identify genes that are correlated with selected treatments or cell types. A new method based on Bayesian statistical theory was developed. Checkpoint function was evaluated as a binary variable to compare lines with checkpoint responses greater and less than the median response, using the quantitative data (Figures 1b and 4b).
For the analysis of G1 checkpoint function in melanocytes and melanomas, SAM identified 26 genes that were significantly different in the two classes (false discovery rate (FDR) <0.1), while the Bayesian analysis identified 166 genes that were significantly different (FDR = 0.05) (the gene lists are given in the Supplementary Table S1). Genes on both lists included the well-established p53 target genes CDKN1A (p21Waf1) and DDB2, which were expressed at lower levels in G1 checkpoint-defective melanomas. Several cell proliferation-associated genes identified in the Bayesian list were expressed at higher levels in G1 checkpoint-defective lines, including CDC7, CKS1, cyclin B1, CDC2, GEMININ, RPA3, and RFC4. Hierarchical clustering of the gene lists indicated that both methods accurately separated the melanoma lines according to G1 checkpoint function (Figure 6).
Figure 6. Signatures of gene expression that were correlated with defective G1 checkpoint function.

Agilent 44K long-oligo microarrays were used to monitor global gene expression in NHMs and melanoma lines. Cells were harvested in log-phase growth and levels of gene expression determined relative to a universal reference. Left panel: unsupervised hierarchical cluster of genes that were determined by SAM to be correlated with G1 checkpoint function (FDR<0.1). Right panel: unsupervised hierarchical cluster of genes that were determined by Bayesian analysis to be correlated with G1 checkpoint function (FDR = 0.05). Heat maps are median-centered. Gene lists are reported in the Supplementary Table S1.
A leave-one-out procedure was employed to determine the predictive efficacy of the two analysis tools. The data set was repeatedly queried with one sample removed using the binary analysis, to generate lists of genes that distinguished checkpoint-effective and -defective lines, and then the withheld sample was tested to see if it segregated with the appropriate centroid (Tibshirani et al., 2002). The Bayesian analysis tool with an FDR of 0.05 assigned the left-out sample to the appropriate centroid in 78% of tests. SAM with an FDR of <0.1 assigned the left-out sample correctly in 69% of tests. Both tools incorrectly classified G1 checkpoint function in the NHM2 lines with expression of HPV16E6 or p53H179Q to inactivate G1 checkpoint function. Even when focused on gene expression that distinguishes G1 checkpoint-effective and -defective lines, the analysis tools placed the genetically modified NHM2 lines with the parental NHM2 strain and the other checkpoint-effective NHMs. Melanoma lines with defects in G1 checkpoint function must display alterations in gene expression beyond simply inactivation of p53. When considering the melanoma lines using the entire data set, the Bayesian analysis tool correctly assigned G1 checkpoint function in 13 out of 16 cases for a correct classification rate of 81%.
A different set of genes was associated with defective DNA damage G2 checkpoint function. When G2 checkpoint function was evaluated as a binary variable (effective versus defective), SAM identified only four genes (FDR<0.1), while the Bayesian method identified 149 genes (FDR = 0.05) (Figure 7; see Supplementary Table S1 for gene lists). In the leave-one-out analysis, the Bayesian method assigned the left-out sample to the appropriate centroid in 76% of tests, while SAM assigned the left-out sample to the appropriate centroid in 69% of tests.
Figure 7. Signature of gene expression that was correlated with defective G2 checkpoint function.
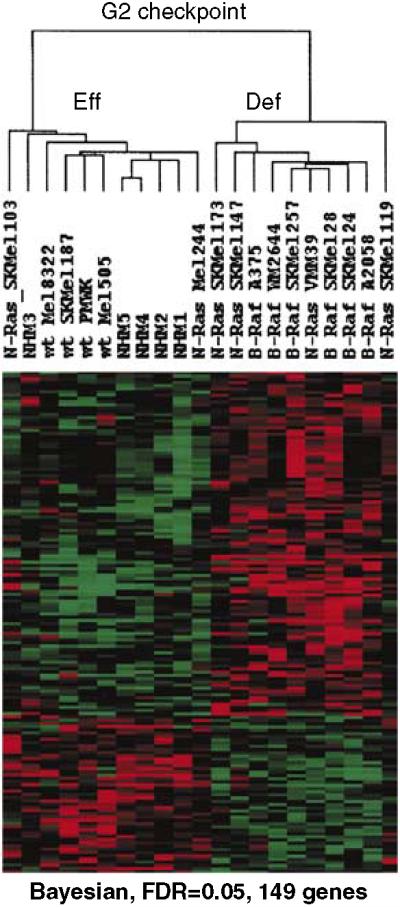
Unsupervised hierarchical cluster of genes that were determined by Bayesian analysis to be correlated with G2 checkpoint function (FDR = 0.05). The gene list is reported in the Supplementary Table S1.
DISCUSSION
We hypothesized that cell lines derived from malignant melanomas would display defects in cell cycle checkpoint function in order for cells to proliferate with the chromosomal instability that characterizes this malignancy. The results indicated that in comparison to NHMs in secondary cultures, the melanoma lines often displayed functional defects in DNA damage cell cycle checkpoints that arrest cell division in G1 or G2. These defects in checkpoint function were associated with distinct patterns of gene expression.
Defective DNA damage checkpoint function in melanoma lines
Recent reports have demonstrated DNA damage and the activation of DNA damage responses in melanomas and dysplastic nevi (Gorgoulis et al., 2005; Warters et al., 2005). While normal melanocyte nuclei displayed very few or no foci of phosphorylated histone H2AX (γ-H2AX), significantly increased frequencies of melanoma and dysplastic nevus cell nuclei displayed γ-H2AX foci. Foci of γ-H2AX expression occur at sites of DNA DSBs (Rogakou et al., 1999; Bekker-Jensen et al., 2006), suggesting that malignant melanocytes and pre-malignant precursors experience increased levels of DNA damage in comparison to normal melanocytes. Expression of γ-H2AX also was associated with the presence of activated forms of ATM, p53, and Chk2, demonstrating activation of the DNA damage response (Bartkova et al., 2005; Gorgoulis et al., 2005). This condition of enhanced DNA damage and activation of the DNA damage response was induced in normal human fibroblasts by expression of gene products such as CDC6, cyclin E, and E2F1 that drive cells into a precocious S phase and by activated oncogenes, including H-RAS (Bartkova et al., 2005, 2006; Di Micco et al., 2006). Normal cell proliferation, as induced by growth factors in medium, did not cause DNA damage, suggesting that the disregulated S phase was responsible (Bartkova et al., 2005). This work forms the basis for a hypothesis that activated oncogenes, such as N-RAS and B-RAF, force cells to enter a precocious S phase, producing a condition of replication stress that leads to the development of DNA DSB. The resulting DNA damage response includes activation of ATM and p53 to arrest the growth of cells with damaged DNA. Cancer emerges under these conditions as checkpoint control mechanisms that limit proliferation of cells with damaged DNA are degraded, permitting the expansion of clones with genetic instability. Activation of the DNA damage response by oncogenes also was associated with a G2 arrest (Bartkova et al., 2005), suggesting that the G2 checkpoint represents a barrier to carcinogenesis and cells with defects in G2 checkpoint function may display a growth advantage at early stages of carcinogenesis.
The DNA damage G1 checkpoint requires ATM signaling to activate p53 and induce expression of the cyclin-dependent kinase inhibitor p21Waf1. This signaling pathway appeared to be intact and effective in secondary cultures of NHMs, as reported previously (Marrot et al., 2005; Sun et al., 2005). While p53 is a central component of this checkpoint, p53 mutations are uncommon in malignant melanomas, being scored in 10-25% of tumors (Daniotti et al., 2004). However, mutations in genes other than p53 also can affect DNA damage G1 checkpoint function. For example, inactivation of RB has been shown to ablate the G1 checkpoint response to DNA damage (Demers et al., 1994). Given the large number of gene products that participate in checkpoint signaling upstream and downstream of p53, a variety of genetic alterations could ablate or attenuate G1 checkpoint function. A study of several uveal melanoma lines found that IR treatment induced the activation of p53, but the p53-dependent induction of p21Waf1 or BAX was defective (Sun et al., 2005). Another study of radioresistant melanoma lines identified a defect in post-translational modification of p53 in response to radiation treatment (Satyamoorthy et al., 2000). The current analysis detected a functional deficit in the G1 checkpoint in 69% of the melanoma lines tested, and in the four epithelial-like melanoma lines with wild-type N-RAS and B-RAF. By global analysis of gene expression, this functional defect was associated with reduced basal levels of expression of two p53-responsive genes, p21Waf1 and DDB2. As radioresistance is often associated with defective signaling in the p53 pathway, it is likely that the lines with defective G1 checkpoint function are radioresistant.
Analysis of checkpoint gene expression using tissue arrays has been used to define a set of protein markers (Ki-67, p16, Bcl-6, and p21Waf1) that predicts clinical outcome for malignant melanoma (Alonso et al., 2004). Poor clinical outcome (shorter survival in patients with vertical-growth-phase melanoma) was associated with reduced expression of p16 protein and elevated expression of Ki-67, Bcl-6, and p21Waf1 proteins in primary tumor samples (Alonso et al., 2004). Our analyses showed that G1 checkpoint-defective melanoma lines displayed reduced expression of p21Waf1 mRNA and protein. Thus, elevated expression of p21Waf1 mRNA or protein may be a biomarker for melanomas with effective p53 signaling and G1 checkpoint function. It is remarkable that enhanced expression of p21Waf1 was a marker of adverse clinical outcome; however, in the same clinical study (Alonso et al., 2004), elevated p21Waf1 expression was associated with increases in proliferation markers, such as cyclin D1 and Ki-67, and the proliferation marker Ki-67 was also found to predict clinical outcome. It is notable from our study that G1 checkpoint-defective melanomas displayed higher levels of expression of CDC7 and CKS1, genes that act to stimulate DNA synthesis. Together, these results suggest a paradox in melanoma prognosis. Enhanced proliferation is a marker of poor prognosis, yet expression of the cyclin-dependent kinase inhibitor and effector of p53-dependent inhibition of proliferation, p21Waf1, also is a marker of poor prognosis. In most cancers examined to date, inactivation of p53 function is associated with poor prognosis. It is possible that in the study of Alonso et al. (2004) expression of p21Waf1 served as a marker of cell proliferation (Michieli et al., 1994) and not p53-dependent checkpoint function.
The DNA damage G2 checkpoint prevents cells with broken chromosomes from entering mitosis, thereby allowing more time for repair of the breaks. Defects in G2 checkpoint function in ataxia telangiectasia cells are associated with significantly enhanced susceptibility to induction of chromosomal aberrations by IR (Zampetti-Bosseler and Scott, 1981) and UV (Kaufmann and Wilson, 1994). Clearly, conditions of in vitro cell culture do not select for defects in the G2 checkpoint as cancer lines with effective checkpoint function can be isolated, as shown here. Defective G2 checkpoint function in melanoma lines was associated with mutations in the B-RAF oncogene but not mutations in N-RAS. As allelic deletions in CDKN2A/INK4A locus are comparatively common in melanoma (Grafstrom et al., 2005), it will be of interest to determine whether defects in G2 checkpoint function sensitize melanocytes to UV-induced chromosomal aberrations and deletion of CDKN2A/INK4A alleles. Enhanced UV clastogenesis could explain the earlier clinical appearance of melanomas with B-RAF mutations (Thomas et al., 2007).
Expression of oncogenic B-RAF in normal melanocytes induces growth arrest through induction of p16 (Michaloglou et al., 2005; Gray-Schopfer et al., 2006). Inactivation of p16 is common in melanomas, and four of six melanoma lines that we examined had no detectable p16 protein (Figure 3). However, factors other than p16 also appeared to contribute to the B-RAF-induced growth arrest (Michaloglou et al., 2005). Melanocyte lines that expressed hTERT and a dominant-negative p53 proliferated with the mutant B-RAF (Chudnovsky et al., 2005), suggesting that p53 also contributes to oncogene-induced growth arrest in melanocytes. It remains to be determined whether the defects in G2 checkpoint function seen in melanoma lines with mutant B-RAF are a direct effect of the mutant oncogene or a consequence of a secondary genetic alteration. Inactivation of p16 or p53 does not appear to explain the defect as a melanoma line with defective p53 signaling, and absence of p16 protein expression (RPMI8332) displayed an effective G2 checkpoint response to DNA damage. One alternative secondary target is PTEN. Mutations in B-RAF are commonly associated with inactivation of PTEN (Tsao et al., 2004), which has been reported to regulate Chk1 and G2 checkpoint function (Puc et al., 2005). It is interesting to note that the B-RAF mutant melanoma line with an effective G2 checkpoint response (A375) was reported to have a wild-type PTEN gene (Tsao et al., 2004). Other potential targets for attenuation of G2 checkpoint function are 14-3-3 proteins and Cdc25C (Peng et al., 1997). Previous studies have demonstrated that expression of mutant Ras oncogenes can attenuate G2 checkpoint function (Santana et al., 2002; Agapova et al., 2004). However, expression of mutant N-RAS in melanoma lines was not associated consistently with a significant defect in G2 checkpoint function.
Signatures of defective DNA damage checkpoint function in melanoma lines
Two methods were used to search for genes whose expression was correlated with DNA damage checkpoint function. SAM has been used to identify a signature of defective p53 signaling in breast cancer (Troester et al., 2006) that included several of the genetic elements in our G1 checkpoint list. The Bayesian method of analysis identified more genes with a lower false discovery rate than SAM. The Bayesian tool also more frequently assigned samples to the correct category of checkpoint function. More studies will be needed to determine whether the Bayesian tool is superior to SAM for identifying genes whose expression is correlated with selected biological states. The Bayesian G1 checkpoint signature included many genes that are associated by gene ontology with cell proliferation (see Supplementary Table S1), as p53-defective melanoma lines grew faster than lines with effective p53 signaling. The Bayesian G2 checkpoint signature included the rather nonspecific gene ontology biological functions of hydrolase activity and catalysis (see Supplementary Table S1). To our knowledge, this was the first attempt to identify a pattern of gene expression that predicts DNA damage G2 checkpoint function. Future studies should establish a minimal gene set for predicting DNA damage checkpoint function in a larger sample of melanomas.
The expression of several p53-responsive genes was correlated with expression of p21Waf1 in melanomas, including DDB2. Germ-line inactivation of DDB2 in humans causes xeroderma pigmentosum (complementation group E) with significantly increased risk of development of skin cancer including melanoma (Itoh et al., 2004). DDB2 encodes a factor that appears to aid in recruitment of other repair factors to sites of damaged DNA in chromatin (Wang et al., 2004). DDB2 also has recently been shown to regulate expression of p53 protein; DDB2-defective cells displayed reduced expression of p53 and concomitant attenuation of p53-dependent apoptosis and signaling in response to UV-induced DNA damage (Itoh et al., 2003). These results suggest that p53-defective melanomas also may express a defect in repair of UV-induced DNA damage through reduced expression of DDB2. Such a defect might enhance melanoma progression in skin exposed to sunlight.
In summary, chromosomal instability in malignant melanoma may be attributed in part to acquired defects in cell cycle checkpoints. Significant defects in DNA damage checkpoint function were observed in 69% of melanoma lines. Such defects may be common in melanomas and identifiable by analysis of patterns of gene expression.
MATERIALS AND METHODS
Conditions of cell culture
Human melanocyte cultures were generated from neonatal foreskin tissues, as described (Scott et al., 2005). Briefly, skin sections were placed in a 35-mm dish containing complete Dulbecco's minimal essential medium with dispase (50 units per ml) and incubated at 41C overnight. After peeling off the dermis, the epidermis was placed in a 15-ml tube containing 2 ml of trypsin (0.25%) and incubated at 37°C for 30 minutes. After briefly vortexing, 1 ml of calf serum was added and cells were collected by sedimentation. The cell pellet was resuspended in 5 ml of Medium 254 (Cascade Biologics, Portland, OR) containing one drop of FCS. Cells were cultured in a T-25 flask and fed three times each week with Medium 254 containing PMA-free Human Melanocyte Growth Supplement (HMGS-2; Cascade Biologics, Portland, OR). Secondary cultures of neonatal melanocytes were obtained by releasing cells from primary culture dishes with trypsin-EDTA, and then replating cells at lower cell density. For storage, melanocytes were frozen in liquid nitrogen at 1 × 107 cells/ml in CS-C serum-free cryopreservation medium (Cell Systems Corp., Kirkland, WA). Four different melanocyte cultures were established for these studies. A fifth foreskin melanocyte culture (NHM-5) was purchased from Cambrex Corporation, Walkersville, MD. One melanocyte culture (NHM-2) was infected with a replication-defective retrovirus that transduced expression of HPV16E6 oncoprotein (NHM2-E6) or a dominant-negative p53 (p53H179Q), along with a neomycin-resistance gene (Simpson et al., 2005). Infected cells were selected by growth for 2 weeks in culture medium containing 400 μg/ml G418.
Melanoma cell lines were obtained from several sources (Table 1) and grown as recommended by the supplier. All melanocytes and melanoma cell lines were checked for mycoplasma contamination using the GenProbe® kit (Gen-Probe Inc., San Diego, CA), according to the manufacturer's recommended protocol. Cell lines with RLU greater than twice background (approximately 4,000 RLU), considered contaminated with mycoplasma, were treated with Plasmocin (Invitrogen Corp., Carlsbad, CA), according the manufacturer's recommended protocol, and then retested. No data are reported for contaminated cultures. The F1-hTERT diploid human fibroblast line was derived from foreskin and used in these studies, as previously described (Heffernan et al., 2002).
The medical ethical committees of the University of North Carolina at Chapel Hill and Rochester University approved all described studies. The study was conducted according to the Declaration of Helsinki Principles.
Mutational Analyses of B-RAF and N-RAS
The N-RAS/B-RAF mutational status was determined from genomic DNA for the set of melanoma cell lines (Table 1). Genomic DNA was isolated from cells using Genomic Tips kits (Qiagen, Valencia, CA) and by following the manufacturer's recommended protocol; the mutational status of B-RAF at exons 11 and 15, and N-RAS at codons 12, 13, 18, and 61 was determined as follows: PCR amplification utilized the following primers: B-RAF at exon 15, for:5′-TCATAATGCTTGCTCTGATAGGA-3′, rev:5′-GGCCAAA-AATTTAA TCAGTGGA-3′; B-RAF at exon 11, for:5′-CTGTTTGGCTTGACTTG AC-3′, rev:5′-GACTTGTCACAATGTCACC-3′ (Thomas et al., 2004); N-RAS at codons 12, 13, and 18, for:5′-GACTGAGTACAAACTG GTGG-3′, rev:5′-GGGCCTCACCTCTATGGTG-3′; N-RAS at codon 61, for:5-GGTGAAACCTGTTTGTTGGA-3′, rev:5′-ATACACAGAGG AAGCCTTC-3′. PCR products were purified from 2% agarose gels and DNA was sequenced using the forward PCR primers at the University of North Carolina at Chapel Hill Automated DNA Sequencing Facility on a 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Quantification of DNA damage G1 and G2 checkpoint functions
Exponentially growing cells in flasks with medium were irradiated with 1.5 Gy 137Cs gamma rays (Gammacell40, MDS Nordion, Ottawa, Canada) at a dose rate of 0.9 Gy per min. Sham-treated controls were subjected to the same movements to and from the irradiation facility but without irradiation. For quantification of G1 checkpoint function, cells were incubated with 10 μM BrdU at 6-8 hours post-irradiation to label S phase cells and then harvested for two-channel flow cytometric analysis of incorporation of BrdU and DNA content (Doherty et al., 2003). The fraction of BrdU-labeled nuclei within the first half of the S phase (2-3N DNA content) in irradiated cell preparations was expressed as a percentage of the equivalent fraction in sham-treated controls as a quantitative measure of DNA damage G1 checkpoint function. For quantification of G2 checkpoint function, cells were incubated for 2 hours post-irradiation, and then harvested for determination of mitotic fraction by flow cytometry, using anti-phospho-histone H3 antibody to label mitotic cells (Doherty et al., 2003). DNA damage G2 checkpoint function was quantified as the mitotic fraction in irradiated cells expressed as a percentage of the mitotic fraction in sham-treated controls. For statistical comparisons between melanoma cell lines and NHMs, data were log-transformed and compared by Student's t-test, with Dunnett's adjustment for multiple comparisons.
Western immunoblot analyses
Logarithmically growing cells were seeded at 106 per 100-mm dish and incubated for 40 hours. Cultures were treated with IR as described above and incubated for 2, 6, and 24 hours at 37°C. Controls were harvested at 6 hours after sham treatment. Cells were harvested by trypsinization, washed once in PBS, and resuspended in 10 mM sodium phosphate buffer, pH 7.2, 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, 1% NP40, supplemented with 1 × protease inhibitor cocktail (Sigma, St Louis, MO). Protein concentrations were determined using the Bio-Rad DC Protein Assay (Bio-Rad Laboratories, Hercules, CA). Samples containing equal amounts of protein were mixed with an equal volume of 2 × Laemmli sample buffer (125 mM Tris-HCl, pH 6.8, 4% SDS, 20% glycerol) containing 5% β-mercaptoethanol, heated in boiling water, and separated by SDS-polyacrylamide gel electrophoresis. Proteins were transferred to nitrocellulose and probed with antibodies against various proteins and phosphorylation epitopes that are associated with cell cycle checkpoint function including p53 (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-ser15-p53 (Cell Signaling Technology, Danvers, MA), Chk2 (Oncogene Research Products, Cambridge, MA), phospho-ser68-Chk2 (Cell Signaling Technology), p21Waf1 (Lab-vision Corp., Fremont, CA), Chk1 (Santa Cruz Biotechnology), phospho-ser317-Chk1 (Cell Signaling Technology), ATM (Bethyl Laboratories, Montgomery, TX), ATR (Bethyl Laboratories), NBS1 (Bethyl Laboratories), and Myt1 (Upstate Biotechnology, Lake Placid, NY). Proteins were detected with horseradish peroxidase-conjugated secondary antibodies, using the Amersham ECL reagent (Amersham Pharmacia Biotech Inc., Piscataway, NJ). Films were scanned using a DuoScan T1200 (Agfa Div., Bayer Corp., Ridgefield Park, NJ) to produce JPEG images. These images were then analyzed for pixel intensity using the AlphaEaseFC TM software (Alpha Innotech Corp., San Leandro, CA). The relative pixel intensity was normalized to the signal detected with extracts (included on every blot) prepared from a sham-treated, human diploid fibroblast line immortalized by ectopic expression of telomerase (F1-hTERT).
Microarray analysis
RNA was isolated from exponentially growing cultures using a commercial (Qiagen) kit. Cellular RNA was labeled with Cy5-dUTP and a pooled reference RNA preparation (Stratagene, La Jolla, CA) was labeled with Cy3-dUTP by the manufacturer's suggested methods, using the Agilent low-RNA input linear amplification kit. The Cy3- and Cy5-labeled samples were quantified, combined, and then hybridized overnight at 65°C to a 44,000-element human genome 60-mer oligonucleotide microarray (G4112A, Agilent Technologies, Palo Alto, CA). Arrays were then washed and scanned using a GenePix 4000B scanner (Axon Instruments, Foster City, CA). Images were gridded and quantified using GenePix Pro 5.1 software (Molecular Devices Corp, Sunnyvale, CA). Scanned, gridded images were uploaded to the UNC microarray database (http://genome.unc.edu/). All primary data from this work are available at that site and have been deposited into the gene expression omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE7469.
The array data were retrieved from the UNC microarray database, under the following criteria: (1) only reliable spots, as determined by the array scanning software and the array manufacturer, were selected; (2) only those spots whose channels 1 and 2 Lowess normalized values were ≥30 were selected; and (3) only genes with greater than 70% complete data were selected. In the end, a total of 23,221 genes were selected for analysis.
SAM (Tusher et al., 2001) was applied to the gene lists using a binary analysis, in which NHMs and melanoma lines were separated into two classes, checkpoint-effective and checkpoint-defective. NHMs and melanoma lines with levels of IR-induced cell cycle arrest that were less than the median were considered checkpoint-defective, those with levels of arrest greater than the median were considered to be checkpoint-effective. SAM was applied to identify genes that were differentially expressed between the two classes.
A second analytical tool based on Bayesian statistical theory was also applied, to identify genes that distinguished the two classes. The log-transformed gene expression values for the effective and defective groups were assumed to follow normal distributions with different group means and a common variance. Common non-informative priors were applied to the unknown group means and variance for each gene. The test for equality of the group means on each gene was performed using the Bayes factor, and the multiple testing adjustments in the Bayesian analysis were conducted as described (Newton et al., 2004) with a 0.05 FDR.
Once supervised gene lists were identified, hierarchical clustering analysis was conducted using the program Cluster (http://rana.lbl.gov/EisenSoftware.htm) to perform median-centered, average-linkage clustering. Clusters were visualized using Treeview.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs Melissa Troester and Charles Perou for helpful discussions. This work was supported in part by PHS grants ES10126, CA16086, and ES11391. KRN was supported by Environmental Pathology Training Grant ES07017.
Glossary
Abbreviations
- ATM
ataxia telangiectasia-mutated
- ATR
ATM- and rad3-related
- DSB
double-strand break
- FDR
false discovery rate
- IR
ionizing radiation
- NHM
normal human melanocyte
- SAM
significance analysis of microarrays
Footnotes
CONFLICT OF INTEREST The authors state no conflict of interest.
SUPPLEMENTARY MATERIAL Table S1. The Supplementary table contains lists of genes that were associated with defects in G1 and G2 checkpoint responses to DNA damage in melanocyte and melanoma lines, detected using either SAM or the Bayesian approach described in Materials and Methods.
REFERENCES
- Agapova LS, Volodina JL, Chumakov PM, Kopnin BP. Activation of Ras-Ral pathway attenuates p53-independent DNA damage G2 checkpoint. J Biol Chem. 2004;279:36382–9. doi: 10.1074/jbc.M405007200. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Ross DT, Perou CM, van de Rijn M. Towards a novel classification of human malignancies based on gene expression patterns. J Pathol. 2005;195:41–52. doi: 10.1002/path.889. [DOI] [PubMed] [Google Scholar]
- Alonso SR, Ortiz P, Pollan M, Perez-Gomez B, Sanchez L, Acuna MJ, et al. Progression in cutaneous malignant melanoma is associated with distinct expression profiles: a tissue microarray-based study. Am J Pathol. 2004;164:193–203. doi: 10.1016/s0002-9440(10)63110-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bauer J, Bastian BC. Distinguishing melanocytic nevi from melanoma by DNA copy number changes: comparative genomic hybridization as a research and diagnostic tool. Dermatol Ther. 2006;19:40–9. doi: 10.1111/j.1529-8019.2005.00055.x. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloethner S, Chen B, Hemminki K, Muller-Berghaus J, Ugurel S, Schadendorf D, et al. Effect of common B-RAF and N-RAS mutations on global gene expression in melanoma cell lines. Carcinogenesis. 2005;26:1224–32. doi: 10.1093/carcin/bgi066. [DOI] [PubMed] [Google Scholar]
- Brown AL, Lee CH, Schwarz JK, Mitiku N, Piwnica-Worms H, Chung JH. A human Cds1-related kinase that functions downstream of ATM protein in the cellular response to DNA damage. Proc Natl Acad Sci USA. 1999;96:3745–50. doi: 10.1073/pnas.96.7.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudnovsky Y, Adams AE, Robbins PB, Lin Q, Khavari PA. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nat Genet. 2005;37:745–9. doi: 10.1038/ng1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RJ, Curtis RE, Inskip PD, Fraumeni JF., Jr The risk of developing second cancers among survivors of childhood soft tissue sarcoma. Cancer. 2005;103:2391–6. doi: 10.1002/cncr.21040. [DOI] [PubMed] [Google Scholar]
- Daniotti M, Oggionni M, Ranzani T, Vallacchi V, Campi V, Di Stasi D, et al. BRAF alterations are associated with complex mutational profiles in malignant melanoma. Oncogene. 2004;23:5968–77. doi: 10.1038/sj.onc.1207780. [DOI] [PubMed] [Google Scholar]
- Demers GW, Foster SA, Halbert CL, Galloway DA. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proc Natl Acad Sci USA. 1994;91:4382–6. doi: 10.1073/pnas.91.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- Doherty SC, McKeown SR, McKelvey-Martin V, Downes CS, Atala A, Yoo JJ, et al. Cell cycle checkpoint function in bladder cancer. J Natl Cancer Inst. 2003;95:1859–68. doi: 10.1093/jnci/djg120. [DOI] [PubMed] [Google Scholar]
- Eskandarpour M, Hashemi J, Kanter L, Ringborg U, Platz A, Hansson J. Frequency of UV-inducible NRAS mutations in melanomas of patients with germline CDKN2A mutations. J Natl Cancer Inst. 2003;95:790–8. doi: 10.1093/jnci/95.11.790. [DOI] [PubMed] [Google Scholar]
- Fletcher O, Easton D, Anderson K, Gilham C, Jay M, Peto J. Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst. 2004;96:357–63. doi: 10.1093/jnci/djh058. [DOI] [PubMed] [Google Scholar]
- Geller AC, Miller DR, Annas GD, Demierre MF, Gilchrest BA, Koh HK. Melanoma incidence and mortality among US whites, 1969-1999. JAMA. 2002;288:1719–20. doi: 10.1001/jama.288.14.1719. [DOI] [PubMed] [Google Scholar]
- Gibbs P, Brady BM, Robinson WA. The genes and genetics of malignant melanoma. J Cutan Med Surg. 2002;6:229–35. doi: 10.1007/s10227-001-0037-6. [DOI] [PubMed] [Google Scholar]
- Giglia-Mari G, Sarasin A. TP53 mutations in human skin cancers. Hum Mutat. 2003;21:217–28. doi: 10.1002/humu.10179. [DOI] [PubMed] [Google Scholar]
- Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340:1341–8. doi: 10.1056/NEJM199904293401707. [DOI] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Grafstrom E, Egyhazi S, Ringborg U, Hansson J, Platz A. Biallelic deletions in INK4 in cutaneous melanoma are common and associated with decreased survival. Clin Cancer Res. 2005;11:2991–7. doi: 10.1158/1078-0432.CCR-04-1731. [DOI] [PubMed] [Google Scholar]
- Gray-Schopfer VC, Cheong SC, Chong H, Chow J, Moss T, Abdel-Malek ZA, et al. Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br J Cancer. 2006;95:496–505. doi: 10.1038/sj.bjc.6603283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haqq C, Nosrati M, Sudilovsky D, Crothers J, Khodabakhsh D, Pulliam BL, et al. The gene expression signatures of melanoma progression. Proc Natl Acad Sci USA. 2005;102:6092–7. doi: 10.1073/pnas.0501564102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffernan TP, Simpson DA, Frank AR, Heinloth AN, Paules RS, Cordeiro-Stone M, et al. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol Cell Biol. 2002;22:8552–61. doi: 10.1128/MCB.22.24.8552-8561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek K, Rimm DL, Williams KR, Zhao H, Ariyan S, Lin A, et al. Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res. 2004;64:5270–82. doi: 10.1158/0008-5472.CAN-04-0731. [DOI] [PubMed] [Google Scholar]
- Itoh T, Cado D, Kamide R, Linn S. DDB2 gene disruption leads to skin tumors and resistance to apoptosis after exposure to ultraviolet light but not a chemical carcinogen. Proc Natl Acad Sci USA. 2004;101:2052–7. doi: 10.1073/pnas.0306551101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, O'Shea C, Linn S. Impaired regulation of tumor suppressor p53 caused by mutations in the xeroderma pigmentosum DDB2 gene: mutual regulatory interactions between p48(DDB2) and p53. Mol Cell Biol. 2003;23:7540–53. doi: 10.1128/MCB.23.21.7540-7553.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Kaufmann WK, Campbell CB, Simpson DA, Deming PB, Filatov L, Galloway DA, et al. Degradation of ATM-independent decatenation checkpoint function in human cells is secondary to inactivation of p53 and correlated with chromosomal destabilization. Cell Cycle. 2002;1:210–9. [PubMed] [Google Scholar]
- Kaufmann WK, Heffernan TP, Beaulieu LM, Doherty S, Frank AR, Zhou Y, et al. Caffeine and human DNA metabolism: the magic and the mystery. Mutat Res. 2003;532:85–102. doi: 10.1016/j.mrfmmm.2003.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WK, Wilson SJ. G1 arrest and cell-cycle-dependent clastogenesis in UV-irradiated human fibroblasts. Mutat Res. 1994;314:67–76. doi: 10.1016/0921-8777(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Knauf JA, Ouyang B, Knudsen ES, Fukasawa K, Babcock G, Fagin JA. Oncogenic RAS induces accelerated transition through G2/M and promotes defects in the G2 DNA damage and mitotic spindle checkpoints. J Biol Chem. 2006;281:3800–9. doi: 10.1074/jbc.M511690200. [DOI] [PubMed] [Google Scholar]
- Kraemer KH, Herlyn M, Yuspa SH, Clark WH, Jr, Townsend GK, Neises GR, et al. Reduced DNA repair in cultured melanocytes and nevus cells from a patient with xeroderma pigmentosum. Arch Dermatol. 1989;125:263–8. [PubMed] [Google Scholar]
- Kumar R, Angelini S, Snellman E, Hemminki K. BRAF mutations are common somatic events in melanocytic nevi. J Invest Dermatol. 2004;122:342–8. doi: 10.1046/j.0022-202X.2004.22225.x. [DOI] [PubMed] [Google Scholar]
- Marrot L, Belaidi JP, Jones C, Perez P, Meunier JR. Molecular responses to stress induced in normal human Caucasian melanocytes in culture by exposure to simulated solar UV. Photochem Photobiol. 2005;81:367–75. doi: 10.1562/2004-10-13-RA-343. [DOI] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Michieli P, Chedid M, Lin D, Pierce JH, Mercer WE, Givol D. Induction of WAF1/CIP1 by a p53-independent pathway. Cancer Res. 1994;54:3391–5. [PubMed] [Google Scholar]
- Nambiar S, Mirmohammadsadegh A, Doroudi R, Gustrau A, Marini A, Roeder G, et al. Signaling networks in cutaneous melanoma metastasis identified by complementary DNA microarrays. Arch Dermatol. 2005;141:165–73. doi: 10.1001/archderm.141.2.165. [DOI] [PubMed] [Google Scholar]
- Newton MA, Noueiry A, Sarkar D, Ahlquist P. Detecting differential gene expression with a semiparametric hierarchical mixture method. Biostatistics. 2004;5:155–76. doi: 10.1093/biostatistics/5.2.155. [DOI] [PubMed] [Google Scholar]
- Noonan DM, Severino A, Morini M, Tritarelli A, Manente L, D'Agnano I, et al. In vitro and in vivo tumor growth inhibition by a p16-mimicking peptide in p16(INK4A)-defective, pRb-positive human melanoma cells. J Cell Physiol. 2005;202:922–8. doi: 10.1002/jcp.20182. [DOI] [PubMed] [Google Scholar]
- Paules RS, Levedakou EN, Wilson SJ, Innes CL, Rhodes N, Tlsty TD, et al. Defective G2 checkpoint function in cell from individuals with familial cancer syndromes. Cancer Res. 1995;55:1763–73. [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–5. doi: 10.1126/science.277.5331.1501. see comments. [DOI] [PubMed] [Google Scholar]
- Pho L, Grossman D, Leachman SA. Melanoma genetics: a review of genetic factors and clinical phenotypes in familial melanoma. Curr Opin Oncol. 2006;18:173–9. doi: 10.1097/01.cco.0000208791.22442.09. [DOI] [PubMed] [Google Scholar]
- Puc J, Keniry M, Li HS, Pandita TK, Choudhury AD, Memeo L, et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005;7:193–204. doi: 10.1016/j.ccr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–16. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana C, Ortega E, Garcia-Carranca A. Oncogenic H-ras induces cyclin B1 expression in a p53-independent manner. Mutat Res. 2002;508:49–58. doi: 10.1016/s0027-5107(02)00172-0. [DOI] [PubMed] [Google Scholar]
- Satyamoorthy K, Chehab NH, Waterman MJ, Lien MC, El-Deiry WS, Herlyn M, et al. Aberrant regulation and function of wild-type p53 in radioresistant melanoma cells. Cell Growth Differ. 2000;11:467–74. [PubMed] [Google Scholar]
- Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BL, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 2003;63:756–9. [PubMed] [Google Scholar]
- Scott G, Jacobs S, Leopardi S, Anthony FA, Learn D, Malaviya R, et al. Effects of PGF2alpha on human melanocytes and regulation of the FP receptor by ultraviolet radiation. Exp Cell Res. 2005;304:407–16. doi: 10.1016/j.yexcr.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Seykora JT, Jih D, Elenitsas R, Horng WH, Elder DE. Gene expression profiling of melanocytic lesions. Am J Dermatopathol. 2003;25:6–11. doi: 10.1097/00000372-200302000-00002. [DOI] [PubMed] [Google Scholar]
- Shields JM, Thomas NE, Cregger M, Berger AJ, Leslie M, Torrice C, et al. Lack of extracellular signal-regulated kinase mitogen-activated protein kinase signaling shows a new type of melanoma. Cancer Res. 2007;67:1502–12. doi: 10.1158/0008-5472.CAN-06-3311. [DOI] [PubMed] [Google Scholar]
- Simpson DA, Livanos E, Heffernan TP, Kaufmann WK. Telomerase expression is sufficient for chromosomal integrity in cells lacking p53 dependent G1 checkpoint function. J Carcinog. 2005;4:18. doi: 10.1186/1477-3163-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley KS, Herlyn M. Loitering with intent: new evidence for the role of BRAF mutations in the proliferation of melanocytic lesions. J Invest Dermatol. 2004;123:xvi–i. doi: 10.1111/j.0022-202X.2004.23430.x. [DOI] [PubMed] [Google Scholar]
- Sun Y, Tran BN, Worley LA, Delston RB, Harbour JW. Functional analysis of the p53 pathway in response to ionizing radiation in uveal melanoma. Invest Ophthalmol Vis Sci. 2005;46:1561–4. doi: 10.1167/iovs.04-1362. [DOI] [PubMed] [Google Scholar]
- Thomas NE, Alexander A, Edmiston SN, Parrish E, Millikan RC, Berwick M, et al. Tandem BRAF mutations in primary invasive melanomas. J Invest Dermatol. 2004;122:1245–50. doi: 10.1111/j.0022-202X.2004.22523.x. [DOI] [PubMed] [Google Scholar]
- Thomas NE, Edmiston SN, Alexander A, Millikan RC, Groben PA, Hao H, et al. Number of nevi and early life ambient UV exposure are associated with BRAF-mutant melanoma. Cancer Epidemiol Biomarkers Prev. 2007;16:991–7. doi: 10.1158/1055-9965.EPI-06-1038. [DOI] [PubMed] [Google Scholar]
- Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA. 2002;99:6567–72. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troester MA, Herschkowitz JI, Oh DS, He X, Hoadley KA, Barbier CS, et al. Gene expression patterns associated with p53 status in breast cancer. BMC Cancer. 2006;6:276. doi: 10.1186/1471-2407-6-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337–41. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QE, Zhu Q, Wani G, Chen J, Wani AA. UV radiation-induced XPC translocation within chromatin is mediated by damaged-DNA binding protein, DDB2. Carcinogenesis. 2004;25:1033–43. doi: 10.1093/carcin/bgh085. [DOI] [PubMed] [Google Scholar]
- Warters RL, Adamson PJ, Pond CD, Leachman SA. Melanoma cells express elevated levels of phosphorylated histone H2AX foci. J Invest Dermatol. 2005;124:807–17. doi: 10.1111/j.0022-202X.2005.23674.x. [DOI] [PubMed] [Google Scholar]
- Zampetti-Bosseler F, Scott D. Cell death, chromosome damage and mitotic delay in normal human, ataxia telangiectasia and retinoblastoma fibroblasts after X-irradiation. Int J Radiat Biol Relat Stud Phys Chem Med. 1981;39:547–58. doi: 10.1080/09553008114550651. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.