

Abstract
During meiotic prophase the repair of self-inflicted DNA double-strand break (DSB) damage leads to meiotic recombination in yeast. We employed a genetic screen to specifically characterize cellular functions that become essential after this DSB formation. As a result a new allele of MRE11, termed mre11S (for Separation of functions) was isolated that allows initiation but not processing and repair of meiotic DSBs similar to the well-characterized rad50S allele. In contrast, the mre11-1 allele blocks initiation of meiotic DSBs as reported previously by others. The mre11S allele, which is mutated in the 5′ part of the gene, can partially complement mre11 alleles disrupted close to the 3′ end that cannot initiate DSBs when homozygous. This suggests homodimerization of the Mre11 protein and the presence of separate domains for DSB initiation and 5′ resection. The fact that two genes, RAD50 and MRE11, required for DSB processing are also essential for DSB initiation dictates a model in which a bifunctional initiation/repair complex is required to initiate meiotic recombination. A subset of mre11S nuclei was shown to perform extensive but partially nonhomologous synapsis. We propose that the unprocessed DSBs present in mre11S allow for synapsis, but that homologous synapsis is only ensured at a later stage of recombination.
Keywords: Meiosis, synaptonemal complex, MRE11, recombination, DNA double-strand breaks, Saccharomyces cerevisiae
All sexually reproducing organisms rely on a specialized cell division called meiosis to ultimately produce gametes for conjugation. During this unique process a single round of DNA replication is followed by two rounds of chromosome segregation, reducing the nuclear content by half. As a result of an elaborate homology search, sorting, and recombination processes, physical connections between homologs are formed. In yeast at least some of the chromosome sorting precedes the initiation of recombination by DNA double-strand breaks (DSBs) because pairing of homologous DNA can be observed before DSBs appear and in mutants unable to induce DSBs (Scherthan et al. 1992; Loidl et al. 1994; Weiner and Kleckner 1994). Similarly, hot spots for recombination interact before DSBs are formed, because the degree of their homology was shown to influence the degree of DSB formation (Rocco and Nicolas 1996; Xu and Kleckner 1995). DSB initiation requires the gene products of at least 10 genes, namely SPO11, MRE11, RAD50, XRS2, MER2, MRE2, MEK1/MRE4, MEI4, REC104, and REC114 and, most likely, REC102 (Alani et al. 1990; Cao et al. 1990; Bhargava et al. 1992; Ivanov et al. 1992; Menees et al. 1992; Johzuka and Ogawa 1995; Ogawa et al. 1995; Rockmill et al. 1995a; Bullard et al. 1996; S. Keeney; N. Leem, N. Satoh, and H. Ogawa, both unpubl.). Recently, evidence has been presented that Spo11p catalyzes DSB formation (Keeney et al. 1997; Bergerat et al. 1997), but the role of the other genes is less clear.
To characterize essential processes in the wake of DSB initiation we performed a mutant hunt for DSB-dependent meiotic lethals. The desired mutants are unable to complete meiosis successfully in a wild-type strain background where meiotic DSBs are initiated. Any meiotic mutant defective in DSB initiation can be rescued by an additional spo13 mutation because such strains undergo only a single mitosis-like meiotic division. spo13 alone, however, cannot rescue a DSB-dependent lethal because the DSBs initiate the defective recombination pathway. In contrast, a spo13 rad50 double mutation blocks both recombination and reductional division and thus restores spore viability to the desired mutants (Malone and Esposito 1981). As a result of this mutant screen the isolation of a new allele of mre11 that unexpectedly caused this phenotype is presented below.
mre11 was originally described as a mutation interfering with meiotic recombination (Ajimura et al. 1993). mre11 mutants produce dead spores because of a lack of meiotic recombination, but an additional spo13 mutation restores spore viability. A hint that MRE11 also has a role after DSB initiation came from the fact that a temperature-sensitive mutant entering meiosis at permissive temperature was unable to repair some of the breaks when shifted to restrictive temperature (Ogawa et al. 1995). Johzuka and Ogawa (1995) have cloned MRE11 and have shown that a disruption mutant (mre11::hisG) cannot initiate meiotic DSBs. Consistent with its essential meiotic function MRE11 transcription is highly up-regulated during meiosis as expected for a recombination enzyme. During vegetative growth MRE11 is not essential but required for full radio resistance and control of mitotic recombination levels, indicating that Mre11p is required for DNA repair. The same functions are dependent on RAD50 and XRS2.
Mre11p was shown to interact with itself physically, as well as with Rad50p (Johzuka and Ogawa 1995) by virtue of the two-hybrid system, suggesting that it may act as a heteromultimere in vivo. Recently, Moore and Haber (1996) and Tsukamoto et al. (1996) showed that Mre11p, Rad50p, and Xrs2p play major roles in nonhomologous end joining in mitotic cells. When repair of a HO-induced DSB was studied in a situation where homologous recombination was precluded, mutants in mre11, rad50, or xrs2 decreased the yield of nonhomologous end joining, whereas most other members of the rad52 epistasis group did not affect the process (Moore and Haber 1996).
Homologs of MRE11 have been identified in various organisms. The human homolog of Mre11p was identified by Petrini et al. (1995) and was shown by coimmunopreciptitation to interact with the human homolog of Rad50p (Dolganov et al. 1996). Homologs to MRE11 have also been found in Caenorhabditis elegans, in Mus musculus, and in Schizosaccharomyces pombe, where it is called RAD32 (Tavassoli et al. 1995). rad32 mutants show defects analogous to mre11 mutants, namely impaired mitotic DSB repair activity, as well as decreased meiotic recombination and spore viability. Interestingly, a prokaryotic homolog of MRE11, called sbcD was identified from Escherichia coli by sequence comparison (Gibson et al. 1992; Sharples and Leach 1995). SbcC and SbcD proteins were shown to interact physically and to have double-strand exonuclease and single-strand endonuclease activity in vitro (Connelly and Leach 1996). Both reactions are dependent on chelatable divalent kations like Mn2+, but only the exonuclease activity relies on ATP while endonucleolytic DNA degradation is ATP independent and can be performed by SbcD alone (J.C. Connolly and D.R.F. Leach, unpubl.). Therefore, SbcD might be the catalytic subunit of the SbcCD complex, which is modulated by SbcC in the presence of ATP. SbcC is homologous to RAD50, both being members of the Smc protein family, which is involved in ATP-dependent chromosome condensation (Sharples and Leach 1995). The homology of the prokaryotic SbcC/SbcD pair with their eukaryotic counterparts Rad50/Mre11 suggests a nuclease function for the latter complex.
Data presented below show that Mre11p, in conjunction with Rad50p, is required for the 5′ resection of meiotic DSBs by a 5′ → 3′ exonucleolytic activity or a single-strand endonucleolytic activity suggesting that Mre11/Rad50 presumably catalyzes this reaction.
Results
A genetic screen to identify new functions required after initiation of recombination
A screen for mutants in new genes and functions required after initiation of meiotic DNA DSBs was carried out. Such mutants are expected to derail meiosis because meiotic DSBs entail a series of events that require specific functions for their completion. For instance, failure to repair recombination intermediates may lead to mis-segregation in both meiotic divisions and/or may possibly cause cell cycle arrest by activating a checkpoint. However, the desired mutation is expected to be rescued by the inactivation of both the RAD50 and the SPO13 gene. Inactivation of the RAD50 gene prevents DSB formation and initiation of meiotic recombination. Therefore chiasmata do not form, which results in subsequent failure of the reductional division. The need for physical connections can be alleviated by an additional spo13 mutation that causes the cells to skip the first meiotic division. The resulting dyads contain diploid spores with viability similar to those from a RAD50 spo13 meiosis (Table 1). Because all potentially lethal DSB-induced events are bypassed in a rad50 spo13 strain, a function of the defined specificity is dispensable.
Table 1.
rad50 is epistatic to mre11S
| Strain
|
Relevant genotype (homozygous)
|
Spore viability (%)
|
|---|---|---|
| Y9Dx2A | spo13 | 31.9 |
| Y401 | rad50 spo13 | 31.9 |
| Y403 | mre11S spo13 | 1.9 |
| Y402 | rad50 mre11S spo13 | 30.0 |
spo13 lowers spore viability in SK1 more than reported for other strains (Wagstaff et al. 1982). No further decrease is found in the spo13 rad50 double mutant, but in a mre11S spo13 double mutant viability is very low in the few spores formed. rad50 is epistatic to mre11S because viability in the rad50 mre11S spo13 is the same as in rad50 spo13, demonstrating that RAD50 acts upstream of MRE11. Eighty dyads were dissected each.
Figure 1 summarizes the mutant screen. The homothallic strain Y136 is rad50, ade2, and spo13 but carries SPO13 on centromere (CEN)–plasmid pTW15 and RAD50 linked to ADE2 on CEN–plasmid p3. Single spores from Y136 were mutagenized with N-methyl-N-nitro-N-nitrosoguanidine (MNNG) and plated on synthetic complete medium lacking adenine (SC–ADE). For mutagenesis, MNNG was preferred over ethylmethane sulfonate (EMS) because mutations are “fixed” immediately (Klein et al. 1990). Shortly after germination HO-induced mating-type switching leads to diploidization, rendering all mutations homozygous and thereby eliminating mutants defective in mating-type switching (such as rad52). During subsequent proliferation on SC–ADE only plasmid p3 was selected for resulting in loss of plasmid pTW15. After 2 days colonies were transferred to sporulation medium (SPM+; see Materials and Methods), where cells grew for several generations without selective pressure for any two plasmids before they sporulated.
Figure 1.

Schematic representation of the mutant screen. Three crucial steps of the protocol for the mutant hunt are illustrated. Spores giving rise to red colonies are shaded, and dead spores are marked by a cross.
For wild-type cells, the presence of the two plasmids does not interfere with spore viability. Colonies derived from spores of such cells are white, owing to the relative stability of the CEN–vectors and the slow growth of ade2 rad50 cells. In a mutant of the required specificity, however, only cells that lack both plasmids are able to complete meiosis successfully, giving rise to red colonies. After 2 days on SPM+, colonies were replica plated to rich medium and were exposed to ether to kill unsporulated cells that would otherwise cause a white background. Predominantly red colonies were scored as potential mutants.
One mutant candidate was chosen for further investigation because it displayed a strong phenotype and cytological examination suggested that synapsis of axial elements was reduced. We cloned the affected gene by complementation of the spore lethality phenotype (see Materials and Methods), identifying MRE11 as the responsible element. Because MRE11 had been described as being required for DSB initiation (Ajimura et al. 1993; Johzuka and Ogawa 1995; Ogawa et al. 1995) we were surprised to find that MRE11 also functions downstream of RAD50. The mre11 null mutant does not initiate meiotic recombination as a rad50 null mutant does and therefore is predicted to ultimately form white colonies in our assay. The allele described here leads to a lethal event in spo13 cells that initiates meiotic recombination. Because this indicates that the newly identified mre11 allele is partially functional and because of the similarity of the phenotypes caused by this allele and by rad50S (Alani et al. 1990) it is referred to as mre11S.
Isolation of mre11S, a new non-null allele of MRE11
We cloned the mre11S allele by gap repair and sequenced it. The sequence of the open reading frame (ORF) recovered from the mutant differed from the sequence of the complementing plasmid at positions +250 and +563 (both C → T transitions), which predicts a proline-to-serine change at position 84 and a threonine-to-isoleucine change at postion 188 of the protein (Fig. 2).
Figure 2.
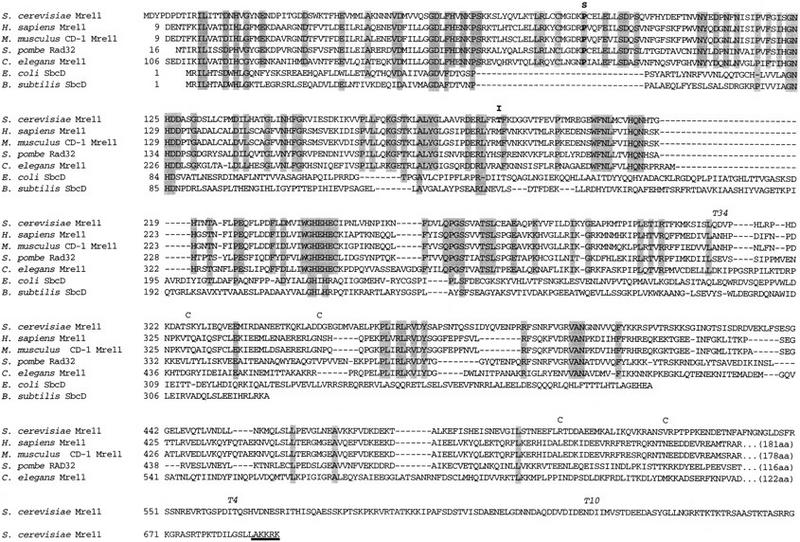
Alignment of predicted Saccharomyces cerevisiae Mre11 protein sequence with eukaryotic and prokaryotic homologs. Identical amino acids are shaded, S indicates the site of the proline 84 to serine, and I the threonine 188 to isoleucine exchange in mre11S. A potential nuclear localization site is underlined in bold. The first and last amino acids of predicted coiled–coil regions are marked with C. T34, T4, and T10 indicate the approximate transposon insertion sites (see also Fig. 3A).
Several other differences to the reported sequence (Johzuka and Ogawa 1995) were found in both the complementing wild-type clone and the mutated allele, including a frameshift rendering the predicted protein 49 amino acids longer (to equal 692 amino acids). This carboxy-terminal tail is especially rich in arginine and lysine, as well as serine and threonine, and has a potential nuclear localization site. Two potential coiled–coil domains that span positions 326–351 and, with less likelihood, 506–526 were detected (see Materials and Methods). As mentioned previously by Sharples and Leach (1995) the amino terminus exhibits homology to the bacterial SbcD recombination enzyme. The region between Pro-63 and Pro-117 contains several amino acids, including Pro-84, which are conserved throughout the eukaryotic homologs known to date. However, it is devoid of any detectable homology to SbcD while being surrounded by modules well conserved from bacteria to human. The change from Thr to Ile at position 188, which is the second mutation contributing to the mre11S phenotype, is also located in a conserved eukaryotic domain.
To confirm the epistatic relationship between rad50 and the newly isolated allele of mre11 suggested by the result of the screen we investigated spore viability in various spo13 strains by dissection of dyads (Table 1). The results clearly verified that rad50 is epistatic to mre11S with respect to the spore viability phenotype.
Genetic evidence for homodimerization of Mre11p
We have generated a set of mre11 alleles of various lengths by transposon mutagenesis in E. coli using the bacterial Tn3 (Seifert et al. 1986). The positions of these insertions were mapped by restriction analysis, and disruptions at different sites within the MRE11–ORF were selected for complementation studies (Fig. 3A). Strains with different combinations of mre11 alleles were constructed by crossing one mre11S strain to various partners harboring MRE11, mre11S, or different mre11 transposon disruption alleles. The resulting diploids (wild type for all other sporulation genes) were sporulated for 2 days on solid medium at 30°C and tested for formation of viable spores either by patch assay (Fig. 3B) or by tetrad dissection (Table 2).
Figure 3.

Intragenic complementation between mre11S and 3′ truncated alleles. (A) Transposon insertion sites at the MRE11 locus. Arrows point in the direction of the lacZ ORF, and the name of the allele is given. Asterisks and S and I indicate the sites of the mre11S mutations. Transposon insertions mre11–T4 and mre11–T10 are in-frame lacZ fusions. (B) Patch assay to study intragenic complementation between mre11S and the various transposon insertion alleles. Complementation is apparent for MRE11/mre11S and to a high extent for heterozygotes involving mre11–T4, mre11–T10, mre11–T40, and mre11–T16. Scarce colonies arising in the mre11S homozygote and other heterozygotes may represent few viable spores or vegetative cells surviving ether treatment.
Table 2.
Different allele combinations of MRE11 fall into three different categories
| Relevant genotype
|
Unsporulated, aberrant (%)
|
Dyads (%)
|
Tetrads (+triads) (%)
|
Spore viability (%) of tetrads (+triads)
|
% viability × % tetrads/100
|
|---|---|---|---|---|---|
| MRE11/mre11S | 39 | 4 | 57 | 96 | 55 |
| mre11S/mre11–T10 | 52 | 22 | 26 | 75 | 20 |
| mre11S/mre11–T4 | 58 | 11 | 31 | 66 | 20 |
| mre11S/mre11–T34 | 90 | 8 | 2 | 30 | 0.6 |
| mre11S/mre11–T24 | 87 | 9 | 4 | 3 | 0.1 |
| mre11S/mre11–T37 | 84 | 11 | 5 | 30 | 1.5 |
| mre11S/mre11–T20 | 84 | 9 | 7 | 28 | 2 |
| mre11S/mre11S | 73 | 18 | 9 | 1 | 0.1 |
| mre11–T4/mre11–T4 | 11 | 24 | 65 | <1 | <0.6 |
| mre11–T10/mre11–T10 | 31 | 13 | 53 | <1 | <0.5 |
| mre11–T20/mre11–T20 | 30 | 18 | 52 | <1 | <0.5 |
The mre11S allele in combination with MRE11 or with the complementing 3′ disrupted alleles mre11–T4 and mre11–T10 exhibited high sporulation as well as high spore viability. Therefore, the sporulation yield [% spore viability in tetrads (triads) × % tetrads (triads)/100] was also relatively high. A value of >20 corresponds to viable patches after ether treatment in Fig. 3. Strains carrying mre11S combined with other mre11 alleles, such as mre11–T34, had a very low frequency of tetrad formation, but the small fraction of tetrads formed contained relatively many living spores. Interestingly, the homozygous mre11S caused lower spore viability than a combination of mre11S and a possible null mutation (mre11–T20). Strains used were the same as in Fig. 3, except for Y324 as mre11S/mre11–T4. mre11S, mre11–T4, mre11–T20, and mre11–T10 homozygotes were Y323, Y327, Y328, and Y442 × Y262, respectively. Cells were sporulated on plates, and 200 cells were counted each. Asci with microspores or only one spore were classified as aberrant. At least 100 spores were dissected each for the determination of spore viability.
Comparison between mre11S/mre11S and MRE11/mre11S confirmed that mre11S is recessive regarding spore formation and spore viability. Alleles disrupted in the 5′ part of the MRE11 gene (mre11–T20 through mre11–T34) could not complement mre11S. Such heterozygotes behave similar to a mre11S homozygote by showing greatly reduced sporulation. When the rare tetrads produced by these heterozygotes were dissected they exhibited higher spore viability than the mre11S homozygote, but because of the low sporulation efficiency the meiotic yield (number of tetrads produced times viable spores) remained low (∼1%). All homozygous mre11–T20, mre11–T4, and mre11–T10 disruption strains sporulated well, but spores were inviable.
In contrast, disruptions at the 3′ end (mre11–T4, mre11–T10) lacking different parts of the evolutionary less-conserved carboxyl terminus complemented mre11S to almost wild-type levels of both sporulation and spore viability (when sporulated on solid medium). Transposons T4 and T10 generate in-frame mre11–lacZ fusions (Materials and Methods). mre11–T40 and mre11–T16, which map to the same positions and are equally oriented as mre11–T4 and mre11–T10, respectively, but are out-of-frame, complemented also (Fig. 3). None of the disrupted mre11 alleles produced a significant number of viable spores when homozygous (Table 2). Thus, there is intragenic complementation between mre11S and the 3′ disruptions.
Two-hybrid studies by Johzuka and Ogawa (1995) have already shown that Mre11p can interact with itself. Our results are compatible with this finding and suggest that there is homodimerization while the proteins are active. Homozygous mre11S strains or strains heterozygous for mre11S and 5′ mre11 disruptions show strongly reduced tetrad formation, which is similar to the situation in rad50S, a partially functional allele of RAD50 that blocks meiotic DSB processing and repair (Alani et al. 1990). When the two point mutations of mre11S were tested separately by dissection, each conferred a strong sporulation and spore viability defect (data not shown).
If the different combinations of mre11 alleles behaved differently in DSB formation, this could result in different levels of induction of meiotic gene conversion. For meiotic mutants induction of meiotic gene conversion can be determined by a return-to-growth experiment (Sherman and Roman 1963), where cells re-enter the mitotic pathway after meiotic induction, but before meiotic chromosome segregation. For some meiotic mutants (e.g., dmc1, zip1), recombination intermediates that would cause a block or even lethal damage to the cells on meiotic medium can be repaired mitotically during return to growth (Bishop et al. 1992; Sym et al. 1993).
We determined meiotic gene conversion between heteroalleles at the artificial his4–LEU2 and the natural ARG4 recombination hot spots (Cao et al. 1990; Storlazzi et al. 1995) during return to growth. Homozygous mre11–T4 and mre11–T20 strains are unable to induce meiotic recombination, whereas mitotic recombination is increased compared with wild type (Fig. 4A,B). Mitotic hyper-recombination has been observed previously in mre11, rad50, and xrs2 null mutants (Malone and Esposito 1981; Ivanov et al. 1992; Ajimura et al. 1993). A rad50S strain is committed to about the same low level of meiotic gene conversion, but the mitotic rate remains almost unchanged compared to wild type. This results in a 5- to 10-fold meiotic induction of gene conversion in rad50S, a phenomenon also seen by Prinz et al. (1997). In the homozygote mre11S, gene conversion is induced considerably stronger than in rad50S (up to 10-fold, Fig. 4C,D). At the ARG4 site almost wild-type levels of gene conversion could be observed in the mre11S/mre11–T4 heterozygote thus supporting the notion of intragenic complementation between mre11S and 3′ disruptions. No intragenic complementation was seen at the artificial his4–LEU2 hot spot under these conditions in two experiments (see Materials and Methods). However, when the mre11S/mre11–T4 heterozygote was sporulated on solid medium, viability was high (71%) and genetic distance between URA3 inserted at his4–LEU2 and MAT was reduced only moderately compared with wild type (Table 3). So this strain clearly is proficient for reciprocal recombination at his4–LEU2 under proper sporulation conditions. (A homozygous mre11S strain could not be tested because of low spore viability).
Figure 4.

Meiotic recombination can be induced to an intermediate level in the mre11S homozygote and to clearly higher levels for mre11S/mre11–T4 at ARG4. Recombinants are counted as prototrophs on SC–ARG (SC–HIS) per 10,000 colony-forming units (CFU) on YPD. Data were pooled from two independent experiments. (A,B) Gene conversions at two different meiotic hot spots in homozygous mre11–T4, mre11–T20, and rad50S strains. Mitotic recombination levels can be read at t = 0 and are elevated for mre11–T4 and mre11–T20. Because mitotic recombination is not altered in the rad50S strain, a 10-fold induction during meiosis is apparent. A similar level of meiotic induction for mre11–T4 and mre11–T20 would be masked by the recombinants of mitotic origin. (C,D) Gene conversions at two different meiotic hot spots in MRE11/mre11S, mre11S/mre11S and mre11S/mre11–T4 strains. Normal commitment was observed for MRE11/mre11S, but was reduced ∼50-fold for the homozygous mre11S. The heterozygous mre11S/mre11–T4 was between wild-type and homozygous mre11S at the ARG4 locus but was similar to mre11S at HIS4. (E,F) Decrease of viability [expressed as CFU(t)/CFU(t0) × 100 during progression through meiosis of all strains shown in A–D.
Table 3.
Genetic distance between MAT and URA3 is significantly reduced in the complementing mre11S/mre11–T4
| Relevant genotype
|
Spore viability (%)
|
Interval MAT–URA3 (cM)
|
|---|---|---|
| MRE11/mre11S (Y325) | 97.5 | 45.3 |
| mre11S/mre11–T4 (Y324) | 71 | 29.8 |
The genetic distance between MAT and URA3 inserted at the his4–LEU2 hot spot was determined in 99 complete tetrads of strain Y324 and 95 complete tetrads of strain Y325. The significance of the decrease in recombination rate was (P < 0.025) when a χ2 test was performed (Bailey 1995). Strains were sporulated on plates.
Because a mre11 null mutant had been shown to be repair deficient (Ajimura et al. 1993) we tested the resistance to methylmethane sulfonate (MMS), a DNA-damaging agent that ultimately causes DNA DSBs, of various mre11 mutants. Although the MRE11/mre11S heterozygote behaves like the isogenic wild type, the homozygous mre11S strain is partially MMS sensitive (Fig. 5). However, compared to the mre11 disruptions (even when disrupted at the very 3′ end as in mre11–T4), it is remarkably resistant. This is reminiscent of the situation in a rad50S mutant, where repair during vegetative growth is much less affected than meiosis (Alani et al. 1990). When the two point mutations in mre11S were tested separately, each exhibited MMS sensitivity between wild type and mre11S (data not shown). As for spore viability, intragenic complementation was also observed for MMS resistance, because the 3′ disrupted mre11–T4 allele improved viability when heterozygous with mre11S. Comparing MMS tolerance of mre11–T4/mre11–T4 and the 5′ disrupted mre11–T20/mre11–T20 suggests that mre11–T4 retains some DNA repair activity, which is in good agreement with its ability to partially complement the mre11S allele. MMS sensitivity and mitotic hyper-recombination (seen at the t = 0 point in Fig. 4) seem to correlate, as the homozygous disruptions with the highest MMS sensitivity also exhibit the highest mitotic level of gene convertants. The same mutants behave like rad50Δ concerning sporulation (see above).
Figure 5.

Residual resistance to MMS varies over several orders of magnitude for different diploid mre11 mutants. Cells from late exponential phase were plated on YPD containing the indicated amount of MMS and scored after 3 days. Resistance decreased strongly in the strains in the following order except for the first two strains, which were similar: MRE11/MRE11, MRE11/mre11S, mre11S/mre11–T4, mre11S/mre11S, mre11–T4/mre11–T4, mre11–T20/mre11–T20. Y328 (mre11–T20/mre11–T20) did not form visible colonies after 3 days on plates containing 0.005% MMS. The symbol at the end of the dashed line therefore marks the lower limit of detection.
mre11S strains accumulate unresected DSBs
The molecular interaction of Rad50p and Mre11p (Dolganov et al. 1996; Johzuka and Ogawa 1995), together with the similarity of rad50S and mre11S sporulation phenotype, suggested that mre11S mutants might not be able to process meiotic DSBs (Alani et al. 1990). Meiotic DSBs were assayed at three different sites, the artificial his4–LEU2 hot spot (Cao et al. 1990; Storlazzi et al. 1995)(Fig. 6A–D,F–H) as well as at the natural THR4 and DED81 hot spots (de Massy and Nicolas 1993; Goldway et al. 1993) (Fig. 6E). The results show clearly that mre11S mutants are defective in DSB processing at all sites (Fig. 6A,D,E). No difference concerning the location of the breakage sites was found for rad50S and mre11S (Fig. 6C,D). mre11S is also recessive to MRE11 concerning the DSB kinetics because one functional allele can restore DSB processing, resulting in a faint, fuzzy band at the 4- and 5.5-hr time points that disappears due to repair (Fig. 6B). No DSBs could be detected in mre11–T4 or mre11–T20 homozygotes at his4–LEU2, as expected. Thus, although both of these genes seem not to be functional on their own, only mre11–T20 is a null mutant because the truncated mre11–T4 still is able to improve viability and recombination when heterozygous with mre11S. However, no difference was observed between mre11S/mre11–T4 and mre11S/mre11S concerning accumulation of DSBs at the his4–LEU2 hotspot (Fig. 6A,D,H), which is consistent with the absence of an effect of mre11–T4 on induction of meiotic gene conversion at this locus. We attribute the apparent contradiction that mre11–T4 can partially complement mre11S when testing for sporulation or MMS sensitivity (solid media), but not when testing for commitment to meiotic recombination and DSB repair at his4–LEU2 (liquid media) to the different conditions used. Also complementation of the spore formation defect only worked when cells were sporulated on plates but not in liquid SPM (data not shown).
Figure 6.

mre11S mutants accumulate meiosis-specific unresected DNA DSBs at recombination hot spots. All experiments shown except for E, were done at the artificial his4–XLEU2-MluI::BamHI/his4–BLEU2–MluI hot spot and blots were hybridized with probe A (pNKY291). DSB-I and DSB-II mark the location of major cleavage sites. (A) Quantification of the amount of broken DNA at site DSB-I at his4–LEU2 relative to the parental fragment is the result of two independent experiments. The symbols represent the mean value, whereas individual results are symbolized by the error bars. (▪) Y329 (rad50S/rad50S); (□) Y324 (mre11S/mre11–T4); (○) Y323 (mre11S/mre11S). One set of data is C, D, and H. (B) For wild-type strain Y325 (MRE11/mre11S) processed breaks can be seen as a smear at 4 and 5.5 hr. Homozygous rad50S (C) and mre11S (D) strains accumulate unresected DSBs visible as sharp bands in a similar way. (E) Quantification of the amount of broken DNA at the major DSB site close to THR4 relative to the parental fragment (representation analogous to A). (▪) Y329 (rad50S/rad50S); (○) Y323 (mre11S/mre11S); (•) Y292×294 (mre11S rad50S/mre11S rad50S). No DSBs were detectable in strains Y328 (mre11–T20/mre11–T20) (F) and Y327 (mre11–T4/mre11–T4) (G). Asterisks indicate signals corresponding to repetitive DNA on the ethidium bromide-stained gel (not shown; see also Storlazzi et al. 1995). (H) In Y324 (mre11S/mre11–T4) DSB formation was not different from that of mre11S homozygote (D).
Intensities of parental and DSB-induced signals were determined for each lane using a PhosphorImager. The ratio of DSB versus parental band was used as the parameter to indicate DSB levels. Such quantification revealed that at both loci examined more broken DNA molecules are present at later time points in rad50S(KI81) than in mre11S mutants, the relative difference depending on the locus (Fig. 6A,E).
Unlike in rad50S the mre11S DSB signals decrease at the 8.5- and 9-hr time points. If this decrease is attributable to residual single-strand degradation in mre11S strains (but to a lesser extent in rad50S) and the broken molecules are finally converted to recombination products, this may explain why mre11S is more inducible for gene conversion than rad50S in return-to-growth experiments.
DSBs also accumulate at another hot spot for recombination—THR4—both in mre11S and rad50S single mutants, as well as in a rad50S mre11S double mutant (Fig. 6E). In addition, a com1 mre11S mutant was tested (data not shown) and found to yield the same breakage pattern at THR4 as mre11S and com1/sae2 (McKee and Kleckner 1997; Prinz et al. 1997) alone.
mre11S mutants are defective in forming homologous synaptonemal complexes
Mutants that can be rescued by spo13 are unable to synapse their axial elements (AEs) (Alani et al. 1990; Engebrecht et al. 1991; Bhargava et al. 1992; Menees et al. 1992; Loidl et al. 1994; Johzuka and Ogawa 1995), whereas mutants that require an additional spo11 mutation to produce viable spores were shown to have reduced synaptonemal complex (SC) formation (Alani et al. 1990; Bishop et al. 1992; Loidl et al. 1994; Rockmill et al. 1995b; Prinz et al. 1997). In such mutants AEs much longer than those in wild type have also been observed. In this study a considerable fraction of nuclei showed extensive, although not wild type-like, synapsis.
To relate DSB formation with a cytological phenotype, synapsis and DSBs were examined in the very same experiment for the MRE11/mre11S, the heterozygous mre11S/mre11–T4, and the homozygous mre11–T4 and mre11S strains. Nuclei of wild-type and mutant cells were classified as belonging to five categories by their content of SC-related structures visualized in the transmission electron microscope (TEM) (Figs. 7A–I and 8A–E). One hundred eighty-one to two hundred seventy-five nuclei were counted per time point per strain. The kinetics of SC formation and degradation in the MRE11/mre11S heterozygote was as reported for a published wild type, and the kinetics of the mre11–T4 strain was similar to rad50Δ (Padmore et al. 1991). mre11S/mre11–T4 behaved like mre11S/mre11S with respect to SC formation, further indicating that intragenic complementation is not very pronounced during sporulation in liquid medium.
Figure 7.
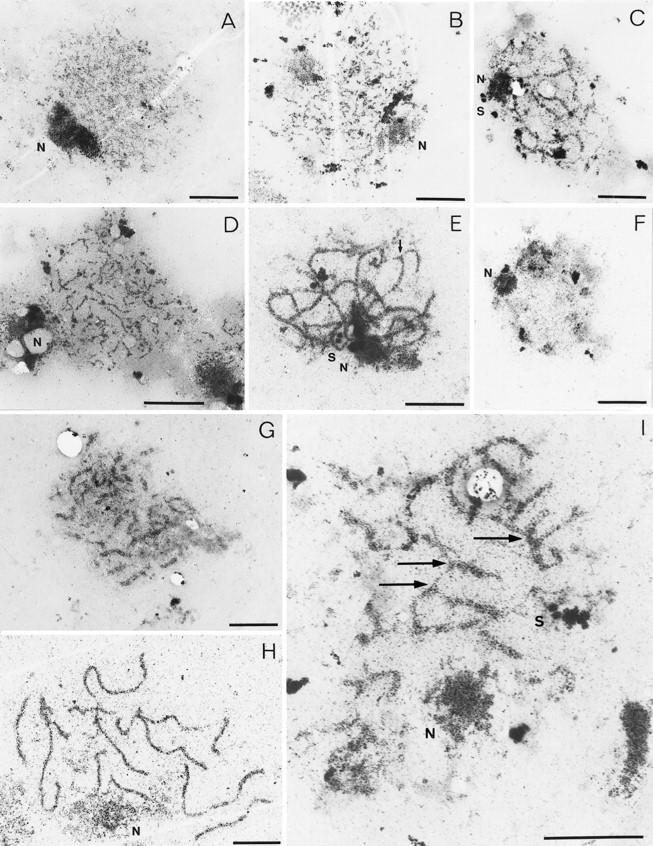
Aberrant SC formation in mre11 mutants shown by electron microscopy. Examples of nuclei classified according to their SC-related morphology are shown. SCs and their precursors are visible as electron dense linear structures, nucleoli (N) as prominent large irregularly shaped dots, and spindle pole bodies (S) as little, round, double spots (bars, 2 μm). (A,B) Class 1 nuclei containing short, unsynapsed axial elements. mre11 nuclei sometimes contain slightly thickened short axial elements as in (B); (C) class 2 nuclei contain both short unsynapsed axial elements and partially synapsed stretches; (D) class 3 nuclei containing elongated unsynapsed axial elements were predominantly observed in mre11S mutants; (E) extensive SC formation (class 4) in a mre11S/mre11S nucleus with only few SC ends visible (the arrow marks a potential partner switch); (F) class 5 nucleus devoid of SC structures possibly past prophase; (G) a rare example of SC formation in mre11–T4 homozygotes; (H) wild-type SCs (class 4) from the MRE11/mre11S strain; (I) partner switches of axial elements confer a network-like appearance to some SCs in the mre11S mutant. Arrows indicate sites of presumed partner switches. A, B, and G are from strain Y327 (mre11–T4/mre11–T4); C–F and I are from Y323 (mre11S/mre11S) and H is from Y325 (MRE11/mre11S).
Short AEs comprise the first class of nuclei observed in wild type and mutants. However, they can differ, as very thick short AEs were found in nuclei of all mre11 mutants but not of wild type. Wild-type nuclei of this class contain typically very faint AEs. Also, the total number of nuclei with short AEs is lower and decreases faster in wild type than in the mutants, indicating that the turnover of these precursors to the next stage is blocked or delayed in the mutants (Fig. 8A–D). The mre11S/mre11S and mre11S/mre11–T4 strains exhibit a second category of unsynapsed AEs (class 3) that are elongated and thickened (Figs. 7D and 8B–D). They also have been described for rad50S/rad50S strains (Alani et al. 1990). This class may either exist as a direct consequence of the cell cycle delay in mre11S cells or, alternatively, represent AEs that were unstably synapsed in vivo, thus belonging to class 2 or 4 (see below), but had been disrupted by the spreading forces. Figure 7C shows an example of a nucleus with both unsynapsed AEs and synapsed stretches (class 2). This class is represented well in mre11S/mre11S and mre11S/mre11–T4 strains, albeit slightly delayed relative to wild type (Fig. 8A,C,D). This also is interpreted as a consequence of a delay in the progression of one transition state (class 2) to the next (class 4). Class 2 nuclei are very rarely found for mre11–T4/mre11–T4, suggesting a role of DSBs for synapsis (see below). Because synapsing and degrading SCs are sometimes morphologically indistinguishable, we propose that class 2 nuclei contain mainly synapsing SCs before class 4 peaks and mainly degrading SCs after that.
Figure 8.

Kinetics of class 1–5 nuclei during progression through meiotic prophase. Quantitative analysis of SC-related structures of the time course experiment presented in Figs. 6 and 7. Cells past meiosis I are bi- and tetranucleate as observed after DAPI staining. (A–D) Symbols corresponding to class 1–3 and class 5 nuclei and meiotic progression are described in the upper right panel. (E) For clarity, class 4 nuclei exhibiting extensive SCs are presented separately. Wild type exhibited up to 30% SC formation, mre11S mutants up to 7%, but the mre11–T4 homozygote only ∼1%.
Class 4 nuclei show extensive SC formation but no unsynapsed AEs (Fig. 7E,G–I). However, whereas in wild type there are usually a number of 10–16 discrete complexes corresponding to the paired chromosomes, the mutants give a more variable picture. Figure 7E depicts a mre11S/mre11S nucleus with extensive SC loops, but SCs are not separated from each other. Figure 7I shows a different mre11S/mre11S nucleus, where a network of SCs connected via AEs that perform partner switches can be observed. This situation is similar to that decribed earlier for haploids undergoing meiosis (Loidl et al. 1991) indicating that synapsed AEs connect, at least in part, nonhomologous chromosomes. A very rare example of a mre11–T4/mre11–T4 nucleus with extensive synapsis, showing far more than 16 synapsed entities is shown in Figure 7G. We also observed very small fractions of nuclei forming elongated AEs and showing extensive synapsis (Fig. 8E). We suspect that rare examples of SC formation arise from nuclei initiating some DSBs that are below the limit of detection in our Southern blot (Fig. 6G) and in return to growth (Fig. 4A). In general, most of the mre11–T4 nuclei were only able to form short AEs (Fig. 7B), as expected from a mutant unable to initiate DSBs.
The parallel monitoring of DSB levels and synapsis allows the correlation of these two meiotic landmarks. For the wild type, DSBs reached their maximum level at 4 hr. At the same time, first synapsis, but no complete SC, was observed. In the mre11S mutants the first major increase in DSBs is at 5.5 hr, which is shortly before or at the time when the first synapsis can be demonstrated.
Both homologous pairing and SC formation are reduced in mre11S
The presence of partner switches in mre11 mutants demonstrated by electron microscopy of silver-stained spreads indicated the occurrence of nonhomologous synapsis. We have combined fluorescence in situ hybridization (FISH) and immunocytology in an effort to study synapsis and homologous pairing—to compare the reduction of SC formation in the mutant with the quantity of homologous DNA pairing. The total amount of SCs in the mutant allows predictions about the minimum level of DNA pairing if synapsis were homologous. If SC formation exceeds DNA pairing, at least some of the SCs must connect nonhomologous chromosomes.
The approach of combining FISH and immunocytology is complicated by two facts: First, it is at present inapplicable to monitor synapsis by immunocytology and pairing by FISH in the same cell because of the different conditions required. Second, not all paired DNA sequences need to be in the context of a SC because pairing precedes SC formation (Scherthan et al. 1992; Weiner and Kleckner 1994). Below, we present statistical evidence that a deficit of homologous pairing relative to the amount of SCs was found, confirming that at least a fraction of the SCs observed by electron microscopy were nonhomologous.
Pairing was monitored in a MRE11/mre11S and a mre11S/mre11S strain at stages corresponding to maximum SC formation, that is, at 5 hr in wild type and at 7 hr in the mutant (see Fig. 8E). Of each same preparation one slide was used for immunostaining with anti-Zip1 antibodies to decorate the SC (Sym et al. 1993) and one was hybridized with a mixture of two differently labeled cosmid probes originating from a small and a large chromosome (chromosomes I and IV) (Fig. 9).
Figure 9.
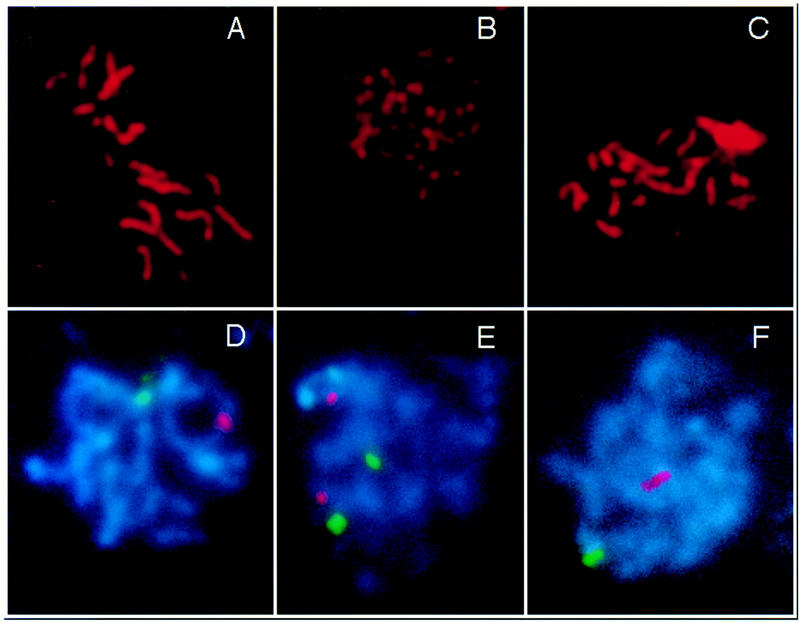
SC formation and homologous pairing are reduced in mre11S. (A–C) Nuclei stained with anti-Zip1 antibody; (D–F) pairing status of chromosome I and IV signals (green and red, respectively). Strains are Y325 (MRE11/mre11S) and Y323 (mre11S/mre11S). (A) Wild-type pachytene SCs; (B) Zip1-positive mre11S nucleus without synapsis; (C) extensive synapsis in mre11S. The upper right patch corresponds to an overexposed Zip1 aggregate. (D) both chromosome I and IV signals are paired in wild type; (E) typical pairing behavior in the mre11S mutant; (F) very rarely both chromosome signals are paired in mre11S.
Distances between the homologous signal pairs as well as between nonhomologous signals were determined. DNA sequences connected by a homologous SC are believed to be within 0.7 μm (Weiner and Kleckner 1994), and such signals will be referred to as being closely paired. Table 4 lists the percentage of closely paired signals for wild-type and mutant. Each single signal was closely paired in only ∼15% of the cases in the mutant compared to 83 % in the wild type. An even stronger difference is apparent when counting nuclei with both signals paired simultaneously. In only 2.9% of the mutant, but in 72% of wild-type nuclei, both signals were paired at once.
Table 4.
In mre11S synapsed nuclei are more frequent than nuclei with both chromosomes I and IV paired
| Relevant genotype
|
Chromosome I paired (%)
|
Chromosome IV paired (%)
|
Both chromosomes I and IV paired (%)
|
Nuclei counted
|
(Nearly) complete synapsis judged by Zip1 (%)
|
Zip1 positive (%)
|
Nuclei counted
|
|---|---|---|---|---|---|---|---|
| mre11S/mre11S | 14.7 | 14.9 | 2.9 | 450 | 7.3 | 43.6 | 300 |
| MRE11/mre11S | 83 | 83 | 72 | 100 | 41 | 60 | 200 |
Spread nuclei from the same experiment were either hybridized with probes for chromosome I and IV or immunostained with anti-Zip1 antibodies. Cells were spread at time points when synapsis was maximal. Zip1-positive nuclei may contain unsynapsed axial elements and degrading SCs. Only continuous Zip1 axes were regarded as synapsed. Fewer nuclei have both chromosomes I and IV signals paired in mre11S than expected if synapsis were homologous.
Synapsis was quantified by determining the fraction of nuclei with long, bright, continuous Zip1-positive axes (corresponding to class 4 nuclei in Figs. 7 and 8). These could be clearly differentiated from other Zip1-positive nuclei, because the Zip1 antibody binds strongly only to the synapsed regions of chromosomes (Sym et al. 1993). In wild type 41% of the nuclei had extensive SCs, with an average total length of 31.8 ± 7.0 μm (data from 37 nuclei). Only 7.3% mre11S nuclei were largely synapsed, whereas the number of Zip1-positive nuclei was similar to wild type (Table 4). In these nuclei the total length of the SC complement was 28.3 ± 7.3 μm (data from 29 nuclei). Therefore, SC length is not significantly different between wild type and mutant in the subpopulations described (class 4), the average mutant being only 3.5 μm or a factor of 0.89 shorter than the average wild-type SC.
If the SCs were homologous in the mre11 mutant the 7.3% of almost completely synapsed nuclei are expected to contribute largely to the fraction of nuclei with both signals paired. Double pairing may also occur in partially synapsed nuclei (class 2) or by presynaptic alignment in nuclei without synapsis. Therefore, the 7.3% contribution of the synapsed subpopulation alone represents a minimum estimate for the percentage of nuclei with both signals closely paired. The observed fraction of 2.9%, however, is clearly smaller than predicted by the amount of SC, indicating that a significant portion of the SCs formed in the mre11S mutant are connecting nonhomologous chromosomes.
Discussion
rad50 is epistatic to mre11S
A screen for new genes and functions that become essential after initiation of meiotic DSBs was carried out which lead to the identification of a new allele of MRE11 with unexpected properties. MRE11 has been described before (Ajimura et al. 1993) as one of the genes required for initiation of meiotic DSBs. The mre11::hisG mutant is epistatic to rad50S, a separation-of-function mutation that is only defective in the processing of the meiotic DSBs, whereas the other function of RAD50, namely formation of DSBs, is unaffected (Alani et al. 1990; Ogawa et al. 1995). The activity of the mre11 allele described in this work mirrors this property in that it also forms DSBs, but the rad50 null mutation is epistatic to the new allele, which is therefore called mre11S. We conclude that the respective proteins that have also been shown to interact physically in a two-hybrid assay (Johzuka and Ogawa 1995) are intimately associated with the processes immediately before and after DSB formation.
Sequencing of the mutant allele identified two changes, Pro to Ser at position 84 and Thr to Ile at position 188 of the predicted amino acid sequence. The identified S mutations thus maps to the amino-terminal part of the protein, which shows strong homologies to both eukaryotic homologs of Mre11 and the prokaryotic SbcD (SbcD has nuclease activity) (Connelly and Leach 1996), whereas the carboxyl terminus does not exhibit strong sequence conservation. This suggests that the core regions and a possible catalytic domain reside in the amino terminal half of the protein and the carboxyl terminus may have a more yeast specific role. Strong homology to the bacterial SbcD is limited to several short blocks throughout the amino terminus. Neither of the two point mutations identified in mre11S maps to such a block but to regions conserved among all eukaryotic Mre11 relatives. This may mean that these are newly evolved regions in eukaryotes to serve meiosis-specific functions. The affected domain may be involved in regulation of Mre11p activity or in conserved protein–protein interactions.
mre11S causes a defect in DSB processing and repair
The mre11S mutant was analyzed for possible defects during vegetative growth and in meiosis. Resistance to MMS in mre11S was between wild-type and homozygous disruptions. No indication for a similar mitotic hyper-recombination as in mre11 disruptions was found at two heteroalleles tested. This implies that mitotic functions of the Mre11Sp are largely intact or only weakened and that a possible catalytic function is not seriously impaired. Commitment to meiotic recombination as assayed by return to growth was also intermediate, suggesting that many of the induced DSBs could be repaired on vegetative medium in a mre11S strain. However, serious defects were found when cells were required to complete meiosis.
Spore formation and spore viability are strongly reduced in mre11S and in the mre11S spo13 mutants. On the other hand, a mre11S rad50 spo13 triple mutation is viable in meiosis. The most obvious explanation is that MRE11 function is dispensable because rad50 prevents DSB formation. In this study meiotic DSBs were shown to accumulate in the mre11S/mre11S mutant; therefore, the defective function is required for processing and repair of DSBs. Quantification of the relative amount of broken DNA revealed a difference between rad50S and mre11S at the his4–LEU2 locus. After a similar initial increase DSBs accumulated to a higher level in rad50S/rad50S than in mre11S/mre11S or in mre11S/mre11–T4. In addition, the latter two leveled off at 7 hr and decreased by 8.5 hr, indicating loss of breaks. Although nonspecific degradation of a fraction of DNA ends cannot be ruled out, DNA repair is suggested to explain the loss of breaks because the bands remained sharp at 8.5 hr. Such repair may cause the observed partial recombination competence in a mre11S strain in the return-to-growth experiment. However, neither heteroduplex DNA nor physical recombinants could be detected at his4–LEU2 in the homozygous mre11S strain under conditions where ∼30% of wild-type levels would have been detected (data not shown). At the natural hot spot close to THR4 as well as at DED81 (data not shown) differences observed in DSB levels between rad50S and mre11S were less clear.
It is not known whether Spo11p, which remains covalently linked to the 5′ ends of DSBs in rad50S and most likely also in com1/sae2 mutants (de Massy et al. 1995; Keeney and Kleckner 1995; Liu et al. 1995; Keeney et al. 1997), also does so in mre11S mutants. It is likely because the DNA ends seem to be blocked from processing. However, a fraction of the DSBs finally disappears, suggesting that the DNA ends have been liberated. The higher instability of DSBs in mre11S versus rad50S observed at some recombination hot spots may be related to the higher yield of gene conversions in mre11S versus rad50S observed in return to growth. The fact that in such an experiment a considerable fraction of cells remains viable after induction of meiosis demonstrates that under vegetative conditions in a fraction of the cells every break can be repaired .
The homology of Mre11p/Rad50p to the SbcC/SbcD nuclease suggests that Mre11p might be the catalytic subunit of a Mre11p/Rad50p complex, being directly responsible for 5′ resection at DSB sites either exo- or endonucleolytically. This is compatible with the results from physical DSB analysis and recombination studies assuming that a different pathway that works more efficiently during mitosis (as seen in return to growth) finally processes the breaks. Taking into account that mitotic functions are largely intact, as discussed above, we propose that a domain that regulates meiosis-specific Mre11p activity is mutated in Mre11Sp. Recent results from studies in vegetatively growing cells relate Mre11, Rad50, and Xrs2 to exonucleolytic function. Ivanov et al. (1996) have studied HO-induced illegitimate recombination and found a retardation of 5′ resection in rad50 and xrs2 mutants. In mre11 strains, repair thought to involve 5′ resection is 10-fold reduced at HO cuts (Moore and Haber 1996) and 5′ degradation is even slower in mre11 than in rad50, but not absent (J. Haber, pers. comm.). These experiments show that there is residual 5′ → 3′ resection in the absence of MRE11. One important difference between vegetative and meiotic repair processes requiring Mre11p is the presumptive protection of 5′ ends by Spo11p in meiosis (Keeney et al. 1997). A role for Mre11p/Rad50p could therefore be to liberate Spo11p by a single strand endonucleolytic step. A compatible activity has also been demonstrated for SbcC/SbcD. In any case, Mre11p should be associated with meiotic chromosomes. When DSB processing was blocked in a rad50S mutant Myc-tagged Mre11p was found clustered in distinct foci of varying intensity within spread nuclei (Nairz 1997).
Intragenic complementation between mre11S and 3′ disrupted alleles of MRE11
The fact that intragenic complementation was observed between mre11S and the 3′ disruptions strongly suggests that DSB initiation and DSB processing functions reside in separate domains.
The regions truncated in the complementing alleles mre11–T4 and mre11–T10 lack strong sequence conservation. They are nevertheless essential for DSB initiation, probably for interaction with yeast-specific factors that allow access to the DNA or that activate Spo11 endonuclease. These interactions as well as binding to Rad50p, which has been shown to involve the amino terminus (Johzuka and Ogawa 1995), are intact in the mre11S mutant. The intragenic complementation is best explained by homotypic interaction of Mre11 proteins. This is in good agreement with the demonstration that Mre11p interacts with itself in two-hybrid studies (Johzuka and Ogawa 1995). Intragenic complementation for mre11 alleles can thus be explained by a model in which a partially functional heteromultimeric complex contains both Mre11–T4(T10)p and Mre11Sp. Whereas Mre11Sp allows Spo11p to interact with the DNA, Mre11–T4(T10)p may contribute the 5′ resecting activity either alone or in conjunction with another component. Less likely the carboxy-terminally truncated alleles reactivate the processing function of Mre11Sp through direct interaction or Mre11Sp and Mre11–T4p work independently and sequentially.
With MRE11 and RAD50 two genes are known to be required both for initiation and repair of DSBs and these functions can be separated in both genes. Furthermore, their products interact with each other physically. Therefore, we propose a model in which a complex, a “recombinosome”, consisting of Rad50p, Mre11p, and other components required for recombinational repair, must be present at a given hot spot to allow Spo11p to cut. The use of such bifunctional complexes would greatly enhance repair efficiency by eliminating the damage recognition step. Others have presented indirect evidence that even the homologous template for DNA repair may already be in place at the time of DSB initiation (Rocco and Nicolas 1996; Xu and Kleckner 1995).
mre11S causes reduced, nonhomologous synapsis and decreases spore viability
The failure to process and repair the DSBs leads to a number of observable consequences. Spore formation is generally reduced in mre11S/mre11S, and the number of dyads rises considerably at the cost of the number of complete tetrads. Of those spores formed, viability is extremely low (1%). The first meiotic division is delayed considerably in the SK1 strain background but not in the disruption mutants (Fig. 8A–D). The delay was also observed for rad50S (Alani et al. 1990) and for com1/sae2 (Mc Kee and Kleckner 1997; Prinz et al. 1997). In other backgrounds these mutations may cause a complete block. In the case of dmc1 the block has been shown to monitor DSBs, utilizing the checkpoint genes MEC1, RAD17, and RAD24 (Lydall et al. 1996).
The spreading of yeast nuclei revealed other consequences of the absent repair of DSBs. SC precursors were enriched relative to wild type, and a new class of nuclei containing long unsynapsed AEs appeared possibly as a consequence of delayed synapsis, most likely caused by the absence of 3′ single-stranded ends for homology search. However, even in the absence of repair of the majority of DSBs there was extensive SC formation in a fraction of mre11S nuclei. Such synapsis has also been observed in rad50S cells (Loidl et al. 1994) and in com1/sae2 nuclei (Prinz et al. 1997). However, in all such cases, synapsis seemed aberrant in that sometimes network-like structures were observed but never the complete chromosome complement with 16 separate bivalents. In this study evidence for partner switches of axial elements is presented, which suggests nonhomologous synapsis. In corroboration of this finding the fraction of nuclei with homologously associated FISH signals was compared to the fraction of nuclei exhibiting extensive SCs. Considerably more nuclei were found with almost the complete chromosome complement synapsed than nuclei with two signals paired concomitantly. We conclude that at least in a fraction of the completely synapsed nuclei homologous signals were not paired, that is, that at least some synapsed regions were nonhomologous. It has been shown earlier for yeast that synapsis of nonhomologous chromosomes is possible in haploid yeast (Loidl et al. 1991). Also nonhomologous synapsis in haploids depends on RAD50 and other DSB initiation genes (F. Klein, unpubl.). We therefore propose that synapsis requires the presence of DSBs but that homologous synapsis is only ensured by at least the formation of the 3′-overhanging single-stranded DNA ends. In DSB negative mutants (e.g., in mre11–T4/mre11–T4) synapsis does normally not take place, but in DSB-accumulating mutants the homologous registration fails while synapsis occurs by default and irrespective of homologies. The few cases where long axial elements and extensive synapsis is found in mre11–T4/mre11–T4 may be explained by initiation of synapsis through DSBs below the limits of physical detection. These DSBs may only occur in a very small subpopulation and may not be sufficient to ensure viability.
A model presenting a unified view of the relation between DNA metabolism and formation of SCs and their precursors is introduced (Fig. 10). Axial elements do form in the absence of DSBs, as observed in many different null mutants. Unprocessed DSBs allow for synapsis which is at least partially nonhomologous, as shown for mre11S. Synapsis induced by unrepaired DSBs may represent a default pathway that comes into effect when no homologies are found representing the uncoupling of synapsis from homology search. Homologous synapsis is ensured only by the recombination processes that follow DSB formation, processing, and strand invasion. These processes are likely to establish homologous interactions that allows synapsis of the proper axial elements.
Figure 10.

Model of the relation between DSBs and synapsis. It is proposed that unprocessed DSBs are sufficient to allow for synapsis. Only when DSBs are repaired successfully is homologous synapsis possible. Rare synapsis observed in the mre11–T4 homozygote, where neither induction of meiotic recombination nor DSBs were detected, could be explained if DSBs below the limit of detection were able to induce SC formation. (<) Increasing amount of either SCs or DSBs.
Materials and Methods
Media, genetic techniques, growth regimen, and ether killing
Yeast was grown in rich medium (YPD), synthetic complete medium lacking factors (SC−) or minimal medium supplemented with specific factors (SD+) as described (Rose et al. 1990). Presporulation medium (YPA) was 1% yeast extract (Difco), 2% peptone (Difco), 2% potassium acetate, and SPM was 2% potassium acetate (pH 7) (optionally solidified with 2% agar). However, sporulation of strain Y136 on solid medium was performed on SPM+ (0.25% yeast extract, 0.1% glucose, 1.5% potassium acetate plus one-fortieth volume of an amino acid and nucleotide mix containing 40 mg of Pro, 80 mg of Tyr, 200 mg of His, Leu, Lys, Met,Trp, and Arg, 400 mg of Ade and Ura, and 1 gram of Phe per 25 ml). MMS (Schuchardt) was added to YPD just prior to pouring plates. Asci were digested with 200 μg/ml of Zymolyase 20T (Seikagaku Kogyo Ltd) for 20 min at 37°C for tetrad dissection.
Sporulation for DSB assays, SC spreading, and return to growth was done as described by Loidl et al. (1994). Commitment to meiotic recombination was monitored in return-to-growth experiments as described (Sherman and Roman 1963; Alani et al. 1990). Samples of synchronized sporulating cells were taken at the specified time after transfer to liquid SPM, sonicated, diluted, and plated on at least two YPD, SC−HIS, and SC−ARG petri dishes. Ether treatment for enrichment of spores by selectively killing unsporulated cells was performed as described recently (Prinz et al. 1997).
Plasmids
p3 was derived from pNKY1070 (a YCP50-based RAD50 plasmid from N. Kleckner, Harvard University, Cambridge, MA) by cutting with SalI and religating it to yield p1. p1 was digested with EcoRI and ligated to a 3.95-kb EcoRI fragment from R990 (S. Roeder, Yale University, New Haven, CT) containing ADE2 (and 460 bp of mer2 sequence). URA3 was removed by deleting a 1.6 kb SmaI–NruI fragment. p21 was constructed by ligating a 2.2-kb BamHI–NruI fragment from pTW15 (R. Esposito, University of Chicago, IL) containing SPO13 coding sequence into pRS315 (Sikorski and Hieter 1989), which was cut with HindIII, blunted by filling in, and then cut with BamHI. p265 originated from YCplac33 (Gietz and Sugino1988) into which a 4.3-kb BamHI fragment containing MRE11 was inserted. p266 is derived from p265 by gap repair (see Cloning and Sequencing, below).
Mutagenesis
Strain Y136 was grown on SC–ADE–URA for 2 days, transferred to YPD for 8 hr, and sporulated on 2% potassium acetate plates for 24 hr. Asci were resuspended in 150 μl of 10 mm DTT with 50 μg of Zymolyase 20T (Seikagaku), incubated for 90 min at 37°C, and sonicated for 5 × 5 sec on ice to obtain single spores. Spores (5 × 107) in 1 ml were mutagenized for 30 min at 30°C with 27 or 32 ng/ml of MNNG (Sigma) to a viability of 86% or 36% (corresponding to a 5- or 20-fold increase in canavanine-resistant colonies as a measure of mutational yield) and frozen in 50% glycerol at −80°C. After determination of viability the mutagenized spores were diluted and plated to a density of 180 or 275 colonies per SC–ADE plate. After 2 days colonies were replica-plated to SPM+. After 3 days of sporulation at 30° the colonies were stamped onto YPD, ether treated to select for spores, and incubated for another 3 days at 30°C. Twenty-two candidates of 18,000 colonies of the first mutagenesis and 16 candidates of 13,000 colonies of the second mutagenesis formed red patches and were isolated.
Cloning and sequencing
Because difficulties were expected in crossing out mutant alleles that prevent successful meiosis from a homothallic diploid strain, the affected gene was cloned by direct complementation of the spore viability defect. Among eight mutant candidates, number 220 was chosen for complementation. The candidate was transformed with a CEN-based genomic URA3 library [a gift of Brian Jensen and Breck Byers (University of Washington, Seattle) made from a A364A derivative], and transformants were selected on SM−URA, SM−ADE, and SM−LEU to ascertain the presence of both the library plasmid and of plasmids p21 (SPO13, LEU2) and p3 (RAD50, ADE2), which allows initiation of recombination in cells with “white” progeny. The resulting colonies were sporulated on SPM+, replica plated to YPD, ether treated, and examined for the appearance of white patches. One such patch was detected, indicating that cells had regained the ability to form viable spores in the presence of RAD50 and SPO13. Such spores were directly mated to strain Y7, and a descendant (ho, RAD50, spo−) was further crossed to strain Y5. In 12 of 12 complete tetrads from this cross, the sporulation defect segregated 2 : 2, indicating Mendelian behavior of a recessive gene. When 104 spores from this cross were analyzed, 51 were spo− and 53 were SPO+.
The complementing plasmid was isolated, the large insert was subcloned into YCplac33, and the resulting plasmid p265, containing a 4.3-kb BamHI fragment of the insert with a central ClaI site, proved sufficient to complement the sporulation defect of Y233. Neither the internal 2.0-kb ClaI–BamHI nor the 2.3-kb ClaI–BamHI fragment alone could complement identifying the ClaI site as mapping within the coding sequence. Both fragments were subcloned into Bluescript SK− and sequenced from the ClaI site by the dideoxy chain-termination method (Sanger et al. 1977) using the T7 sequencing kit (Pharmacia) and 35S-labeled dATP (New England Nuclear). The result identified the sequence as a meiotic recombination gene, MRE11, that had just been published by Johzuka and Ogawa (1995).
The cloning and sequencing of mre11S
mre11S was recovered by gap repair (Orr-Weaver et al. 1983): A haploid strain (Y227 mre11S, ura3) was transformed to URA+ with the gel-purified 7.9-kb AflII–NruI fragment of p265 devoid of the complete MRE11 coding and of 170-bp MRE11 upstream sequence (Fig. 3A). The gap-paired plasmid, p266, was recovered from a transformant, amplified in E. coli, and transformed into Y233 (mre11S/mre11S) for complementation of the mre11S-dependent sporulation defect. p266 did not restore sporulation in nine of nine transformants tested.
For sequencing of mre11S (p266) and MRE11 (p265) various restriction fragments were subcloned into Bluescript (SK−). In addition, a set of exonuclease III-generated deletions were derived from SphI–AflII double-digested p266 (Sambrook et al. 1989) and sequenced using universal primers MJ3 and MJ4 (MJ3, 5′-AGCGGATAACAATTTCACACAGGA-3′, MJ4, 5′-CGCCAGGGTTTTCCCAGTCACGAC-3′). The SK1-derived mre11S sequence differed from the published sequence (Johzuka and Ogawa 1995) at two positions in the promotor (−28 and −4), 4 redundant sites (+219, +786, +843, +1155), one site close to the stop codon leading to a frame shift (+1928), which renders the protein 49 amino acids longer [previously mentioned by J. Skelton and C.M. Churcher (unpubl.), GenBank accession no. S57592], one nonconservative change in this tail (+1975), as well as three nonconservative changes in the 3′ part of the gene (+250, +563, +1138). To clarify which of the base differences was responsible for the mre11S phenotype, MRE11 from p265 was sequenced as well. MRE11 on p265 and mre11S on p266 differ at one redundant and two nonconservative sites, namely at positions +250 and +563. In addition, wild-type MRE11 was recovered from SK1 by gap repair and sequenced as well. It proved identical to the MRE11 sequence of p265 for both position +250 and + 563. The SK1-derived sequence has been submitted to GenBank (accession no. U60829).
Transposon mutagenesis and complementation analysis
A 2.0-kb BamHI–ClaI fragment containing the 5′ part of MRE11 and a 2.3-kb ClaI–BamHI fragment harboring the 3′ part were cloned into pHSS6 separately and subjected to in vivo transposon mutagenesis in E. coli using mTn3(LEU2, lacZ without promotor, and ATG) as described (Seifert et al. 1986). Orientation and location of inserted transposons was determined by restriction analysis, taking advantage of a BamHI site at the 5′ end of lacZ on the end of the 6.6-kb transposon. Appropriate clones were digested with NotI and used to construct MRE11 disruptions in strain NKY857 by single-step gene replacement (Rothstein 1983) selecting for LEU prototrophs. Yeast transformation was done according to Gietz et al. (1992). Correct integration of disrupted DNA fragments was confirmed both by complementation analysis with a mre11S strain (Y224) and by Southern blotting probing with a mixture of the 2.0-kb and 2.3-kb BamHI–ClaI fragments (not shown).
The transposon in mre11–T20 mapped within 50 bp of the start codon of MRE11 and the lacZ ORF is oriented opposite to the MRE11 ORF (Fig. 3A). In mre11–T4 and mre11–T10, lacZ is fused in-frame to the remaining mre11 fragment: Patches of sporulated and unsporulated mre11S/mre11–T4 and mre11S/mre11–T10 heterozygotes were transferred to nitrocellulose filters (Schleicher & Schuell) from YPD and sporulation plates, respectively, frozen at −80°C for 30 min and thawed to break up the cells. Ten microliters of X-gal solution (2 mg/ml in dimethylformamide) was added to the cells, which turned blue in sporulated, but not in unsporulated, patches, indicating expression of β-galactosidase activity from the MRE11 promoter. mre11–T40 and mre11–T16, which are disrupted at roughly the same positions as mre11-T4 and mre11–T10, respectively, by equally oriented transposons do not show meiotically induced lacZ activity, suggesting that integration was out of frame.
DSB
Meiotic DNA DSBs were monitored at three hot spots, one close to THR4 (Goldway et al. 1993), one at DED81 (de Massy and Nicolas 1993), and one at the artificial his4–LEU2 construct (Cao et al. 1990; Storlazzi et al. 1995) Physical maps of the hot spots are given there. Yeast DNA was isolated as described by Cao et al. (1990) and modified by Bishop et al. (1992), except that the ether extraction after phenol/chloroform/isoamyl alcohol (25:24:1) was replaced by a chloroform/isoamyl alcohol (24:1) extraction. DNA was taken at the time indicated, digested appropriately, separated on 0.8% TAE-buffered agarose gels, and transferred to Hybond-N nylon membranes (Amersham) by alkaline blotting. Probes were gel purified by Elu-Quick (Schleicher & Schuell) according to the manufacturer’s protocol and radioactively labeled by random priming using [α-32P]-dATP (NEN).
A 0.89-kb HindIII fragment from pMJ338 (M. Lichten, National Cancer Institute, Bethesda, MD) mapping close to the THR4 hotspot was used to probe BglII-digested genomic DNA. For examination of the artificial his4–LEU2 hot spot, DNA was digested with PstI and probed with a combination of a 0.7-kb and a 0.9-kb PstI–EcoRI fragment from pNKY155 (probe B) or a 1.8-kb PstI–EcoRI fragment from pNKY291 (probe A) (Storlazzi et al. 1995; Xu and Kleckner 1995). To probe for the hot spot close to DED81, an EcoRV–BglII fragment of the ARG4 gene was used (de Massy and Nicolas 1993).
Quantification of radioactive label was done on a PhosphorImager (Storm 840 from Molecular Dynamics). For each lane, a two-dimensional intensity profile was generated and peaks corresponding to parental and DSB bands were integrated after removing background noise. Signal intensity was calculated as a ratio of DSB value versus parental value.
Spreading, immunocytology, FISH, and silver staining for electron microscopy
All cytological techniques used are described in detail elsewhere (Engebrecht et al. 1997). Spreads were prepared as described originally (Loidl et al. 1991). In brief, 10 ml of sporulation culture was centrifuged and resuspended in 1 ml of spheroplasting solution (2% potassium acetate, 0.8 m sorbitol, 10 mm DTT), including 7 μl of Zymolyase 20T (10 mg/ml), and digested at 37°C until cells lysed in 1% sodium Sarkosyl. The reaction was stopped by adding 10 ml of ice-cold solution II [0.1 m 2-(N-morpholino) ethane sulfonic acid, 1 m sorbitol, 1 mm EDTA, 0.5 mm MgCl2], and the spheroplasts were spun down at low speed and resuspended in 1 ml of solution II. On a clean slide 20 μl of cell suspension was prefixed with 40 μl of fixative (4% paraformaldehyde, 3.4% sucrose), lysed with 80 μl of 1% lipsol, and fixed with 80 μl of fixative.
For silver staining slides were dryed overnight, washed with water, and air-dried. Slides were incubated in a humid chamber with 100 μl of a silver nitrate solution (5 grams plus 5 ml of H2O) under a nylon mesh at 60°C for 40 min. For electron microscopy silver-stained spreads were transferred on a plastic film (from 1% Formvar in chloroform) to microscopical grids and examined in a Zeiss EM 900 at 50 kV and 3000- or 7000-fold magnification. For immunostaining, drying of the preparations was prevented during all of the procedure. Slides were washed with PBS (10× PBS: 75.97 grams of NaCl, 9.94 grams Na2HPO4, 4.14 grams of NaH2PO4/1 liter of H2O), blocked for 10 min with 100 μl of blocking buffer (0.5% BSA and 0.2% gelatin in 1× PBS) under a coverslip, and incubated with 40 μl of a 1:100 dilution of mouse anti-Zip1 antibody (in blocking buffer) at 4°C in a humid chamber overnight. After washing in PBS the slide was incubated with 40 μl of FITC-conjugated goat anti-rabbit secondary antibody (Sigma) for 2 hr at room temperature, washed again in PBS, and mounted in antifade buffer (Vectashield) with DAPI (0.4 μg/ml).
For FISH we followed a variation of the protocol of Scherthan et al. (1992) described in Engebrecht et al. (1997). A mixture of Cy3-labeled cosmid ATCC 70929 (34-kb insert from the right arm of chromosome IV) and biotin-labeled cosmid ATCC 70893 (33-kb insert from the left arm of chromosome I) was used to hybridize the respective loci in the spread nuclei. Extravidin–FITC [Sigma 1:200 in blocking solution (3% bovine serum albumin (BSA)/4× SSC/0.1% Tween 20)], biotin (Sigma, 1:200 in blocking solution), and again extravidin–FITC (1:200) were applied in consecutive rounds to create the signal for the biotin-labeled probe. DNA was stained for 5 min with DAPI-containing buffer [40 ml of McIlvaine (9 mm citric acid, 80 mm NaH2PO4, 2.5 mm MgCl2), 5 ml of 10× PBS, 5 ml 40% paraformaldehyde (pH 7.5), 250 μl of DAPI (0.2 mg/ml)]. Samples were first dehydrated for 2 min in 70% EtOH, 2 min in 96% EtOH, and then mounted in antifade buffer.
Fluorescent microscopical examination was performed on a Zeiss Axioplan; pictures were digitalized by a Photometrics CCD camera and processed by IPlab software.
Sequence analysis
Sequence analysis was done via internet on the BCM search launcher (http://kiwi.imgen.bcm.tmc.edu:8088/search-launcher/launcher.html) using BLAST and BEAUTY programs for sequence comparison (Altschul et al. 1990; Smith et al. 1996). Coiled–coils were predicted according to Lupas et al. (1991) via BCM using window sizes 21 and 14, and sequence motives were found with PROSITE (Bairoch et al. 1995; http://expasy.hcuge.ch/sprot/prosite.html).
Strains
All strains are isogenic derivatives of SK1 (Kane and Roth 1974) and are listed in Table 5. For intragenic complementation analysis, isogenic diploid strains were derived from the MATα parent Y224 (mre11S) and the following MATa strains: Y253 (mre11–T20), Y254 (mre11–T37), Y255 (mre11–T24), Y256 (mre11–T34), Y257 (mre11–T4), Y258 (mre11–T40), Y261 (mre11–T16), Y262 (mre11–T10), and NKY857 (MRE11). For DSB assays, cytological analysis, test of MMS resistance, and return-to-growth experiments, the following strains were used: Y325 (MRE11/mre11S), Y327 (mre11–T4/mre11–T4), Y328 (mre11–T20/mre11–T20), Y329 (rad50S/rad50S), Y323 (mre11S/mre11S), and Y324 (mre11S/mre11–T4). They are descendants of strains NKY1303 and NKY1543 described in Storlazzi et al. (1995) and Xu and Kleckner (1995) carrying arg4 heteroalleles (arg4–Bgl or arg4–Nsp) and his4 heteroalleles (his4X–LEU2–MluI::BamHI or his4B–LEU2–MluI).
Table 5.
Strains used in this study
| Strain (SK1)
|
Genotype (MATa/MATα, HO)
|
|---|---|
| NKY857 | MATa, ho::LYS2, lys2, leu2::hisG, his4X, ura3 |
| Y5 | MATα, ho::LYS2, lys2, leu2::hisG, his4B, ura3, ade2::hisG::URA3::hisG |
| Y7 | MATa, ho::LYS2, ade2::hisG, ura3 |
| Y136 | MATa/MATα, HO, rad50::hisG, leu2::hisG, his4B, ade2::hisG, ura3, pTW15 (SPO13, URA3), p3 (RAD50, ADE2) |
| Y224 | MATα, ho::LYS2, ura3, ade2::hisG, leu2::hisG, mre11S |
| Y227 | MATα, ho::LYS2, ura3, ade2::hisG, mre11S |
| Y233 | MATa/MATα, HIS4/his4B, ura3/ura3, ADE2/ade2::hisG, LEU2/leu2::hisG, SPO13/spo13::hisG, mre11S/mre11S |
| Y253 | MATa, ho::LYS2, lys2, leu2::hisG, his4X, ura3, mre11–T20(–LEU2) |
| Y254 | MATa, ho::LYS2, lys2, leu2::hisG, his4X, ura3, mre11–T37(–LEU2) |
| Y255 | MATa, ho::LYS2, lys2, leu2::hisG, his4X, ura3, mre11–T24(–LEU2) |
| Y256 | MATa, ho::LYS2, lys2, leu2::hisG, his4X, ura3, mre11–T34(–LEU2) |
| Y257 | MATa, ho::LYS2, lys2, leu2::hisG, his4X, ura3, mre11–T4(–LEU2) |
| Y258 | MATa, ho::LYS2, lys2, leu2::hisG, his4X, ura3, mre11–T40(–LEU2) |
| Y261 | MATa, ho::LYS2, lys2, leu2::hisG, his4X, ura3, mre11–T16(–LEU2) |
| Y262 | MATa, ho::LYS2, lys2, leu2::hisG, his4X, ura3, mre11–T10(–LEU2) |
| Y292×294 | MATa/MATα, ho::LYS2/ho::LYS2, rad50S–KI81::URA3/rad50S–KI81::URA3, ura3/ura3, LEU2/leu2R, arg4Δ10/arg4Δ10, ADE2/ade2::hisG, mre11S/mre11S |
| Y323 | MATa/MATα, ho::LYS2/ho::LYS2, his4B–LEU2–MluI/his4X–LEU2–MluI::BamHI–URA3, leu2::hisG/leu2::hisG, ura3/ura3, ADE2/ade2::hisG, arg4–Bgl/arg4–Nsp, mre11S/mre11S |
| Y324 | MATa/MATα, ho::LYS2/ho::LYS2, his4B–LEU2–MluI/his4X–LEU2–MluI::BamHI–URA3, leu2::hisG/leu2::hisG, ura3/ura3, ADE2/ade2::hisG, arg4–Bgl/arg4–Nsp, mre11S/mre11–T4(–LEU2) |
| Y325 | MATa/MATα, ho::LYS2/ho::LYS2, his4B–LEU2–MluI/his4X–LEU2–MluI::BamHI–URA3, leu2::hisG/leu2::hisG, ura3/ura3, ADE2/ade2::hisG, arg4–Bgl/arg4–Nsp, mre11S/MRE11 |
| Y327 | MATa/MATα, ho::LYS2/ho::LYS2, his4B–LEU2–MluI/his4X–LEU2–MluI::BamHI–URA3, leu2::hisG/leu2::hisG, ura3/ura3, ADE2/ade2::hisG, arg4–Bgl/arg4–Nsp, mre11–T4–(LEU2)/mre11–T4–(LEU2) |
| Y328 | MATa/MATα, ho::LYS2/ho::LYS2, his4B–LEU2–MluI/his4X–LEU2–MluI::BamHI–URA3, leu2::hisG/leu2::hisG, ura3/ura3, ADE2/ade2::hisG, arg4–Bgl/arg4–Nsp, mre11–T20–(LEU2)/mre11–T20–(LEU2) |
| Y329 | MATa/MATα, ho::LYS2/ho::LYS2, lys2/lys2, his4B–LEU2–MluI/his4X–LEU2–MluI::BamHI–URA3, leu2::hisG/leu2::hisG, ura3/ura3, ADE2/ade2::hisG, arg4–Bgl/arg4–Nsp, rad50S::URA3/rad50S::URA3 |
| Y401 | MATa/MATα, ho::LYS2/ho::LYS2, rad50::hisG::URA3::hisG/rad50::hisG::URA3::hisG, ura3/ura3, LEU2/leu2::hisG, ADE2/ade2::hisG, HIS4/his4B, spo13::hisG/spo13::hisG |
| Y402 | MATa/MATα, ho::LYS2/ho::LYS2, rad50::hisG::URA3::hisG/rad50::hisG::URA3::hisG, ura3/ura3, LEU2/leu2::hisG, ADE2/ade2::hisG, HIS4/his4B, spo13::hisG/spo13::hisG, mre11S/mre11S |
| Y403 | MATa/MATα, ho::LYS2/ho::LYS2, ADE2/ade2::hisG, ura3/ura3, LEU2/leu2::hisG, HIS4/his4B, spo13::hisG/spo13::hisG, mre11S/mre11S |
| Y442 | MATα, ho::LYS2, leu2::hisG, ade2::hisG, ura3, mre11–T10(–LEU2) |
| Y9Dx2A | MATa/MATα, ho::LYS2/ho::LYS2, ura3/ura3, LEU2/leu2::hisG, ADE2/ade2::hisG, HIS4/his4B, spo13::hisG/spo13::hisG |
Acknowledgments
Many thanks go to Rochelle E. Esposito, Shirleen Roeder, Mike Lichten, and especially Liuzhong Xu and Nancy Kleckner for sharing plasmids and strains. We are indebted to Brian Jensen and Breck Byers for providing the library, to Quan-wen Jin for his expert advice in “fishing” and for providing probes, to Rainer de Martin for printing the color figure and help in sequencing, to Peter Moens for sending us anti-Zip1 antibody, and to Susanne Prinz for close cooperation. The ideas in this work were born during the stimulating time we spent in the laboratory of Breck Byers. We are grateful to Jim Haber, Hideyuki Ogawa, and Dieter Schweizer for critical comments on the manuscript. This work was supported by grants S5815-BIO and in part S5807-BIO of the Austrian Science Foundation (FWF) and Jubiläumsfondsprojekt nr. 5955 of the Österreichische Nationalbank.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL fklein@s1.botanik.univie.ac.at; FAX +43(1)79794/131.
References
- Ajimura M, Leem SH, Ogawa H. Identification of new genes required for meiotic recombination in Saccharomyces cerevisiae. Genetics. 1993;133:51–66. doi: 10.1093/genetics/133.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alani E, Padmore R, Kleckner N. Analysis of wild-type and rad50 mutants of yeast suggests an intimate relationship between meiotic chromosome synapsis and recombination. Cell. 1990;61:419–436. doi: 10.1016/0092-8674(90)90524-i. [DOI] [PubMed] [Google Scholar]
- Altshul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bailey NTJ. Statistical methods in biology. Cambridge, UK: Cambridge University Press; 1995. [Google Scholar]
- Bairoch A, Bucher P, Hofmann K. The PROSITE database, its status in 1995. Nucleic Acids Res. 1995;24:189–191. doi: 10.1093/nar/24.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergerat A, de Massy B, Gadelle D, Varoutas P-C, Nicolas A, Forterre P. An atypical topoisomerase II from archaea with implication for meiotic recombination. Nature. 1997;386:414–417. doi: 10.1038/386414a0. [DOI] [PubMed] [Google Scholar]
- Bhargava J, Engebrecht J, Roeder GS. The rec102 mutant of yeast is defective in meiotic recombination and chromosome synapsis. Genetics. 1992;130:59–69. doi: 10.1093/genetics/130.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop D K, Park D, Xu L, Kleckner N. DMC1: A meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell. 1992;69:439–456. doi: 10.1016/0092-8674(92)90446-j. [DOI] [PubMed] [Google Scholar]
- Bullard SA, Kim S, Galbraith AM, Malone RE. Double strand breaks at the HIS2 recombination hot spot in Saccharomyces cerevisiae. Proc Natl Acad Sci. 1996;93:13054–13059. doi: 10.1073/pnas.93.23.13054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Alani E, Kleckner N. A pathway for generation and processing of double-strand breaks during meiotic recombination in S. cerevisiae. Cell. 1990;61:1089–1101. doi: 10.1016/0092-8674(90)90072-m. [DOI] [PubMed] [Google Scholar]
- Connelly JC, Leach DRF. The sbcC and sbcD genes of Escherichia coli encode a nuclease involved in palindrome inviability and genetic recombination. Genes Cells. 1996;1:285–291. doi: 10.1046/j.1365-2443.1996.23024.x. [DOI] [PubMed] [Google Scholar]
- de Massy B, Nicolas A. The control in cis of the position and the amount of the ARG4 meiotic double-strand break of Saccharomyces cerevisiae. EMBO J. 1993;12:1459–1466. doi: 10.1002/j.1460-2075.1993.tb05789.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Massy B, Rocco V, Nicolas A. The nucleotide mapping of DNA double-strand breaks at the CYS3 initiation site of meiotic recombination in Saccharomyces cerevisiae. EMBO J. 1995;14:4589–4598. doi: 10.1002/j.1460-2075.1995.tb00138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolganov GM, Maser RS, Novikov A, Tosto L, Chong S, Bressan DA, Petrini JH. Human Rad50 is physically associated with human Mre11: Identification of a conserved multiprotein complex implicated in recombinational DNA repair. Mol Cell Biol. 1996;16:4832–4841. doi: 10.1128/mcb.16.9.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engebrecht JA, Voelkel-Meiman K, Roeder GS. Meiosis-specific RNA splicing in yeast. Cell. 1991;66:1257–1268. doi: 10.1016/0092-8674(91)90047-3. [DOI] [PubMed] [Google Scholar]
- Engebrecht, J., F. Klein, and J. Loidl. 1997. Genetic and morphological approaches for the analysis of meiotic chromosomes in yeast. Methods Cell Biol. (in press). [DOI] [PubMed]
- Gibson FP, Leach DRF, Lloyd RG. Identification of sbcD mutations as cosuppressors of recBC that allow propagation of DNA palindromes in Escherichia coli K-12. J Bacteriol. 1992;174:1222–1228. doi: 10.1128/jb.174.4.1222-1228.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. New yeast—Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Gietz D, St. Jean A, Woods RA, Schiestl R. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldway M, Sherman A, Zenvirth D, Arbel T, Simchen G. A short chromosomal region with major roles in yeast chromosome III meiotic disjunction, recombination and double strand breaks. Genetics. 1993;133:159–169. doi: 10.1093/genetics/133.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Korolev VG, Fabre F. XRS2, a DNA repair gene of Saccharomyces cerevisiae, is needed for meiotic recombination. Genetics. 1992;132:651–664. doi: 10.1093/genetics/132.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, Fishman-Lobell J, Haber JE. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics. 1996;142:693–704. doi: 10.1093/genetics/142.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johzuka K, Ogawa H. Interaction of Mre11 and Rad50: Two proteins required for DNA repair and meiosis-specific double-strand break formation in Saccharomyces cerevisiae. Genetics. 1995;139:1521–1532. doi: 10.1093/genetics/139.4.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane SM, Roth R. Carbohydrate metabolism during ascospore development in yeast. J Bacteriol. 1974;118:8–14. doi: 10.1128/jb.118.1.8-14.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Kleckner N. Covalent protein-DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci. 1995;92:11274–11278. doi: 10.1073/pnas.92.24.11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Giroux CN, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell. 1997;88:375–384. doi: 10.1016/s0092-8674(00)81876-0. [DOI] [PubMed] [Google Scholar]
- Klein F, Karwan A, Wintersberger U. Pedigree analyses of yeast cells recovering from DNA damage allow assignment of lethal events to individual post-treatment generations. Genetics. 1990;124:57–65. doi: 10.1093/genetics/124.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wu TC, Lichten M. The location and structure of double-strand DNA breaks induced during yeast meiosis: Evidence for a covalently linked DNA-protein intermediate. EMBO J. 1995;14:4599–4608. doi: 10.1002/j.1460-2075.1995.tb00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loidl J, Nairz K, Klein F. Meiotic chromosome synapsis in a haploid yeast. Chromosoma. 1991;100:221–228. doi: 10.1007/BF00344155. [DOI] [PubMed] [Google Scholar]
- Loidl J, Klein F, Scherthan H. Homologous pairing is reduced but not abolished in asynaptic mutants of yeast. J Cell Biol. 1994;125:1191–1200. doi: 10.1083/jcb.125.6.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A, van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Lydall D, Nikolsky Y, Bishop DK, Weinert T. A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature. 1996;383:840–843. doi: 10.1038/383840a0. [DOI] [PubMed] [Google Scholar]
- Malone RE, Esposito RE. Recombinationless meiosis in Saccharomyces cervisiae. Mol Cell Biol. 1981;1:891–901. doi: 10.1128/mcb.1.10.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee A, Kleckner N. A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics. 1997;146:797–816. doi: 10.1093/genetics/146.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menees T M, Ross-MacDonald PB, Roeder GS. MEI4, a meiosis-specific yeast gene required for chromosome synapsis. Mol Cell Biol. 1992;12:1340–1351. doi: 10.1128/mcb.12.3.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairz, K. 1997. “mre11S—A new yeast mutation that blocks double-strand break processing and leads to nonhomologous synapsis,” Ph.D. thesis. University of Vienna, Austria. [DOI] [PMC free article] [PubMed]
- Ogawa H, Johzuka K, Nakagawa T, Leem SH, Hagihara AH. Functions of the yeast meiotic recombination genes, MRE11 and MRE2. Adv Biophys. 1995;31:67–76. doi: 10.1016/0065-227x(95)99383-z. [DOI] [PubMed] [Google Scholar]
- Orr-Weaver TL, Szostak JW, Rothstein RJ. Genetic applications of yeast transformation with linear and gapped plasmids. Methods Enzymol. 1983;101:228–245. doi: 10.1016/0076-6879(83)01017-4. [DOI] [PubMed] [Google Scholar]
- Padmore R, Cao L, Kleckner N. Temporal comparison of recombination and synaptonemal complex formation during meiosis in S. cerevisiae. Cell. 1991;66:1239–1256. doi: 10.1016/0092-8674(91)90046-2. [DOI] [PubMed] [Google Scholar]
- Petrini JH, Walsh ME, Di Mare C, Chen XN, Korenberg JR, Weaver DT. Isolation and characterization of the human MRE11 homologue. Genomics. 1995;29:80–86. doi: 10.1006/geno.1995.1217. [DOI] [PubMed] [Google Scholar]
- Prinz S, Amon A, Klein F. Isolation of COM1, a new gene required to complete meiotic double-strand break induced recombination in S. cerevisiae. Genetics. 1997;146:781–795. doi: 10.1093/genetics/146.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocco V, Nicolas A. Sensing of DNA non-homology lowers the initiation of meiotic recombination in yeast. Genes Cells. 1996;1:645–661. doi: 10.1046/j.1365-2443.1996.00256.x. [DOI] [PubMed] [Google Scholar]
- Rockmill B, Engebrecht JA, Scherthan H, Loidl J, Roeder GS. The yeast MER2 gene is required for chromosome synapsis and the initiation of meiotic recombination. Genetics. 1995a;141:49–59. doi: 10.1093/genetics/141.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockmill B, Sym M, Scherthan H, Roeder GS. Roles for two RecA homologs in promoting meiotic chromosome synapsis. Genes & Dev. 1995b;9:2684–2695. doi: 10.1101/gad.9.21.2684. [DOI] [PubMed] [Google Scholar]
- Rose MD, Winston F, Hieter P. Methods in yeast genetics. A laboratory course manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- Rothstein RJ. One step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci. 1977;83:735–739. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherthan H, Loidl J, Schuster T, Schweizer D. Meiotic chromosome condensation and pairing in Saccharomyces cerevisiae studied by chromosome painting. Chromosoma. 1992;101:590–595. doi: 10.1007/BF00360535. [DOI] [PubMed] [Google Scholar]
- Seifert HS, Chen EY, So M, Heffron F. Shuttle mutagenesis: A method of transposon mutagenesis for Saccharomyces cerevisiae. Proc Natl Acad Sci. 1986;83:735–739. doi: 10.1073/pnas.83.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples GJ, Leach DR. Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast [letter] Mol Microbiol. 1995;17:1215–1217. doi: 10.1111/j.1365-2958.1995.mmi_17061215_1.x. [DOI] [PubMed] [Google Scholar]
- Sherman F, Roman H. Evidence for two types of allelic recombination in yeast. Genetics. 1963;48:255–261. doi: 10.1093/genetics/48.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorsky RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RF, Wiese BA, Wojzynski MK, Davison DB, Worley KC. BCM Search Launcher—An integrated interface to molecular biology data base search and analysis services available on the World Wide Web. Genome Res. 1996;6:454–462. doi: 10.1101/gr.6.5.454. [DOI] [PubMed] [Google Scholar]
- Storlazzi A, Xu L, Cao L, Kleckner N. Crossover and noncrossover recombination during meiosis: Timing and pathway relationships. Proc Natl Acad Sci. 1995;92:8512–8516. doi: 10.1073/pnas.92.18.8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sym M, Engebrecht JA, Roeder GS. ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell. 1993;72:365–378. doi: 10.1016/0092-8674(93)90114-6. [DOI] [PubMed] [Google Scholar]
- Tavassoli M, Shayeghi M, Nasim A, Watts FZ. Cloning and characterisation of the Schizosaccharomyces pombe rad32 gene: A gene required for repair of double strand breaks and recombination. Nucleic Acids Res. 1995;23:383–388. doi: 10.1093/nar/23.3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto Y, Kato J, Ikeda H. Effects of mutations of RAD50, RAD51, RAD52, and related genes on illegitimate recombination in Saccharomyces cerevisiae. Genetics. 1996;142:383–391. doi: 10.1093/genetics/142.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff JE, Klapholz S, Esposito RE. Meiosis in haploid yeast. Proc Natl Acad Sci. 1982;79:2986–2990. doi: 10.1073/pnas.79.9.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner BM, Kleckner N. Chromosome pairing via multiple interstitial interactions before and during meiosis in yeast. Cell. 1994;77:977–991. doi: 10.1016/0092-8674(94)90438-3. [DOI] [PubMed] [Google Scholar]
- Xu L, Kleckner N. Sequence non-specific double-strand breaks and interhomolog interactions prior to double-strand break formation at a meiotic recombination hot spot in yeast. EMBO J. 1995;14:5115–5128. doi: 10.1002/j.1460-2075.1995.tb00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]