

Abstract
Determinants of the Fe-CO and C-O stretching frequencies in (imidazole) heme-CO adducts have been investigated via Density Functional Theory (DFT) analysis, in connection with puzzling characteristics of the heme sensor protein CooA, and of the H-NOX (Heme-Nitric Oxide and/or OXygen binding) family of proteins, including soluble guanylate cyclase (sGC). The computations show that two mechanisms of Fe-histidine bond weakening have opposite effects on the νFeC/νCO pattern. Mechanical tension is expected to raise νFeC with little change in νCO, while weakening of H-bond donation from the imidazole ligand has the opposite effect. Data on CooA indicate imidazole H-bond weakening associated with heme displacement, as part of the activation mechanism. The computations also reveal that protein-induced distortion of the porphyrin ring, a prominent structural feature of the H-NOX protein TtTar4H (Thermoanaerobacter tengcongensis Tar4 protein), has surprisingly little effect on νFeC or νCO. However, another structural feature, strong H-bonding to the propionates, is suggested to account for the weakened backbonding that is evident in sGC. TtTar4H-CO itself has an elevated νFeC, which is successfully modeled as a compression effect, resulting from steric crowding in the distal pocket. νFeC/νCO data, in conjunction with modeling, can provide valuable insight into mechanisms for heme-protein modulation.
Introduction
The stretching frequencies of Fe-CO and CO bonds in heme proteins, observable via resonance Raman spectroscopy, have emerged as useful probes of electrostatic and mechanical influences in the heme binding pocket (1). When νFeC is plotted against νCO, many heme proteins and model adducts fall on a single line, of negative slope, reflective of backbonding. Donation of Fe(II) dπ electrons into the empty CO π* orbitals strengthens the Fe-C bond while weakening the C-O bonds. The variable position of the heme-CO adducts along this line reflects variations in the extent of backbonding. In heme proteins these variations are largely controlled by electrostatic effects in the vicinity of the bound CO (2–4). Distal sidechains that are H-bond donors enhance backbonding, while those that are electron pair donors diminish backbonding. In model compounds, variations along the backbonding line can be induced with peripheral substituents of differing electron donating propensity (5, 6). The effect has been modeled via Density Functional Theory (DFT)1 computation of the frequencies (5), and improvements in the wavefunctions utilized have yielded good agreement between calculated and observed slopes of the backbonding line (6).
In addition, variations in the donor strength of the ligand trans to the CO have differing effects on the FeC and CO bonds, and shift the backbonding line. Stronger donors weaken the Fe-CO bond via σ competition, and shift the line to lower νFeC values, while weaker donors have the opposite effect (1, 3). Thus the νFeC/νCO plot can be a valuable aid in assessing various protein interactions with the heme group.
However, there are puzzling aspects to the data, which suggest that additional factors can come into play. A case in point is the heme sensor protein CooA, which responds to CO by activating a set of genes in CO-metabolizing bacteria (7). Although the proximal ligand is histidine, νFeC/νCO is displaced above the standard backbonding line defined by myoglobin variants, suggesting a weakened Fe-His bond in CooA-CO (8, 9). This weakening was attributed to displacement of the heme further into the interior of the protein, as part of the activation mechanism. The question arises whether this weakening results from mechanical tension on the Fe-His bond, or from weakening of an H-bond from the His ligand to a nearby asparagine side-chain.
Another puzzle involves the H-NOX (Heme-Nitric Oxide and/or OXygen binding) class of heme sensor proteins (10). The best known of these is soluble guanylate cyclase (sGC), the mammalian transducer of NO signaling. sGC catalyzes GTP conversion to cGMP when NO binds to its heme (11). νFeC/νCO data indicate weakened backbonding in sGC (12), but the reason for this effect has not been clear. On the other hand, the first structurally characterized member of the class, the bacterial protein TtTar4H (Thermoanaerobacter tengcongensis Tar4 protein) (13), gives a νFeC/νCO point that falls significantly above the standard backbonding line (14). Yet there is no evidence for a weakened Fe-His bond in this case.
In the present study we investigate these issues by DFT modeling of a series of potential influences, both axial and equatorial to the heme, other than the well-studied effects of distal polarity in the heme pocket (15). These influences include proximal histidine H-bonding and tension, heme distortion and H-bonding to the propionate substituents, and steric compression of the FeCO unit (Figure 1). The modeling results yield significant insight into the structure and mechanisms of heme proteins.
Figure 1.
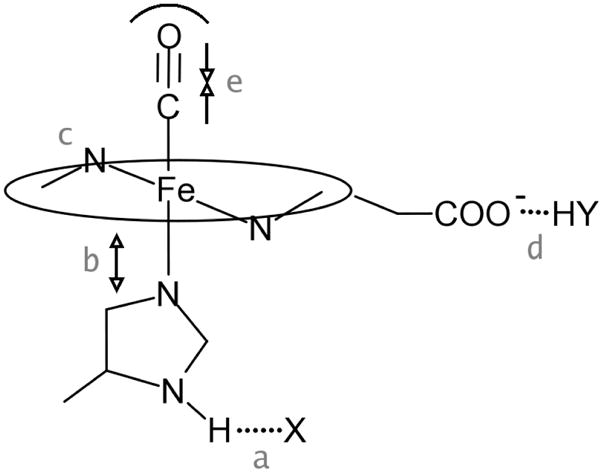
Schematic representation of influences investigated via DFT modeling, a. H-bonding from the proximal imidazole, b. tension on the Fe-ImH bond, c. porphyrin distortion, d. propionate H-bonding, e. steric compression
Methods
Computations were performed with the Gaussian 03 program (16), using the B3LYP functional and standard 6-31G* basis set for all the atoms except Fe, for which Ahlrichs’ valence triple-ζ (VTZ) (17) basis set was employed. The CO-heme model was (ImH)FeP(CO) (ImH = imidazole, P = porphine), except in the study of H-bonding effects on propionates where protoporphyrin IX instead of simple porphine was used. The structures were optimized with minimal or no constraints when various effects were examined, as described in each section below.
We also performed Quantum Mechanical/Molecular Mechanical (QM/MM) calculations on the O2- bound crystal structure TtTar4H (PDB ID: 1U55) (13). The full protein was solvated in a SPC water box, defining a 15Å solvation layer around the protein. The waters were equilibrated by molecular dynamics simulation (18). The system was then reduced to protein and waters within 10Å distance of any protein atoms. The heme ring, O2, and the proximal ligand (His102) were defined as the QM region with the rest of the protein and waters defined as the MM region. The QM calculation used the B3LYP functional and LACVP*/6-31G* basis functions. The classical (MM) region employed parameters from the OPLS 2001 force field (19). The simulations were performed using QSite from the Schrodinger suite of programs (20). After the O2-bound structure was optimized, O2 was replaced by CO and the CO-bound form was re-optimized using the same QM/MM protocol.
Results and Discussion
CooA: proximal histidine tension vs H-bonding
CooA is a dimeric transcription factor that binds to its target DNA sequence, once CO binds to its heme (7). The mechanism of activation has been proposed to involve displacement of the heme and of the adjacent C-helix (8), which connects the heme and DNA-binding domains (21). These displacements are propelled by hydrophobic forces once CO replaces the endogenous ligand, which is the N-terminus of the opposite subunit in the homodimer (21).
Evidence for heme displacement in CooA-CO came from resonance Raman (RR) measurement of νFeC and νCO positions, identifying, via residue replacements, which side-chains help form the CO binding pocket (8). The data also indicated weakening of the proximal ligand donor strength, since the νFeC/νCO points fell above the standard backbonding line (Figure 2 – see Table 1 for list of frequencies), although, like Myoglobin (Mb), CO-free CooA has a histidine proximal ligand, with a H-bond of moderate strength (21). H-bond donation from the ImH ligand (Figure 1) increases imidazolate character, and is a well-recognized mechanism for modulating the Fe-ImH bond (1). The strength of this H-bond correlates well with the Fe-ImH stretching frequency in 5-coordinate Fe(II) hemes, for which this vibrational mode is detectable via RR spectroscopy (22). The frequency is 220 cm−1 for Mb, but ~240 cm−1 for peroxidases, in which the ImH ligand is strongly H-bonded to a negatively charged aspartate side-chain, and only 205 cm−1 for a peroxidase variant in which this H-bond is abolished (23).
Figure 2.

νFeC/νCO backbonding line for variants of Mb. Mb points (square) show positions with (H64) and without (H64L) distal H-bond donors. Horizontal displacements are shown for proximal imidazole deprotonation (PPDMeIm−) and for stronger (CCPMI) or weaker (CCPMI(D235N)) H-bonding of the proximal imidazole. CooA deviations are attributed to imidazole H-bond weakening.
Table 1.
νFeC and νCO data for Fe(II) heme proteins and models
| Sample | νFeC (cm−1) | νCO (cm−1) | Ref |
|---|---|---|---|
| WT Mb | 508 | 1941 | (56–58) |
| H64L Mb | 490 | 1965 | (56–60) |
| H64V/V68T | 479 | 1984 | (35), (36) |
| WT CooA | 487 | 1982 | (9) |
| WT CooA + DNA | 487 | 1985 | (9) |
| L120F CooA | 486 | 1975 | (9) |
| CCPMI | 503 | 1922 | (27) |
| CCPMI(D253N) | 531 | 1933 | (27) |
| PPDMeImH | 495 | 1960 | (26) |
| PPDMe(Im−) | 490 | 1942 | (26) |
| Cyt ox (α) | 519 | 1966 | (52), (53) |
| Cyt ox (β) | 493 | 1955 | (52), (53) |
| C2Cap(NMeIm) | 497 | 2002 | (3) |
| TPP(NMeIm) (MC) | 495 | 1962 | (6) |
| TPP(NMeIm) (Bz) | 486 | 1972 | (50, 51) |
| SGC | 472 | 1987 | (12), (30), (61) |
| sGC1 | 497 | 1964 | (41) |
| sGC+YC-1 | 488 | 1972 | (62) |
| sGC+BAY | 489 | 1972 | (45) |
| sGC1 β1(1-385) H105G(ImH) | 495 | 1964 | (12), (44) |
| sGC1 β1(1-385) | 478 | 1985 | (12), (44) |
| VCA0720 | 491 | 1985 | (14) |
| TtTar4H | 490 | 1989 | (14) |
The H-bond in CooA is to the amide side-chain of Asn42 (Fig 2), a residue upstream from the His77 ligand (21). Heme displacement was suggested to weaken the Fe-His77 bond either via mechanical tension, or by weakening the His77··· Asn42 H-bond as a result of His77 movement (8, 9). Support for Fe-His77 bond weakening came from picosecond RR spectroscopy on the prompt CooA-CO photolysis product, which revealed Fe-ImH stretching frequency, 216 cm−1, appreciably below the Mb value (24). (We note, however, that the H-bond status can change upon CO binding via structural rearrangement in the heme pocket – see below.)
The H-bond explanation was clouded because replacement of Asn42 with Ala, which cannot form a H-bond, had no effect, either on the νFeC and νCO positions (8) or on the 5-coordinate Fe-His stretching frequency in a variant engineered to have a significant 5-coordinate heme population (9). However, it was recognized that this negative result might be explained if a water molecule substituted for the Asn amide group as a H-bond acceptor. Nevertheless, mechanical tension resulting from the heme displacement seemed a viable alternative explanation (9).
Imidazole H-bonding
To address the mechanism of Fe-His weakening computationally, we modeled His H-bonding by placing the H-bond acceptors H2O, NH3 and HCOO− (formate) near the NH group of (ImH)FeP(CO) (P = porphine), a well-studied heme analog (5, 6). The limiting case of imidazolate ligation was also investigated. Structural parameters from the optimized geometries are listed in Table 2, as are the computed νFeC and νCO values. It can be seen that while νCO steadily decreases with increasing imidazolate character, νFeC changes very little. A similar result was reported by Franzen (25), who examined the effect of H-bond donation to H2O and acetate from (ImH)FeP(CO). The CO bond strength decreases because backbonding increases as a result of increased electron donation from imidazolate to the Fe, but the backbonding-induced increase in the Fe-C bond strength is counterbalanced by diminished σ bonding due to increased σ competition from imidazolate.
Table 2.
Effect of H-bond donation or deprotonation on computed parameters for (ImH)FeP(CO)
| Distances | Frequencies | |||||
|---|---|---|---|---|---|---|
| H-bond acceptor (X) | Fe-N(ImH) (Å) | ImH···X (Å) | Fe-C (Å) | C-O (Å) | νFeC (cm−1) | νCO (cm−1) |
| None | 2.082 | -- | 1.796 | 1.150 | 474 | 2111 |
| H2O | 2.076 | 1.871 | 1.797 | 1.150 | 474 | 2108 |
| NH3 | 2.075 | 1.904 | 1.797 | 1.150 | 475 | 2107 |
| HCOO− | 2.055 | 1.554 | 1.798 | 1.153 | 478 | 2086 |
| (-H+) | 2.028 | -- | 1.805 | 1.155 | 475 | 2070 |
When the νFeC and νCO positions are plotted, they describe an essentially horizontal line (Figure 3), because of the νFeC invariance. In contrast, as described previously (6), a standard backbonding line is obtained when electron donating and withdrawing substituents, X, are added in silico to the porphine ring (P-X line in Figure 3), with a negative slope close to that found experimentally for Mb variants.
Figure 3.

DFT modeling of vibrational shifts due to proximal ImH H-bonding (see structures) and of Fe-ImH bond tension (X) for (ImH)FeP(CO). The computed backbonding lines are for 6- and 5-coordinate CO adducts of Fe-P-Xs, where X are electron donating and withdrawing substituents.
A horizontal displacement below the Mb line has been observed experimentally (Figure 2) when the model complex (ImH)FePPDME(CO) (PPDME = protoporphyrin dimethyl ester) is deprotonated (26). νCO was markedly lowered, while νFeC showed little change. In the case of cytochrome c peroxidase (CCPMI – a genetic variant), the effect of the strong His-Asp H-bond is evident in a large negative displacement from the Mb line (27) (Figure 2). If one draws a horizontal line through this point, it intercepts the Mb line slightly lower than the position of wild-type Mb, in which the distal histidine (H64) provides positive polarity to the bound CO. The CCP-CO crystal structure shows a water molecule H-bonded to a distal Arg residue, and in turn polarizing the bound CO (28), an interaction somewhat weaker than that of H64 in Mb. Abolition of the proximal H-bond via Asp replacement in CCPMI (CCPMI(D235N)) (27) produces a positive displacement from the Mb line, consistent with moderate proximal His H-bonding (to a Ser hydroxyl group and to a backbone carbonyl) in Mb (diagram in Figure 2). A horizontal line through this point intersects the Mb line well above the WT protein, suggesting that the bound CO interacts directly with the distal Arg in CCPMI(D235N), producing a stronger distal interaction (27). This change might result from loss of the constraining proximal H-bond.
Thus the positions of CCPMI and CCPMI(D235N) on the νFeC and νCO plot are consistent with horizontal deviations below and above the Mb line, the directions expected from the proximal H-bond strengths, at νCO positions reflecting the character of likely distal interactions in the CO binding pocket. (We note, however, that there are CCP variants whose CO adducts fall on the Mb line) (29), indicating structural rearrangements in the heme pocket that attenuate the proximal His H-bonding to the level of Mb).
CooA variants show displacements above the Mb line (Figure 2), as expected if a moderate proximal H-bond is weakened. They describe a horizontal line, reflecting variable extents of heme displacement. Thus the L120F variant obstructs heme displacement sterically, due to the bulky Phe side-chain located near the heme (9). On the other hand, heme displacement is augmented by DNA binding which pulls CooA-CO into the fully active state (9). The horizontal line for CooA intersects the Mb line close to the position of Mb variants in which the distal His is replaced by hydrophobic residues e.g. H64L. This behavior is consistent with evidence that the CO binding pocket is indeed hydrophobic in CooA (8).
Mechanical tension
We asked the question whether the same effect could be produced by mechanical tension imposed by the protein on the proximal His residue. This would also weaken the Fe-His bond, but would it have the same effect as charge variation due to H-bonding changes? To model the effect of tension, we stretched the Fe-His bond by constraining the Fe-N distance to increasing values, while re-optimizing the remaining atomic coordinates. The results are listed in Table 3, and plotted in Figure 3. In contrast to diminished H-bonding, stretching the Fe-His bond steadily increases νFeC, until the 5-coordinate FeP(CO) position is reached. νCO decreases slightly as Fe-His is stretched, but the main effect is the increase in νFeC. This increase reflects diminished σ competition from the more distant His ligand. However, there is no compensating decrease in back-donation, as happens when the negative charge on the imidazole is diminished via weakened H-bonding.
Table 3.
Computed effect of Fe-ImH length on selected parameters for (ImH)FeP(CO)
| Distances | Frequencies | |||
|---|---|---|---|---|
| Fe-ImH(Å) | Fe-C(Å) | C-O(Å) | νFeC (cm−1) | νCO (cm−1) |
| 2.082 | 1.796 | 1.150 | 474 | 2111 |
| 2.132 | 1.789 | 1.150 | 480 | 2109 |
| 2.182 | 1.783 | 1.150 | 485 | 2108 |
| 2.232 | 1.778 | 1.151 | 490 | 2107 |
| 2.332 | 1.769 | 1.151 | 500 | 2106 |
| 2.432 | 1.762 | 1.151 | 509 | 2105 |
| 2.532 | 1.756 | 1.151 | 518 | 2104 |
| 2.632 | 1.751 | 1.152 | 525 | 2103 |
| 2.732 | 1.747 | 1.152 | 531 | 2102 |
Thus tension on the proximal His is expected to produce a nearly vertical displacement of νFeC/νCO from the backbonding line, while weakening the His H-bond is expected to produce a horizontal displacement. Since CooA displays the latter pattern, it is clear that diminished H-bonding, and not mechanical tension is the mechanism of Fe-His bond weakening when the heme moves upon CO binding.
We also carried out a calculation to gauge the effect of tilting the imidazole ring relative to the Fe-ImH bond. The effect was very small, a 20° tilt producing νFeC and νCO shifts of 2 and 0.3 cm−1.
H-NOX: propionate H-bonding, porphyrin distortion and steric crowding
The mammalian heme sensor protein, sGC, generates the second messenger, cGMP, in response to NO binding, thereby using the NO signal to regulate a host of critical physiological responses (11). It belongs to a recently discovered class of H-NOX proteins, that have similar heme-binding motifs, and exhibit preference for NO or for O2 binding to the heme (10). The mechanism of sGC activation remains uncertain, despite extensive studies.
One of the unexplained features of sGC is the unusually high νCO, 1987 cm−1, of the CO adduct. Unlike CooA-CO, which also has a high νCO, νFeC is unusually low, 472 cm−1, and the νFeC/νCO point falls squarely on the Mb back-bonding line (Figure 4), indicating a similarly moderate proximal His H-bond.
Figure 4.

Variable backbonding in sGC constructs, and direction of displacements from the Mb line expected from proposed compression effects in a benzene-capped model compound C2Cap(NMeIm), the H-NOX proteins TtTar4H and VCA0720, and the α form of cytochrome c oxidase. The C2Cap parent compound, TPP, falls on the backbonding line, but at positions which are solvent dependent (MC = methylene chloride, Bz = benzene).
This inference implies a significant conformation change upon CO binding to sGC, since the νFe-His frequency of 5-coordinate, unligated sGC, 204 cm−1 (30, 31), is much lower than that of Mb, 220 cm−1. Thus the Fe-His bond is weakened substantially when CO dissociates. Evidence for a conformation change can also be seen in the elevation of νFe-His, to 213 cm−1, after CO photodissociation from the truncated heme domain; the mode relaxes to the 204 cm−1 equilibrium value in 50 nanoseconds (32). In the native protein, however, relaxation to 204 cm−1 occurs within the 10 ns time resolution of the experiment. While CO binding strengthens the Fe-His bond, NO bonding induces Fe-His dissociation (33, 34).
A low position on the νFeC/νCO backbonding line implies weakened backbonding. The sGC position is lower than that of Mb variants with hydrophobic replacements of the distal histidine (Figure 4), and is near that of the H64V/V68T (35, 36) variant. In this double mutant, the introduced threonine sidechain is oriented (via H-bonding to a backbone carbonyl) so that the O atom lone pair points at the bound CO, thereby weakening backbonding via negative polarity. It had been thought that a similarly positioned lone pair or negative charge would account for the νFeC/νCO position of sGC (12). However, homology modeling with TtTar4H, a bacterial H-NOX heme domain of known structure, renders this explanation unlikely (37). The only candidate residue is Cys78, which replaces Phe78 in TtTar4H. The O2 affinity of TtTar4H is abolished when the distal H-bond donor, Tyr140, is replaced by Leu, but the affinity is restored in the double mutant Y140L/F78Y (38), implying that Tyr78 is close enough to H-bond with bound O2. Conceivably, the homologous residue in sGC, Cys78, might interact with bound CO via the –SH lone pair, but this would require a unique lone-pair orientation, as is found for Thr68 in the H64V/V68T variant of Mb. This ad hoc explanation seems unlikely, and it cannot be generalized to other members of the H-NOX family, all of whom display high values of νCO (14)
Porphyrin distortion
A striking feature of the TtTar4H crystal structure (13) is that the porphyrin ring is highly distorted (Figure 5). The ring atoms are displaced from the mean heme plane, and describe a combination of saddling and ruffling distortions (39). Could these distortions, which might be expected to misalign the Fe and porphyrin orbitals, account for the diminished back-bonding in the H-NOX proteins?
Figure 5.
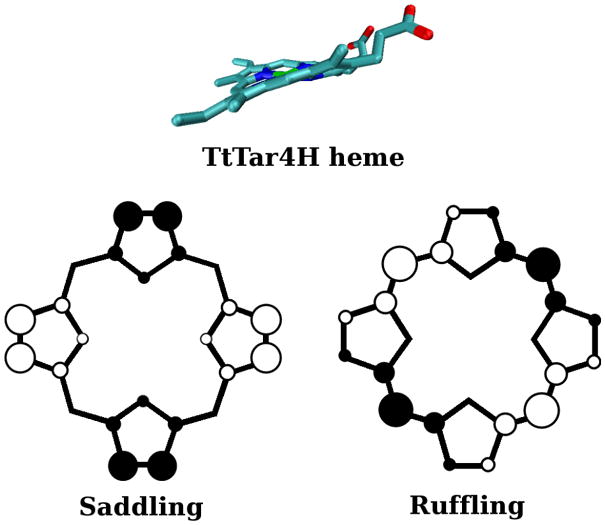
Side-on view of the distorted heme in TtTar4H (13), and top view of the porphine ring, showing up (○) and down (●) atomic displacements along saddling and ruffling coordinates.
To examine this question, we constrained the geometry of (ImH)FeP(CO) in silico via unit displacement (1 Å) of the porphine atoms along the normal coordinates for saddling and for ruffling (39), the dominant distortion coordinates found in TtTar4H (Figure 5). This extent of distortion is close to that seen in the TtTar4H crystal structure (in molecule A, the more distorted of the two molecules found in the unit cell) (13). The results (Table 4) show surprisingly little change in the FeCO geometry or vibrational frequencies. Expected shifts are no more than ~ 2 cm−1. Clearly, the extent of backbonding is essentially unchanged by the porphyrin distortion.
Table 4.
Computed effects of porphyrin distortiona in (ImH)FeP(CO)
| Distances | Frequencies | |||||
|---|---|---|---|---|---|---|
| Fe-N(ImH) (Å) | Fe-N(p) (Å) | Fe-C (Å) | C-O (Å) | νFeC (cm−1) | νCO (cm−1) | |
| unconstrained | 2.082 | 2.025 | 1.796 | 1.1497 | 474 | 2111 |
| Saddled | 2.083 | 2.025 | 1.796 | 1.1497 | 475 | 2111 |
| Ruffled | 2.076 | 2.025 | 1.795 | 1.1500 | 477 | 2108 |
| Sad+Ruf | 2.076 | 2.025 | 1.796 | 1.1498 | 475 | 2110 |
constrained by applying 1Å displacement along saddling or ruffling eigenvectors, or both.
Propionate H-bonding
An alternative explanation for diminished backbonding, which appears not to have been considered previously, is neutralization of the negative charge on the two propionate substituents on the protoporphyrin ring (Figure 1). The TtTar4H crystal structure (13) shows the propionate groups to be buried, and tightly bound by positively charged and H-bond donor side-chains. In contrast the propionates are exposed to solvent in Mb and in many other heme proteins. We note that Harada et al (40) found insignificant shifts in νFeC and νCO when Mb was reconstituted with heme in which one or the other propionate was replaced by a methyl group. In TtTar4H, Arg135 forms H-bonds to both propionates, while Tyr131 and Ser133 donate H-bonds to one of the propionates. These three residues form a “YxSxR” motif that is common to all H-NOX proteins, including sGC (10). Since electron donating and withdrawing substituents are known to influence FeCO backbonding in model porphyrins (6), we reasoned that charge neutralization at the periphery of protoporphryin might similarly modulate backbonding.
To model propionate neutralization, we carried out DFT calculations on (ImH)FePP(CO) (PP = protoporphyrin) and protonated one or both propionates. Computed bond distances and vibrational frequencies are listed in Table 5, while the νFeC/νCO values are plotted in Figure 6. Also shown is the back-bonding line computed for (ImH)FeP-X(CO) species. The three (ImH)FePP(CO) points fall very close to this line, indicating that neutralization of the propionates does indeed modulate back-bonding in the same way as electron withdrawing substituents on porphine do.
Table 5.
Computed effects of protonating one or both propionate groups on protoporphyrin
| Distances | Frequencies | ||||
|---|---|---|---|---|---|
| Fe-ImH (Å) | Fe-C (Å) | C-O (Å) | νFeC (cm−1) | νCO (cm−1) | |
| FePP | 2.081 | 1.785 | 1.153 | 486 | 2087 |
| FePP+H+ | 2.082 | 1.790 | 1.152 | 481 | 2098 |
| FePP+2H+ | 2.080 | 1.796 | 1.150 | 475 | 2109 |
Figure 6.

DFT modeling of Fe-C-O compression (X, Fe---O distances) in (ImH)FeP(CO), and propionate protonation in (ImH)FePP(CO). The PX backbonding line is computed for various electron accepting and donating substituents, X, on porphine.
Protonation of both propionates shifts νCO up ~20 cm−1. This is the same elevation seen for sGC, relative to Mb variants with hydrophobic binding pockets (e.g. H64L) (Figure 4). Since even strong H-bonds are not as neutralizing as protons, and since the Mb reference state involves water-exposed propionates, we surmise that the effect of propionate neutralization is underestimated in the calculations. Nevertheless these results make it plausible that strong H-bonds to propionate can reduce FeCO backbonding substantially, and can account for the high νCO seen for H-NOX proteins.
Interestingly, the low νFeC/νCO position of sGC-CO on the backbonding line can be altered by various modifications. One of these is reconstituted sGC (labeled sGC1 in Figure 4), in which heme is added back to protein that has lost heme during isolation (41). The νFeC/νCO point for sGC1 is close to that of Mb(H64L), suggesting that the reconstitution did not establish the strong propionate H-bonds of the native protein (although it did support activation by NO (41)). Another modification is replacement of the proximal His residue with Gly, and ligation instead by exogenous ImH, (H105G(ImH)), in an expressed sGC fragment containing the heme domain, β1(1-385) (42, 43). The νFeC/νCO point for H105G(ImH)-CO is likewise close to that of Mb(H64L), although the point for β1(1-385)-CO is close to that of sGC itself (Figure 4) (44). Schelvis et al conjectured that loosening of the proximal connection in H105G(ImH)-CO allows formation of a distal H-bond to the bound CO (44). Another possibility is that in this construct, as in sGC1, strong propionate H-bonds are absent. Since heme is not incorporated when H105G itself is expressed (42), but is recruited to the protein by addition of ImH to the expression medium, it is possible that the re-incorporated heme does not form native contacts.
The most significant instance of a νFeC/νCO shift, again to the Mb(H64L) region (Figure 4), is when ‘YC-1’ (3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole) (12), or BAY 41-2272 (5-Cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine) (45), complex heterocyclic activators, are added to sGC-CO. Although CO promotes sGC enzyme activity only slightly, adding YC-1 to sGC-CO brings activity to the same level as adding NO. This was a surprising finding, since dissociation of the proximal ligand is thought to be critical to the mechanism of activation by NO (46). In the presence of these activators sGC-CO RR spectra indicate a small population of 5-coordinate CO-heme (small νFeC peak at 530 cm−1), but the main population remains 6-coordinate.
The YC-1- or BAY- induced νFeC/νCO shift (Figure 4) suggests that, as in sGC1 and H105G(ImH)-CO, the native propionate H-bonds are abolished. How this happens is unclear, because the activator binding site is unknown. YC-1 or BAY might bind in the heme pocket (replacement of the proximal His has even been suggested (47)), or they might bind elsewhere, and lock the protein in its active formation, influencing the heme indirectly. In either case it is possible that strong propionate H-bonding restrains the protein in an inactive conformation until activation, by NO or by CO + YC-1 or BAY, tilts the energetics to the active conformation. Relevant to this proposal is the finding that the two molecules in the TtTar4H unit cell have distinctly different conformations (13). Comparison of the two molecules revealed a ~11° rotation of the entire distal half, relative to the proximal half; in addition the extent of heme distortion differed, as did the propionate conformations and the H-bond contacts with the Y×S×R residues. Although these differences were attributed to crystal contacts, they serve to suggest how the propionate H-bonds might be coupled to protein conformation change in the H-NOX proteins.
Crowding and compression
Like sGC, TtTar4H-CO displays a high νCO, 1989 vs 1987 cm−1, presumably reflecting the same propionate neutralization mechanism (14). However, νFeC is much higher, 490 cm−1 for TtTar4H vs 472 cm−1 for sGC (Table 1 and Figure 4). Thus, unlike sGC-CO, TtTar4H-CO falls well above the Mb back-bonding line. So does the CO adduct of another H-NOX protein VCA0720 (from Vibrio cholerae) (14) (Figure 4). Could these H-NOX representatives have weakened Fe-His bonding, as in CooA-CO? This possibility can be discounted on the basis of the TtTar4H-O2 crystal structure, which reveals a Mb-like H-bond from the proximal ImH ligand to a backbone carbonyl (N···O = 2.80 Å). Also the unligated proteins have Mb-like νFe-His values, 218 and 224 cm−1 for VCA0720 and TtTar4H (whereas, as mentioned above, it is sGC that has a low value, 204 cm−1) (14).
The TtTar4H crystal structure suggests another possibility, namely steric crowding, leading to Fe-C bond compression. There is precedent for such an effect in the model compound C2Cap (5,10,15,20- [pyromellitoyl(tetrakis-o-oxyethoxy-phenyl)]porphyrin), in which a benzene ring is strapped across a porphyrin by short covalent tethers (48). With N-methylimidazole as trans ligand, the Fe(II)CO adduct of C2Cap gave high νCO and νFeC values, 2002 and 497 cm−1 (Figure 4) (3), while the crystal structure revealed a close contact between the CO and the benzene ring (49). The parent complex, (NMeIm)FeTPP(CO) (TPP = tetraphenylporphyrin), which lacks the distal strap, has νCO and νFeC values, 1972 and 486 cm−1 (50, 51), which place it on the Mb backbonding line (Figure 4).
Bond compression was also proposed to account for the high νFeC/νCO position for the dominant form, α, of cytochrome oxidase (cyt ox) (Figure 4) (52, 53). The heme binding site in this case is part of a binuclear site, involving Cu+. The positively charged Cu+, which is close enough to interact directly with the Fe-bound CO, increases the Fe-CO backdonation, consistent with the lowered νCO (1966 cm−1) relative to C2Cap, but the high νFeC, 497 cm−1, implies Fe-C compression. A minor form of cyt ox, β, becomes prevalent at low or high pH, and has νFeC/νCO frequencies falling close to the Mb line (Figure 4), suggesting movement of the Cu+ away from contact with the CO. Consistent with this interpretation is the observation that mutations of the Cu ligands shift νFeC/νCO toward the Mb line (52, 53).
The TtTar4H crystal structure reveals a markedly crowded distal heme pocket (13). To gauge the effect on FeCO structure, we modeled CO into the O2 site (of molecule A, which has the more distorted heme), using a QM/MM algorithm to minimize the energy of the protein. The same procedure was then used on the heme group and its ligands after extracting them from the protein. Table 6 shows that the computed Fe-C and C-O bond lengths, especially the former, are shortened in the protein. Thus an increase in νFeC is expected. We note that sGC, in contrast to TtTar4H, appears to have a large hydrophobic binding pocket (54), consistent with νFeC/νCO not being displaced from the backbonding line.
Table 6.
QM/MM computation of FeCO geometry in TtTar4H
| Fe-C (Å) | C-O (Å) | ∠FeCO (°) | |
|---|---|---|---|
| TtTar4H (QM/MM) | 1.743 | 1.154 | 175.4 |
| Extracted heme (QM) | 1.785 | 1.153 | 179.3 |
To model the effect of compression on the vibrations, we constrained the distance between the Fe and O atoms of FeCO to values shorter than the equilibrium distance in (ImH)FeP(CO), while optimizing all other structure parameters. The results (Table 7) show that the main effect of compression is to shorten the Fe-C bond, with a smaller contraction of the CO bond; as expected, νFeC increases rapidly, accompanied by a small increase in νCO. The trend is shown Figure 6, where it can be seen that the equilibrium structure of (ImH)FeP(CO) places νFeC/νCO close to the value for (ImH)FePP(CO) with two protons on the propionate groups, as expected for a neutral complex. (The effect of angular displacement of the bound CO was previously modeled (55) and found to be quite small up to 0.6 Å displacement of the O atom from the heme normal).
Table 7.
Computed effect of Fe-C-O compression in (ImH)FeP(CO)
| Distances | Frequencies | |||
|---|---|---|---|---|
| |Fe···O| (Å) | Fe-C (Å) | C-O (Å) | νFeC (cm−1) | νCO (cm−1) |
| 2.946 | 1.796 | 1.150 | 474 | 2111 |
| 2.943 | 1.794 | 1.150 | 477 | 2113 |
| 2.940 | 1.791 | 1.149 | 481 | 2115 |
| 2.937 | 1.788 | 1.149 | 485 | 2118 |
| 2.934 | 1.785 | 1.149 | 489 | 2120 |
| 2.930 | 1.782 | 1.148 | 494 | 2124 |
When |Fe···O| is compressed, the computed values of νFeC/νCO describe a line with a positive slope of 1.5. If this slope is applied to the displaced experimental points of Figure 4, νFeC/νCO for C2Cap(NMeIm) is seen to connect with the backbonding line at the sGC position. Since C2Cap(NMeIm) is a neutral pophyrin, this connection supports the suggestion that the propionate groups are effectively neutralized in sGC. (As noted above the C2Cap(NMeIm) parent, (NMeIm)FeTPP(CO), falls on the backbonding line, but its position along it is variable, and depends on solvent interactions of the exposed CO; quite different positions are seen for benzene and methylene chloride solutions – Figure 4).
The TtTar4H displacement connects to a somewhat higher position on the Mb line. TtTar4H has a distal Tyr residue that H-bonds to bound O2, stabilizing the O2 complex (13). It would provide positive polarity for bound CO as well, moving νFeC/νCO up from the sGC position. However the interaction is not as effective as that of His64 in Mb, which is located at the side of the bound CO, a position shown to be optimal for H-bonding (15), whereas the Tyr OH in TtTar4H-O2 is directly above the distal O atom. Meanwhile TtTar4H does have strong propionate H-bonds, which shift the extrapolated νFeC/νCO point down from that of Mb(H64L). The extrapolated point for VCA0720 is slightly higher than for TtTar4H, although its sequence does not indicate a distal tyrosine; no crystal structure is available.
Conclusions
The νFeC and νCO frequencies of heme-CO adducts, which are readily monitored via RR spectroscopy, are sensitive to a variety of interactions with the surrounding protein environment. Distal interactions with polar residues, which directly modulate back-bonding, and give rise to well-recognized back-bonding anti-correlations, have previously been recognized. Another important influence is the donor strength of the proximal ligand, which shifts the back-bonding anti-correlations to lower or higher νFeC.
The most common proximal ligand is from a His residue, and the strength of the Fe-His bond depends on the H-bond status of the imidazole side-chain. Weakening this H-bond weakens the Fe-His bond, but it has not been clear how to disentangle this effect from Fe-His weakening due to tension generated by the protein. DFT modeling now shows that the expected vibrational effects are quite different. H-bond weakening increases νCO with little change in νFeC, while the opposite pattern is predicted for tension. The data for CooA variants conform to the first pattern, confirming the previous conjecture that the νFeC/νCO values monitor heme displacement because of H-bond weakening in the heme sensor protein.
DFT modeling also establishes a back-bonding influence other than distal polarity, namely neutralization of the equatorial negative charge by H-bond donation to the heme propionate substituents. This is the likely explanation for the weak backbonding in sGC, judging from the crystal structure of another member of the H-NOX family, TtTar4H. However, the νFeC/νCO point for TtTar4H-CO itself is displaced above the back-bonding line, although there is no evidence of a weak Fe-His bond. In this case, DFT modeling suggests that the displacement arises from compression of the FeCO unit, due to steric crowding in the distal pocket.
Out-of-plane distortion of the heme group, which is a striking feature of the TtTar4H structure, has negligible effects on the FeCO vibrations.
Since νFeC and νCO are only two parameters, they are manifestly unable to specify all the possible influences on the heme group. However, if structural features are known, or can be inferred, then comparison with the wealth of data on well-characterized adducts, together with DFT modeling, can discriminate among the possible effects and lead to fresh insights into heme protein mechanisms.
Footnotes
Abbreviations: BAY 41-2272, 5-Cyclopropyl-2-[1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-pyrimidin-4-ylamine; C2Cap, 5,10,15,20-[pyromellitoyl(tetrakis-o-oxyethoxy-phenyl)]porphyrin; CCP, cytochrome c peroxidase; cyt ox, cytochrome oxidase; DFT, Density Functional Theory; H-NOX, Heme-Nitric Oxide and/or OXygen binding; ImH, imidazole; Mb, myoglobin; P, porphine; PP, protoporphyrin; PPDME, protoporphyrin dimethyl ester; QM/MM, Quantum Mechanical/Molecular Mechanical; RR, resonance Raman; sGC, soluble guanylate cyclase; TPP, tetraphenylporphyrin; TtTar4H, Thermoanaerobacter tengcongensis Tar4 protein; VCA0720, an H-NOX protein from Vibrio cholerae; YC-1, 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole.
This work was supported by NIH grant GM 33576 from the National Institute of General Medical Sciences.
References
- 1.Spiro TG, Wasbotten IH. CO as a vibrational probe of heme protein active sites. J Inorg Biochem. 2005;99:34–44. doi: 10.1016/j.jinorgbio.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 2.Li XY, Spiro TG. Is Bound Co Linear or Bent in Heme-Proteins - Evidence from Resonance Raman and Infrared Spectroscopic Data. J Am Chem Soc. 1988;110:6024–6033. doi: 10.1021/ja00226a017. [DOI] [PubMed] [Google Scholar]
- 3.Ray GB, Li XY, Ibers JA, Sessler JL, Spiro TG. How Far Can Proteins Bend the Feco Unit - Distal Polar and Steric Effects in Heme-Proteins and Models. J Am Chem Soc. 1994;116:162–176. [Google Scholar]
- 4.Phillips GN, Teodoro ML, Li TS, Smith B, Olson JS. Bound CO is a molecular probe of electrostatic potential in the distal pocket of myoglobin. J Phys Chem B. 1999;103:8817–8829. [Google Scholar]
- 5.Vogel KM, Kozlowski PM, Zgierski MZ, Spiro TG. Role of the axial ligand in heme-CO backbonding; DFT analysis of vibrational data. Inorg Chim Acta. 2000;297:11–17. [Google Scholar]
- 6.Ibrahim M, Xu CL, Spiro TG. Differential sensing of protein influences by NO and CO vibrations in heme adducts. J Am Chem Soc. 2006;128:16834–16845. doi: 10.1021/ja064859d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts GP, Kerby RL, Youn H, Conrad M. CooA, a paradigm for gas sensing regulatory proteins. J Inorg Biochem. 2005;99:280–292. doi: 10.1016/j.jinorgbio.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 8.Coyle CM, Puranik M, Youn H, Nielsen SB, Williams RD, Kerby RL, Roberts GP, Spiro TG. Activation mechanism of the CO sensor CooA - Mutational and resonance Raman spectroscopic studies. J Biol Chem. 2003;278:35384–35393. doi: 10.1074/jbc.M301000200. [DOI] [PubMed] [Google Scholar]
- 9.Ibrahim M, Kerby RL, Puranik M, Wasbotten IH, Youn H, Roberts GP, Spiro TG. Heme displacement mechanism of CooA activation - Mutational and Raman spectroscopic evidence. J Biol Chem. 2006;281:29165–29173. doi: 10.1074/jbc.M605568200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boon EM, Marletta MA. Ligand specificity of H-NOX domains: from sGC to bacterial NO sensors. J Inorg Biochem. 2005;99:892–902. doi: 10.1016/j.jinorgbio.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 11.Denninger JW, Marletta MA. Guanylate cyclase and the (NO)-N-./cGMP signaling pathway. Biochim Biophys Acta - Bioenerg. 1999;1411:334–350. doi: 10.1016/s0005-2728(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 12.Denninger JW, Schelvis JPM, Brandish PE, Zhao Y, Babcock GT, Marletta MA. Interaction of soluble guanylate cyclase with YC-1: Kinetic and resonance Raman studies. Biochemistry. 2000;39:4191–4198. doi: 10.1021/bi992332q. [DOI] [PubMed] [Google Scholar]
- 13.Pellicena P, Karow DS, Boon EM, Marletta MA, Kuriyan J. Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc Natl Acad Sci USA. 2004;101:12854–12859. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karow DS, Pan DH, Tran R, Pellicena P, Presley A, Mathies RA, Marletta MA. Spectroscopic characterization of the soluble guanylate cyclase-like heme domains from Vibrio cholerae and Thermoanaerobacter tengcongensis. Biochemistry. 2004;43:10203–10211. doi: 10.1021/bi049374l. [DOI] [PubMed] [Google Scholar]
- 15.Franzen S. An electrostatic model for the frequency shifts in the carbonmonoxy stretching band of myoglobin: Correlation of hydrogen bonding and the Stark tuning rate. J Am Chem Soc. 2002;124:13271–13281. doi: 10.1021/ja017708d. [DOI] [PubMed] [Google Scholar]
- 16.Frisch MJea. Gaussian 03 [Google Scholar]
- 17.Bauernschmitt R, Ahlrichs R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem Phys Lett. 1996;256:454–464. [Google Scholar]
- 18.Guallar V, Jarzecki AA, Friesner RA, Spiro TG. Modeling of ligation-induced helix/loop displacements in myoglobin: Toward an understanding of hemoglobin allostery. J Am Chem Soc. 2006;128:5427–5435. doi: 10.1021/ja057318h. [DOI] [PubMed] [Google Scholar]
- 19.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 20.Schrodinger I. Qsite 2005 [Google Scholar]
- 21.Lanzilotta WN, Schuller DJ, Thorsteinsson MV, Kerby RL, Roberts GP, Poulos TL. Structure of the CO sensing transcription activator CooA. Nat Struct Biol. 2000;7:876–880. doi: 10.1038/82820. [DOI] [PubMed] [Google Scholar]
- 22.Spiro TG, Li XY. Resonance Raman Spectroscopy of Metalloporphyrins. In: Spiro TG, editor. Biological Applications of Raman Spectroscopy. John Wiley & Sons; New York: 1988. pp. 1–38. [Google Scholar]
- 23.Smulevich G, Mauro JM, Fishel LA, English AM, Kraut J, Spiro TG. Heme Pocket Interactions in Cytochrome-C Peroxidase Studied by Site-Directed Mutagenesis and Resonance Raman-Spectroscopy. Biochemistry. 1988;27:5477–5485. doi: 10.1021/bi00415a014. [DOI] [PubMed] [Google Scholar]
- 24.Uchida T, Ishikawa H, Ishimori K, Morishima I, Nakajima H, Aono S, Mizutani Y, Kitagawa T. Identification of histidine 77 as the axial heme ligand of carbonmonoxy CooA by picosecond time-resolved resonance Raman spectroscopy. Biochemistry. 2000;39:12747–12752. doi: 10.1021/bi0011476. [DOI] [PubMed] [Google Scholar]
- 25.Franzen S. Effect of a charge relay on the vibrational frequencies of carbonmonoxy iron porphine adducts: The coupling of changes in axial ligand bond strength and porphine core size. J Am Chem Soc. 2001;123:12578–12589. doi: 10.1021/ja0108988. [DOI] [PubMed] [Google Scholar]
- 26.Evangelistakirkup R, Smulevich G, Spiro TG. Alternative Carbon-Monoxide Binding Modes for Horseradish-Peroxidase Studied by Resonance Raman-Spectroscopy. Biochemistry. 1986;25:4420–4425. doi: 10.1021/bi00363a037. [DOI] [PubMed] [Google Scholar]
- 27.Smulevich G, Mauro JM, Fishel LA, English AM, Kraut J, Spiro TG. Cytochrome-C Peroxidase Mutant Active-Site Structures Probed by Resonance Raman and Infrared Signatures of the Co Adducts. Biochemistry. 1988;27:5486–5492. doi: 10.1021/bi00415a015. [DOI] [PubMed] [Google Scholar]
- 28.Edwards SL, Poulos TL. Ligand-Binding and Structural Perturbations in Cytochrome-C Peroxidase - a Crystallographic Study. J Biol Chem. 1990;265:2588–2595. [PubMed] [Google Scholar]
- 29.Feis A, Rodriguez-Lopez JN, Thorneley RNF, Smulevich G. The Distal Cavity Structure of Carbonyl Horseradish Peroxidase As Probed by the Resonance Raman Spectra of His 42 Leu and Arg 38 Leu Mutants. Biochemistry. 1998;37:13575–13581. doi: 10.1021/bi981399v. [DOI] [PubMed] [Google Scholar]
- 30.Deinum G, Stone JR, Babcock GT, Marletta MA. Binding of nitric oxide and carbon monoxide to soluble guanylate cyclase as observed with resonance Raman spectroscopy. Biochemistry. 1996;35:1540–1547. doi: 10.1021/bi952440m. [DOI] [PubMed] [Google Scholar]
- 31.Pal B, Kitagawa T. Interactions of soluble guanylate cyclase with diatomics as probed by resonance Raman spectroscopy. J Inorg Biochem. 2005;99:267–279. doi: 10.1016/j.jinorgbio.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 32.Schelvis JPM, Kim SY, Zhao YD, Marletta MA, Babcock GT. Structural dynamics in the guanylate cyclase heme pocket after CO photolysis. J Am Chem Soc. 1999;121:7397–7400. [Google Scholar]
- 33.Stone JR, Marletta MA. Soluble Guanylate-Cyclase from Bovine Lung -Activation with Nitric-Oxide and Carbon-Monoxide and Spectral Characterization of the Ferrous and Ferric States. Biochemistry. 1994;33:5636–5640. doi: 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- 34.Yu AE, Hu SZ, Spiro TG, Burstyn JN. Resonance Raman-Spectroscopy of Soluble Guanylyl Cyclase Reveals Displacement of Distal and Proximal Heme Ligands by NO. J Am Chem Soc. 1994;116:4117–4118. [Google Scholar]
- 35.Biram D, Garratt CJ, Hester RE. In: Spectroscopy of Biological Molecules. Hester RE, Girling RB, editors. Royal Society of Chemistry; Cambridge: 1991. pp. 433–434. [Google Scholar]
- 36.Cameron AD, Smerdon SJ, Wilkinson AJ, Habash J, Helliwell JR, Li TS, Olson JS. Distal Pocket Polarity in Ligand-Binding to Myoglobin - Deoxy and Carbonmonoxy Forms of a Threonine(68)(E11) Mutant Investigated by X-Ray Crystallography and Infrared-Spectroscopy. Biochemistry. 1993;32:13061–13070. doi: 10.1021/bi00211a016. [DOI] [PubMed] [Google Scholar]
- 37.Karow DS, Pan DH, Davis JH, Behrends S, Mathies RA, Marletta MA. Characterization of functional heme domains from soluble guanylate cyclase. Biochemistry. 2005;44:16266–16274. doi: 10.1021/bi051601b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boon EM, Huang SH, Marletta MA. A molecular basis for NO selectivity in soluble guanylate cyclase. Nat Chem Biol. 2005;1:53–59. doi: 10.1038/nchembio704. [DOI] [PubMed] [Google Scholar]
- 39.Jentzen W, Song XZ, Shelnutt JA. Structural characterization of synthetic and protein-bound porphyrins in terms of the lowest-frequency normal coordinates of the macrocycle. J Phys Chem B. 1997;101:1684–1699. [Google Scholar]
- 40.Harada K, Makino M, Sugimoto H, Hirota S, Matsuo T, Shiro Y, Hisaeda Y, Hayashi T. Structure and Ligand Binding Properties of Myoglobins Reconstituted with Monodepropionated Heme: Functional Role of Each Heme Propionate Side Chain. Biochemistry. 2007;46:9406–9416. doi: 10.1021/bi7007068. [DOI] [PubMed] [Google Scholar]
- 41.Vogel KM, Hu SZ, Spiro TG, Dierks EA, Yu AE, Burstyn JN. Variable forms of soluble guanylyl cyclase: protein-ligand interactions and the issue of activation by carbon monoxide. J Biol Inorg Chem. 1999;4:804–813. doi: 10.1007/s007750050354. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Y, Schelvis JPM, Babcock GT, Marletta MA. Identification of histidine 105 in the beta 1 subunit of soluble guanylate cyclase as the heme proximal ligand. Biochemistry. 1998;37:4502–4509. doi: 10.1021/bi972686m. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Y, Marletta MA. Localization of the heme binding region in soluble guanylate cyclase. Biochemistry. 1997;36:15959–15964. doi: 10.1021/bi971825x. [DOI] [PubMed] [Google Scholar]
- 44.Schelvis JPM, Zhao Y, Marletta MA, Babcock GT. Resonance Raman characterization of the heme domain of soluble guanylate cyclase. Biochemistry. 1998;37:16289–16297. doi: 10.1021/bi981547h. [DOI] [PubMed] [Google Scholar]
- 45.Martin E, Czarnecki K, Jayaraman V, Murad F, Kincaid J. Resonance Raman and Infrared Spectroscopic Studies of High-Output Forms of Human Soluble Guanylyl Cyclase . J Am Chem Soc. 2005;127:4625–4631. doi: 10.1021/ja0440912. [DOI] [PubMed] [Google Scholar]
- 46.Marti MA, Capece L, Crespo A, Doctorovich F, Estrin DA. Nitric oxide interaction with cytochrome c ‘ and its relevance to guanylate cyclase. Why does the iron histidine bond break? J Am Chem Soc. 2005;127:7721–7728. doi: 10.1021/ja042870c. [DOI] [PubMed] [Google Scholar]
- 47.Sharma VS, Magde D, Kharitonov VG, Koesling D. Soluble guanylate cyclase: Effect of YC-1 on ligation kinetics with carbon monoxide. Biochem Biophys Res Commun. 1999;254:188–191. doi: 10.1006/bbrc.1998.9812. [DOI] [PubMed] [Google Scholar]
- 48.Hashimoto T, Dyer RL, Crossley MJ, Baldwin JE, Basolo F. Ligand, Oxygen, and Carbon-Monoxide Affinities of Iron(II) Modified Capped Porphyrins. J Am Chem Soc. 1982;104:2101–2109. [Google Scholar]
- 49.Kim K, Ibers JA. Structure of a Carbon-Monoxide Adduct of a Capped Porphyrin- Fe(C2-Cap)(CO)(1-Methylimidazole) J Am Chem Soc. 1991;113:6077–6081. [Google Scholar]
- 50.Kerr EA, Mackin HC, Yu NT. Resonance Raman studies of carbon monoxide binding to iron “picket fence” porphyrin with unhindered and hindered axial bases. An inverse relationship between binding affinity and the strength of iron-carbon bond. Biochemistry. 1983;22:4373–4379. doi: 10.1021/bi00288a005. [DOI] [PubMed] [Google Scholar]
- 51.Silvernail NJ, Roth A, Schulz CE, Noll BC, Scheidt WR. Heme carbonyls: environmental effects on ν(C-O) and Fe-C/C-O bond length correlations. J Am Chem Soc. 2005;127:14422–14433. doi: 10.1021/ja053148x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang JL, Takahashi S, Hosler JP, Mitchell DM, Fergusonmiller S, Gennis RB, Rousseau DL. 2 Conformations of the Catalytic Site in the Aa(3)-Type Cytochrome-C-Oxidase from Rhodobacter-Sphaeroides. Biochemistry. 1995;34:9819–9825. doi: 10.1021/bi00031a001. [DOI] [PubMed] [Google Scholar]
- 53.Das TK, Tomson FL, Gennis RB, Gordon M, Rousseau DL. pH-dependent structural changes at the heme-copper binuclear center of cytochrome c oxidase. Biophys J. 2001;80:2039–2045. doi: 10.1016/S0006-3495(01)76177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Derbyshire ER, Tran R, Mathies RA, Marletta MA. Characterization of nitrosoalkane binding and activation of soluble guanylate cyclase. Biochemistry. 2005;44:16257–16265. doi: 10.1021/bi0515671. [DOI] [PubMed] [Google Scholar]
- 55.Kozlowski PM, Vogel KM, Zgierski MZ, Spiro TG. Steric contributions to CO binding in heme proteins: a density functional analysis of FeCO vibrations and deformability. J Porphyrins Phthalocyanines. 2001;5:312–322. [Google Scholar]
- 56.Li TS, Quillin ML, Philips GN, Olson JS. Structural Determinants of the Stretching Frequency of CO Bound to Myoglobin . Biochemistry. 1994;33:1433–1146. doi: 10.1021/bi00172a021. [DOI] [PubMed] [Google Scholar]
- 57.Balasubramanian S, Lambright DG, Marden MC, Boxer SG. Perturbations of the distal heme pocket in human myoglobin mutants probed by infrared spectroscopy of bound CO: correlation with ligand binding kinetics. Biochemistry. 1993;32:2202–2212. doi: 10.1073/pnas.90.10.4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ling JH, Li TS, Olson JS, Bocian DF. Identification of the iron-carbonyl stretch in distal histidine mutants of carbonmonoxymyoglobin. Biochim Biophys Acta -Bioenerg. 1994;1188:417–421. doi: 10.1016/0005-2728(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 59.Unno M, Christian JF, Olson JS, Sage JT, Champion PM. Evidence for Hydrogen Bonding Effects in the Iron Ligand Vibrations of Carbonmonoxy Myoglobin. J Am Chem Soc. 1998;120:2670–2671. [Google Scholar]
- 60.Anderton CL, Hester RE, Moore JN. A chemometric analysis of the resonance Raman spectra of mutant carbonmonoxy-myoglobins reveals the effects of polarity. Biochim Biophys Acta - Prot Str Mol Enzym. 1997;1338:107–120. doi: 10.1016/s0167-4838(96)00194-x. [DOI] [PubMed] [Google Scholar]
- 61.Tomita T, Ogura T, Tsuyama S, Imai Y, Kitagawa T. Effects of GTP on Bound Nitric Oxide of Soluble Guanylate Cyclase Probed by Resonance Raman Spectroscopy. Biochemistry. 1997;36:10155–10160. doi: 10.1021/bi9710131. [DOI] [PubMed] [Google Scholar]
- 62.Makino R, Obayashi E, Homma N, Shiro Y, Hori H. YC-1 facilitates release of the proximal His residue in the NO and CO complexes of soluble guanylate cyclase. J Biol Chem. 2003;278:11130–11137. doi: 10.1074/jbc.M209026200. [DOI] [PubMed] [Google Scholar]