


Abstract
Bloom’s syndrome (BS) which associates genetic instability and predisposition to cancer is caused by mutations in the BLM gene encoding a RecQ family 3′–5′ DNA helicase. It has been proposed that the generation of genetic instability in BS cells could result from an aberrant non-homologous DNA end joining (NHEJ), one of the two main DNA double-strand break (DSB) repair pathways in mammalian cells, the second major pathway being homologous recombination (HR). Using cell extracts, we report first that Ku70/80 and the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs), key factors of the end-joining machinery, and BLM are located in close proximity on DNA and that BLM binds to DNA only in the absence of ATP. In the presence of ATP, BLM is phosphorylated and dissociates from DNA in a strictly DNA-PKcs-dependent manner. We also show that BS cells display, in vivo, an accurate joining of DSBs, reflecting thus a functional NHEJ pathway. In sharp contrast, a 5-fold increase of the HR-mediated DNA DSB repair in BS cells was observed. These results support a model in which NHEJ activation mediates BLM dissociation from DNA, whereas, under conditions where HR is favored, e.g. at the replication fork, BLM exhibits an anti-recombinogenic role.
INTRODUCTION
Bloom’s syndrome (BS) is a rare human autosomal recessive disorder characterized by a predisposition to the development of all types of cancer and by a chromosomal instability including an increase in chromosome breakage, sister chromatid exchanges (SCEs) and symmetric quadriradial chromatid interchanges between homologous chromosomes (1). BS results from mutations in both copies of the BLM gene located on chromosome 15 at 15q26.1. This gene encodes the BLM protein that belongs to the DExH box-containing RecQ helicase subfamily (2). The BLM protein displays an ATP- and Mg2+-dependent 3′–5′ DNA helicase activity (3), but its function is still unclear.
Several studies have suggested a role for BLM in double-strand break (DSB) repair. DSBs occur under physiological conditions, but can also be induced by a number of exogenous agents, constituting a major threat to genome integrity. In mammalian cells, DSBs can be repaired by two major pathways: non-homologous end joining (NHEJ) that permits the joining, precisely or not, of broken DNA ends containing little or no homology, and homologous recombination (HR) that requires a homologous sequence provided by either a sister chromatid or a homologous chromosome (4,5). HR includes several mechanisms such as gene conversion, single-strand annealing and break-induced replication (4,6). NHEJ plays a dominant role during G1 to early S phase of the cell cycle, whereas HR is preferentially used in late S to G2 phases (7). Furthermore, a competition may exist between NHEJ and HR (8–11), and an inhibition of the HR pathway by components of NHEJ has been reported (12,13).
Using an in vivo host cell end-joining assay, Rünger and Kraemer (14) were the first to describe a reduced and error-prone joining of linear plasmid DNA in BS cells. Recently, controversial results differing from those of Rünger and Kraemer were published by two independent groups that analyzed the efficiency and fidelity of NHEJ in vitro in cell-free nuclear extracts prepared from BS cells (15,16). Langland et al. (16) showed that the efficiency and fidelity of DSB repair by BS extracts were comparable with those of normal extracts when ligatable ends were present. In contrast, Gaymes et al. (15) showed an increase in the activity of the NHEJ pathway in BS extracts, associated with repair infidelity and the formation of large deletions. Thus, no clear conclusions on the role of BLM in DSB repair via the NHEJ pathway can be drawn from these three studies. However, these studies only tested the NHEJ pathway that plays an important role in G1, whereas we have previously shown that the endogenous BLM protein is undetectable in G1. We and others have shown that BLM accumulated to high levels in S phase, that persisted in G2, which is consistent with a role for BLM in a replicative and/or post-replicative process (17,18). Several lines of evidence are in favor of an increase in DSB repair via the HR pathway in BLM-deficient cells (19). Indeed, BS cells are characterized by an increase in interchange between homologous chromosomes at homologous sites and by a 10-fold increase in SCEs (1). HR mediates SCEs in vertebrate cells (20), and a large fraction of SCEs in BS cells depend on Rad54 function, and thus are formed via HR (21). Moreover, BLM recombinant protein promotes an ATP-dependent branch migration of Holliday junctions (22). A model has been proposed in which a BLM defect would result in the annealing of free ends of newly synthesized daughter strands at sites of blocked replication forks, forming Holliday junctions. If these Holliday junctions cannot be resolved by a potential reverse branch migration activity of BLM, their resolution would generate DNA breaks that are repaired mainly by HR (22,21). Finally, Luo et al. (23), who generated the only viable Bloom mice, showed that the gene targeting frequency obtained in the BLM-deficient embryonic stem (ES) cells was 3.5-fold higher than that obtained in wild-type ES cells, and proposed that this accounts for the mechanism of elevation in SCEs.
These data, associated with our results showing that BLM is phosphorylated via the ATM pathway in response to ionizing radiation, known to generate DSBs (24,25), prompted us to explore the possible relationship between BLM and the key components of the NHEJ pathway, Ku70/80 and the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs), and to analyze the capacity of BS cells to repair in vivo DSBs by NHEJ or HR.
MATERIALS AND METHODS
Cell lines and extracts
The Epstein–Barr virus (EBV)-transformed lymphoblastoid B-cell lines GM03403D and D1, and HeLa cells were cultured as previously described (17,24).
HeLa nuclear extracts from the Computer Cell Culture Center (Seneffe, Belgium), and D1 and GM03403D nuclear extracts were prepared according to Dignam et al. (26).
DNA-PKcs-deficient and -complemented cell lines (Fus9, alias M059J, and Fus1, respectively) (27) were kindly donated to B. Salles’s group by Dr C. Kirchgessner (Stanford University School of Medicine, CA). Nuclear protein extracts were prepared as previously described (28) except that the final dialysis was performed for 3 h at 4°C in an excess volume of 50 mM HEPES KOH pH 7.9, 10% glycerol, 100 mM potassium glutamate, 1 mM EDTA, 1 mM dithiothreitol (DTT). Extracts were immediately frozen and stored at –80°C.
Antibodies
Anti-β-actin, anti-Ku70 (N3H10), anti-Ku80 (111), anti Ku70/80 (162) and anti-p460 (18.2 and 25.4) monoclonal antibodies were from Neomarkers (USA). The rabbit anti-Ku70 (C19) was from Santa Cruz (CA). The rabbit anti-BLM antiserum 1340 was generated and used as described (17). Peroxidase-conjugated goat anti-mouse or anti-rabbit secondary antibodies were from Jackson Immunoresearch Laboratories (USA).
Oligonucleotides
DNA binding assay. The 80mer oligonucleotide 5′-GATTCGGATCCGATCATGTATTGTATTATTGTGTATGACACACAATGTGCATAATGTTAATGTGAACGAAGCTTCCCGGG-3′ carrying a 5′ biotin moiety was annealed with the unmodified 77mer oligonucleotide 5′-GGGAAGCTTCGTTCACATTAACATTATGCACATTGTGTGTCATACACAATAATACAATACATGATCGGATCCGAATC-3′ in order to construct a 5′-biotinylated double-stranded (ds) DNA with a 3′-protruding end. Digestion of this double-stranded oligonucleotide with AluI restriction enzyme generated a blunt-ended 71 bp biotinylated DNA fragment. The 5′-protruding 30 bp biotinylated DNA fragment was constructed by annealing the oligomer 5′-TAAAGGGAACAAAAGCTGGGTACCGGTGTT-3′ biotinylated at the 5′ end with the complementary 32mer or 34mer non-biotinylated oligonucleotide 5′-CCAACACCGGTACCCAGCTTTTGTTCCCTTTA-3′ and 5′-GGCCAACACCGGTACCCAGCTTTTG TTCCCTTTA-3′, respectively. A blunt-ended 30 bp biotinylated DNA fragment was constructed by annealing the above 30mer with the complementary 30mer oligonucleotide.
Electrophoretic mobility shift assay (EMSA). A 5′-protruding 22 bp double-stranded oligonucleotide was constructed by annealing the 26mer 5′-AATTCCGTCCCAGCTGGGCTCTTCCC-3′ with the complementary 22mer 5′-GGGAAGAGCCCAGCTGGCACGG-3′.
PCR analysis. PCR analysis was performed by using two 21mer primers: 5′-ATT CAG GCT ACG CAA CTG TTG-3′ and 5′-GTG AGT TAC CTC ACT CAT TAG-3′, corresponding to positions 450–471 and 1213–1234, respectively, on plasmid pHRecSJ, and to positions 450–471 and 1266–1287, respectively, on plasmid pHRecCJI.
Plasmids
The pHRecSJ plasmid [Fig. 5A and Smith et al. (29)] is based on a pBluescript plasmid (Stratagene, CA) and contains the prokaryotic ColE1 replication origin and the β-lactamase gene. The plasmid is autonomously replicated in human cells due to the presence of the SV40 large T-antigen-coding sequence and its replication origin. This plasmid contains a target LacZ gene interrupted by a 376 bp fragment flanked by a pair of the consensus V(D)J recombination signal sequences RSS12 and RSS23 because it was initially designed to analyze V(D)J recombination activity in human cells (29). EcoRI and NotI restriction sites are located at positions 647 and 978, respectively.
Figure 5.
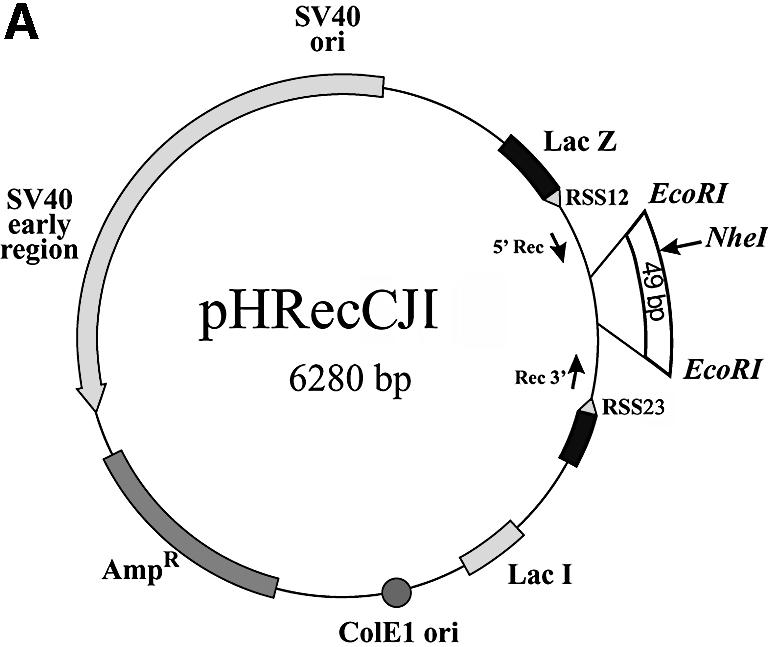

Assessing the fidelity of in vivo DNA-end joining in BLM- deficient cells. (A) Plasmid pHRecCJI was used to analyze in vivo host cell end joining. ColE1 is the prokaryotic replication origin. AmpR is the β-lactamase gene conferring ampicillin resistance in E.coli. The SV40 large T-antigen-coding sequence and its replication origin (SV40 ori) confer on pHRecCJI the capacity to replicate in human cells. The target is a modified LacZ gene containing a unique restriction site for NheI between two EcoRI restriction sites (31). (B) The percentage of plasmids accurately rejoined recovered from wild-type D1 cells and GM03403D BS cells. The input plasmid digested with EcoRI contains a DSB with complementary ligatable ends (5′EcoRI). The input plasmid digested with EcoRI and treated with phosphatase contains non-ligatable complementary ends without terminal phosphates (5′EcoRI + PPase). The reported values are the average from three (5′EcoRI) and two (5′EcoRI + PPase) independent transfections, respectively. Bars and error bars represent the means and standard deviations, respectively, of the percentage of error-free rejoined plasmids.
The pRSJ plasmid [Fig. 5B and Baldeyron et al. (30)] is a derivative of pHRecSJ (Fig. 5A) in which the SV40 large T-antigen-coding sequence and its replication origin were deleted.
The pHRecCJI plasmid (Fig. 4A) is a derivative of pHRecCJ (29) modified by the insertion of a 49 bp EcoRI fragment containing a unique NheI site at the EcoRI site (31). The EcoRI sites are located at positions 978 and 1027, respectively.
Figure 4.
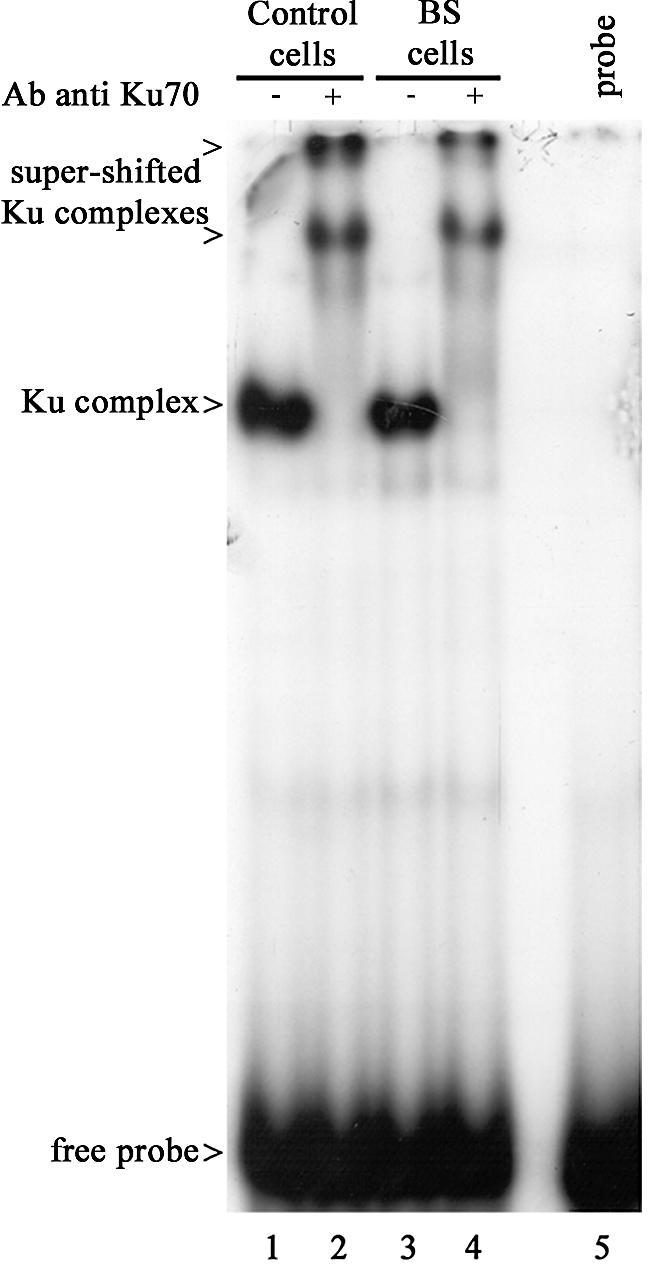
Gel shift analysis of Ku DNA-end binding activity in wild-type and BS cells. EMSA was performed using nuclear extracts from either wild-type D1 cells (lanes 1 and 2) or GM03403D BS cells (lanes 3 and 4) and a 32P-labeled double-stranded oligonucleotide as a probe. Supershift assays were carried out using C19 anti-Ku antibody (lanes 2 and 4).
Co-immunoprecipitation assay
Five picomoles of 77 bp double-stranded oligonucleotides were incubated with 60 µg of HeLa nuclear extracts for 30 min at 30°C in 20 µl of reaction buffer [40 mM HEPES-KOH pH 7.8, 5 mM MgCl2, 60 mM KCl, 0.5 mM DTT, 0.4 mM EDTA, 3.4% glycerol, 6 µg of bovine serum albumin, 10 mM NaF, 0.2 mM sodium orthovanadate, 1 mM cantharidin (Sigma)] in the presence of 2 mM glucose and 0.2 U of hexokinase (Sigma) in order to remove traces of ATP. In some reactions, ethidium bromide (EtBr) was added to a final concentration of 0.3 mg/ml to the reaction mixture and maintained throughout the immunoprecipitation procedure. Then, the reaction volume was adjusted to 200 µl with the IP buffer [25 mM HEPES-KOH pH 7.5, 100 mM NaCl, 20% glycerol, 5 mM EDTA, 1 mM DTT, 0.05% NP-40, 10 mM NaF, 0.2 mM sodium orthovanadate, 1 mM cantharidin and a protease inhibitor cocktail tablet (Roche Molecular biochemicals)]. Then 10 µl of magnetic anti-mouse IgG or anti-rabbit IgG immunobeads suspension coated with primary anti-β-actin, anti-Ku70/80 (162) or anti-p460 (25.4) antibodies according to the manufacturer’s protocol (Dynal) were added. The beads were mixed gently on a wheel for 4 h at 4°C. The beads were pulled down over a magnet and the supernatants were removed. After two washes with 1 ml of the IP buffer, proteins in the immunoprecipitates and in the supernatants were heated in SDS sample buffer and separated by 6% SDS–PAGE.
DNA binding assay
Five picomoles of biotinylated double-stranded oligonucleotide were immobilized on 10 µl of streptavidin paramagnetic beads (Dynabeads M280 streptavidin, Dynal) in 50 µl of 5 mM Tris–HCl, pH 7.5, 0.5 mM EDTA, 1 M NaCl for 30 min at 20°C under agitation. After washes with the same buffer, DNA- or mock-treated beads were incubated in a 50 µl reaction mixture containing 60 µg of nuclear protein extracts in the reaction buffer either supplemented with 2 mM glucose and 0.2 U of hexokinase (Sigma) (conditions –ATP) or in the presence of 0.2 mM ATP, for 30 min at 30°C under agitation. The supernatant was removed and the beads were washed twice with 250 µl of the IP buffer; then the supernatant or the beads were heated in SDS sample buffer and proteins were separated by 6% SDS–PAGE. For phosphatase treatment, reactions were performed without phosphatase inhibitors and then 30 U of calf intestinal alkaline phosphatase (CIAP; New England Biolabs) was added or not to the reaction supernatant mixtures and incubated at 37°C for 60 min.
Immunoblotting
Protein samples resolved by SDS–PAGE were electro-transferred onto polyvinylidene difluoride membrane (Amersham Pharmacia Biotech). Membranes were blocked with 5% milk in phosphate-buffered saline (PBS) for 1 h at room temperature and proteins were immunoblotted with primary antibodies overnight at 4°C, and then probed with secondary antibody conjugated to horseradish peroxidase. Specific proteins were visualized as immunoreactive bands by the enhanced chemiluminescence detection system (Supersignal West Pico, Pierce). Providing extensive washing and probing first with polyclonal antibodies had been carried out, successive immunoblottings were performed on the same membranes without stripping. For data presentation, films were scanned and processed with Adobe Photoshop 3.0 software.
EMSA
Assays were carried out essentially as previously described (32). Briefly, 5 µg of nuclear extracts were diluted in buffer containing 10 mM Tris–HCl pH 8, 150 mM NaCl, 1 mM DTT, 10% glycerol and 20 µg/ml circular pUC19 in a total volume of 20 µl. γ-32P-labeled double-stranded 22mer (0.25 ng; [γ-32P]ATP, Amersham, France) was added to the mixture, and the reaction was incubated for 20 min at room temperature. A 1 µg aliquot of anti-Ku70 antibody (C19) was added or not, and the mixture was then subjected to an additional 15 min incubation at room temperature. Samples were run on a 6% non-denaturating polyacrylamide gel in 0.5× Tris borate/EDTA buffer. Following electrophoresis, the gel was dried and autoradiographed.
Host cell DSBs repair assays
Linear plasmids (EcoRI-digested pHRecCJI and EcoRI–NotI-digested pHRecSJ) were separated from the deleted fragment and any uncut molecules by agarose gel electrophoresis followed by an electroelution (Biotrap, Schleicher and Schuell, France). Terminal phosphates were removed from the electroeluted EcoRI fragments by CIAP treatment (0.1 U/µg DNA) (Biolabs, Ozyme, France) for 1 h at 37°C. The fragments were recovered by ethanol precipitation after phosphatase heat inactivation (10 min at 75°C) and phenol/chloroform extraction, and assayed by bacterial transformation.
Transfections of lymphoblastoid cells were performed by electroporation. Briefly, 2 µg of linear and/or supercoiled plasmid DNA were introduced into 3 × 106 exponentially growing cells by double-pulse electroporation (pulse 1: 800 V, 25 µF, 99 Ω; pulse 2: 100 V, 1500 µF, 99 Ω; Easy CellJect, Eurogentec). Immediately after transfection, cells were diluted into 10 ml of RPMI medium containing 15% fetal calf serum and incubated at 37°C. The transfection efficiency, measured by the use of a green fluorescent protein (GFP)-based plasmid (Stratagene, USA) was similar (4–6%) for all cell lines studied. At 48 h after transfection, the cells were harvested, washed twice with PBS and stored at –20°C. Plasmids were extracted by an alkaline lysis procedure (33). Plasmids that were not replicated were eliminated by digestion with DpnI. Following dialysis against distilled water, plasmids were further electroporated into XL1-blue competent bacteria (Stratagene, CA) and plated on LB agar plates containing ampicillin (50 µg/ml). PCR analysis of the plasmids was carried out as described in the Results.
RESULTS
BLM, Ku70/80 and DNA-PKcs assemble on the same dsDNA molecule
The Ku heterodimer (Ku70/Ku80) is an essential component of DSB repair via NHEJ and represents the major dsDNA end-binding protein in human cells (34). A functional interaction between Ku70 and the Drosophila Dmblm, homolog of the human BLM, was shown in vivo (35), and Gaymes et al. (15) reported that BS cells displayed an aberrant in vitro end joining of EcoRI-induced DSB dependent upon the presence of the Ku70/80 heterodimer. On the basis of these data, we examined whether BLM could interact with the DNA-PK complex that consisted of the heterodimeric Ku protein and a large catalytic subunit (DNA-PKcs). We were unable to directly co-immunoprecipitate BLM and Ku70 from HeLa protein extracts (data not shown). Then we studied whether DNA mediated BLM interaction with the DNA-PK complex, as is known to occur for Ku and DNA-PKcs (36,37). Immunoprecipitates were prepared from HeLa nuclear protein extracts supplemented with 77 bp double-stranded oligonucleotides, using either anti-Ku (162) or anti-DNA-PKcs (Ab25.4) antibodies, in the presence or absence of EtBr at a concentration known to disrupt most of the DNA–protein interactions. As shown in Figure 1, BLM was detected on both anti-Ku and anti-DNA-PKcs immunobeads (lanes 2 and 4), but not on anti-actin beads (lane 1). However, addition of EtBr completely disrupted the interaction of both Ku and DNA-PKcs with BLM (lanes 3 and 5). The DNA-dependent Ku–DNA-PKcs assembly was also destabilized in the presence of EtBr (lanes 3 and 5), as previously described (36).
Figure 1.
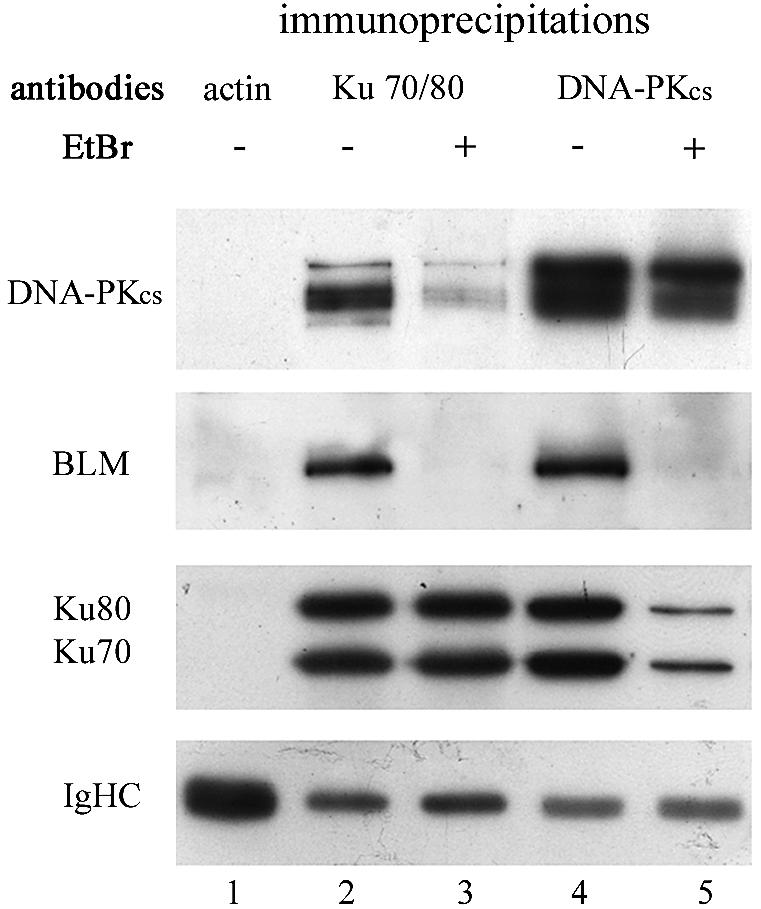
Co-immunoprecipitation of protein–DNA complexes. HeLa nuclear extracts were incubated with 5 pmol of double-stranded 77 bp oligonucleotides in the presence of glucose and hexokinase in order to remove trace ATP. The mixture was then mixed with magnetic beads coated with anti-β-actin (lane 1), anti-Ku (lanes 2 and 3) or anti-DNA-PKcs (lanes 4 and 5) primary antibodies. In some reactions, EtBr was added to the reaction mixture and maintained throughout the immunoprecipitation procedure (lanes 3 and 5). After washing, proteins in the immunoprecipitate were heated in SDS sample buffer and separated by 6% SDS–PAGE. Successive immunoblottings with anti-DNA-PKcs, anti-BLM and anti-Ku were performed on the same membranes without stripping. The lower panel (IgHC) corresponds to the immunoglobulin heavy chains of each antibody used in the immunoprecipitation.
Our results clearly show that BLM, Ku and DNA-PKcs can assemble on the same dsDNA molecule, but do not support interaction between these proteins in the absence of DNA.
The BLM protein is retained with Ku70/80 and DNA-PKcs on DNA, but only in the absence of ATP
To further characterize the DNA-mediated assembly of BLM with Ku and DNA-PKcs, we performed an assay where double-stranded oligonucleotides were coupled to paramagnetic streptavidin beads and used as a target to retain proteins (37). The associated proteins were then analyzed by western blotting. Double-stranded oligonucleotides exhibiting different termini structures [30 bp, blunt; 30 bp, 5′ protuding 2 nt single-strand (ss); 30 bp, 5′ protuding 4 nt ss; 77 bp, 3′ protuding 3 nt ss; 71 bp blunt] were coupled to beads. DNA-treated beads were incubated with HeLa nuclear protein extracts, in the presence or absence of ATP, which is crucial for DNA-PKcs function (38–41). No non-specific binding to the paramagnetic beads was detectable in the absence of DNA (Fig. 2A, lane 1). BLM was present together with DNA-PKcs and Ku70/80 in the protein fractions retained on DNA, independently of the type of DNA termini, but only in the absence of ATP (Fig. 2A, lanes 2, 4, 6, 8 and 10). Indeed, the BLM protein did not bind to any type of oligonucleotide in the presence of ATP (Fig. 2A, lanes 3, 5, 7, 9 and 11). Furthermore, analysis of the supernatants of the binding reactions clearly showed that in the presence of ATP, the unbound BLM protein was shifted to a slightly slower migrating form (compare in Fig. 2B, lanes 3, 5, 7 and 9 with lanes 1, 2 and 8). One can note that in the presence of ATP, the amount of DNA-PKcs retained on DNA beads was reduced, and Ku80 was shifted to a slower migrating form (Fig. 2A, lanes 3, 5, 7, 9 and 11), as already reported (37).
Figure 2.

Recovery of DNA-binding proteins on DNA beads and effect of ATP. (A) HeLa nuclear extracts supplemented with glucose and hexokinase (–ATP) (lanes 2, 4, 6, 8 and 10) or in the presence of ATP (lanes 3, 5, 7, 9 and 11) were incubated with free beads (lane 1) or beads bound to double-stranded oligonucleotides exhibiting different termini as indicated (lanes 2–11), in the standard reaction buffer. After incubation, the beads were washed and the bound proteins were analyzed by western blotting. (B) Unbound proteins from the reactions loaded in lanes 1–3, 5 and 7–9 in (A) were subjected to western blot analysis with anti-BLM antibody.
Hence, BLM, Ku70/80 and DNA-PKcs are present together in the protein fraction retained on DNA in the absence of ATP. In the presence of ATP, the BLM protein is shifted to a slightly slower migrating form that is not retained on DNA.
BLM is phosphorylated and dissociates from DNA in a DNA-PKcs-dependent manner
It has been shown that in the presence of DNA and ATP, DNA-PK underwent rapid autophosphorylation of each of the three subunits, DNA-PKcs, Ku70 and Ku80, which correlated with a loss of DNA-PK kinase activity and resulted in dissociation of DNA-PKcs from the Ku–DNA complex (37,38,42). In this context, our results suggest that in the presence of DNA and ATP, BLM could be phosphorylated by DNA-PKcs, which would result in its dissociation from DNA. To test this, DNA-treated (30 bp, 5′ + 4 nt) beads were incubated in the absence or presence of ATP, with nuclear protein extracts from either M059J cells that do not express DNA-PKcs (DNA-PKcs-deficient cells, Fus9) or M059J-complemented cells that contain an extra copy of the human gene coding for DNA-PKcs (DNA-PKcs-complemented cells, Fus1) (27). As shown in Figure 3A, BLM from DNA-PKcs-deficient cells was retained similarly on DNA in both the absence and presence of ATP (lanes 2 and 3). In contrast, BLM from DNA-PKcs-complemented cells was retained on DNA only in the absence of ATP (Fig. 3A, lanes 5 and 6), as observed in HeLa extracts (Fig. 2A). Analysis of the supernatants of the binding reactions showed that in the presence of ATP, BLM protein from DNA-PKcs- complemented cells was shifted to a slightly slower migrating form (compare in Fig. 3B, lane 12 with lane 11). In order to determine if this shift was due to phosphorylation, the corresponding supernatant of the binding reaction was treated with CIAP. As shown in Figure 3C, phosphatase treatment resulted in the recovery of a band that migrated similarly to unbound BLM in the absence of ATP.
Figure 3.

Effect of ATP on DNA-binding activity of BLM from protein extracts from either M059J DNA-PKcs-deficient or DNA-PKcs-complemented cells. (A) M059J or M059J-complemented nuclear extracts supplemented with glucose and hexokinase (–ATP) (lanes 2 and 5) or in the presence of ATP (lanes 3 and 6) were incubated with free beads (lanes 1 and 4) or beads bound to double-stranded oligonucleotides (30 bp 5′ + 4 nt) (lanes 2, 3, 5 and 6), in the standard reaction buffer. After incubation, the beads were washed and the bound proteins were analyzed by western blotting. (B) Unbound proteins from the reactions loaded in lanes 1–6 in (A) were subjected to western blot analysis (lanes 7–12). (C) Unbound proteins from the reaction loaded in lane 6 in (A) were incubated in the absence (lane 2) or presence (lane 3) of calf intestine alkaline phosphatase and subjected to western blot analysis with anti-BLM antibody.
Notably, part of DNA-PKcs from DNA-PKcs-complemented cells dissociated from DNA in the presence of ATP (Fig. 3B, lane 12), as expected (37).
These results demonstrate that in the presence of ATP, BLM is phosphorylated and dissociates from DNA in a strictly DNA-PKcs-dependent manner.
Ku DNA ends-binding activity is not altered in BS cells
To further investigate the relationship between BLM and Ku, we tested the Ku DNA ends-binding activity in BLM-deficient cells by using EMSA. A 32P-labeled double-stranded oligonucleotide was incubated with nuclear protein extracts from BS GM3403D or wild-type D1 EBV-transformed lymphoblastoid cells, in the presence or absence of C19 anti-Ku70 antibody. As shown in Figure 4, the same shifts (lanes 1 and 3) and supershifts (lanes 2 and 4) were observed with the protein extracts prepared from either BS or control cells. These results indicate that Ku DNA ends-binding activity is not affected in BS cells.
DSB rejoining efficiency and fidelity are not affected in BS cells
The above results and the controversial data on efficiency and fidelity of EcoRI-induced DSB repair in BS cells (14–16) prompted us to examine in vivo end joining in BS cells by using a plasmid-based host cell end-joining assay (43). A cohesive-ended DSB was generated by restriction endonuclease EcoRI digestion of an autoreplicating plasmid pHRecCJI that contained two EcoRI sites separated by 49 bp containing a unique NheI site (Fig. 5A). Precise end-joining repair of such a substrate would lead to restoration of a unique EcoRI site associated with a loss of the NheI site. Experiments were carried out in the BS EBV-transformed lymphoblastoid cell line GM03403D, homozygous for the blmAsh allele which contains a frameshift mutation commonly found in patients from Ashkenazi Jewish ancestry (2), leading to production of an undetectable truncated protein (17). We used a wild-type EBV-transformed lymphoblastoid B cell line D1, which displays a growth rate comparable with that of BS GM03403D cells, as a control (data not shown). Expo nentially growing BS and control cells were transiently transfected in parallel with the circular supercoiled plasmid and with the EcoRI-digested plasmid. After 48 h, the plasmids were extracted and non-replicated plasmids were eliminated by DpnI digestion which cleaves DNA that was not replicated in human cells and thus retained the bacterial dam methylation (43). In our experiments, only recircularized plasmids could be replicated, ensuring that the recovered molecules were repaired in human cells. Replicated DpnI-resistant plasmids harvested from human cells were used to transform bacteria.
The DSB end-joining efficiency in BS and control cells was estimated on the basis of three independent experiments by calculating the ratio between the number of bacterial colonies obtained after transformation with repaired plasmids versus the control circular plasmids. We observed a high variability between each experiment in both BS and control cells, and the survival rate of linear plasmid relative to uncut plasmid varied from 13 to 39% in BS cells, and from 15 to 49% in control cells. These non-significant differences led us to conclude that BS lymphoblasts are as efficient as normal cells in rejoining EcoRI DNA ends.
The DSB end-joining fidelity in BS and control cells was analyzed by performing PCR on 1782 bacterial colonies obtained after transformation with repaired plasmids using primers located on either side of the DSB. PCR products from accurately repaired molecules were up to 788 bp in length, thus migrating faster than PCR products amplified from the native pHRecCJI plasmid (837 bp) used as the control, since they lack at least 49 bp. PCR products were subjected to EcoRI digestion to detect the error-free repaired molecules. We have found by analyzing three independent experiments that 644 of 939 plasmids (68.5%) and 577 of 843 plasmids (68.4%) were accurately joined in the control and BS cell lines, respectively (Fig. 5B). Thus, BS cells are not defective in the fidelity of rejoining of EcoRI-induced DSBs, which is in agreement with the results published by Langland et al. (16). Interestingly, the authors observed a 5-fold increase in mutation rate with BS extracts when terminal phosphates were removed from EcoRI-digested DNA substrate (16). In order to determine the efficiency and fidelity of the repair of such a substrate in our system, EcoRI-digested plasmid was treated with CIAP before transfection into BS and control cells. The efficiency of the phosphatase treatment was confirmed by an in vitro ligation followed by transformation. The end-joining efficiency of dephosphorylated EcoRI ends varied between 16.4 and 47% in the control cells, and between 23 and 42% in BS cells. The error-free joining of dephosphorylated EcoRI ends was 63% (357/567 repaired plasmids) in the control cells and 64.6% (370/572 repaired plasmids) in BS cells (Fig. 5B).
These results demonstrate that the joining efficiency and fidelity of EcoRI ends without terminal phosphates is similar in BS and control cells.
The repair of DSBs by HR is significantly increased in BS cells
To analyze the DNA DSB repair by the HR pathway in BS cells, we developed a two-plasmid assay. One plasmid, pHRecSJ (Fig. 6A), is autoreplicating in human cells due to the presence of the SV40 large T-antigen-coding sequence and the replication origin (29). The other plasmid, pRSJ, is a derivative of pHRecSJ in which the SV40 large T-antigen-coding sequence and the replication origin have been deleted (Fig. 6B). Thus, pRSJ is non-replicating but homologous to pHRecSJ, and can serve as a substrate for HR. The autoreplicating pHRecSJ plasmid was digested by the restriction endonucleases EcoRI and NotI, generating a substrate with a deleted 331 bp fragment and exhibiting non-complementary termini. The linear EcoRI–NotI pHRecSJ plasmid and the circular non-replicating pRSJ plasmid, containing the 331bp fragment missing in the pHRecSJ plasmid, were transiently co-transfected into GM03403D BS cells and D1 control cells. After 48 h, the plasmids were extracted and unreplicated plasmids were eliminated by DpnI digestion, as described above. In these experiments, all unreplicated pRSJ molecules were destroyed by DpnI digestion, and only repaired and replicated pHRecSJ plasmids were DpnI resistant. EcoRI–NotI end rejoining was globally less efficient than EcoRI–EcoRI end rejoining in both BS and control cells, but we again observed a high variability between each of the three experiments in both BS and control cells, and the survival rate of linear plasmid relative to uncut plasmid varied from 7.7 to 12% in BS cells, and from 1.5 to 20% in control cells. The EcoRI–NotI DSB introduced into the pHRecSJ plasmid can be repaired by NHEJ or by HR. In the latter case, the missing 331 bp EcoRI–NotI fragment would be restored. The pathway by which the EcoRI–NotI pHRecSJ plasmid was repaired was analyzed by performing PCR on bacterial colonies from the above experiments using the same primers as mentioned above. PCR products from the molecules repaired by HR were 784 bp in length (Fig. 6C, lane 8) and displayed the same migration pattern as PCR products amplified from the native pHRecSJ plasmid used as the control (Fig. 6C, lane 4) since they contained the 331 bp fragment. PCR products of 453 bp or less were repaired by NHEJ (Fig. 6C, lanes 1–3, 5–7 and 9–11).
Figure 6.



Assessing the HR of DSBs in BLM-deficient cells. (A) pHRecSJ plasmid contains the SV40 large T-antigen-coding sequence and its replication origin (SV40 ori) conferring on it the capacity to replicate in human cells. The target is a modified LacZ gene containing unique restriction sites for EcoRI and NotI separated by 331 bp. (B) pRSJ, a derivative from pHRecSJ in which the SV40 large T-antigen-coding sequence and its replication origin have been deleted (30). (C) The EcoRI–NotI DSB introduced into the pHRecSJ plasmid (A) can be repaired by NHEJ or by HR. In the latter case, the missing 331 bp EcoRI–NotI fragment would be restored. The pathway by which the EcoRI–NotI pHRecSJ plasmid was repaired was analyzed by performing PCR on bacterial colonies using primers located on either side of the DSB (see Materials and Methods). PCR products were migrated in a 1.2% agarose gel. PCR products from the molecules repaired by HR were 784 bp in length (lane 8) and displayed the same migration pattern as PCR products amplified from the native pHRecSJ plasmid used as the control (lane 4) since they contained the 331 bp fragment. PCR products of 453 bp or less were repaired by NHEJ (lanes 1–3, 5–7 and 9–11). (D) The percentage of plasmids repaired by HR recovered from wild-type D1 cells and GM03403D BS cells. The input pHRecSJ plasmid was digested by EcoRI and NotI restriction endonucleases, generating a substrate containing non-complementary termini, and lacking a 331 bp fragment. Both the Eco-RI–NotI pHRecSJ plasmid and the circular pRSJ were co-transfected into the control and BS cells. The pHRecSJ plasmids repaired by the HR pathway contain the EcoRI–NotI 331 bp fragment. The reported values are the average from three independent transfections. Bars and error bars represent the means and standard deviations, respectively, of the percentage of HR repair events.
To determine the percentage of EcoRI–NotI DSB molecules repaired via the HR pathway, we first analyzed the PCR products from 883 rejoined plasmids from D1 control cells, randomly chosen from three independent experiments. As shown in Figure 6D, eight molecules in 883 (0.9%) were repaired via the HR pathway in these cells. Concerning BS cells, the analysis of only 386 rejoined plasmids, again randomly chosen from three independent experiments, was enough to show a significant difference since 18 molecules in 386 were repaired via the HR pathway in BS cells (4.66%) (Fig. 6D). Sequence analysis of 10 of these 784 bp PCR products from either BS or control cells confirmed that each molecule recovered precisely the 331bp EcoRI–NotI fragment (data not shown).
Moreover, PCR products (43 BS and 46 controls) repaired by NHEJ were also subjected to sequence analysis. Termini rearrangements produced during the processing of the non-complementary ends by EcoRI–NotI, which are mainly deletions varying from 1 to 358 bp (see Supplementary Material), were comparable in the control and BS cells. Only two out of 43 clones derived from BS cells and one out of 46 clones derived from control cells were accurately repaired, probably by filling-in of the 3′-recessed end by a DNA polymerase to create a ligatable blunt end (see Supplementary Material).
Altogether, these results clearly show that the DSB repair by HR is increased more than five times in BS cells compared with wild-type cells, whereas NHEJ remains unchanged.
DISCUSSION
In this study, we have first shown that BLM, Ku and DNA-PKcs assemble on the same DNA molecules only in the presence of added DNA. In our experimental conditions, EtBr destabilized the Ku–DNA-PKcs complex, as previously described (36), and also disrupted the interaction between BLM and Ku or DNA-PKcs. Our results also suggest that BLM and DNA-PK complex bind to DNA independently, and are co-immunoprecipitated even if they do not physically interact, since we were unable to co-immunoprecipitate these proteins in the absence of DNA. However, we cannot formally exclude that binding to DNA facilitates their physical interaction, which does not resist the EtBr exposure. Another possibility is that other DNA-binding proteins are required to mediate the assembly of BLM and DNA-PK complex on DNA. Future experiments with BLM, Ku and DNA-PKcs purified proteins will help address these questions. Altogether, our results indicate that BLM and DNA-PK complex can be located in close proximity on DNA. Since BLM does not unwind DNA double-stranded substrates (44), it is unlikely that it is recruited with DNA-PK complex to process DNA ends. Moreover, we showed that BS cells display an accurate NHEJ (see below), indicating that BLM does not play a major role in this pathway. Thus, our results suggest that NHEJ and HR compete for DSB sites, which can potentially result in the simultaneous recruitment of proteins specific for each pathway, as discussed below. Interestingly, we also found that in the presence of ATP, BLM protein is phosphorylated and dissociates from DNA via a DNA-PKcs-dependent pathway. Indeed, BLM phosphorylation and dissociation from DNA are not observed in M059J DNA-PKcs-deficient cells, whereas they are restored in M059J DNA-PKcs-complemented cells. These results suggest that once the NHEJ pathway is activated, BLM is phosphorylated by the DNA-PKcs, which mediates its dissociation from DNA, possibly through the alteration of its substrate specificity.
Our results that BS cells display an accurate NHEJ pathway are consistent with the data that BLM-deficient cells are not radiosensitive (45) and are not defective in V(D)J recombination (46–49), and also fit with the BLM expression during the cell cycle. Indeed, we have previously shown that BLM is undetectable in G1 phase cells, whereas it is strongly expressed in S and G2/M cells (17). These data do not support an important role for BLM during the G1 phase of the cell cycle and, in addition, it is now admitted that NHEJ is essential in G1 (7). Our results are in agreement with those reported by Langland et al. (16) showing that efficiency and fidelity of DSB repair by BS extracts are comparable with those of normal extracts when ligatable ends (EcoRI) are present. However, when terminal phosphates were removed, we did not observe the increase in mutation rate in BS cells that these authors reported. In contrast, we found that 5′-OH termini were joined accurately in BS cells, which is in agreement with the recent data showing that phosphate addition by human polynucleotide kinase is coupled to functional NHEJ (50). Interestingly, mouse cell lines containing targeted mutations in Ku70, DNA-PKcs or XRCC4 do not exhibit an elevation of the rate of SCEs (9), which indicates that the NHEJ defect is not accompanied by an increase in the generation of SCEs, and thus the high level of SCEs in BS cells does not reflect an impaired NHEJ. It is interesting to note that all the experiments showing an error-prone NHEJ in BS cells have been performed in the SV40-transformed BS cells, GM08505 (14–16). It is well known that the SV40 T antigen inactivates p53, and inactivation of p53 results in high rates of HR (51,52), indicating that wild-type p53 suppresses HR (53). Moreover, p53 and BLM functionally interact in apoptosis (45), BLM cooperates with p53 in regulation of transcription and cell growth control (54), and p53 modulates BLM’s ability to disrupt Holliday junctions (55). It has recently been demonstrated that p53 and BLM functionally interact during resolution of stalled DNA replication forks (56). These data suggest that the SV40-transformed BS fibroblasts lack not only BLM but also functional p53, which was shown to interfere with the BS phenotype (57). The EBV-transformed BS cells that we used in the present study exhibit a normal p53 response to ionizing radiation (14,57), and are not impaired in the rejoining of linear plasmid [Rünger and Kraemer (14); this study]. Hence, differences in p53 status could explain the apparent discrepancy between our results and those reported by other groups (14–16), suggesting that under certain conditions (e.g. p53 inactivation), BS cells can exhibit an NHEJ defect.
Finally, we demonstrated that the repair of DSBs by HR was five times higher in BS cells than in control cells. These results are consistent with the high rate of SCEs and the increase in interchange between homologous chromosomes at homologous sites in BS cells (1), and the 4- to 5-fold increase of spontaneous HR events induced by dominant-negative mutants of BLM (56). HR is an important process in restarting broken replication forks through the break-induced replication (BIR) mechanism (58). Richardson and Jasin (59) reported that following an I-SceI endonuclease-induced DSB, almost all recombination events were initiated by only one end of the DSB, suggesting that BIR plays an important role in DSB repair in mammalian cells. Furthermore, BIR has been proposed to be involved in mammalian telomere maintenance in the absence of telomerase (ALT cells) (60), and it has recently been shown that BLM promotes telomeric DNA synthesis in ALT cells, suggesting that it facilitates recombination-driven amplification of telomeres in these cells (61). Our data are consistent with a role for BLM in BIR, as recently proposed by other groups (62–64).
Several studies suggest that the NHEJ and HR pathways overlap, compete or act sequentially at sites of DSBs (8–13). Our results also suggest that NHEJ and HR compete for DSB sites, resulting in a simultaneous recruitment of the proteins specific for each pathway. We propose a model (Fig. 7) in which during S and G2 phases, BLM stays at the site of the DSB during the time interval required for NHEJ to take place. Once the NHEJ pathway is activated, BLM is phosphorylated by the DNA-PKcs, and this phosphorylation mediates its dissociation from DNA. If the NHEJ fails to repair the DSB, the recombination machinery will replace it, as proposed by Delacôte et al. (11). Then the BIR process takes place, creating a single Holliday junction that will be resolved by the reverse branch migration activity of BLM, probably in association with topoisomerase IIIα (65) and, in some cases, with p53 (56), leading to the restoration of intact molecules. During replication, when a fork encounters a nicked template and gives rise to a one-ended DSB and an intact chromosome, the one-ended DSB invades the intact sister chromatid leading to the formation of a single Holliday junction (9). BLM’s branch migration activity may resolve the Holliday junction, leading to the restoration of the replication fork. In the absence of a functional BLM protein, the Holliday junction will be resolved by either gene conversion (SCE) or by crossing-over between the homologous chromatids (symmetric quadriradial chromatid interchange). Alternatively, the Holliday junction will migrate to the end of the chromosome arm as a result of extensive DNA synthesis, leading to loss of a chromosom arm (loss of heterozygosity) (63). This model fully explains why SCE, symmetric quadriradial chromatid interchange and loss of heterozygosity are hallmarks of BS cells.
Figure 7.

Model for BLM’s role in HR. NHEJ and HR compete at sites of DSB, leading to the simultaneous recruitment of the proteins specific for each pathway, including BLM. Once NHEJ is activated, BLM phosphorylation mediates its dissociation from DNA (A). If the NHEJ fails (B), or during replication when nicks are converted into DSBs (C), break-induced replication (BIR) gives rise to recombination intermediates that are resolved by the reverse branch migration activity of BLM.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Dr Yegor Vassetzky for stimulating discussions and critical reading of the manuscript, and Dr Marc Lipinski for advice and helpful discussions. B.S. warmly acknowledges Cordula Kirchgessner (Stanford University School of Med-cine, CA) for the kind gift of Fus1 and Fus9 cell lines. The laboratory of M.A.-G. is supported by grants from the Centre National de la Recherche Scientifique, the Fondation de France, the Ligue Nationale contre le Cancer, the Fondation pour la Recherche Médicale and the Association pour la Recherche sur le Cancer (ARC 4722). The laboratory of B.S. is supported by the Association pour la Recherche sur le Cancer (ARECA program to B.S.) and a Radiobiology grant of Electricité de France (EDF) to P.C. The laboratory of D.P. is supported by the Association pour la Recherche sur le Cancer (ARC 4333), Conseil de Radioprotection (EDF) and the Commission of the Europeean Community (grant FIGH-CT 1999-00010).
REFERENCES
- 1.German J. (1993) Bloom syndrome: a mendelian prototype of somatic mutational disease. Medicine, 72, 393–406. [PubMed] [Google Scholar]
- 2.Ellis N.A., Groden,J., Ye,T.Z., Straughen,J., Lennon,D.J., Ciocci,S., Proytcheva,M. and German,J. (1995) The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell, 83, 8655–8666. [DOI] [PubMed] [Google Scholar]
- 3.Karow J.K., Chakraverty,R.K. and Hickson,I.D. (1997) The Bloom’s syndrome gene product is a 3′–5′ DNA helicase. J. Biol. Chem., 272, 30611–30614. [DOI] [PubMed] [Google Scholar]
- 4.Haber J.E. (2000) Partners and pathways repairing a double-strand break. Trends Genet., 16, 259–264. [DOI] [PubMed] [Google Scholar]
- 5.van Gent D.C., Hoeijmakers,J.H. and Kanaar,R. (2001) Chromosomal stability and the DNA double-stranded break connection. Nature Rev. Genet., 2, 196–206. [DOI] [PubMed] [Google Scholar]
- 6.Haber J.E. (1999) DNA repair. Gatekeepers of recombination. Nature, 398, 665–667. [DOI] [PubMed] [Google Scholar]
- 7.Takata M., Sasaki,M.S., Sonoda,E., Morrison,C., Hashimoto,M., Utsumi,H., Yamaguchi-Iwai,Y., Shinohara,A. and Takeda,S. (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J., 17, 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Dyck E., Stasiak,A.Z., Stasiak,A. and West,S.C. (1999) Binding of double-strand breaks in DNA by human Rad52 protein. Nature, 398, 728–731. [DOI] [PubMed] [Google Scholar]
- 9.Pierce A.J., Hu,P., Han,M., Ellis,N. and Jasin,M. (2001) Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev., 15, 3237–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saintigny Y., Delacôte,F., Vares,G., Petitot,F., Lambert,S., Averbeck,D. and Lopez,B.S. (2001) Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J., 20, 3861–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delacôte F., Han,M., Stamato,T.D., Jasin,M. and Lopez,B.S. (2002) An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res., 30, 3454–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukushima T., Takata,M., Morrison,C., Araki,R., Fujimori,A., Abe,M., Tatsumi,K., Jasin,M., Dhar,P.K., Sonoda,E., Chiba,T. and Takeda,S. (2001) Genetic analysis of the DNA-dependent protein kinase reveals an inhibitory role of Ku in late S–G2 phase DNA double-strand break repair. J. Biol. Chem., 276, 44413–44418. [DOI] [PubMed] [Google Scholar]
- 13.Allen C., Kurimasa,A., Brenneman,M.A., Chen,D.J. and Nickoloff,J.A. (2002) DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination. Proc. Natl Acad. Sci. USA, 99, 3758–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rünger T.M. and Kraemer,K.H. (1989) Joining of linear plasmid DNA is reduced and error-prone in Bloom’s syndrome cells. EMBO J., 8, 1419–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaymes T.J., North,P.S., Brady,N., Hickson,I.D., Mufti,G.J. and Rassool,F.V. (2002) Increased error-prone non homologous DNA end-joining—a proposed mechanism of chromosomal instability in Bloom’s syndrome. Oncogene, 21, 2525–2533. [DOI] [PubMed] [Google Scholar]
- 16.Langland G., Elliott,J., Li,Y., Creaney,J., Dixon,K. and Groden,J. (2002) The BLM helicase is necessary for normal DNA double-strand break repair. Cancer Res., 62, 2766–2770. [PubMed] [Google Scholar]
- 17.Dutertre S., Ababou,M., Onclercq,R., Delic,J., Chatton,B., Jaulin,C. and Amor-Gueret,M. (2000) Cell cycle regulation of the endogenous wild type Bloom’s syndrome DNA helicase. Oncogene, 19, 2731–2738. [DOI] [PubMed] [Google Scholar]
- 18.Kawabe T., Tsuyama,N., Kitao,S., Nishikawa,K., Shimamoto,A., Shiratori,M., Matsumoto,T., Anno,K., Sato,T., Mitsui,Y., Seki,M., Enomoto,T., Goto,M., Ellis,N.A., Ide,T., Furuichi,Y. and Sugimoto,M. (2000) Differential regulation of human RecQ family helicases in cell transformation and cell cycle. Oncogene, 19, 4764–4772. [DOI] [PubMed] [Google Scholar]
- 19.Thompson L.H. and Schild,D. (2002) Recombinational DNA repair and human disease. Mutat. Res., 509, 49–78. [DOI] [PubMed] [Google Scholar]
- 20.Sonoda E., Sasaki,M.S., Morrison,C., Yamaguchi-Iwai,Y., Takata,M. and Takeda,S. (1999) Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol. Cell. Biol., 19, 5166–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang W., Seki,M., Narita,Y., Sonoda,E., Takeda,S., Yamada,K., Masuko,T., Katada,T. and Enomoto,T. (2000) Possible association of BLM in decreasing DNA double strand breaks during DNA replication. EMBO J., 19, 3428–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karow J.K., Constantinou,A., Li,J.L., West,S.C. and Hickson,I.D. (2000) The Bloom’s syndrome gene product promotes branch migration of Holliday junctions. Proc. Natl Acad. Sci. USA, 97, 6504–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo G., Santoro,I.M., McDaniel,L.D., Nishijima,I., Mills,M., Youssoufian,H., Vogel,H., Schultz,R.A. and Bradley,A. (2000) Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nature Genet., 26, 424–429. [DOI] [PubMed] [Google Scholar]
- 24.Ababou M., Dutertre,S., Lecluse,Y., Onclercq,R., Chatton,B. and Amor-Gueret,M. (2000) ATM-dependent phosphorylation and accumulation of endogenous BLM protein in response to ionizing radiation. Oncogene, 19, 5955–5963. [DOI] [PubMed] [Google Scholar]
- 25.Dutertre S., Sekhri,R., Tintignac,L.A., Onclercq-Delic,R., Chatton,B., Jaulin,C. and Amor-Gueret,M. (2002) Dephosphorylation and subcellular compartment change of the mitotic Bloom’s syndrome DNA helicase in response to ionizing radiation. J. Biol. Chem., 277, 6280–6286. [DOI] [PubMed] [Google Scholar]
- 26.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res., 11, 1475–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoppe B.S., Jensen,R.B. and Kirchgessner,C.U. (2000) Complementation of the radiosensitive M059J cell line. Radiat. Res., 153, 125–130. [DOI] [PubMed] [Google Scholar]
- 28.Jessberger R. and Berg,P. (1991) Repair of deletions and double-strand gaps by homologous recombination in a mammalian in vitro system. Mol. Cell. Biol., 11, 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith J., Andrau,J.C., Kallenbach,S., Laquerbe,A., Doyen,N. and Papadopoulo,D. (1998) Abnormal rearrangements associated with V(D)J recombination in Fanconi anemia. J. Mol. Biol., 281, 815–825. [DOI] [PubMed] [Google Scholar]
- 30.Baldeyron C., Jacquemin,E., Smith,J., Jacquemont,C., De Oliveira,I., Gad,S., Feunteun,J., Stoppa-Lyonnet,D. and Papadopoulo,D. (2002) A single mutated BRCA1 allele leads to impaired fidelity of double strand break end-joining. Oncogene, 21, 1401–1410. [DOI] [PubMed] [Google Scholar]
- 31.Smith J., Riballo,E., Kysela,B., Baldeyron,C., Manolis,K., Masson,C., Lieber,M.R., Papadopoulo,D. and Jeggo,P. (2003) Impact of DNA ligase IV on the fidelity of end-joining in human cells. Nucleic Acids Res., 31, 2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minc E., de Coppet,P., Masson,P., Thiery,L., Dutertre,S., Amor-Gueret,M. and Jaulin,C. (1999) The human copper–zinc superoxide dismutase gene (SOD1) proximal promoter is regulated by Sp1, Egr-1 and WT1 via non-canonical binding sites. J. Biol. Chem., 274, 503–509. [DOI] [PubMed] [Google Scholar]
- 33.Birnboim H.C. and Doly,J. (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res., 7, 1513–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gottlieb T.M. and Jackson,S.P. (1993) The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell, 72, 131–142. [DOI] [PubMed] [Google Scholar]
- 35.Kusano K., Johnson-Schlitz,D.M. and Engels,W.R. (2001) Sterility of Drosophila with mutations in the Bloom syndrome gene—complementation by Ku70. Science, 291, 2600–2602. [DOI] [PubMed] [Google Scholar]
- 36.Suwa A., Hirakata,M., Takeda,Y., Jesch,S.A., Mimori,T. and Hardin,J.A. (1994) DNA-dependent protein kinase (Ku protein–p350 complex) assembles on double-stranded DNA. Proc. Natl Acad. Sci. USA, 91, 6904–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calsou P., Delteil,C., Frit,P., Drouet,J. and Salles,B. (2003) Coordinated assembly of Ku and p460 subunits of the DNA-dependent protein kinase on DNA ends is necessary for XRCC4-ligase IV recruitment. J. Mol. Biol., 326, 93–103. [DOI] [PubMed] [Google Scholar]
- 38.Chan D.W. and Lees-Miller,S.P. (1996) The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J. Biol. Chem., 271, 8936–8941. [DOI] [PubMed] [Google Scholar]
- 39.Calsou P., Frit,P., Humbert,O., Muller,C., Chen,D.J. and Salles,B. (1999) The DNA-dependent protein kinase catalytic activity regulates DNA end processing by means of Ku entry into DNA. J. Biol. Chem., 274, 7848–7856. [DOI] [PubMed] [Google Scholar]
- 40.Frit P., Li,R.Y., Arzel,D., Salles,B. and Calsou,P. (2000) Ku entry into DNA inhibits inward DNA transactions in vitro. J. Biol. Chem., 275, 35684–35691. [DOI] [PubMed] [Google Scholar]
- 41.Chan D.W., Chen,B.P., Prithivirajsingh,S., Kurimasa,A., Story,M.D., Qin,J. and Chen,D.J. (2002) Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev., 6, 2333–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merkle D., Douglas,P., Moorhead,G.B., Leonenko,Z., Yu,Y., Cramb,D., Bazett-Jones,D.P. and Lees-Miller,S.P. (2002) The DNA-dependent protein kinase interacts with DNA to form a protein–DNA complex that is disrupted by phosphorylation. Biochemistry, 41, 12706–12714. [DOI] [PubMed] [Google Scholar]
- 43.Escarceller M., Buchwald,M., Singleton,B.K., Jeggo,P.A., Jackson,S.P., Moustacchi,E. and Papadopoulo D. (1998) Fanconi anemia C gene product plays a role in the fidelity of blunt DNA end-joining. J. Mol. Biol., 279, 375–385. [DOI] [PubMed] [Google Scholar]
- 44.Mohaghegh P., Karow,J.K., Brosh Jr,R.M., Bohr,V.A. and Hickson,I.D. (2001) The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res., 29, 2843–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X.W., Tseng,A., Ellis,N.A., Spillare,E.A., Linke,S.P., Robles,A.I., Seker,H., Yang,Q., Hu,P., Beresten,S., Bemmels,N.A., Garfield,S. and Harris,C.C. (2001) Functional interaction of p53 and BLM DNA helicase in apoptosis. J. Biol. Chem., 276, 32948–32955. [DOI] [PubMed] [Google Scholar]
- 46.Hsieh C.L., Arlett,C.F. and Lieber,M.R. (1993) V(D)J recombination in ataxia telangiectasia, Bloom’s syndrome and a DNA ligase I-associated immunodeficiency disorder. J. Biol. Chem., 268, 20105–20109. [PubMed] [Google Scholar]
- 47.Petrini J.H., Donovan,J.W., Dimare,C. and Weaver,D.T. (1994) Normal V(D)J coding junction formation in DNA ligase I deficiency syndromes. J. Immunol., 152, 176–183. [PubMed] [Google Scholar]
- 48.Sack S.Z., Liu,Y., German,J. and Green,N.S. (1998) Somatic hypermutation of immunoglobulin genes is independent of the Bloom’s syndrome DNA helicase. Clin. Exp. Immunol., 112, 248–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaneko H., Matsui,E., Fukao,T., Kasahara,K., Morimoto,W. and Kondo,N. (1999) Expression of the BLM gene in human haematopoietic cells. Clin. Exp. Immunol., 118, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chappell C., Hanakahi,L.A., Karimi-Busheri,F., Weinfeld,M. and West,S.C. (2002) Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J., 21, 2827–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bertrand P., Rouillard,D., Boulet,A., Levalois,C., Soussi,T. and Lopez,B.S. (1997) Increase of spontaneous intrachromosomal homologous recombination in mammalian cells expressing a mutant p53 protein. Oncogene, 14, 1117–1122. [DOI] [PubMed] [Google Scholar]
- 52.Mekeel K.L., Tang,W., Kachnic,L.A., Luo,C.M., DeFrank,J.S. and Powell,S.N. (1997) Inactivation of p53 results in high rates of homologous recombination. Oncogene, 14, 1847–1857. [DOI] [PubMed] [Google Scholar]
- 53.Buchhop S., Gibson,M.K., Wang,X.W., Wagner,P., Sturzbecher,H.W. and Harris,C.C. (1997) Interaction of p53 with the human Rad51 protein. Nucleic Acids Res., 25, 3868–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garkavtsev I.V., Kley,N., Grigorian,I.A. and Gudkov A.V. (2001) The Bloom syndrome protein interacts and cooperates with p53 in regulation of transcription and cell growth control. Oncogene, 20, 8276–8280. [DOI] [PubMed] [Google Scholar]
- 55.Yang Q., Zhang,R., Wang,X.W., Spillare,E.A., Linke,S.P., Subramanian,D., Griffith,J.D., Li,J.L., Hickson,I.D., Shen,J.C., Loeb,L.A., Mazur,S.J., Appella,E., Brosh Jr.,R.M., Karmakar,P., Bohr,V.A. and Harris,C.C. (2002) The processing of Holliday junctions by BLM and WRN helicases is regulated by p53. J. Biol. Chem., 277, 31980–31987. [DOI] [PubMed] [Google Scholar]
- 56.Sengupta S., Linke,S.P., Pedeux,R., Yang,Q., Farnsworth,J., Garfield,S.H., Valerie,K., Shay,J.W., Ellis,N.A., Wasylyk,B. and Harris,C.C. (2003) BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. EMBO J., 22, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu X. and Lane,D.P. (1993) Differential induction of transcriptionally active p53 following UV or ionizing radiation: defects in chromosome instability syndromes? Cell, 75, 765–778. [DOI] [PubMed] [Google Scholar]
- 58.Michel B. (2000) Replication fork arrest and DNA recombination. Trends Biochem. Sci., 4, 173–178. [DOI] [PubMed] [Google Scholar]
- 59.Richardson C. and Jasin,M. (2000) Coupled homologous and nonhomologous repair of a double-strand break preserves genomic integrity in mammalian cells. Mol. Cell. Biol., 20, 9068–9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kraus E., Leung,W.Y. and Haber,J.E. (2001) Break-induced replication: a review and an example in budding yeast. Proc. Natl Acad. Sci. USA, 98, 8255–8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stavropoulos D.J., Bradshaw,P.S., Li,X., Pasic,I., Truong,K., Ikura,M., Ungrin,M. and Meyn M.S. (2002) The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum. Mol. Genet., 11, 3135–44. [DOI] [PubMed] [Google Scholar]
- 62.Nakayama H. (2002) RecQ family helicases: roles as tumor suppressor proteins. Oncogene, 21, 9008–9021. [DOI] [PubMed] [Google Scholar]
- 63.Stark J.M. and Jasin M. (2003) Extensive loss of heterozygosity is suppressed during homologous repair of chromosomal breaks. Mol. Cell. Biol., 23, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hickson I.D. (2003) RecQ helicases: caretakers of the genome. Nature Rev. Cancer, 3, 169–178. [DOI] [PubMed] [Google Scholar]
- 65.Wu L. and Hickson,I.D. (2002) The Bloom’s syndrome helicase stimulates the activity of human topoisomerase IIIα. Nucleic Acids Res., 30, 4823–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.