

Immobilization of biomolecules onto surfaces is important in many of the biological and physical sciences, including cell and molecular biology, analytical chemistry, and in applied and interdisciplinary fields such as medical diagnostics, tissue engineering, and bioprocess engineering.[1-4] Strategies for biomolecule immobilization onto surfaces generally exploit either noncovalent or covalent reactions. Noncovalent methods allow reversible immobilization of biomolecules under specific conditions, and include physical adsorption and affinity immobilization. Some widely adapted examples are (strep)avidin-biotin, nitriloacetic acid (NTA)-histidine, and DNA-DNA interactions.[5-8] In contrast, covalent immobilization of molecules onto surfaces typically relies on conjugation reactions between ‘active’ functional groups, such as N-hydroxysuccinimide (NHS)[9] or maleimide,[10] and companion target moieties, such as amines and sulfhydryls. For reactions involving biomolecules performed in aqueous solvents, susceptibility of NHS, maleimide, and other activating groups to hydrolysis during storage and reaction can lead to low efficiency of surface bioconjugation.[11,12] In this study, we report a facile two-step aqueous approach to immobilization of biomolecules onto surfaces. The approach involves simple dip-coating of a biomimetic polymer thin film onto a substrate, followed by conjugation of biomolecules to the biomimetic polymer film. The method exploits the latent reactivity of the biomimetic polymer thin film towards nucleophiles, is unaffected by water, and allows for discrimination between nucleophiles on the basis of pKa.
Substrates were first coated with a thin adherent polydopamine film by immersion in an alkaline dopamine · HCl solution (2 mg of dopamine · HCl dissolved in 10 mM Tris buffer, pH 8.5) for up to 12-18 h.[13] Dopamine, a biomolecule that contains catechol and amine functional groups, found also in high concentration in mussel adhesive proteins,[14,15] polymerizes at alkaline pHs to form thin adherent polydopamine films that exhibit latent reactivity toward amine and thiol groups.[13,16] In the second step of the approach, the reactivity of the polydopamine films is exploited to covalently immobilize biomolecules onto surfaces through a reaction between nucleophiles and the polydopamine surface (Scheme 1).
Scheme 1.
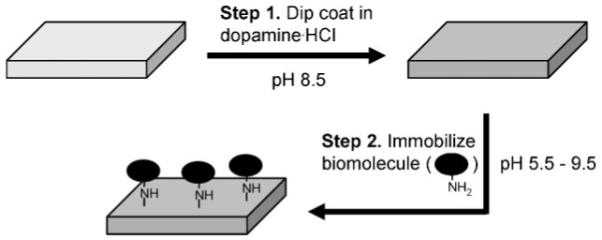
Schematic description of polydopamine coating and subsequent biomolecule immobilization through reaction between amines and polydopamine-coated substrates. The approach works also for imidazoles, such as those found in side chains of native histidine residues in proteins or in histidine tags of engineered proteins.
As an example of this strategy, we demonstrated the immobilization of the protein enzyme trypsin onto a variety of substrates. To illustrate the substrate versatility, several organic and inorganic substrates were first modified by polydopamine: copper, titanium oxide (TiOx), polycarbonate (PC), and cellulose. X-ray photoelectron spectroscopic (XPS) analysis of the polydopamine-modified substrates yielded virtually indistinguishable XPS spectra, with nitrogen-to-carbon (N/C) ratios of 0.118-0.128, similar to the theoretical N/C of 0.125 for dopamine (Fig. 1 and Table 1). Furthermore, complete suppression of photoelectron peaks unique to Cu (932 eV for Cu2p3/2 and 953 eV for Cu2p½) and Ti (458 eV for Ti2p3/2 and 464eV for Ti2p½), along with emergence of a nitrogen peak (N1s, 399.5 eV) on organic substrates (PC and cellulose) that do not contain nitrogen, confirm formation of the polydopamine thin film.
Figure 1.

Facile surface immobilization of trypsin on polydopamine-coated substrates. A) XPS spectra of (top to bottom) unmodified copper, TiOx, polycarbonate, and cellulose. B) Representative XPS spectrum of polydopamine-coated substrate. The spectrum shown is for polydopamine on TiOx; XPS spectra of polydopamine on other substrates were found to be very similar. C) XPS spectrum of trypsin-immobilized polydopamine-coated TiOx. D) Enzymatic activity of immobilized trypsin on cellulose at 45 °C (circle) and 25 °C (triangle). The squares show the activity of unmodified cellulose. The results were averaged by activity assays from two independent surface preparations.
Table 1.
Surface chemical composition of polydopamine-coated TiOx before and after trypsin immobilization. The data was obtained from peak intensities of XPS spectra shown in Figure 1B and C
| Surface composition | C1s(%) | N1s(%) | O1s(%) |
|---|---|---|---|
| Polydopamine/TiOx | 72.7 | 8.8 | 18.5 |
| Trypsin/Polydopamine/TiOx | 66.7 | 13.7 | 19.6 |
Polymerization of dopamine occurs in a manner that is reminiscent of melanin formation, involving oxidation of catechol to quinone, which further reacts with amines and other catechols/quinones to form an adherent polymer film.[13] The deposited polydopamine coating is chemically heterogeneous, and its chemical composition is not precisely known. Nevertheless, catechol and quinone functional groups are believed present in the polydopamine coating, the latter of which are capable of covalent coupling to nucleophiles. For example, conjugation of trypsin to polydopamine-coated substrates was afforded through simple immersion in a trypsin solution for several hours at pH 7.8.
Trypsin immobilization on polydopamine altered the surface chemical composition as measured by increases in the peaks corresponding to nitrogen and oxygen (531.5 eV) and decreases in the carbon peak (285 eV, Fig. 1C and Table 1).
Trypsin conjugation on polydopamine-modified cellulose paper was easily achieved, as shown in Figure 1D and S1. The surface-immobilized trypsin remained enzymatically active, as evidenced by its ability to convert the substrate N-α-benzoyl-DL-arginine p-nitroanilide (BAPNA) into a chromophoric product easily monitored by absorbance at 420 nm (Fig. 1D). The enzymatic activity of immobilized trypsin exhibited the expected temperature dependence of activity, as shown by an increase in rate of product formation when the temperature was increased from 25 to 45 °C. Although cellulose and other natural fibrous materials are inexpensive and abundant, bioconjugation to such materials has proven challenging, often requiring specific functional groups, present on the native fiber surface or introduced through chemical or physical pre-treatment of the fiber surface.[17] This result therefore illustrates one of the advantages of polydopamine-assisted protein conjugation - it’s the possibility of application on many types of materials without surface preparation.
The latent reactivity of polydopamine coatings toward nucleophiles is a function of both catechol/quinone chemical equilibrium and of the pKa of the nucleophile, and is largely resistant to the hydrolytic effects of water, which are common-place in other bioconjugation strategies such as the NHS-ester system. A previous study involving a surface-tethered catechol revealed that the catechol and quinone states are in equilibrium in aqueous media, with the equilibrium shifted toward the quinone at alkaline pH (Fig. 2A).[16] Thus, one should expect that bioconjugation reactions on polydopamine-coated surfaces be relatively enhanced at alkaline pH. This behavior is in stark contrast with NHS esters, which hydrolyze at increasing rates under alkaline conditions (Fig. 2B).
Figure 2.
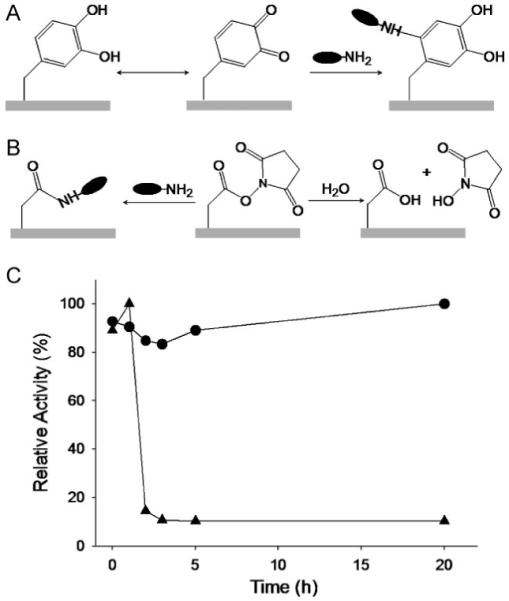
Comparison of chemical reactivity and hydrolysis of polydopamine and NHS surfaces. A) Aqueous chemical equilibrium between catechol (left) and quinone (middle). The equilibrium shifts toward quinone under alkaline conditions, conferring latent reactivity toward amine groups of proteins (right). B) Schematic description of amine coupling (left) and hydrolysis (right) reactions of NHS esters. Consumption of the activated ester by hydrolysis is enhanced at alkaline pHs, and decreases availability for amine coupling. C) Comparison of chemical reactivity of polydopamine (circles) and NHS ester (triangles) after preincubation for up to 20 h in 50 mM Na-phosphate, 100 mM NaCl, pH 7.5. The rapid drop in NHS activity is due to hydrolysis of the NHS ester.
To illustrate the chemical reactivity and stability to hydrolysis, polydopamine-coated silicon substrates were preincubated in sodium phosphate buffer (pH 7.5) for up to 20 h, and were subsequently transferred to trypsin solution. XPS analysis of chemical changes associated with trypsin conjugation to the surface indicated that the reactivity of polydopamine toward trypsin bioconjugation did not diminish during the period of time investigated, and even increased slightly after 5 h of preincubation in alkaline buffer (circles, Fig. 2C). In contrast, XPS analysis of NHS-functionalized glass under the same conditions revealed significant NHS hydrolysis after only 2 h preincubation (triangles, Fig. 2C), as indicated by a decrease in nitrogen-to-silicon ratio, the appearance of glass-substrate signals (102.5 eV for Si2p and 153.5 eV for Si2s), and reduction of nitrogen signal (Fig. S2). Furthermore, atomic force microscopy images clearly confirmed a decrease in trypsin immobilization after preincubation of NHS-functionalized glass surfaces (Fig. S3).
Finally, we demonstrated that bioconjugation reactions on polydopamine surfaces can be modulated by pH, affording discrimination among nucleophiles. First we synthesized the model peptide N-acetyl-Histidine-(ethylene glycol)3-Lysine-CO2H (His-EG3-Lys) by solid-phase peptide synthesis; the peptide was purified by high-performance liquid chromatography (HPLC) and confirmed by matrix-assisted laser desorption and ionization time-of-flight mass spectrometry (MALDI-TOF MS). The compound contained two different nucleophiles, lysine and histidine, at opposite ends of the molecule, allowing in principle coupling to polydopamine through either residue. The orientation of immobilized His-EG3-Lys was detected via a peroxidaze/streptavidin conjugate, as follows. In the case of His-EG3-Lys-conjugation to polydopamine through the histidine residue, the free (unbound) lysine side chain allows for subsequent binding of peroxidase-streptavidin molecules via biotinylation of the lysine side-chain amine using a NHS-biotin intermediate (Fig. 3B). On the other hand, conjugation of His-EG3-Lys to polydopamine via the lysine side-chain amine should reduce biotinylation of the surface due to the low reactivity of histidine with NHS-biotin, suppressing the subsequent streptavidin/enzyme reaction.
Figure 3.
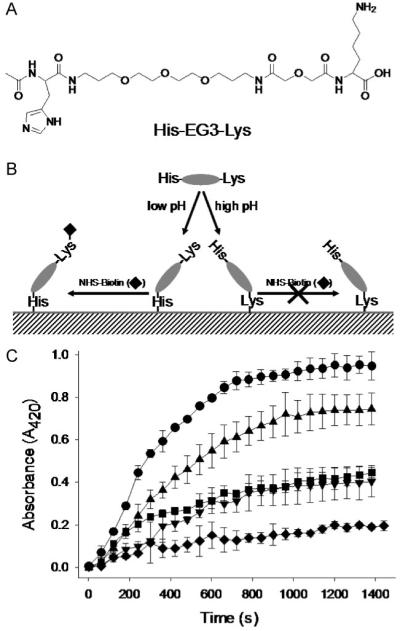
Amino-acid-selective immobilization of biomolecules on polydopamine surfaces through pH control. A) Molecular structure of model peptide His-EG3-Lys. B) Schematic illustration of pH-based control of His-EG3-Lys conjugation to polydopamine. Under acidic conditions, the imidazole side chain of His preferentially reacts with polydopamine, whereas under alkaline pH reaction to amine side chains of Lys is favored. As a result, biotinylation through reaction between free Lys and NHS-biotin, and the subsequent streptavidin-horseradish peroxidase linking, occurs pre-dominantly at acidic and neutral pH. C) Peroxidase activity traces as a function of pH (from top to bottom: 7.4 (circle), 6.4 (triangle), 8.4 (square), 5.5 (inverse triangle), and 9.5 (diamond)). Results shown are averages of two independent experiments.
As shown in Figure 3C, the immobilization of His-EG3-Lys occurred in a pH-dependent manner, due to the large difference in pKa values of histidine (pKa ∼6) and ε-amines (pKa∼10). At low pH (5.5), peroxidaze activity was low, due to the low reactivity of protonated His and Lys residues toward polydopamine below the pKa. At neutral or slightly acidic pH, activity increased significantly, with maximum peroxidase activity being observed at pH 7.4, indicating preferred coupling of His-EG3-Lys via imidazole-polydopamine reaction. Similar coupling reactions between histidine (imidazole) and quinones have been found in biological tissues such as in insect cuticles.[18,19] Peroxidase activity decreased again at high pH (pH 8.4 and 9.5), likely due to an increased preference for coupling of His-EG3-Lys via amine-polydopamine reaction as the pKa of Lys is approached. The resulting free His residue is poorly reactive with NHS under aqueous conditions, thus reducing the biotinylation and subsequent peroxidase immobilization. Furthermore, the decreasing trend in peroxidase activity as the immobilization pH increases implies that the primary amine (Lys) is more reactive than the secondary amine (His) when both species are deprotonated. Given the complexities of the polydopamine surface, and its own pH-dependent behavior, a complete understanding of the reactivity of even model biomolecules, such as His-EG3-Lys, toward polydopamine will require further study.
In summary, we have shown that bioconjugation to polydopamine is easily adapted for a variety of materials without surface pretreatment, is unaffected by incubation in water, and through pH control it offers selectivity of reaction with amine or imidazole functional groups of biomolecules. The method may prove useful for surface immobilization of a variety of biomolecules, such as proteins and DNA. Control of orientation of immobilized biomolecules may even be possible through the use of terminally modified DNA or His containing proteins,[20] including both native His residues as well as those in His tags of engineered proteins.
Experimental
Polydopamine Coating and Trypsin Immobilization
Substrates were immersed in 2 mg of dopamine.HCl per 1 mL of 10 mM Tris buffer (pH 8.5) typically overnight (12-18 h) and rinsed with water for further use. The polydopamine-coated substrates were transferred into trypsin solution (50 mM sodium phosphate buffer, pH 7.8) for overnight reaction (12-18 h). Hydrolysis tests of polydopamine-coated silicon and NHS-modified glass (μsurfaces Inc. Minneapolis, MN) were performed in 50 mM sodium phosphate buffer (pH 7.5) with 100 mM NaCl.
Surface Characterization
Surface topography was measured in air using an MFP-3D atomic force microscope (Asylum Research, San Diego, CA) operated in AC and contact modes. X-ray photoelectron spectroscopy (Omicron ESCALAB, Omicron, Taunusstein, Germany) was performed using a monochromated Al Kα (1486.8 eV) 300 W X-ray source with an ultrahigh vacuum (<10 -8 Torr). The takeoff angle was fixed at 45°, and all spectra were calibrated using the hydrocarbon C 1s peak (284.5 eV).
Synthesis of His-EG3-Lys
N-acetyl-His-EG3-Lys-CO2H was synthesized by traditional Fmoc solid-state peptide synthesis strategy. Briefly, Fmoc-Lys-Wang resin (EMD Chemicals, Germany) was activated by deprotection of Fmoc groups and then oligo-ethylene(glycol) and His(Trt) were sequentially reacted. The N-terminal amine was acetylated using acetic anhydride. Deprotection and cleavage from resin was performed by adding a volume ratio of 95/2.5/2.5 of trifluoroacetic acid(TFA)/tri-isopropylsilane(TIS)/water for 3 h. The synthesized product was purified using HPLC (water-acetonitrile gradient, C18 column), and the molecular mass was confirmed by MALDI-TOF MS ([M + H]+=645.1, theoretical mass=643.2 Da).
XPS Analysis of Hydrolysis and Trypsin Immobilization on NHS and Polydopamine Surfaces
XPS C1s spectra of polypeptides typically show a strong carbonyl-carbon (C=O) peak at 286.5 eV. However, C=O was found to be negligible in polydopamine alone. Thus, C=O intensity with respect to a total carbon signal was used to follow the extent of trypsinization on the surface (Fig. S4). The ratio of carbonyl to total carbon for polydopamine was found to be relatively invariant with time of incubation in water (0.22 (0 h), 0.21 (1 h), 0.20 (2 h), 0.20 (3 h), 0.21 (5 h), and 0.24 (20 h)). The polydopamine data shown in Figure 2C (circles) were determined by normalization by the largest value (0.24, 20 h hydrolysis). For NHS-glass surfaces, the relative reactivities plotted in Figure 2C (triangles) were determined as the ratio of nitrogen to silicon in the XPS spectra of surfaces incubated under identical conditions.
Histidine-Selective Immobilization of His-EG3-Lys on Polydopamine-Coated Surfaces
Polydopamine-coated silicon substrates were immersed in 10 mM acetate/phosphate/Tris co-buffers (pH 5.5, 6.4, 7.4, 8.4, and 9.5) with 0.1 mM of His-EG3-Lys for 5 h. Subsequently, 10 mM NHS-Biotin (PIERCE, Rockford, IL) was reacted with the surface-immobilized His-EG3-Lys in 10 mM sodium phosphate buffer (pH 7.8) for 4 h. The substrates were transferred into 10 mM sodium phosphate buffer (pH 7.0), 100 mM NaCl with 1 μg mL-1 streptavidin-horseradish peroxidase for biotin-streptavidin linkage (1 h). Surface-immobilized horseradish peroxidase activity was monitored at absorbance 420 nm using a UV-vis spectrophotometer (HP-8453, Agilent). The substrate was placed inside a UV-vis cuvette, and then solution was added: 2 mL of sodium phosphate buffer (100 mM, pH 6.0), 0.3 mL of pyrogallol solution (10 mg mL-1 dissolved in the same phosphate buffer), and 0.2 mL of 0.4% (v/v) hydrogen peroxide.
Supplementary Material
Acknowledgements
This work is supported by grant DE14192 from NIH. Portions of this work were performed in the Keck-II facility of NUANCE at Northwestern University, which is supported by NSF-NSEC, NSF-MRSEC, the Keck foundation, the state of Illinois, and Northwestern University. Supporting Information is available online from Wiley InterScience or from the authors.
References
- [1].MacBeath G. Nat. Biotechnol. 2001;19:828. doi: 10.1038/nbt0901-828. [DOI] [PubMed] [Google Scholar]
- [2].Hodneland CD, Lee Y-S, Min D-H, Mrksich M. Proc. Natl. Acad. Sci. U.S.A. 2002;99:5048. doi: 10.1073/pnas.072685299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hodneland CD, Lee Y-S, Min D-H, Mrksich M. Adv. Drug Delivery Rev. 2007;59:360. [Google Scholar]
- [4].van Beilen JB, Li Z. Curr. Opin. Biotechnol. 2002;13:338. doi: 10.1016/s0958-1669(02)00334-8. [DOI] [PubMed] [Google Scholar]
- [5].Luo S, Walt DR. Anal. Chem. 1989;61:174. [Google Scholar]
- [6].Schimid EL, Keller TA, Dienes Z, Vogel H. Anal. Chem. 1997;69:1979. doi: 10.1021/ac9700033. [DOI] [PubMed] [Google Scholar]
- [7].Kozlov IA, Melnyk PC, Stromsborg KE, Chee MS, Marker DL, Zhao C. Biopolymers. 2004;73:1024. doi: 10.1002/bip.20009. [DOI] [PubMed] [Google Scholar]
- [8].Glynou K, Ioannou PC, Christopoulos TK. Bioconjugate Chem. 2003;14:1024. doi: 10.1021/bc0341021. [DOI] [PubMed] [Google Scholar]
- [9].Yan M, Cai SX, Wybourne MN, Keana JFW. J. Am. Chem. Soc. 1993;115:814. [Google Scholar]
- [10].Viitala T, Vikholm I, Peltonen J. Langmuir. 2000;16:4952. [Google Scholar]
- [11].Gong P, Grainger DW. Surf. Sci. 2004;570:67. [Google Scholar]
- [12].Su X, Zong Y, Richter R, Knoll W. J. Colloid Interface Sci. 2005;287:35. doi: 10.1016/j.jcis.2005.01.089. [DOI] [PubMed] [Google Scholar]
- [13].Lee H, Dellatore SM, Miller WM, Messersmith PB. Science. 2007;318:426. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Papov VV, Diamond TV, Biemann K, Waite JH. J. Biol. Chem. 1995;270:20183. doi: 10.1074/jbc.270.34.20183. [DOI] [PubMed] [Google Scholar]
- [15].Waite JH, Qin XX. Biochemistry. 2001;40:2887. doi: 10.1021/bi002718x. [DOI] [PubMed] [Google Scholar]
- [16].Lee H, Scherer NF, Messersmith PB. Proc. Natl. Acad. Sci. U.S.A. 2006;103:12999. doi: 10.1073/pnas.0605552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen H, Hsieh Y-L. Biotechnol. Bioeng. 2005;90:405. doi: 10.1002/bit.20324. [DOI] [PubMed] [Google Scholar]
- [18].Merritt ME, Christensen AM, Kramer KJ, Hopkins TL, Schaefer J. J. Am. Chem. Soc. 1996;118:11278. [Google Scholar]
- [19].Hopkins TL, Kramer KJ. Annu. Rev. Entomol. 1992;37:273. [Google Scholar]
- [20].Becker CFW, Hunter CL, Seidel RP, Kent SBH, Goody RS, Engelhard M. Chem. Biol. 2001;8:243. doi: 10.1016/s1074-5521(01)00003-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.