


Abstract
There is little information on chromatin structure that allows access of trans-acting transcription factors. Logically, the target DNA elements become accessible by either exposing themselves towards the environment on the surface of the nucleosome, or making the regulatory region free of the nucleosome. Here, we demonstrate that curved DNA that mimics a negative supercoil can play both roles in the promoter region. By constructing 35 reporter plasmids and using in vivo assay systems, we scrutinized the relationships between upstream DNA geometry, nucleosome positioning and promoter activity. When the left-handedly curved DNA was linked to the herpes simplex virus thymidine kinase (HSV tk) promoter at a specific rotational phase and distance, the curved DNA attracted the nucleosome and the TATA box was thereby left in the linker DNA with its minor groove facing outwards, which led to the activation of transcription. Neither planar curving, nor right-handedly curved DNA nor straight DNA had this effect. Our results seem to provide a clue for solving the problem of why curved DNA is often located near transcriptional control regions.
INTRODUCTION
DNA having a curved trajectory of the helix-axis is called bent or curved DNA. It has been found in a wide variety of genomes, often in or around the origins of DNA replication, transcriptional control regions, DNA recombination loci, matrix attachment regions and satellite DNA repeating units (reviewed in 1–5). Thus, it has long been suspected that DNA curvature is of functional importance in a wide variety of biological processes. Many studies have been carried out, and the function of curved DNA in prokaryotic transcription and the mechanism by which it functions have been gradually revealed (1,5 and references therein). Interestingly, the three-dimensional architecture of curved DNA appears to be important. In Escherichia coli promoters, curved DNA with a right-handed superhelical writhe stimulates not only RNA polymerase binding to the promoter, but also the promoter complex’s transition from closed to open. Curved DNA with a left-handed superhelical writhe negatively influences these processes (4,6).
Despite many reports showing the presence of curved DNA structure in transcriptional control regions of eukaryotic genes (7–12), the function of curved DNA in eukaryotic transcription is still obscure. Computational analysis suggests that intrinsic DNA curvature may be one important criterion for the recognition of the TATA box by the TATA-binding protein (TBP) (11). However, in several TATA-box-containing promoters, the curved DNA is upstream of the TATA box. For example, the promoters of the human β-actin gene (13), Xenopus A2 vitellogenin gene (14), scorpion AaH I′ toxin gene (15), pea rbcS-3A and rbcS-3.6 (16) and yeast GAL1 (17) are in this category. Furthermore, many promoters other than RNA polymerase II (Pol II) promoters and TATA-less Pol II promoters also frequently contain a curved DNA structure (8,10,18). This indicates that curved DNA may play a role common to many promoters, irrespective of the promoter type. Experimental attempts have also been made to clarify the function of curved DNA. Kim et al. (19) showed that an A6CGTG repeat could activate eukaryotic transcription. Subsequently, the evidence showing that DNA conformation per se affects transcription was obtained in 1996 (20). The mechanism by which curved DNA can activate transcription in the nucleus, however, remains unclear. Considering that (i) in the nucleus, regulatory regions of transcription must also be packaged into chromatin keeping definite cis-DNA elements accessible and (ii) curved DNA structure has been suggested to influence nucleosome positioning (12,17,21), it is hypothesized that the significant function of curved DNA is to organize local chromatin infrastructure in order to help the very first interaction between cis-acting DNA elements and trans-acting transcription factors (1).
In order to substantiate the hypothesis, we synthesized various curved DNA structures, experimentally assessing the relationship between upstream DNA geometry, transcription efficiency and local chromatin structure. We now report that a curved DNA with a close resemblance to a part of a negative supercoil attracts a histone core. We show that this curved DNA can activate eukaryotic promoters when it is introduced into the upstream region of the promoter with appropriate combination of distance and spatial positioning. We discuss the mechanistic role of the curved DNA in chromatin organization and transcription.
MATERIALS AND METHODS
Plasmid constructions
The constructs pST0/TLN, pST4/TLN, pLHC4/TLN, pTDC4/TLN and pRHC4/TLN (basic constructs) were prepared as follows. The multiple cloning site (mcs) of the pUC19 was replaced with that of Bluescript II SK(–) (Stratagene) by exchanging the PvuII–PvuII region of each plasmid to produce pUBlmcs. Then, the EcoRI–PstI fragment of the tk promoter was inserted between corresponding sites of pUBlmcs to generate pUBlparttk. Subsequently, a synthetic double-stranded oligonucleotide, obtained by annealing oligos 5′-GACGTCATATGGGCCCTCGAGGGCCCTCGC GACACAAACCCCGCCCAGCGTCTTGTCATTGGCG-3′ and 5′-AATTCGCCAATGACAAGACGCTGGGCGGGG TTTGTGTCGCGAGGGCCCTCGAGGGCCCATATGAC GTC-3′, was inserted between EcoRI and HincII sites of pUBlparttk to generate pUBlcomptk. Then, the synthetic double-stranded oligonucleotides, each with unique DNA configuration (Fig. 1), were cloned between KpnI and HincII sites of pUBlcomptk. Finally, the KpnI–BamHI fragment containing the tk promoter and the synthetic DNA segment was cloned into pGL2 Basic vector (Promega). By manipulating the structure of the mcs of the basic constructs, the variant plasmids were constructed. All constructs were sequenced for verification.
Figure 1.


The reporter plasmids used in this study. (A) Structure of the plasmids. The symbols ‘mcs’ and ‘luc’ indicate the multiple cloning site and the luciferase gene, respectively. Positions of the (T/A)5 tracts are indicated with bold bars. The deleted nucleotides in the mcs are indicated by lines. The nucleotides inserted into the mcs are shaded. The centers of curved DNA and the TATA box are marked with asterisks. (B) Three-dimensional architecture of the curved and straight DNA segments in the plasmids. They were drawn by a combination of softwares DIAMOD (50) and RASMOL (51). The modeling algorithm was based on that of Bolshoy et al. (52) with the twist angles from Kabsch et al. (53). For left-handedly and right-handedly curved DNA structures, the superhelical axis is drawn.
Luciferase assay
Simian COS-7 cells were grown in Eagle’s minimal essential medium containing 5% fetal bovine serum at 37°C in 5% CO2. Each construct was introduced into the cells by electroporation at 300 V/300 µF, in cuvettes containing 1.25 × 106 cells and 5 µg of construct in a 260 µl volume. After electroporation, the cells were cultured for 21 h. The cells were then washed and lysed with lysis buffer from the Promega luciferase assay system and luciferase activity was measured according to the manufacturer’s instructions.
Nuclei preparation
The introduction of plasmid DNAs into COS-7 cells and culture of the cells were carried out as for the luciferase assay. Nuclei were isolated as described by Masumoto et al. (22) with a modified isolation buffer. Our buffer contained, unless otherwise indicated, 0.5% (v/v) NP-40 (Sigma) instead of 0.1% digitonin and the buffer was supplemented with 0.1 µg/ml pepstatin, 0.2 µg/ml leupeptin and 1 mM NaHCO3. Nuclei were finally suspended in the washing buffer (22) supplemented with 1 mM NaHCO3 (for hydroxyl radical footprinting), with 1 mM NaHCO3 and 3 mM CaCl2 (for MNase digestion), or with 1 mM NaHCO3 and 5 mM MgCl2 (for DNase I digestion).
Digestion of chromatin with MNase
Aliquots of 100 µl (6 × 106 nuclei/ml) were prepared from the pLHC4/TLN-6-harboring nuclei suspension. They were pre-incubated at 37°C for 1 min. MNase (Worthington Biochemical Corporation) was added at 0.1 or 1.0 U/ml for indirect end-labeling analysis, and at 2.4, 3.2 or 6.4 U/ml for the analysis of nucleosomal arrays. The digestion was done at 37°C for 2 min. For indirect end-labeling analysis, using 0.008 or 0.02 U of MNase, naked DNA (supercoiled plasmid DNA) was digested in the same way. The reaction was stopped by the addition of 3 µl of 0.1 M EGTA and placing on ice. Subsequently, 200 µl of the washing buffer without PMSF and DTT, 6 µl of 20 mg/ml Proteinase K (TaKaRa) and 6 µl of a solution containing 5% (w/v) SDS and 60 mM EDTA were added to each sample. The resulting mixture was incubated at 37°C for 30 min. DNAs in the mixture were extracted three times with phenol, once with phenol/chloroform (1/1, v/v) and once with chloroform/isoamyl alcohol (24/1, v/v), precipitated, dried and resuspended in 4 or 8 µl of TE (10 mM Tris–HCl, pH 7.5; 1 mM EDTA, pH 8.0). In the detection of nucleosomal arrays, the untreated aliquot was used as an undigested control sample. We added 3 µl of 0.1 M EGTA to it instead of adding MNase. The subsequent steps were carried out as described above.
Digestion of chromatin with DNase I
Digestions of the chromatin DNA with DNase I were performed in parallel with the digestions of protein-free DNA controls (supercoiled plasmid DNA). Four aliquots of 100 µl (3 × 106 nuclei/ml) were prepared from the nuclei suspension. Three were pre-incubated at 37°C for 1 min and subsequently 3.5 U of DNase I was added to each aliquot. Then, one aliquot was incubated for 2 min, another for 8 min, and the third for 32 min. Using 0.35 U of DNase I, naked DNA was digested in the same way. The reaction was stopped by the addition of 3 µl of 0.5 M EDTA and placing on ice. The subsequent steps were performed using the same procedure as described in the above section. The recovered sample was resuspended in 5 µl of water. The fourth aliquot, to which we added 3 µl of 0.5 M EDTA instead of DNase I, was used as an undigested control sample.
Hydroxyl radical footprinting
Aliquots of 140 µl (2 × 106 nuclei/ml) were prepared from the pLHC4/TLN-6- or pST0/TLN-7-harboring nuclei suspension. The reaction was carried out using 0.5 mM Fe(EDTA)2, 1 mM sodium ascorbate and 0.06% (v/v) hydrogen peroxide (final concentrations). For the digestion of supercoiled DNA alone, 50 µM Fe(EDTA)2, 1 mM sodium ascorbate and 0.03% (v/v) hydrogen peroxide were used. The reaction proceeded for 1 min and was quenched by the addition of glycerol to 5%. DNA recovery was carried out as described above.
Analysis of cutting sites
MNase cleavage sites were detected as follows. Purified DNAs were used directly to detect nucleosomal arrays or restricted to completion with Eco81I to determine nucleosome positions in the tk promoter region. Samples and marker fragments (the latter were prepared by digesting pST0/TLN-7 variously) were electrophoresed on 1% agarose gels in Tris-borate–EDTA buffer, and the DNAs were transferred to nylon membranes. In the detection of nucleosomal arrays, the membrane was hybridized with the 171 bp probe that spanned from –137 to +34 relative to the transcription start site of the tk gene. In the indirect end-labeling analysis, the membranes were hybridized with the 250 bp probe that spanned from the Eco81I site to the position +427. The probes were labeled with [α-32P]dCTP (3000 Ci/mmol) by random priming.
Both of the DNase I cleavage sites and hydroxyl radical cleavage sites were detected by PCR-based primer extension, using a CircumVent™ Thermal Cycle Dideoxy Sequencing Kit (New England BioLabs). To detect the sites, the (5′-32P)-labeled primer (5′-GGAATGCCAAGCTTACTTAG-3′) was used and the reaction contained 208 µM each of dATP, dGTP, dCTP and TTP instead of the sequencing mixes in the kit. Sequence ladders for the DNase I footprinting were prepared by sequencing plasmid DNAs recovered from DNase I-untreated nuclei. The reaction products were resolved in 6% polyacrylamide–7 M urea gels.
RESULTS
Plasmid constructs
The constructs employed in this study are shown in Figure 1 and Table 1. We constructed 35 luciferase reporter plasmids each containing a HSV tk promoter, a piece of synthetic DNA with a defined geometry, and a mcs of variable length in between. It is known that left-handedly curved DNA (intrinsic DNA curvature with left-handed superhelical configuration), right-handedly curved DNA (intrinsic DNA curvature with right-handed superhelical configuration) and two-dimensional DNA curvature can be prepared by manipulating the periodicity of the A-tracts (6,23). In addition, in the study of Brukner et al. (24), it was experimentally proved that these curvatures were actually formed in solution. Based on this knowledge, synthetic DNAs with different geometries were constructed by changing the number of base pairs between each of four (T/A)5 repeat tracts (Fig. 1A). The mcs allowed us to manipulate the distance and the rotational phase between the curved DNA and promoter, by removing up to 20 bp or alternatively inserting up to 89 bp. Further variations of 1 or 2 bp were due to random deletions, which occurred during the construction of the plasmids. For the study, plasmids were grouped according to the shape of synthetic DNA (Table 1). In the constructs ST0, ST4, LHC4, TDC4 and RHC4 the nomenclature is derived thus: straight, straight (zigzag), left-handedly curved, two-dimensionally curved and right-handedly curved, respectively (Fig. 1B). The number of (T/A)5 tracts in the construct is indicated by ‘0’ and ‘4’. Finally, the number at the end of each plasmid name indicates the number of base pairs that were deleted from or inserted into the mcs.
Table 1. Structural characteristics of the reporter constructs.
| Construct | Helix axisa | Rotational orientationb (°) | Distancec (bp) |
|---|---|---|---|
| pST0/TLN | Straight | – | 131 |
| pST0/TLN-7 | Straight | – | 124 |
| pST0/TLN-10 | Straight | – | 121 |
| pST0/TLN-17 | Straight | – | 114 |
| pST0/TLN-21 | Straight | – | 110 |
| pST0/TLN+46 | Straight | – | 177 |
| pST0/TLN+87 | Straight | – | 218 |
| pST4/TLN | Straight (zigzag) | – | 131 |
| pST4/TLN-6 | Straight (zigzag) | – | 125 |
| pST4/TLN-10 | Straight (zigzag) | – | 121 |
| pST4/TLN-16 | Straight (zigzag) | – | 115 |
| pST4/TLN-20 | Straight (zigzag) | – | 111 |
| pST4/TLN+47 | Straight (zigzag) | – | 178 |
| pST4/TLN+89 | Straight (zigzag) | – | 220 |
| pLHC4/TLN | Left-handedly curved | –17.1 | 131 |
| pLHC4/TLN-6 | Left-handedly curved | 137.1 | 125 |
| pLHC4/TLN-10 | Left-handedly curved | 0 | 121 |
| pLHC4/TLN-16 | Left-handedly curved | 154.3 | 115 |
| pLHC4/TLN-20 | Left-handedly curved | 17.1 | 111 |
| pLHC4/TLN+47 | Left-handedly curved | 154.3 | 178 |
| pLHC4/TLN+89 | Left-handedly curved | 154.3 | 220 |
| pTDC4/TLN | Two-dimensionally curved | 34.3 | 131 |
| pTDC4/TLN-7 | Two-dimensionally curved | 154.3 | 124 |
| pTDC4/TLN-10 | Two-dimensionally curved | 51.4 | 121 |
| pTDC4/TLN-17 | Two-dimensionally curved | 171.4 | 114 |
| pTDC4/TLN-21 | Two-dimensionally curved | 34.3 | 110 |
| pTDC4/TLN+47 | Two-dimensionally curved | –154.3 | 178 |
| pTDC4/TLN+89 | Two-dimensionally curved | –154.3 | 220 |
| pRHC4/TLN | Right-handedly curved | 137.1 | 131 |
| pRHC4/TLN-7 | Right-handedly curved | –102.9 | 124 |
| pRHC4/TLN-10 | Right-handedly curved | 154.3 | 121 |
| pRHC4/TLN-17 | Right-handedly curved | –85.7 | 114 |
| pRHC4/TLN-21 | Right-handedly curved | 137.1 | 110 |
| pRHC4/TLN+47 | Right-handedly curved | –51.4 | 178 |
| pRHC4/TLN+89 | Right-handedly curved | –51.4 | 220 |
aTrajectory of the helical axis of the curved DNA or straight DNA region.
bThe rotational angle of the curved DNA center relative to the center of the TATA box. The angle is calculated as [(D/10.5) – n]360, where D is the distance between the centers (bp) and n is the integer nearest to the value of D/10.5. The minus sign corresponds to the leftwards rotation relative to the center of the TATA box.
cDistance between the center of the TATA box and the nucleotide at the downstream end of the synthetic curved DNA or straight DNA.
Left-handedly curved DNA activates transcription
Promoter activity was studied by electroporating each construct into COS-7 cells and performing a luciferase assay after 21 h in culture. The hybrid tk promoter was most activated in the pLHC4/TLN-6 that contained a left-handedly curved DNA and this activity was ∼9-fold higher than the corresponding straight controls in pST0/TLN-7 and pST4/TLN-6 (Fig. 2A). The promoter of pLHC4/TLN-16 was also activated. Thus, the left-handedly curved DNA seems to be important for activation of transcription. Although distances between the left-handedly curved DNA and the TATA box are similar among the constructs pLHC4/TLN-6, pLHC4/TLN-16, pLHC4/TLN, pLHC4/TLN-10 and pLHC4/TLN-20, the latter three constructs showed low promoter activities. This reason is indicated in Figure 2B. These three constructs were quite different from the pLHC4/TLN-6 and pLHC4/TLN-16 for the parameter of the rotational orientation (the spatial positioning of the curved DNA is reversed between two groups). Thus, the importance of the rotational orientation is indicated. However, despite the orientations being the same among pLHC4/TLN-16, pLHC4/TLN+47 and pLHC4/TLN+89, promoter was not activated in the latter two constructs (Fig. 2B), indicating that the distance between the curved DNA and the promoter is also important. For the two parameters, the pTDC4/TLN-7, pRHC4/TLN, pRHC4/TLN-10 and pRHC4/TLN-21 are similar to the activated constructs. However, the tk promoter was not activated in these four constructs. This strengthens the importance of the shape of the curved DNA again. In conclusion, activation of the promoter seems to be optimized when the left-handedly curved DNA is introduced with appropriate combination of distance and spatial positioning.
Figure 2.

Effect of DNA architecture on the activity of the tk promoter. (A) Activities plotted against distance of the synthetic DNA from the TATA box. The data are grouped according to the shape of the upstream DNA. The distance is measured between the center of the TATA box and the nucleotide at the downstream end of the curved DNA, or straight DNA (Table 1). Values represent the mean ± SD (n = 7 or 8). Constructs are indicated with the last numbers that appear in the plasmid names. The constructs with the complete mcs are indicated with ‘±0’. (B) Activities plotted against the rotational orientation of the curved DNA’s center relative to the center of the TATA box.
Nucleosomes are formed in vivo on the region containing the tk promoter
To know why pLHC4/TLN-6 showed high tk promoter activity, the in vivo structure of pLHC4/TLN-6 was investigated by electroporating the plasmid into COS-7 cells, isolating nuclei and performing MNase digestions and electrophoretic analyses. This enzyme cuts the linker regions between nucleosomes. A Southern blot of the digest was probed with a promoter DNA fragment (Fig. 3). The large part of the DNA molecules stayed at positions of nicked and linearized forms in the gel (lanes 2–4), indicating that the majority of the pLHC4/TLN-6 molecules were protected against the digestion. A small fraction was, however, fragmented and regularly spaced DNA bands of sizes ∼200, ∼360, ∼530, ∼710 and ∼880 bp were generated. The distance between these bands ranged from 160 to 180 bp, consistent with the presence of nucleosomal arrays. When we used nuclei that were prepared in the presence of 5% NP-40, the digestion proceeded more efficiently and the ladder of the bands emerged more clearly (lanes 5 and 6). The linker DNA regions of the plasmid chromatins were presumably covered with various proteins in cells, such as transcription factors, chromatin modifying enzymes, DNA topoisomerases, HMG proteins and non-specific DNA binding proteins. We speculate that at low concentrations of NP-40 they protected the plasmid chromatins against the digestion. However, presumably, high concentrations of the detergent could considerably remove such proteins and allow the MNase access to the linker DNA regions. Aside from the speculation, it seems safe to conclude that the nucleosomal arrays were formed, at least, on the promoter region.
Figure 3.
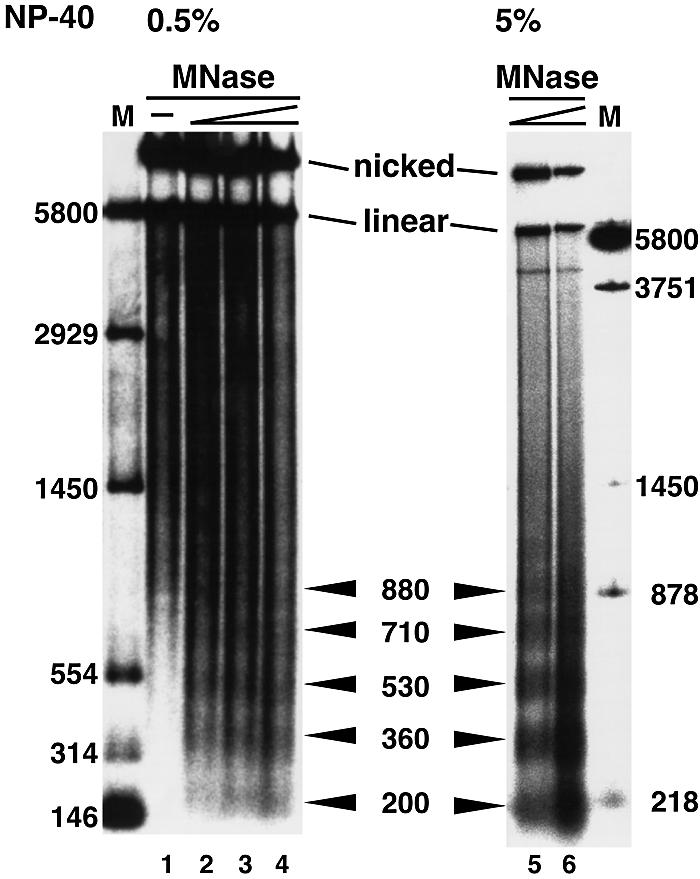
Agarose gel electrophoresis of pLHC4/TLN-6 DNA recovered from MNase-digested COS-7 nuclei. The nuclei were isolated in the presence of 0.5 or 5% NP-40 and subjected to the digestion. A DNA fragment spanning from –137 to +34 was used for the probe; lane 1, no MNase; lanes 2 and 5, 2.4 U/ml; lanes 3 and 6, 3.2 U/ml; lane 4, 6.4 U/ml. Label ‘M’ indicates the size marker.
Translational and rotational positionings of nucleosomes play an important role in transcription (25–28). Subsequently, potential sequence-specific localization (translational positioning) of the nucleosomes on pLHC4/TLN-6 was investigated by indirect end-labeling analysis (29,30). Some bands in the chromatin lanes also emerged in DNA lanes (Fig. 4). This is often the case (31,32), and is caused by the high selectivity of MNase for some sequences (33,34). However, the intensity of bands is strikingly different in chromatin and in DNA. If the nucleosomes were not formed, we would observe the same bands with a similar intensity in chromatin and in naked DNA. Therefore, we conclude that some of the MNase-specific sites in naked DNA are protected by histones in nuclei. The regions between –470 (relative to the tk Cap site) and –670, and between –250 and –470, were clearly protected from the MNase attack in nuclei (position number indicates the center of each band). This result indicates that translationally positioned nucleosomes were formed in these regions. We named these nucleosomes α1 and α2, respectively. A smear pattern was observed in the promoter region. However, the position –80 was considerably protected in chromatin. Considering the result that the promoter is involved in the nucleosome structure (Fig. 3), these results indicate that multiple populations of nucleosomes with different positioning were present on this region. Among them, we may recognize two relatively major populations, one located between approximately –30 and –250 and the other between approximately +30 and –140, which were more clearly detected in DNase I footprinting (described later). The same analysis was carried out using the control constructs such as pST0/TLN-7, pST4/TLN-6, pTDC4/TLN-7 and pRHC4/TLN-7. Each has a different DNA segment in the same locus where the pLHC4/TLN-6 has the left-handedly curved DNA. However, almost the same patterns as observed for the pLHC4/TLN-6 were obtained (data not shown). Thus, we could not clarify the reason why pLHC4/TLN-6 showed high promoter activity in this low resolution analysis.
Figure 4.

Analysis of translational positions of nucleosomes formed on the pLHC4/TLN-6. Plasmid-containing nuclei and naked supercoiled plasmid DNA were subjected to MNase digestion. After deproteinization, DNAs were digested with Eco81I and MNase-sensitive DNA sequences were detected with the indirect end-labeling technique. The relative extent of MNase digestion is indicated on the top of the autoradiogram. Labels ‘DNA’, ‘Ch’ and ‘M’ indicate naked DNA, chromatin DNA and the size marker, respectively. The distinctive cleavage sites and the protected sites in chromatin are indicated with black and white arrowheads, respectively. The sites are numbered relative to the transcription start site. α1 and α2 are translationally positioned nucleosomes.
The left-handedly curved DNA can expose the TATA box in chromatin
Using the plasmid-containing nuclei, then, the rotational setting of the pLHC4/TLN-6 promoter on a histone core was investigated by in vivo DNase I footprinting (Fig. 5A). The pST0/TLN-7, pST4/TLN-6, pTDC4/TLN-7, pRHC4/TLN-7 and pLHC4/TLN-16 were used as controls. The pLHC4/TLN-16 harbors the same left-handedly curved DNA as the pLHC4/TLN-6 in the locus 10 bp closer to the TATA box. This construct showed the second highest promoter activity (Fig. 2). A small fraction of each construct recovered from the nuclei had been already nicked or cleaved by endogenous DNases (‘–’ lane within ‘Ch’ lanes in each autoradiogram), though most was intact (see the gel top). Interestingly, most of these cleavage sites coincided with those generated by DNase I. Thus, the endogenous DNases seem to have attacked the chromatin either in the nuclei or during the DNA recovery process. The chromatin-specific DNase I cleavages formed two 10 bp ladders in the promoter region of each construct, indicating that each promoter was rotationally positioned on the surface of a histone core in two different ways. In one ladder, cleavages emerged within each of four cis-DNA elements (also see Fig. 5B), indicating that they all faced outwards from the nucleosome surface. We named the nucleosomes of this type the active (AC)-form nucleosomes. In the other ladder, the cleavages occurred outside (or at the edge of) the cis-DNA elements. These elements were thought to face inwards, towards the histone core. Thus, we named the nucleosomes of this type as the inactive (IA)-form. Using the nucleosomes reconstituted on the pLHC4/TLN-6 fragment in vitro, we also performed DNase I footprinting. Although the IA-form ladder was short, these two ladders were also observed clearly (Supplementary Material, Fig. S1). Thus, it seems safe to conclude that the ladders shown in Figure 5 were actually derived from nucleosomes.
Figure 5.


Rotational positioning of the nucleosomes formed in vivo on the promoters. (A) Footprints of the pLHC4/TLN-6, pLHC4/TLN-16, pST0/TLN-7, pST4/TLN-6, pTDC4/TLN-7 and pRHC4/TLN-7 chromatins. The nuclei harboring each construct were isolated and treated with DNase I. The cleavage sites were detected by linear amplification with a radiolabeled primer and a thermostable DNA polymerase. Lanes labeled ‘DNA’ and ‘Ch’ show DNase I cleavage sites in naked DNA (supercoiled plasmid) and in chromatin DNA, respectively. The lanes labeled ‘–’ indicate DNase I-untreated samples. A sequence ladder was obtained by PCR-based cycle sequencing of part of the sample recovered from DNase I-untreated nuclei. The major cleavage sites in chromatin DNA, and lesser sites of cleavage that did not appear in the plasmid DNA (chromatin-specific DNase I cleavage sites), are indicated on the right of each autoradiogram. The distances between the cleavage sites (bp) are indicated where they were between 8 and 12 bp, or near multiples of 10 bp. ‘AC’ and ‘IA’ indicate active and inactive forms of nucleosomes, respectively. The small filled triangles on the left of each autoradiogram indicate overlap points of the signals. In the sequence of pST0/TLN-7, the region containing the 5′-TCAG-3′ repeat apparently lacked one repeat unit. However, we propagated the same sample in E.coli cells and sequenced it again, verifying that it has 10 repeat units. (B) The DNase I cleavage sites in the tk promoter. These sites are indicated by black arrows or arrowheads. The cleavage sites in the CCAAT and distal GC boxes on the active form nucleosomes were slightly different between pLHC4/TLN-6 and the other constructs, and those of pLHC4/TLN-6 are shown with black arrowheads alone.
A notable difference was observed for the AC-form ladders. The lengths of the ladders differed: i.e. pLHC4/TLN-6, 165 bp; pLHC4/TLN-16, 155 bp; pST0/TLN-7, 135 bp; pST4/TLN-6, 126 bp; pTDC4/TLN-7, 167 bp; pRHC4/TLN-7, 136 bp (Fig. 5A). The difference in the ladder length originates in the curved/straight DNA region. The lengths of 165, 155 and 167 bp indicate that multiple nucleosomes with the same rotational but different translational positioning were implicated in the formation of the respective ladders. The ladders of pLHC4/TLN-6 and pTDC4/TLN-7 measure 147 and 149 bp from the upstream end to the third step from the bottom, respectively. Importantly, their TATA boxes are out of these spans. Although this is a common feature between pLHC4/TLN-6 and pTDC4/TLN-7, the cleavage signals in the curved DNA region of the former were considerably stronger than the corresponding signals of the latter. This indicates that the population of the AC-form nucleosomes that formed on the curved DNA region was less abundant in the case of pTDC4/TLN-7. In contrast, the constructs pLHC4/TLN-6 and pLHC4/TLN-16 showed very similar 10 bp ladders (Fig. 5A). It seems that the populations of the curved-DNA-containing nucleosomes were almost the same between pLHC4/TLN-6 and pLHC4/TLN-16. This was confirmed by a densitometric analysis (data not shown). The AC-form ladder of pLHC4/TLN-16, however, measures 146 bp from the upstream edge to the second step from the bottom. Since this step corresponds to the center of the TATA box, it seems that the TATA box was located at the edge of the relevant nucleosome. Concerning the IA-form nucleosomes, no clear qualitative or quantitative difference was observed among the constructs.
We also performed hydroxyl radical footprinting to map contacts between proteins and pLHC4/TLN-6 more finely. Nuclei harboring the construct and the plasmid alone (supercoiled form) were treated with hydroxyl radicals, respectively. In this experiment, pST0/TLN-7 was used as a control. Except for the loci of synthetic DNA and the TATA box, no notable difference was observed between them. The cleavage patterns in and around the TATA box are shown in Figure 6 (for the whole data set, see Supplementary Material, Fig. S2). The hydroxyl radical footprint clearly indicated that the TATA box region was considerably open in the pLHC4/TLN-6 chromatin. Most nucleosomes were presumably located in the region upstream of the TATA box and only a minor population covered the TATA box region. On the other hand, the TATA sequence was incorporated into nucleosomes in the case of pST0/TLN-7 chromatin. The DNase I footprints indicated that the pLHC4/TLN-6 chromatin and the other chromatins somewhat differed as to the DNA conformations of the distal GC box and the CCAAT box (Fig. 5B). However, this point was not clear in the hydroxyl radical footprints (Supplementary Material, Fig. S2). Even a very minor change occurring in nucleosomal DNA may have influenced the accessibility in the DNase I footprinting because the enzyme is much larger than the chemical reagent.
Figure 6.

Densitometric analysis of hydroxyl radical cleavage patterns of pLHC4/TLN-6 and pST0/TLN-7 chromatin. COS-7 nuclei harboring each construct were subjected to the hydroxyl radical cleavage. Electrophoretic detection of the cleavage sites (data not shown) was performed as described in Materials and Methods. Only the cleavage patterns for each TATA box region are presented. In each panel, the upper tracing corresponds to the hydroxyl radical footprint of the chromatin, and the lower tracing is the pattern of hydroxyl radical cleavage of the same DNA (supercoiled form) when free in solution. AC′ and IA′ correspond to active and inactive forms of nucleosomes in Figure 5, respectively. Plus signs indicate sites protected from the cleavage.
DISCUSSION
In the present study, we scrutinized the relationship between upstream DNA geometry, nucleosome positioning and promoter strength. Curved DNA with a left-handed superhelical writhe activated promoters at a specific rotational phase and distance. It was found that the left-handedly curved DNA could expose the TATA box in chromatin by influencing nucleosome positioning.
The mechanism underlying curved DNA-mediated transcriptional activation observed in this study
The pLHC4/TLN-6 and pLHC4/TLN-16 showed the highest and the second highest promoter activities among the 35 constructs, respectively. Judging from the lengths of their AC-form ladders (Fig. 5), the left-handedly curved DNA of the former certainly generated a chromatin population in which the TATA box was located in the linker region (Fig. 7). We named the nucleosome adjacent to the TATA box α3. In the pLHC4/TLN-6 chromatin, all nucleotides of the TATA sequence were well cleaved by hydroxyl radicals (Fig. 6), indicating that the α3-containing population was not small. On the other hand, DNase I could not adequately cleave them, except one center nucleotide (Fig. 5). This is explained as follows. Rotational setting of the TATA box was presumably regulated by the α3 to expose its minor groove towards the environment even in the linker DNA region, for the TATA box was located in close vicinity to this nucleosome. This structure and large size of the enzyme presumably limited the cleavage. The TBP recognizes the minor groove of the TATA box (35,36). Therefore, it is thought that the access of TBP to this sequence was facilitated by these structural advantages. Viewed in this light, the pLHC4/TLN-6 chromatin containing the α3 seems to be the very chromatin that generated a large quantity of the transcripts. Also, the pLHC4/TLN-16 generated a similar chromatin population. In this case, however, the TATA box was most probably located at the edge of the nucleosome (Fig. 7). In other words, it was less accessible. Concerning the population of the α3-containing chromatins, there was no difference between pLHC4/TLN-6 and pLHC4/TLN-16 (Fig. 5A). Thus, the position of the TATA box presumably made the latter promoter less active.
Figure 7.

Putative local chromatin structures of the pLHC4/TLN-6 and pLHC4/TLN-16 that functioned advantageously in transcription. In this chromatin population, the nucleosome α3 was positioned on the left-handedly curved DNA. The TATA box was located outside (pLHC4/TLN-6) or at the edge (pLHC4/TLN-16) of the α3. The distance between the centers of the distal GC box and the TATA box of the tk promoter is 74 bp and the CCAAT box lies close to this GC box (separated by only one helical turn). Thus, these three elements must be spatially very close to each other. For α1 and α2, see Figure 4.
It seems that the pTDC4/TLN-7 molecules also formed the α3-type nucleosomes. However, judging from the result that the cleavage signals in the curved DNA region were faint (Fig. 5A), their population was presumably small. In the cases of pST0/TLN-7, pST4/TLN-6 and pRHC4/TLN-7, the presence of the α3-type nucleosomes was not obvious (Fig. 5A). It is thought that in major populations of the pTDC4/TLN-7, pST0/TLN-7, pST4/TLN-6 and pRHC4/TLN-7 chromatins, the TATA box was located in nucleosomes. This point was clarified for the pST0/TLN-7 chromatin (Fig. 6). All these things make it clear that the presence of the α3-type nucleosomes and their population played a key role in transcription. Actually, the transcription level of pTDC4/TLN-7 was considerably lower than those of the pLHC4/TLN-6 and pLHC4/TLN-16, but it was slightly higher than those of pST0/TLN-7, pST4/TLN-6 and pRHC4/TLN-7 (Fig. 2).
The general observation is that trans-acting factors often have access to promoters that are organized into chromatin in living cells, even when cis-DNA elements are adjacent to nucleosomes or actually incorporated into them (37–40). Compared with the TATA box, the other cis-DNA elements were presumably less accessible in the pLHC4/TLN-6 chromatin because they were completely incorporated into α3. Furthermore, two GC boxes displayed only their minor grooves towards the environment on the surface of the nucleosome (Fig. 6). A zinc-finger protein Sp1 interacts with the major groove of the GC box (41). Thus, it seems that they were not involved in the very first step of transcription. The efficient interaction of Sp1 with CTF (CCAAT box transcription factor) and TFIID is, however, indispensable for optimal transcription from the tk promoter (42). It is speculated that this interaction occurred after the chromatin was locally remodeled.
Structural basis in the formation of the nucleosome α3
It appears that the α3-type nucleosomes were generated based on the geometrical continuity between the following two supercoils: the promoter DNA on the nucleosome and the intrinsic DNA curvature in the upstream region. However, the nucleosomes assembled on the pLHC4/TLN-6 may have adopted a slightly altered nucleosome structure. Helical axis trajectory of the tk promoter per se is not perfectly straight (data not shown). Thus, in order to let two structures (curved DNA and tk promoter) get along with each other on the same nucleosome, a slightly altered nucleosome structure may have been constructed. The minor difference in the DNase I footprints that was observed between the pLHC4/TLN-6 chromatin and the other chromatins (Fig. 5B) may suggest this ‘adaptation’. A similar example has been reported (43). In the case of pTDC4/TLN-7, it seems that a certain geometrical discontinuity existed between the two, which presumably lowered generation of the α3-type nucleosomes. For pRHC4/TLN-7, pST0/TLN-7 and pST4/TLN-6, the more pronounced geometrical discontinuity and/or a difference in DNA bendability were presumably present. In the low resolution analysis of translational positioning of nucleosomes, one signal was observed at approximately position–30 for pLHC4/TLN-6 (Fig. 4). Certainly, it corresponds to one border (downstream border) of the AC-form nucleosomes detected by in vivo DNase I footprinting (Fig. 5). In these analyses, however, we could not obtain any information on the α3-type nucleosomes from the cleavage signals that emerged in and around the downstream border. This seems simply due to the resolution of the enzymatic analyses. Actually, a high-resolution chemical method showed a difference between the TATA box regions as described above (Fig. 6).
Then, what was the driving force to position a histone core on that locus? It is known that nucleosomes often preferentially associate with curved DNA structures (21,44–47). Also, short range sliding of nucleosome seems to be a general phenomenon that is dependent on the underlying DNA sequence (48). Thus, two possibilities are raised: (i) the core had a high affinity for the left-handedly curved DNA or (ii) nucleosome sliding occurred from the promoter towards the curved DNA. TBP binding to DNA can induce nucleosome sliding (49). Thus, the left-handedly curved DNA segment may have functioned as an acceptor of the sliding nucleosome and helped a dynamic process of transcription. We speculate that the two effects raised as possibilities worked synergistically to activate transcription in the case of pLHC4/TLN-6 and pLHC4/TLN-16.
Curved DNA with a left-handed superhelical writhe activated eukaryotic transcription. Interestingly, it is a rightwards superhelical writhe that activates transcription in prokaryotes (6). It seems that the principal role of curved DNA structures in prokaryotes is to modulate the interaction between a promoter and RNA polymerase (1,5), while in eukaryotes it may organize local chromatin infrastructures appropriate for transcription initiation as suggested by this study.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Y. Kadokawa for the plasmid pEKS, T. Yoshioka, J. Kishida and T. Kawashima for technical assistance, and J. Ohyama for help in preparing the manuscript. This work was supported in part by a Grant-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan, NIG Cooperative Research Program and the Hirao Taro Foundation for Academic Research (to T.O.).
REFERENCES
- 1.Ohyama T. (2001) Intrinsic DNA bends: an organizer of local chromatin structure for transcription. Bioessays, 23, 708–715. [DOI] [PubMed] [Google Scholar]
- 2.Travers A.A. (1989) Curves with a function. Nature, 341, 184–185. [DOI] [PubMed] [Google Scholar]
- 3.Hagerman P.J. (1990) Sequence-directed curvature of DNA. Annu. Rev. Biochem., 59, 755–781. [DOI] [PubMed] [Google Scholar]
- 4.Travers A.A. (1990) Why bend DNA? Cell, 60, 177–180. [DOI] [PubMed] [Google Scholar]
- 5.Pérez-Martín J., Rojo,F. and de Lorenzo,V. (1994) Promoters responsive to DNA bending: a common theme in prokaryotic gene expression. Microbiol. Rev., 58, 268–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirota Y. and Ohyama,T. (1995) Adjacent upstream superhelical writhe influences an Escherichia coli promoter as measured by in vivo strength and in vitro open complex formation. J. Mol. Biol., 254, 566–578. [DOI] [PubMed] [Google Scholar]
- 7.Ohyama T. and Hashimoto,S. (1989) Upstream half of adenovirus type 2 enhancer adopts a curved DNA conformation. Nucleic Acids Res., 17, 3845–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schroth G.P., Siino,J.S., Cooney,C.A., Th’ng,J.P.H., Ho,P.S. and Bradbury,E.M. (1992) Intrinsically bent DNA flanks both sides of an RNA polymerase I transcription start site: both regions display novel electrophoretic mobility. J. Biol. Chem., 267, 9958–9964. [PubMed] [Google Scholar]
- 9.Cress W.D. and Nevins,J.R. (1996) A role for a bent DNA structure in E2F-mediated transcription activation. Mol. Cell. Biol., 16, 2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marilley M. and Pasero,P. (1996) Common DNA structural features exhibited by eukaryotic ribosomal gene promoters. Nucleic Acids Res., 24, 2204–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schätz T. and Langowski,J. (1997) Curvature and sequence analysis of eukaryotic promoters. J. Biomol. Struct. Dyn., 15, 265–275. [DOI] [PubMed] [Google Scholar]
- 12.Angermayr M., Oechsner,U., Gregor,K., Schroth,G.P. and Bandlow,W. (2002) Transcription initiation in vivo without classical transactivators: DNA kinks flanking the core promoter of the housekeeping yeast adenylate kinase gene, AKY2, position nucleosomes and constitutively activate transcription. Nucleic Acids Res., 30, 4199–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawamoto T., Makino,K., Orita,S., Nakata,A. and Kakunaga,T. (1989) DNA bending and binding factors of the human β-actin promoter. Nucleic Acids Res., 17, 523–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Döbbeling U., Roß,K., Klein-Hitpaß,L., Morley,C., Wagner,U. and Ryffel,G.U. (1988) A cell-specific activator in the Xenopus A2 vitellogenin gene: promoter elements functioning with rat liver nuclear extracts. EMBO J., 7, 2495–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delabre M.-L., Pasero,P., Marilley,M. and Bougis,P.E. (1995) Promoter structure and intron–exon organization of a scorpion α-toxin gene. Biochemistry, 34, 6729–6736. [DOI] [PubMed] [Google Scholar]
- 16.Cacchione S., Savino,M. and Tufillaro,A. (1991) Different superstructural features of the light responsive elements of the pea genes rbcS-3A and rbcS-3.6. FEBS Lett., 289, 244–248. [DOI] [PubMed] [Google Scholar]
- 17.Bash R.C., Vargason,J.M., Cornejo,S., Ho,P.S. and Lohr,D. (2001) Intrinsically bent DNA in the promoter regions of the yeast GAL1-10 and GAL80 genes. J. Biol. Chem., 276, 861–866. [DOI] [PubMed] [Google Scholar]
- 18.Nair T.M. (1998) Evidence for intrinsic DNA bends within the human cdc2 promoter. FEBS Lett., 422, 94–98. [DOI] [PubMed] [Google Scholar]
- 19.Kim J., Klooster,S. and Shapiro,D.J. (1995) Intrinsically bent DNA in a eukaryotic transcription factor recognition sequence potentiates transcription activation. J. Biol. Chem., 270, 1282–1288. [DOI] [PubMed] [Google Scholar]
- 20.Ohyama T. (1996) Bent DNA in the human adenovirus type 2 E1A enhancer is an architectural element for transcription stimulation. J. Biol. Chem., 271, 27823–27828. [PubMed] [Google Scholar]
- 21.Piña B., Barettino,D., Truss,M. and Beato,M. (1990) Structural features of a regulatory nucleosome. J. Mol. Biol., 216, 975–990. [DOI] [PubMed] [Google Scholar]
- 22.Masumoto H., Masukata,H., Muro,Y., Nozaki,N. and Okazaki,T. (1989) A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J. Cell Biol., 109, 1963–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calladine C.R., Drew,H.R. and McCall,M.J. (1988) The intrinsic curvature of DNA in solution. J. Mol. Biol., 201, 127–137. [DOI] [PubMed] [Google Scholar]
- 24.Brukner I., Belmaaza,A. and Chartrand,P. (1997) Differential behavior of curved DNA upon untwisting. Proc. Natl Acad. Sci. USA, 94, 403–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simpson R.T. (1990) Nucleosome positioning can affect the function of a cis-acting DNA element in vivo. Nature, 343, 387–389. [DOI] [PubMed] [Google Scholar]
- 26.Li Q. and Wrange,Ö. (1993) Translational positioning of a nucleosomal glucocorticoid response element modulates glucocorticoid receptor affinity. Genes Dev., 7, 2471–2482. [DOI] [PubMed] [Google Scholar]
- 27.Li Q. and Wrange,Ö. (1995) Accessibility of a glucocorticoid response element in a nucleosome depends on its rotational positioning. Mol. Cell. Biol., 15, 4375–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong J., Li,Q., Levi,B.-Z., Shi,Y.-B. and Wolffe,A.P. (1997) Structural and functional features of a specific nucleosome containing a recognition element for the thyroid hormone receptor. EMBO J., 16, 7130–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nedospasov S.A. and Georgiev,G.P. (1980) Non-random cleavage of SV40 DNA in the compact minichromosome and free in solution by micrococcal nuclease. Biochem. Biophys. Res. Commun., 92, 532–539. [DOI] [PubMed] [Google Scholar]
- 30.Wu C. (1980) The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature, 286, 854–860. [DOI] [PubMed] [Google Scholar]
- 31.Thoma F., Bergman,L.W. and Simpson,R.T. (1984) Nuclease digestion of circular TRP1ARS1 chromatin reveals positioned nucleosomes separated by nuclease-sensitive regions. J. Mol. Biol., 177, 715–733. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka S., Livingstone-Zatchej,M. and Thoma,F. (1996) Chromatin structure of the yeast URA3 gene at high resolution provides insight into structure and positioning of nucleosomes in the chromosomal context. J. Mol. Biol., 257, 919–934. [DOI] [PubMed] [Google Scholar]
- 33.Hörz W. and Altenburger,W. (1981) Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res., 9, 2643–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dingwall C., Lomonossoff,G.P. and Laskey,R.A. (1981) High sequence specificity of micrococcal nuclease. Nucleic Acids Res., 9, 2659–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim Y., Geiger,J.H., Hahn,S. and Sigler,P.B. (1993) Crystal structure of a yeast TBP/TATA-box complex. Nature, 365, 512–520. [DOI] [PubMed] [Google Scholar]
- 36.Kim J.L., Nikolov,D.B. and Burley,S.K. (1993) Co-crystal structure of TBP recognizing the minor groove of a TATA element. Nature, 365, 520–527. [DOI] [PubMed] [Google Scholar]
- 37.Piña B., Brüggemeier,U. and Beato,M. (1990) Nucleosome positioning modulates accessibility of regulatory proteins to the mouse mammary tumor virus promoter. Cell, 60, 719–731. [DOI] [PubMed] [Google Scholar]
- 38.Archer T.K., Lefebvre,P., Wolford,R.G. and Hager,G.L. (1992) Transcription factor loading on the MMTV promoter: a bimodal mechanism for promoter activation. Science, 255, 1573–1576. [DOI] [PubMed] [Google Scholar]
- 39.Svaren J. and Hörz,W. (1997) Transcription factors vs nucleosomes: regulation of the PHO5 promoter in yeast. Trends Biochem. Sci., 22, 93–97. [DOI] [PubMed] [Google Scholar]
- 40.Sekinger E.A. and Gross,D.S. (2001) Silenced chromatin is permissive to activator binding and PIC recruitment. Cell, 105, 403–414. [DOI] [PubMed] [Google Scholar]
- 41.Gidoni D., Dynan,W.S. and Tjian,R. (1984) Multiple specific contacts between a mammalian transcription factor and its cognate promoters. Nature, 312, 409–413. [DOI] [PubMed] [Google Scholar]
- 42.Jones K.A., Yamamoto,K.R. and Tjian,R. (1985) Two distinct transcription factors bind to the HSV thymidine kinase promoter in vitro. Cell, 42, 559–572. [DOI] [PubMed] [Google Scholar]
- 43.Zhu Z. and Thiele,D.J. (1996) A specialized nucleosome modulates transcription factor access to a C. glabrata metal responsive promoter. Cell, 87, 459–470. [DOI] [PubMed] [Google Scholar]
- 44.Pennings S., Muyldermans,S., Meersseman,G. and Wyns,L. (1989) Formation, stability and core histone positioning of nucleosomes reassembled on bent and other nucleosome-derived DNA. J. Mol. Biol., 207, 183–192. [DOI] [PubMed] [Google Scholar]
- 45.Costanzo G., Di Mauro,E., Salina,G. and Negri,R. (1990) Attraction, phasing and neighbour effects of histone octamers on curved DNA. J. Mol. Biol., 216, 363–374. [DOI] [PubMed] [Google Scholar]
- 46.Shrader T.E. and Crothers,D.M. (1990) Effects of DNA sequence and histone–histone interactions on nucleosome placement. J. Mol. Biol., 216, 69–84. [DOI] [PubMed] [Google Scholar]
- 47.Widlund H.R., Cao,H., Simonsson,S., Magnusson,E., Simonsson,T., Nielsen,P.E., Kahn,J.D., Crothers,D.M. and Kubista,M. (1997) Identification and characterization of genomic nucleosome-positioning sequences. J. Mol. Biol., 267, 807–817. [DOI] [PubMed] [Google Scholar]
- 48.Meersseman G., Pennings,S. and Bradbury,E.M. (1992) Mobile nucleosomes—a general behavior. EMBO J., 11, 2951–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lomvardas S. and Thanos,D. (2001) Nucleosome sliding via TBP DNA binding in vivo. Cell, 106, 685–696. [DOI] [PubMed] [Google Scholar]
- 50.Dlakic M. and Harrington,R.E. (1998) DIAMOD: display and modeling of DNA bending. Bioinformatics, 14, 326–331. [DOI] [PubMed] [Google Scholar]
- 51.Sayle R.A. and Milner-White,E.J. (1995) RASMOL: biomolecular graphics for all. Trends Biochem. Sci., 20, 374–376. [DOI] [PubMed] [Google Scholar]
- 52.Bolshoy A., McNamara,P., Harrington,R.E. and Trifonov,E.N. (1991) Curved DNA without A-A: experimental estimation of all 16 DNA wedge angles. Proc. Natl Acad. Sci. USA, 88, 2312–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kabsch W., Sander,C. and Trifonov,E.N. (1982) The ten helical twist angles of B-DNA. Nucleic Acids Res., 10, 1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.