

Abstract
Type I interferons (IFNs) play a key role in linking the innate and adaptive arms of the immune system. Although produced rapidly in response to pathogens, IFNs are also produced at low levels in the absence of infection. In the present study we demonstrate that constitutively produced IFNs are necessary in vivo to maintain dendritic cells (DCs) in an “antigen presentation competent” state. Conventional dendritic cells (cDCs) isolated from spleens of IFN-β or IFNs receptor (IFNAR) deficient mice exhibit a highly impaired ability to present antigen and activate naive T cells. Microarray analysis of mRNA isolated from IFN-β-/- and IFNAR-/- cDCs revealed diminished expression of two genes that encoded members of the heat shock protein 70 (Hsp70) family. Consistent with this observation, pharmacological inhibition of Hsp70 in cDCs from wild type (WT) mice impaired their T cell stimulatory capacity. Similarly, the antigen presentation ability of splenic cDCs isolated from Hsp70.1/3-/- mice was also severely impaired in comparison to WT cDCs. Thus, constitutive IFN-β expression regulates Hsp70 levels in order to help maintain DCs in a competent state for efficient priming of effector T cells in vivo.
Keywords: Dendritic cells, T cells, MHC, Antigen Presentation/Processing, Tolerance
Introduction
Dendritic cells (DC)2 are essential for the induction of specific immune responses and represent the most important cellular link between the innate and the adaptive immune system (1-5). DCs are found in most tissues where they capture and transport antigen to draining lymph nodes. During this migration, DCs mature and become highly stimulatory for T as well as B cells (6). In addition to DCs that emigrate from peripheral tissues, resident DCs can also be found in lymphoid organs such as spleen. These DCs are crucial for the sampling of blood borne antigens or pathogens (2).
DCs are generally considered as “professional antigen presenting cells”, in which two principal antigen presentation pathways can be distinguished. Endogenous antigens like self- or viral components are presented via MHC class I (MHC I) molecules to CD8+ T cells, while exogenous antigens are presented via MHC class II (MHC II) to CD4+ T cells. In addition, DCs have the unique capacity to deliver exogenous antigens into the MHC I presentation pathway – a process known as cross presentation. This enables CD8+ T cells to respond against antigens that are not directly expressed within DCs (7-9). The development, migration, maturation and function of DCs are critically influenced by cytokines produced in their surroundings (10, 11), including type I interferons (IFNs).
IFNs encompass a large family of closely related cytokines comprising at least 13 IFN-α isotypes and a single IFN-β. Both IFN-α and IFN-β exert their activity through a common receptor IFNAR (12). IFN-β is thought to be the master regulator in that it is rapidly induced and can, in turn, induce the other IFNs isotypes (13, 14). Furthermore, even in the absence of infection spontaneous IFN-β production, albeit at low level, is known to occur (12, 15, 16). These spontaneously produced IFNs contribute to host defense and cell growth in a manner similar to those induced by pathogens. In addition, constitutive production of IFNs is crucial for maintaining cells in a “primed” state and thus enabling them to mount a rapid and robust response upon encounter of external stimuli. It has thus been proposed that the absence or dysregulation of the basal constitutive IFNs signaling could be the reason for development of certain diseases (12, 15-18).
In the present study, we addressed the question whether spontaneously produced IFNs play a role in the development of cell mediated immunity. Comparing the function of splenic conventional DCs (cDCs) from WT mice and mice deficient in either IFN-β or IFNAR, we found that IFN-β serves as a crucial factor for maturation of the T cell stimulatory capacity of cDCs via MHC I and MHC II. In its absence, we detected a lower number of specific MHC/peptide complexes at the surface of splenic cDCs. We also found that the diminished T cell stimulatory capacity of splenic cDCs occasioned by the in vivo absence of IFN-β is due to low expression of heat shock protein 70 (Hsp70), which is required for efficient generation of stable MHC/peptide complexes expressed on the cell surface of cDCs. Consistent with these findings, cDCs from Hsp70 deficient mice (Hsp70.1/3-/-) were impaired in their capacity to present soluble antigens to naive T cells.
Materials and Methods
Mice
Female IFN-β-/-(14), IFN-β+/+, IFNAR-/- (19) C57BL/6 mice, OT I and OT II mice (20, 21) were bred at the animal facility of the Helmholtz Centre for Infection Research (HZI). Female C57BL/6 mice were obtained from the Harlan-Winkelmann (Borchem, Germany and AN Venvay, The Netherlands). The initial generation of Hsp70.1/3 knockout mice has been previously described (22). The C57BL/6 Hsp70.1/3-/- mice were derived by transfer from a 129 background into the C57BL/6 background and were raised at Washington University School of Medicine in St. Louis. All mice were used between 8 to 12 weeks of age. Mice were bred and maintained in specific pathogen free conditions. Mouse care and experimental procedures were performed under approval of local authority LAVES.
Cell lines
The B3Z T cell hybridoma (23) specific for the H-2·Kb/SIINFEKL complex was maintained in IMDM supplemented with 10% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin. The hybridoma 25-D1.16 secreting an IgG1κ mAb specific for the pOV8·H-2Kb (24) was kindly provided by Dr Ronald Germain (NIAID). Antibody was purified and conjugated with FITC according to standard procedures.
Isolation of splenocytes
Spleen cells were prepared by gentle flushing out the splenocytes with IMDM supplemented with antibiotics (penicillin 100 U/ml and streptomycin 100 μg/ml) and 10% FCS, 50 μM 2-mercaptoethanol, 2 mM L-glutamine. Erythrocytes were lysed for 2 minutes in ACK buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) and washed two or three times in PBS. Cell clumps were removed by passage through a 50 μm nylon filter. Splenocyte preparation was carried out strictly on ice. Splenic cells were than stained with appropriate antibodies and conventional dendritic cells were sorted (see below).
Flow cytometric analysis and cell sorting
Single cell suspensions were treated with anti-mouse CD16/CD32 BD Fc Block (2.4G2, Becton Dickinson, NJ, USA) for 10 min followed by staining with appropriate mAbs for 20 minutes on ice. Abs used in this work included CD11c (clone N418) conjugated with alophycocyanin (APC) or phycoerithrin-Cy7 (PE-Cy7), CD11b (M1/70) PE-Cy7, CD8α (53-6.7) PacificBlue, fluoresecein isothiocyanate (FITC) or phycoerythrin (PE), B220 (RA3-6B2) APC-Alexa Fluor 750, CD4 (GK1.5) PE, CD86 (GL1) FITC, CD40 (HM40-3) FITC, CD80 (16-10A1) PE, CD1d (1B1) PE, ICAM-1 (YN1/1.7.4) PE, B7-H1 (MIH5) PE, B7-H4 (MIH29) PE, B7-RP1 (HK5.3) PE (all purchased from eBioscience, San Diego, USA). H-2Kb (Y-3) FITC, I-Ab (M5/114.15.2) FITC. Anti H-2Kb/SIINFEKL complexes antibody (clone 25-D1.16) FITC was purified and conjugated with FITC in our laboratory. cDCs were sorted as cells exhibiting low side scatter (SSClo) in addition of being CD11chiCD8α+CD11b-, CD11chiCD8α-CD11b+. Flow cytometric analysis and sorting was performed using FACSCanto, LSRII and FACSAria (Becton Dickinson, NJ, USA), respectively. Final purity of cDCs was always >97%. All samples during the sorting procedure were kept at 4°C. The data were analyzed using FACSDiva (Becton Dickinson) software.
Preparation of T cells
OT I (OVA specific CD8+ T cells) and OT II (OVA specific CD4+ T cells) were isolated from lymph nodes (subcutaneous and mesenteric) and spleen. Single cell suspensions were further purified using the CD8 or CD4 negative isolation kits (Dynal, Oslo, Norway) containing mAb against B220, CD11b, Ter-119, CD16/32 and CD4 (for OT I isolation) or CD8 (for OT II isolation) following the protocol provided by the manufacturer. Cell preparations contained more than 90% of the desired cell population and were essentially free of CD11chi cells as determined by flow cytometry using Ab's specific for CD4 or CD8 and CD11c, respectively. For antigen presentation assays OT I or OT II cells were stained with 1μM CFDA(SE) (Molecular Probes, Oregon, USA) for 10 minutes at 37°C according to the manufacturer's protocol.
Analysis of antigen presentation in vitro and ex vivo
For the experiments using soluble OVA or peptides, individual APCs populations were plated in 96-well plates (Nunc, Denmark) at 2×104 cells per well with the indicated amount of soluble EndoGrade OVA (Profos, Regensburg, Germany), OVA257-264 (SIINFEKL, Ana Spec Inc. San Jose, CA) or OVA323-339 (kindly provided by Dr. W. Tegge, HZI) for 1h at 37°C in complete medium. The cells were further washed three times and resuspended in complete medium containing 2×105 CFSE labeled OT I or OT II cells. Proliferation of T cells was analyzed by flow cytometry after 1.5 (OT I peptide) or 2.5 days of culture. Cells were stained with anti-CD4 or anti-CD8 mAbs for 20 minutes, washed and resuspended in 200 μl of PBS containing Cy5-labeled 0.6 μm latex beads. Samples were analyzed until 2×104 beads were collected. The number of divided cells (propidium iodidelow, CFSElow, CD4+ or CD8+) was determined as described (25). For ex vivo experiments mice were injected intravenously with 1 mg of OVA or 1 mg of OVA together with 200 μg poly I:C (Fluka, Steinheim). 24h later mice were sacrificed, APCs were sorted and incubated with OT I or OT II cells for 2.5 days. In some experiments murine recombinant IFN-β (R&D Systems, Minneapolis, USA) was added to the cultures of T cells and APCs.
Determination of antigen uptake and processing
Sorted DCs were incubated with 62.5 μg/ml of DQ-OVA (Molecular Probes, Oregon, USA) for 45 min at 37°C or on ice. DCs were then washed and analyzed by flow cytometry.
B3Z colorimetric assay
Sorted DCs (104 cells/well) were pulsed for 1 h with various concentrations of SIINFEKL peptide, washed twice and resuspended in phenol-red free RPMI (Gibco, Karlsruhe, Germany) containing 100 U/ml penicillin and 100 μg/ml streptomycin, 1% FCS and 2mM L-Glutamine. DCs were then co-cultured in a 96-well U bottomed plate with 5×104 B3Z cells/well overnight at 37°C. The next day 150 μl of supernatant was taken from each well and replaced with 150 μl of PBS containing 5 mM ONPG (Sigma, St. Louis, USA) and 0.5% IGEPAL-20 (Sigma, St. Louis, USA). The plate was than incubated at 37°C for 2h and optical density was measured at 450 nm with wavelength correction set at 650 nm.
Microarray Studies
RNA isolation, cDNA preparation andDNA microarray analysis of gene expression was performed at the gene array facility of the HZI. Fluorescent images of hybridized microarrays (Affymetrix, MOE-430 version 2.0) were obtained using an Affymetrix Genechip Scanner. Microarray data were analyzed using BioConductor Suite 2.1 software. All samples were repeated two times with individually sorted cells and averaged. Data discussed here have been deposited in NCBI's Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO series accession number GSE12392.
Quantitative real-time PCR
Total RNA was extracted from sorted APCs using the RNeasy mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. DNA contamination in the total RNA preparation was eliminated using DNase I (Qiagen, Hilden, Germany). Oligo(dT)18 primers and RevertAid™ First Strand cDNA Synthesis kit (Fermentas, Ontario) were used for reverse transcription of purified RNA. All gene transcripts were quantified by quantitative PCR with Power SYBR Green qPCR Master Mix (Applied Biosystems, Foster City, USA) and Light Cycler apparatus (ABI PRISM Cycler Applied Biosystems, Foster City, USA). Primers specific for Hsp70.1 were synthesized as described before (26).
Intracellular staining of Hsp70
Splenic cDCs were sorted out and stained intracellularly using Cytofix/Cytoperm kit (Becton Dickinson, NJ, USA) according to manufacturer's protocol with anti-Hsp70 (C92F3A-5) PE conjugated Ab (Stressgen, Canada).
Inhibition of Hsp70 by 15-deoxyspergualin (DSG)
DSG was a generous gift of Nippon Kayaku Co. Ltd. (Tokyo, Japan). Animals were injected intraperitoneally daily with 10 mg/kg of DSG or PBS for 6 days before splenic cDCs were sorted out and tested for their antigen presentation capacity with OT I or OT II cells in CFSE dilution assay.
Results
Splenic dendritic cells from IFN-β-/- and IFNAR-/- mice are impaired in T cell stimulation
IFNs are known to be constitutively produced at low levels under non-inflammatory conditions (12). To study the influence of IFNs on antigen presentation under physiological conditions we decided to focus on freshly isolated splenic cDCs. These cells are representative of typical non migratory DCs found in vivo at steady state (1-3). Analysing IFN-β and IFNAR deficient mice we detected no differences with regard to percentage of various splenic cDCs subpopulations in mice with and without either IFN-β or IFNAR (Fig. 1A). Furthermore, we determined the overall number of leukocytes in several lymph nodes and spleen. Consistently, there was no significant difference observed in comparison to WT mice (Fig. 1B).
Figure 1. Similar percentage and frequency of different splenic cDCs populations, macrophages and granulocytes in spleens of WT, IFN-β-/- and IFNAR-/- mice.
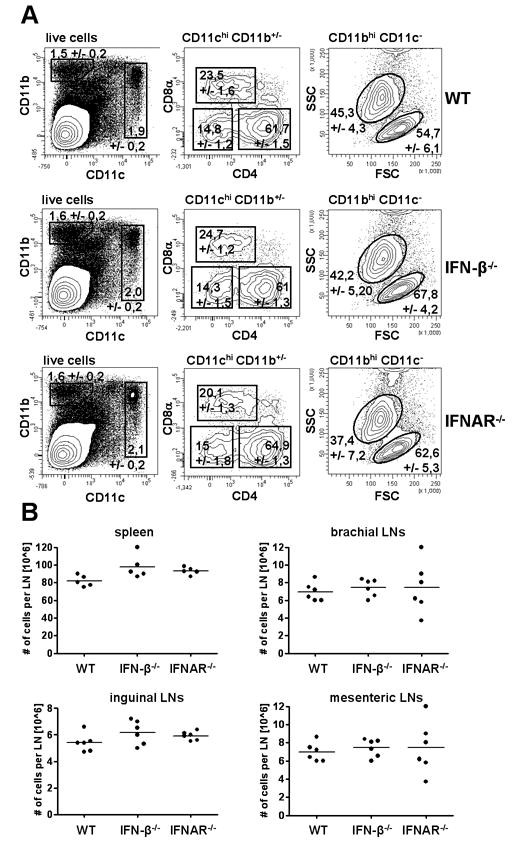
(A) Splenocytes from WT, IFN-β-/- and IFNAR-/- were isolated, stained for following markers: CD11c, CD11b, CD4, CD8α and B220 and analyzed by flow cytometry. Data are representative of three independent experiments with at least 5 mice per group. (B) Similar number of leukocytes per lymph node or spleen in WT, IFN-β-/- and IFNAR-/- mice.
Thus, we analyzed the ability of cDCs exhibiting the markers CD11chi, CD11b+/-, CD8α+/-, B220- from spleens of WT, IFN-β-/- and IFNAR-/- mice to present ovalbumin (OVA) protein to CFSE labeled OT I or OT II T cells. When compared to WT, cDCs from IFN-β-/- and IFNAR-/- mice were severely impaired in their ability to activate such CD8+ and CD4+ T cells (Fig. 2A and 2C).
Figure 2. Splenic conventional dendritic cells (cDCs) from IFN-β-/- and IFNAR-/- mice are impaired in their antigen presentation capacity of soluble OVA as well as OVA derived peptides in the context of both MHC I and MHC II.
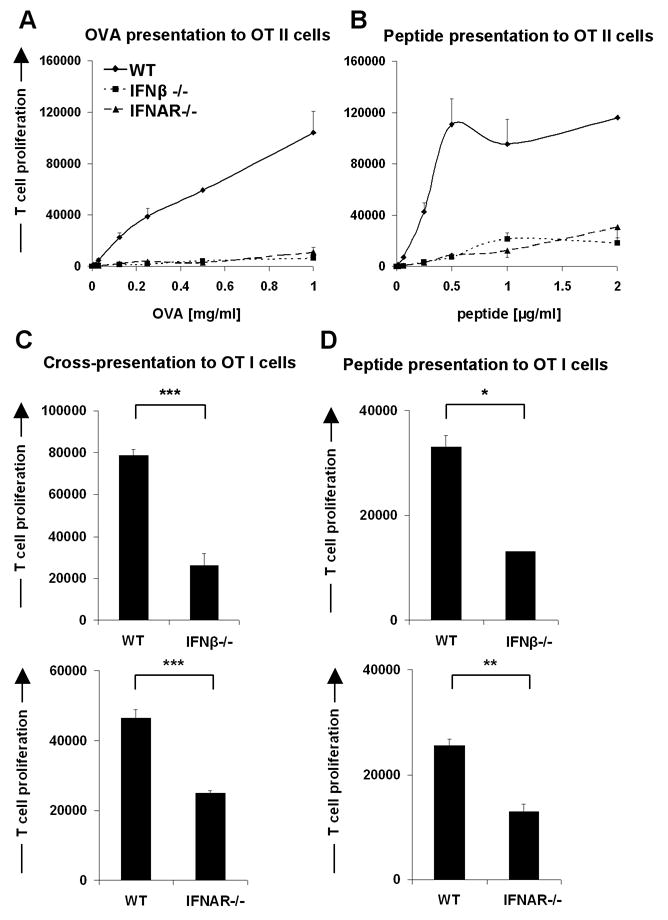
The purified OT I or OT II transgenic T cells were labeled with CFDA(SE) and incubated for 1.5 days (OT I peptide) or 2.5 days with splenic cDCs (CD11chigh, CD11b+/-, CD8α+/-, B220-) in ratio 10:1. cDCs from C57BL/6 WT, IFN-β-/- and from IFNAR-/- mice were preloaded with OVA257-264 (class I-restricted), OVA323-339 (class II-restricted) peptides or whole OVA protein for 1h and further were washed intensively. (A and B) antigen presentation in the context of MHC II, (C and D) antigen presentation in the context of MHC I. OVA concentration 250 μg/ml, OVA257-264 peptide concentration 10 ng/ml. The proliferative response of T cells was enumerated by flow cytometry. Data are representative of at least three mice for WT, IFN-β-/- and IFNAR-/- in multiple independent experiments. Statistical significance was determined using the paired Student's t test. * P<0.05; ** P<0.01; *** P<0.005
To test whether the anomaly associated with IFN-β or IFNAR deficiency affects also the presentation of preprocessed antigen, i.e. peptides, cDCs from WT, IFN-β-/- and IFNAR-/- mice were loaded with MHC class I or MHC class II specific peptides (OVA257-264 (SIINFEKL) and OVA323-339, respectively) and then incubated with CFSE labeled OT I and OT II T cells. As shown in Fig. 2B and 2D, T cell stimulation was also highly impaired when peptide loaded cDCs from IFN-β-/- and IFNAR-/- mice were used.
Throughout most of the experiments we used bulk sorted splenic cDCs because during in vitro co-cultures with T cells the two distinct populations - CD8α+ DCs and CD8α- DCs (myeloid DCs) from IFN-β-/- and IFNAR-/- mice were similarly impaired in their T cell stimulatory capacity compared to WT DCs (Fig. S1).
Deficiency in IFNs does not impair survival of cDCs in vitro
As IFNs provide cellular survival signals under certain conditions (27, 28), we first wanted to test whether the reduced ability to stimulate T cells might be due to lower survival of cDCs from IFN-β-/-or IFNAR-/- mice during the in vitro T cell stimulation assay. Splenic cDCs sensitized with OVA protein were incubated with OT I or OT II cells. After 16 and 32 hours the percentage of live cDCs was assessed by propidium iodide (PI) exclusion. WT, IFN-β-/- and IFNAR-/- cDCs were equally viable under these conditions (Fig. S2A, S2B). Thus, the reduced ability to stimulate T cells in vitro was not due to lower survival of IFN-β-/- cDCs.
Impaired stimulatory capacity of cDCs can be restored by supplementation with recombinant IFN-β in vitro or induction of IFNs with poly I:C in vivo
Next, we asked whether exogenous administration of IFNs could restore the impaired T cell stimulatory capacity of IFN-β-/- cDCs in vitro. Titration of murine recombinant IFN-β (rIFN-β) into co-cultures of IFN-β-/- cDCs and T cells showed that low amounts (0.1 U/ml) could completely restore the impaired T cell stimulatory function (Fig. 3A). However, probably due to activation of negative feedback mechanisms, addition of higher concentrations of rIFN-β (5-500 U/ml) to the co-cultures failed to restore T cell stimulatory ability of cDCs (Fig. 3A and data not shown). These results support the argument that the low levels of IFN-β produced at steady state are well optimized for maintaining of cDCs in antigen presentation competent state.
Figure 3. IFN-β is required in vitro during cDC-T cell contact as well as in vivo during cDCs development.
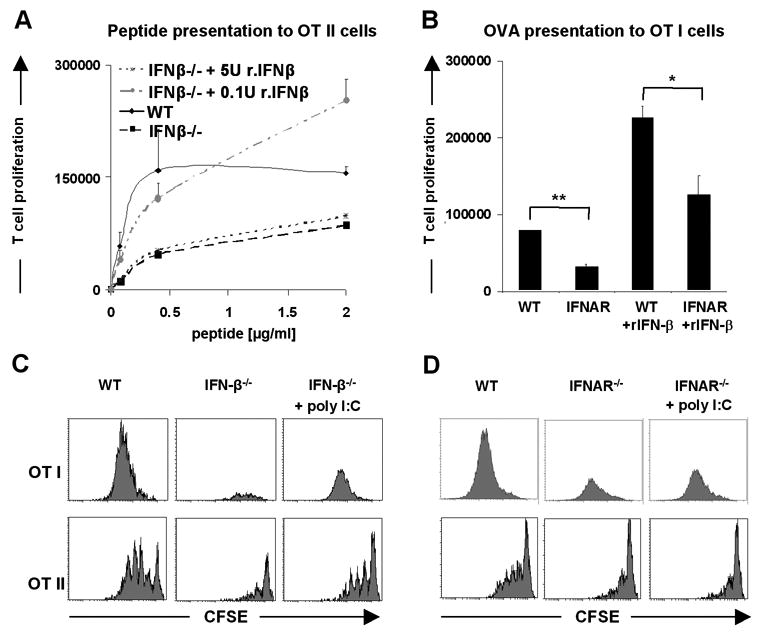
(A) Addition of low amounts of murine recombinant IFN-β (0.1 U/ml) leads to efficient recovery of impaired ability to present antigen by splenic cDCs from IFN-β-/- mice. OT I and OT II cells after purification were labeled with CFDA(SE). Splenic cDCs from WT and IFN-β-/- mice were sorted out from spleens of at least three mice, loaded with indicated concentration of OVA peptide for 1h, washed intensively and cocultured with T cells for 2.5 days in presence of recombinant murine IFN-β. (B) Enhanced proliferation of T cells accompanying impaired function of IFNAR-/- cDCs in the presence of exogenously added low amounts of rIFN-β. Splenic cDCs from WT and IFNAR-/- mice were sorted out from spleens of at least three mice per group, loaded with 250 μg/ml of OVA protein for 1h, washed intensively and cocultured with OT I T cells for 2.5 days in presence of 0.1 U/ml of recombinant murine IFN-β. (C and D) In vivo induction of IFNs by poly I:C leads to partial restoration of function of splenic cDCs from IFN-β-/- mice. WT, IFN-β-/- and IFNAR-/- mice were treated with poly I:C together with OVA or OVA alone, 24h later splenic cDCs were sorted out and incubated in vitro with CFDA(SE) labeled OT I or OT II cells for 2.5 days. The proliferative response of T cells was enumerated by flow cytometry. Data are representative of two independent experiments.
Nevertheless, in such a situation it is difficult to exclude that exogenous rIFN-β influenced T cell proliferation. A direct effect of IFNs on T cells has been well documented (28, 29) although it only partially could explain our results. We could show that IFNAR-/- cDCs which are able to produce IFN-β (data not shown), are still inefficient in activating a T cell response (Fig. 2). In addition, when such co-cultures are complemented with recombinant IFN-β the inefficiency of T cell activation remained (Fig. 3B). This clearly demonstrates that steady state production of IFN-β is required for maintenance of proper cDC function.
Furthermore, we tested whether triggering IFNs-α in vivo could compensate the impaired development of T cell stimulatory capacity of cDCs from IFN-β-/- mice. WT, IFN-β-/- and IFNAR-/- mice were intravenously injected with OVA alone or OVA together with polyinosinic-polycytidylic acid (poly I:C). After 24h, splenic cDCs were sorted and tested for their ability to activate the proliferation of OT I or OT II T cells. Data depicted in Fig. 3C show that IFNs-α induction by poly I:C compensated for the lack of IFN-β during cDC development in vivo and partially recovered their function. As expected, in vivo administration of poly I:C did not improve the stimulatory function of splenic cDCs from IFNAR-/- mice as they are completely unresponsive to IFNs signaling (Fig. 3D).
We also tested the T cell stimulatory capacity of DCs differentiated in vitro by incubating bone marrow cells with IL-4 and GM-CSF (BMDCs). After 8 days of culture we obtained around 80% CD11c positive cells from WT, IFN-β-/- and IFNAR-/- mice. Here, WT and IFN-β-/- or IFNAR-/- BMDCs were comparable in their ability to stimulate the proliferation of T cells (data not shown). This suggests that the influence of IFNs observed ex vivo is greatly dependent on the overall stimulatory context under which the DCs develop.
IFN-β-/- and IFNAR-/- cDCs display normal antigen capture and processing
A differential ability to acquire and process soluble antigen could account for the diminished stimulatory capacity of cDCs in the absence of the IFNs system. Therefore, the efficiency of splenic cDCs from WT, IFN-β-/- and IFNAR-/- mice to take up and degrade soluble OVA was assessed. We used DQ-OVA, which generates fluorescent byproducts upon degradation. As shown in Fig. 4, splenic cDCs from all groups acquired and generated comparable amounts of fluorescent DQ-OVA products. This was true for different DQ-OVA concentrations tested (data not shown). Thus, changes in antigen uptake and degradation could not account for the impaired T cell stimulatory capacity of cDCs from IFN-β-/- or IFNAR-/- mice.
Figure 4. Lack of IFN-β and IFNs signaling has no influence on proper uptake and processing of soluble antigen by splenic cDCs.
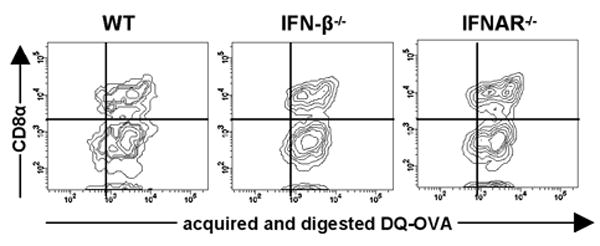
Splenic cDCs from WT, IFN-β-/- and IFNAR-/- mice were sorted out and incubated for 1h with 62.5 μg/ml of DQ-OVA. Further cells were washed and BODIPY fluorescence was measured using flow cytometry. Density plots show DQ-OVA proteolysis by splenic cDCs. Data are representative of three independent experiments and of at least three mice per group.
Similar expression of MHC and co-stimulatory molecules on cDCs from WT, IFN-β-/- and IFNAR-/- mice
The maturation status of DCs is known to be a fundamental factor for proper T cell stimulation. One of the mechanisms by which constitutive IFNs signaling could influence T cell stimulation is to enhance expression of MHC or adhesion and co-stimulatory molecules on the surface of DCs (18). However, analysis of splenic CD8α+ and CD8α- cDCs for MHC I and MHC II as well as co-stimulatory or adhesion molecules like CD86, CD80, CD40 and ICAM-1 indicated no significant differences between WT, IFN-β-/- and IFNAR-/- mice in both cDC populations (Fig. 5, Fig. S3). Therefore, the impaired function of splenic cDCs from IFN-β and IFNAR deficient mice was not due to lower expression of such surface molecules.
Figure 5. Similar surface phenotype of splenic cDCs from WT, IFN-β-/- and IFNAR-/- mice.
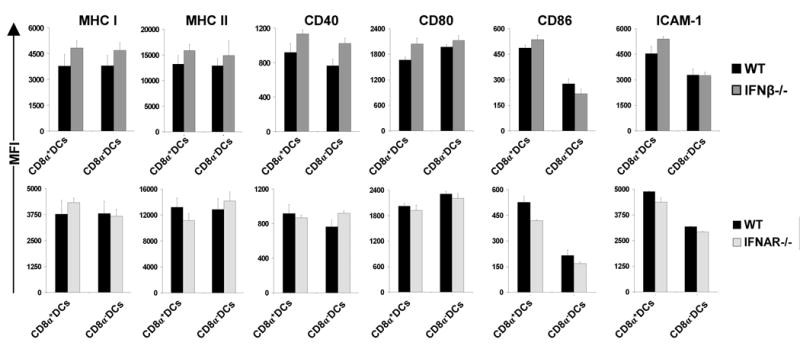
Splenocytes isolated from C57BL/6 WT, IFN-β-/- or IFNAR-/- mice were stained and gated on the basis of CD11c, CD11b, CD8α and B220 on two populations of cDCs. Graphs show MFI (Mean Fluorescence Intensity) values for expression of indicated markers. No major differences between DCs from WT, IFN-β-/- and IFNAR-/- mice were observed. MFI of each marker was measured for at least five mice per group. Data are representative of 5 independent experiments.
An intact IFNs system is required for the efficient formation of stable MHC/peptide complexes at the surface of splenic cDCs
The fact that an impaired T cell stimulatory capacity of cDCs from IFN-β-/- and IFNAR-/- mice was also found for the presentation of peptides, not requiring further processing, as well as unimpaired DQ-OVA degradation suggested that the phenotype of IFN-β-/- and IFNAR-/- cDCs was most likely due to a defect in peptide presentation rather than antigen processing steps. We therefore decided to study the MHC/peptide complexes on the surface of cDCs. We employed B3Z cells, a H-2·Kb restricted T cell hybridoma specific for the OVA epitope SIINFEKL (OVA257-264), which upon T cell receptor (TCR) activation expresses β-galactosidase (23). The activation of B3Z cells, being a hybridoma, is independent of co-stimulation (30) thus, their activation should only be dependent on the concentration of MHC I/peptide complexes recognizable by the TCR. Therefore, splenic cDCs from WT, IFN-β-/- and IFNAR-/- mice were loaded with OVA257-264 peptide and tested for their ability to activate B3Z cells. As shown in Fig. 6A and 6B, cDCs from mice deficient in IFN-β or IFNAR exhibited lower stimulatory capacity. These results confirmed that the impaired function of splenic cDCs from such mice is not due to lower levels of co-stimulatory molecules, but strongly suggested that the defect was in the process of MHC I/peptide complex formation. By using the 25-D1.16 antibody which recognizes SIINFEKL bound to the H-2·Kb molecule (24), we confirmed our interpretation. DCs from either IFN-β-/- or IFNAR-/- mice had lower levels of MHC I/SIINFEKL complexes compared to cDCs from WT mice (Fig. 6C).
Figure 6. Splenic cDCs from IFN-β-/- and IFNAR-/- mice form lower levels of MHC I/SIINFEKL complexes.
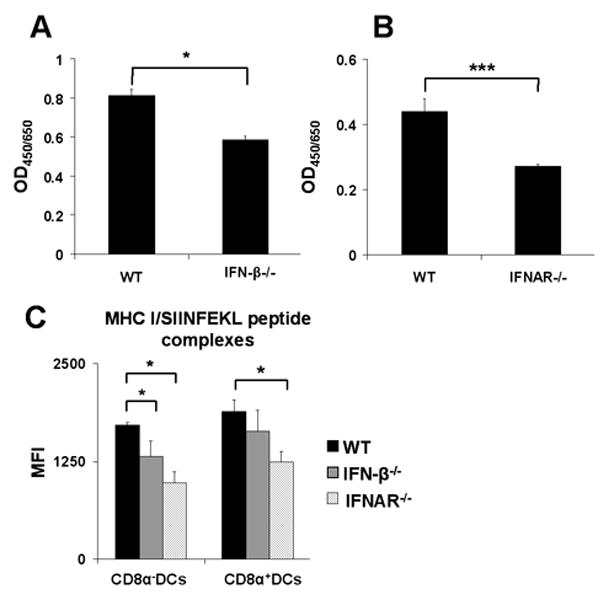
Splenic cDCs were sorted out from spleens of WT, IFN-β-/- or IFNAR-/- mice, pulsed with SIINFEKL peptide (OVA257-264) for 1h, washed intensively and (A, B) co-cultured with the SIINFEKL/H-2Kb restricted B3Z hybridoma T cells for 24h. Cells were then lysed and monitored for LacZ expression by the introduction of ONPG substrate. Optical density was measured at 450 nm with wavelength correction set at 650 nm. (C) cDCs were stained with 25-D1.16 antibody, MFI was measured by flow cytometry, graphs show values for 100 ng/ml of SIINFEKL peptide, cells were gated on CD8α+DCs and myeloid DCs (CD8α-). Results are representative of at least three mice for WT, IFN-β-/- and IFNAR-/- in three independent experiments. Statistical significance was determined using the paired Student's t test. * P<0.05; *** P<0.005
Deficiencies in the IFNs system leads to decreased expression of Hsp70 in splenic cDCs
To understand the molecular basis for the decreased formation of MHC/petide complexes, splenic DCs RNA from WT, IFN-β-/- and IFNAR-/- mice was analyzed by microarrays for expression of genes known to be involved in antigen processing and presentation, co-stimulation or IFNs response. Extensive analysis of the microarrays indicated that most of these genes were unaltered in cDCs from the knockout mice (Fig. S4A-C). The only dramatic difference found was in the expression of the heat shock protein Hsp70.1 and Hsp70.3 genes. The expression of these two genes was significantly lower in cDCs from IFN-β-/- (approximately 15-20 fold down-regulated in comparison to WT) and IFNAR-/- (approximately 75-150 fold down-regulated in comparison to WT) mice (Fig. 7A).
Figure 7. Microarray analysis of splenic cDCs from WT, IFN-β-/- and IFNAR-/- mice showed down-regulation of heat shock protein 70.1 and 70.3 (Hsp70.1 and Hsp70.3) in knockout mice.
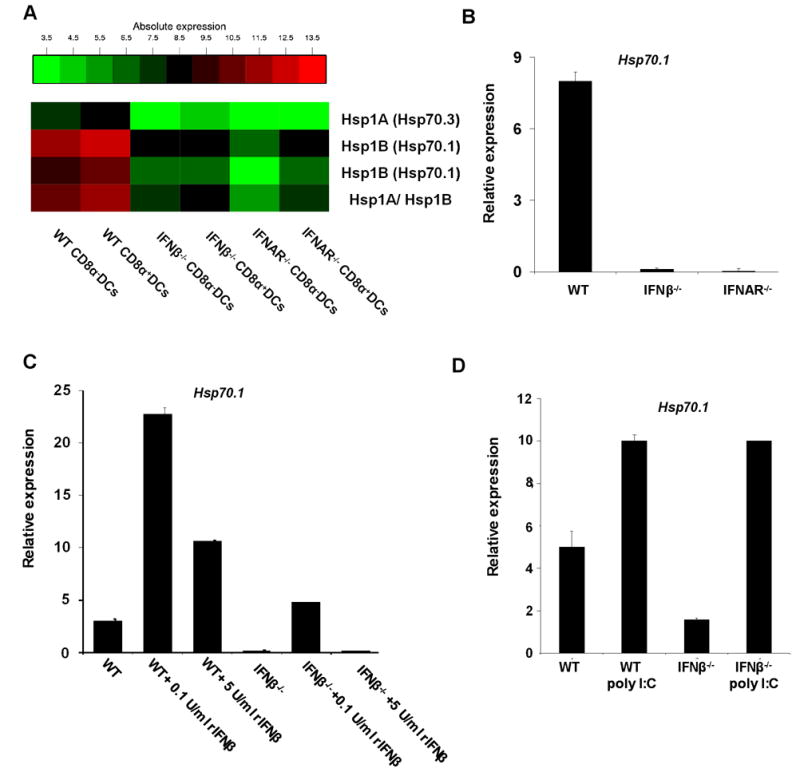
(A) Affymetrix gene array analysis of splenic cDCs. RNA was prepared from sorted splenic CD8α+DCs and myeloid DCs (CD8α-) from WT, IFN-β-/- and IFNAR-/- mice. All samples were repeated twice with individually sorted cells and averaged. (B) Quantitative real-time PCR (qRT-PCR) of RNA from splenic cDCs sorted from WT, IFN-β-/- and IFNAR-/- mice. (C) qRT-PCR of RNA from splenic cDCs untreated and in vitro treated with 0.1 U/ml and 5 U/ml of murine recombinant IFN-β for 3h. (D) qRT-PCR of RNA from splenic cDCs 24h after administration of poly I:C. Results are representative of at least three samples from WT, IFN-β-/- and IFNAR-/- mice in three independent experiments.
To verify the above findings, we first stained for intracellular Hsp70 protein. As shown in Fig. S5 levels of Hsp70 were indeed lower in IFN-β-/- and IFNAR-/- cDCs compared to WT cDCs. Most likely due to presence of other highly homologous members of the Hsp70 family and the low sensitivity of the antibody, differences were not very pronounced. However, in confirmation of the microarray data, transcriptional levels of Hsp70.1 were severely decreased in DCs from IFN-β-/- and IFNAR-/- mice in comparison to WT (Fig. 7B). Moreover, treatment with low amounts of rIFN-β (0.1 U/ml) increased Hsp70 levels in both WT and IFN-β-/- DCs, whereas treatment with 5 U/ml of rIFN-β did not markedly change the Hsp70 levels (Fig. 7C). This correlates well with the functional restoration of IFN-β-/- cDCs at low, but not at high concentrations of rIFN-β. Consistent with this finding, 24h of poly I:C administration up-regulated Hsp70.1 levels in WT as well as in IFN-β-/- cDCs (Fig. 7D).
Inhibition of Hsp70 by 15-deoxyspergualin leads to impairment of antigen presentation
To test for a possible causative link between Hsp70 expression and antigen presentation, we used 15-deoxyspergualin (DSG), a pharmacological inhibitor of Hsp70. DSG is a synthetic derivative of spergualin from Bacillus laterosporus and binds to Hsp70 and Hsp90 (31-33). Therefore, we treated mice for 6 days with DSG and then tested the splenic cDCs of such mice for Hsp70 expression. Intracellular staining revealed that DSG treatment led to partial reduction of Hsp70 level in WT cDCs (Fig. 8A). In contrast, expression of surface molecules involved in T cell stimulation was not affected by this treatment (Fig. S6).
Figure 8. In vivo inhibition of Hsp70 by 15-deoxyspergualin or knockout of Hsp70.1 and Hsp70.3 leads to impaired antigen presentation by splenic cDCs.
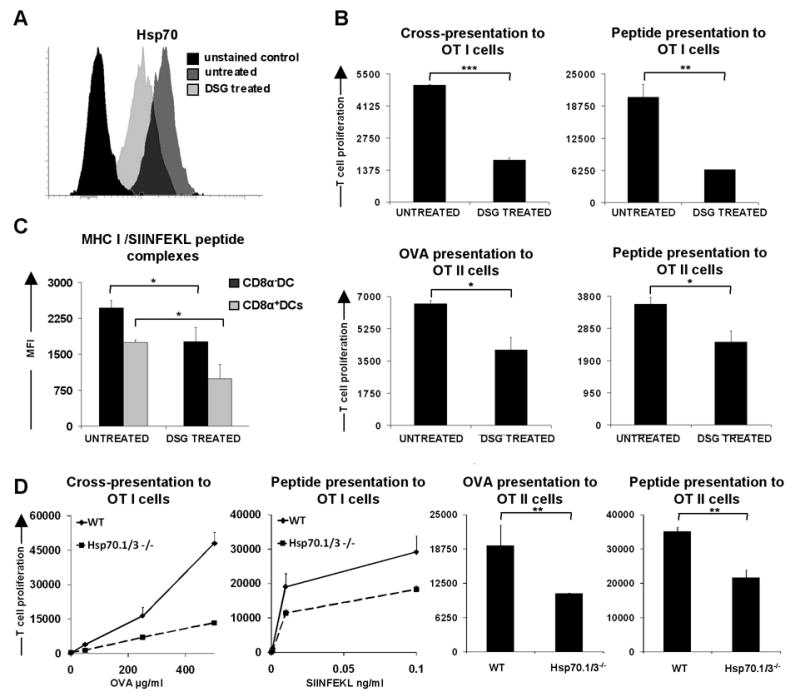
(A) Intracellular staining of Hsp70 in splenic cDC from mice untreated and treated with 15-deoxyspergualin. (B) Antigen presentation assay with splenic cDCs from untreated mice and mice treated with 15-deoxyspergualin. Splenic cDCs were incubated with purified, CFDA(SE) labeled OT I or OTII cells for 1.5 days (class I restricted peptide) and 2.5 days. OVA257-264 peptide concentration 10 ng/ml, OVA323-339 2 μg/ml, OVA protein 1mg/ml (class II presentation) or 500 μg/ml (class I presentation). The proliferative response of T cells was enumerated by flow cytometry. Data are representative of at least five mice for WT untreated and DSG treated in three independent experiments. (C) DCs from untreated and DSG treated mice were loaded for 1h with 100 ng/ml of SIINFEKL peptide, washed and stained with 25-D1.16 antibody recognizing MHC I/SIINFEKL peptide complexes. Graph shows MFI values for five mice per group. Data are representative of two independent experiments. (D) Antigen presentation assay with splenic cDCs sorted from Hsp70.1/3-/- and WT mice. Cells were loaded with appropriate OVA derived peptides or whole OVA, washed and incubated in vitro with CFDA(SE) labeled OT I or OTII cells for 1.5 days (class I restricted peptide) and 2.5 days. OVA323-339 peptide concentration 2 μg/ml, OVA protein 1mg/ml (MHC II presentation). The proliferative response of T cells was enumerated by flow cytometry. Data are representative of 3 mice for Hsp70.1/3-/- and WT in two independent experiments. Statistical significance was determined using the paired Student's t test. * P<0.05; ** P<0.01; *** P<0.005
We then tested cDCs isolated from DSG treated mice for their capacity to stimulate CD4+ and CD8+ T cells. Clearly, cDCs from DSG treated mice exhibited a reduced ability to stimulate OT I or OT II T cells compared to cDCs from untreated mice, independent of whether protein or peptides were used as an antigen (Fig. 8B). This was consistent with the claims that DSG abrogates the ability to present antigen in the context of both MHC I and MHC II (31-34).
In addition, compared to untreated control, cDCs from DSG treated mice revealed lower surface levels of MHC/peptide complexes (Fig. 8C) suggesting that Hsp70 is necessary for efficient formation of MHC/peptide complexes. Thus, by supporting the expression of Hsp70, constitutive IFN-β expression in vivo helps to maintain cDCs in a primed and competent state for antigen presentation.
Splenic dendritic cells from Hsp70.1/3-/- mice are impaired in T cell stimulation
In order to explicitly demonstrate the involvement of the Hsp70.1 and Hsp70.3 proteins in antigen presentation and thus confirm that in the absence of IFN-β or IFNs signaling down-regulation of Hsp70 result in impaired antigen presentation we utilized Hsp70.1/3 double knockout mice (22). The surface phenotype of cDCs from Hsp70.1/3 -/- mice appeared to be very similar to the surface phenotype of cDCs from WT (data not shown). To test the antigen presentation capacity of Hsp70.1/3 -/- cDCs, cells were sorted and loaded with appropriate OVA peptides or whole protein and incubated with OT I or OT II transgenic T cells. The results clearly show that cDCs from Hsp70.1/3-/- are impaired in their ability to present OVA derived peptides as well as whole protein to naive T cells when compared to WT cDCs (Fig. 8D). This further substantiates our finding that down-regulation of Hsp70 in the absence of IFN-β or IFNs can alter antigen presentation. The discovery that IFNs signaling regulates MHC/peptide complex formation by Hsp70 proteins highlights a hitherto unrecognized mechanism via which IFNs might regulate presentation of self antigens in the steady state and has, therefore, important consequences for our understanding of how regular homeostatic conditions are maintained in the immune system.
Discussion
IFNs, found in high amounts in cells exposed to viruses, were first characterized and named as such on the basis of their antiviral activity. It is now well known that IFNs have widely overlapping, pleiotropic and immunomodulatory effects and their production is not the sole preserve of viral infections but they are also induced in response to bacterial and parasitic infections (12, 15, 16). IFNs represent important immunomodulators for the innate as well as the adaptive arm of the immune system (16, 18). They exert broad regulatory effects and various subtypes of DCs are affected by these cytokines (12, 15-18). For instance, IFN-α can promote antigen cross-presentation by enhancing endosomal processing, up-regulating the expression of co-stimulatory molecules and augmenting dendritic cell viability in settings of viral infection (18, 27, 29, 35-37). Additionally, direct stimulation of T cells by IFN-α has been shown to be essential for efficient induction of cross-priming (29). Furthermore, IFN-α/β were described as crucial survival factors for activated T cells (28). Importantly apart from that, even in the absence of infection spontaneous low level production of IFN-β has been shown to occur (12).
The host response elicited by IFNs is largely dependent on signal strength. Most of the studies carried out to date have focused on the cellular effects induced by the high levels of IFNs elicited under inflammatory conditions. However, whether the low levels of IFNs produced under non-inflammatory conditions have an important housekeeping immune function is not known. We now show that the low but constitutive production of IFN-β is necessary for maintaining DCs in a state competent for antigen presentation. Compared to those from WT mice, DCs freshly isolated from spleens of IFN-β-/- and IFNAR-/- mice were found to be highly impaired in antigen presentation to CD4+ and CD8+ T cells. This defect could in part be rectified with exogenous rIFN-β or through in vivo induction of IFNs-α using synthetic dsRNA. Interestingly, restoration of function was possible with extremely low amounts of rIFN-β, probably mimicking amounts produced constitutively. Failure of IFN-β-/- DCs cultured with high rIFN-β levels to activate T cell proliferation might be attributed to negative feedback mechanisms activated by a strong IFN-β signal.
The function of DCs is not only influenced by cytokines present in their environment, but also by other cells of the immune system, particularly T cells. Therefore, splenic cDCs isolated from IFN-β deficient mice theoretically could be altered due to an effect of IFN-β deficiency on T cells. It was shown before that IFN-α and IFN-β have direct effect on activated T cells and prevent their death during inflammatory conditions (28). Activated T cells can provide feedback signals to DCs and induce their maturation. This can be mediated by cytokines produced by T cells as well as by cell-cell interactions, including CD40L-CD40 interactions (38). However, this phenomenon is unlikely to play a significant role in steady state conditions because of the lack of activated T cells. T cell mediated conditioning of DCs via B7-H1 in homeostasis has recently been shown to play an important role in inducing DC maturation (39). However, as surface expression of B7-H1 was not altered on IFN-β-/- and IFNAR-/- cDCs in comparison to WT cDCs, one may assume that this is not the case in our situation. Moreover, we demonstrated that rIFN-β treatment in vitro can regulate cDCs function and Hsp70 levels.
Potential roles of IFNs in antigen presentation have previously been postulated. However, such effects have only been observed for high levels of IFNs reminiscent of inflammation. For instance, induction of IFNs in DCs by dsRNA and LPS in vitro was shown to up-regulate costimulatory and MHC molecules hence enhancing their ability to activate CD8+ T cells (17). Conversely, our results show that the antigen presentation defects in splenic IFN-β-/- and IFNAR-/- DCs are neither due to low expression of co-stimulatory and MHC molecules nor a block in antigen capture and processing. We demonstrate that the defect is due to a blockade in a step downstream of antigen processing: MHC/peptide complex formation. Furthermore, the data presented here strongly suggest that impairment in the MHC/peptide complex formation step caused by the absence of IFNs signaling is due to down-regulation of Hsp70.
The 70 kD Hsps Hsp70.1 and Hsp70.3 belong to a larger, highly homologous and conserved gene family whose expression can be significantly induced in response to a number of pathophysiological conditions including pathogen exposure (40). In addition to a generalized role in protein folding and transport, the proteins also have distinct functions in the promotion of antigen processing and presentation (31, 41-43). Hsp70 proteins are involved in chaperoning proteins/peptides during degradation and during antigen presentation via MHC I as well as via MHC II. Hsp70 has been shown to physically associate with the transporter associated with antigen processing (TAP) hence enabling efficient loading of chaperoned peptides onto MHC I molecules (31, 41, 44). In addition, a role for Hsp70 in antigen presentation by MHC II molecules has also been described (31, 45).
By microarray and quantitative real-time PCR (qRT-PCR) analysis we found that in the absence of a functional IFNs system in vivo, the expression of Hsp70.1 and Hsp.70.3 in splenic cDCs was significantly down-regulated. In addition, partial blockade of Hsp70 protein in WT cDCs using the pharmacological inhibitor DSG resulted in a diminished ability to activate T cells. This correlated with a decrease in the surface MHC/peptide complexes on WT splenic DCs after treatment with DSG. These results were further substantiated when we analyzed the ability of cDCs from Hsp70.1/3-/- mice to present soluble OVA to naive CD4+ and CD8+ T cells. The function of such cDCs was highly impaired when compared to WT cDCs, suggesting that indeed altered levels of Hsp70 in IFN-β-/- and IFNAR-/- cDCs are responsible for impaired antigen presentation in the steady state.
From data presented here, one could expect that in the absence of chaperones, like Hsp70, when formation of antigenic MHC/peptide complexes is impaired, total levels of surface MHC molecules should be diminished. Nevertheless, our experiments show no differences between WT, IFN-β-/- and IFNAR-/- cDCs in total surface MHC I and MHC II expression. Likewise, the analysis of the surface phenotype of Hsp70.1/3-/- cDCs showed no significant difference in comparison to WT cDCs. Possibly, Hsp70 might be required for efficient presentation of particular peptides, like the ones employed in our study, but not for others. Therefore, lack of Hsp70 might not reflect in changes of total surface MHC levels. The exact molecular reason for this phenomenon remains to be elucidated.
The discovery that basal IFNs production regulates efficiency of MHC/peptide complex formation via the expression of Hsp70 is a novel and important finding not only in the context of pathogen recognition but also in homeostasis. Not all pathogens are associated with robust IFNs production. Therefore by sustaining Hsp70 expression, constitutively produced IFNs probably ensure that DCs are kept in a primed state for efficient presentation of antigens from such pathogens. An equally or more important function could be in the maintenance of tolerance to self antigens. DCs that capture and present antigen under non-inflammatory conditions are generally believed to acquire tolerogenic properties and generate regulatory T lymphocytes that potentiate tolerogenic responses.
Supplementary Material
Acknowledgments
We thank Dr Andreas Krueger from Hannover Medical School for critically reading this manuscript, Regina Lesch and Susanne zur Lage for their excellent technical assistance.
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
This work was supported in part by the Kultusministerium of Niedersachsen via the Lichtenberg PhD program, the German Research Counsil (DFG), the Marie Curie Action Miditrain MEST-CT-2004-504990, the Helmholtz Gemeinschaft via HIRSIB, the Deutsche Krebshilfe and the National Institutes of Health (CA10445 and CA123232).
Abbreviations used in this paper: DC, dendritic cells; IFNs, type I interferons; IFNAR, type I IFNs receptor; cDCs, conventional dendritic cells; Hsp, heat shock protein; rIFN-β, recombinant murine IFN-β; poly I:C, polyinosinic-polycytidylic acid; DSG, 15-deoxyspergualin;
This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
Reference List
- 1.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 2.Liu K, Waskow C, Liu XT, Yao KH, Hoh J, Nussenzweig M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nature Immunology. 2007;8:578–583. doi: 10.1038/ni1462. [DOI] [PubMed] [Google Scholar]
- 3.Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. 2007;7:543–555. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 4.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, Steinman RM, Nussenzweig MC. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315:107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 5.Geissmann F. The origin of dendritic cells. Nature Immunology. 2007;8:558–560. doi: 10.1038/ni0607-558. [DOI] [PubMed] [Google Scholar]
- 6.Ardavin C. Origin, precursors and differentiation of mouse dendritic cells. Nat Rev Immunol. 2003;3:582–590. doi: 10.1038/nri1127. [DOI] [PubMed] [Google Scholar]
- 7.Lin ML, Zhan Y, Villadangos JA, Lew AM. The cell biology of cross-presentation and the role of dendritic cell subsets. Immunol Cell Biol. 2008;86:353–362. doi: 10.1038/icb.2008.3. [DOI] [PubMed] [Google Scholar]
- 8.Guermonprez P, Amigorena S. Pathways for antigen cross presentation. Springer Seminars in Immunopathology. 2005;26:257–271. doi: 10.1007/s00281-004-0176-0. [DOI] [PubMed] [Google Scholar]
- 9.Wilson NS, Behrens GMN, Lundie RJ, Smith CM, Waithman J, Young L, Forehan SP, Mount A, Steptoe RJ, Shortman KD, Koning-Ward TF, Belz GT, Carbone FR, Crabb BS, Heath WR, Villadangos JA. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nature Immunology. 2006;7:165–172. doi: 10.1038/ni1300. [DOI] [PubMed] [Google Scholar]
- 10.Sriram U, Biswas C, Behrens EM, Dinnall JA, Shivers DK, Monestier M, Argon Y, Gallucci S. IL-4 suppresses dendritic cell response to type I interferons. J Immunol. 2007;179:6446–6455. doi: 10.4049/jimmunol.179.10.6446. [DOI] [PubMed] [Google Scholar]
- 11.Young LJ, Wilson NS, Schnorrer P, Mount A, Lundie RJ, La Gruta NL, Crabb BS, Belz GT, Heath WR, Villadangos JA. Dendritic cell preactivation impairs MHC class II presentation of vaccines and endogenous viral antigens. Proc Natl Acad Sci U S A. 2007;104:17753–17758. doi: 10.1073/pnas.0708622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taniguchi T, Takaoka A. A weak signal for strong responses: Interferon-alpha/beta revisited. Nature Reviews Molecular Cell Biology. 2001;2:378–386. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- 13.Samuelsson CV, Lienenklaus S, Muller PP, Zawatzky R, Hauser H, Weiss S. Transformation of mouse fibroblasts alters the induction pattern of type I IFNs after virus infection. Biochem Biophys Res Commun. 2005;335:584–589. doi: 10.1016/j.bbrc.2005.07.124. [DOI] [PubMed] [Google Scholar]
- 14.Erlandsson L, Blumenthal R, Eloranta ML, Engel H, Alm G, Weiss S, Leanderson T. Interferon-beta is required for interferon-alpha production in mouse fibroblasts. Curr Biol. 1998;8:223–226. doi: 10.1016/s0960-9822(98)70086-7. [DOI] [PubMed] [Google Scholar]
- 15.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nature Reviews Immunology. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 16.Le Bon A, Tough DF. Links between innate and adaptive immunity via type I interferon. Current Opinion in Immunology. 2002;14:432–436. doi: 10.1016/s0952-7915(02)00354-0. [DOI] [PubMed] [Google Scholar]
- 17.Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nature Immunology. 2004;5:971–974. doi: 10.1038/ni1004-971. [DOI] [PubMed] [Google Scholar]
- 18.Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, Tough DF. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood. 2002;99:3263–3271. doi: 10.1182/blood.v99.9.3263. [DOI] [PubMed] [Google Scholar]
- 19.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 20.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 21.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 22.Hunt CR, Dix DJ, Sharma GG, Pandita RK, Gupta A, Funk M, Pandita TK. Genomic instability and enhanced radiosensitivity in Hsp70.1-and Hsp70.3-deficient mice. Molecular and Cellular Biology. 2004;24:899–911. doi: 10.1128/MCB.24.2.899-911.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karttunen J, Sanderson S, Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc Natl Acad Sci U S A. 1992;89:6020–6024. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 1997;6:715–726. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- 25.Wilson NS, El Sukkari D, Belz GT, Smith CM, Steptoe RJ, Heath WR, Shortman K, Villadangos JA. Most lymphoid organ dendritic cell types are phenotypically and functionally immature. Blood. 2003;102:2187–2194. doi: 10.1182/blood-2003-02-0513. [DOI] [PubMed] [Google Scholar]
- 26.Ostling P, Bjork JK, Roos-Mattjus P, Mezger V, Sistonen L. Heat shock factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J Biol Chem. 2007;282:7077–7086. doi: 10.1074/jbc.M607556200. [DOI] [PubMed] [Google Scholar]
- 27.Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, Tough DF. Cross-priming of CD8(+) T cells stimulated by virus-induced type I interferon. Nature Immunology. 2003;4:1009–1015. doi: 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 28.Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. Journal of Experimental Medicine. 1999;189:521–529. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, Kalinke U, Tough DF. Direct stimulation of T cells by type IIFN enhances the CD8(+) T cell response during cross-priming. Journal of Immunology. 2006;176:4682–4689. doi: 10.4049/jimmunol.176.8.4682. [DOI] [PubMed] [Google Scholar]
- 30.Wells JW, Cowled CJ, Darling D, Guinn BA, Farzaneh F, Noble A, Galea-Lauri J. Semi-allogeneic dendritic cells can induce antigen-specific T-cell activation, which is not enhanced by concurrent alloreactivity. Cancer Immunol Immunother. 2007;56:1861–1873. doi: 10.1007/s00262-007-0328-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li ZH, Menoret A, Srivastava P. Roles of heat-shock proteins in antigen presentation and cross-presentation. Current Opinion in Immunology. 2002;14:45–51. doi: 10.1016/s0952-7915(01)00297-7. [DOI] [PubMed] [Google Scholar]
- 32.Nadler SG, Tepper MA, Schacter B, Mazzucco CE. Interaction of the immunosuppressant deoxyspergualin with a member of the Hsp70 family of heat shock proteins. Science. 1992;258:484–486. doi: 10.1126/science.1411548. [DOI] [PubMed] [Google Scholar]
- 33.Diegel ML, Nadler SG, Kiener PA. In vivo administration of 15-deoxyspergulin inhibits antigen-presenting cell stimulation of T cells and NF-kappaB activation. Int Immunopharmacol. 2002;2:1451–1464. doi: 10.1016/s1567-5769(02)00090-5. [DOI] [PubMed] [Google Scholar]
- 34.Hoeger PH, Tepper MA, Faith A, Higgins JA, Lamb JR, Geha RS. Immunosuppressant deoxyspergualin inhibits antigen processing in monocytes. J Immunol. 1994;153:3908–3916. [PubMed] [Google Scholar]
- 35.Le Bon A, Tough DF. Type I interferon as a stimulus for cross-priming. Cytokine Growth Factor Rev. 2008;19:33–40. doi: 10.1016/j.cytogfr.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 36.Beignon AS, Skoberne M, Bhardwaj N. Type I interferons promote cross-priming: more functions for old cytokines. Nature Immunology. 2003;4:939–941. doi: 10.1038/ni1003-939. [DOI] [PubMed] [Google Scholar]
- 37.Cho HJ, Hayashi T, Datta SK, Takabayashi K, Van Uden JH, Horner A, Corr M, Raz E. IFN-alpha beta promote priming of antigen-specific CD8(+) and CD4(+) T lymphocytes by immunostimulatory DNA-based vaccines. Journal of Immunology. 2002;168:4907–4913. doi: 10.4049/jimmunol.168.10.4907. [DOI] [PubMed] [Google Scholar]
- 38.Sporri R, Reis e Sousa Newly activated T cells promote maturation of bystander dendritic cells but not IL-12 production. J Immunol. 2003;171:6406–6413. doi: 10.4049/jimmunol.171.12.6406. [DOI] [PubMed] [Google Scholar]
- 39.Talay O, Shen CH, Chen L, Chen J. B7-H1 (PD-L1) on T cells is required for T-cell-mediated conditioning of dendritic cell maturation. Proc Natl Acad Sci U S A. 2009;106:2741–2746. doi: 10.1073/pnas.0813367106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Javid B, MacAry PA, Lehner PJ. Structure and function: heat shock proteins and adaptive immunity. J Immunol. 2007;179:2035–2040. doi: 10.4049/jimmunol.179.4.2035. [DOI] [PubMed] [Google Scholar]
- 41.Binder RJ, Srivastava PK. Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat Immunol. 2005;6:593–599. doi: 10.1038/ni1201. [DOI] [PubMed] [Google Scholar]
- 42.Kuppner MC, Gastpar R, Gelwer S, Nossner E, Ochmann O, Scharner A, Issels RD. The role of heat shock protein (hsp70) in dendritic cell maturation: Hsp70 induces the maturation of immature dendritic cells but reduces DC differentiation from monocyte precursors. European Journal of Immunology. 2001;31:1602–1609. doi: 10.1002/1521-4141(200105)31:5<1602::AID-IMMU1602>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 43.Manara GC, Sansoni P, Badiali-De Giorgi L, Gallinella G, Ferrari C, Brianti V, Fagnoni FF, Ruegg CL, De Panfilis G, Pasquinelli G. New insights suggesting a possible role of a heat shock protein 70-kD family-related protein in antigen processing/presentation phenomenon in humans. Blood. 1993;82:2865–2871. [PubMed] [Google Scholar]
- 44.Chen D, Androlewicz MJ. Heat shock protein 70 moderately enhances peptide binding and transport by the transporter associated with antigen processing. Immunol Lett. 2001;75:143–148. doi: 10.1016/s0165-2478(00)00294-7. [DOI] [PubMed] [Google Scholar]
- 45.Panjwani N, Akbari O, Garcia S, Brazil M, Stockinger B. The HSC73 molecular chaperone: Involvement in MHC class II antigen presentation. Journal of Immunology. 1999;163:1936–1942. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.