

The crystal structure of PCNA from the halophilic archaeon H. volcanii reveals specific features of the charge distribution on the protein surface that reflect adaptation to a high-salt environment and suggests a different type of interaction with DNA in halophilic PCNAs.
Keywords: PCNA–DNA interactions, sliding clamps, halophilic environment
Abstract
The sliding clamp proliferating cell nuclear antigen (PCNA) plays vital roles in many aspects of DNA replication and repair in eukaryotic cells and in archaea. Realising the full potential of archaea as a model for PCNA function requires a combination of biochemical and genetic approaches. In order to provide a platform for subsequent reverse genetic analysis, PCNA from the halophilic archaeon Haloferax volcanii was subjected to crystallographic analysis. The gene was cloned and expressed in Escherichia coli and the protein was purified by affinity chromatography and crystallized by the vapour-diffusion technique. The structure was determined by molecular replacement and refined at 3.5 Å resolution to a final R factor of 23.7% (R free = 25%). PCNA from H. volcanii was found to be homotrimeric and to resemble other homotrimeric PCNA clamps but with several differences that appear to be associated with adaptation of the protein to the high intracellular salt concentrations found in H. volcanii cells.
1. Introduction
Proliferating cell nuclear antigen (PCNA), also known as a sliding clamp, plays vital roles in many aspects of DNA replication, repair and recombination in eukaryotic and archaeal cells (Johnson & O’Donnell, 2005 ▶). PCNA is a trimeric ringed-shaped oligomer that encircles double-stranded DNA (dsDNA), along which it can freely slide. The loading of PCNA onto DNA requires that the ring is first opened and then re-closed in an ATP-dependent reaction catalysed by the replication factor C (RFC) complex. When loaded onto dsDNA, PCNA acts as a platform on which other protein factors assemble. Many of these proteins interact with PCNA via a short peptide sequence (called the PCNA-interacting protein peptide or PIP peptide) that binds tightly to a hydrophobic pocket on the surface of the PCNA trimer (Warbrick, 1998 ▶). These interactions are conserved in both eukaryotic and archaeal systems (Johnson & O’Donnell, 2005 ▶).
Structures of eukaryotic PCNA complexes have been determined for the budding yeast Saccharomyces cerevisiae (Krishna et al., 1994 ▶) and human (Gulbis et al., 1996 ▶; Kontopidis et al., 2005 ▶). In each case, the structures revealed a pseudo sixfold-symmetrical architecture with each PCNA monomer comprising two topologically identical domains. The central cavity of the ring, through which the dsDNA passes, is predominantly positively charged. Additional available structures show the interactions of yeast PCNA with the pentameric RFC complex (Bowman et al., 2004 ▶), as well as with the PIP peptide derived from the DNA ligase I protein Cdc9 (Vijayakumar et al., 2007 ▶), and the interactions of human PCNA with PIP peptides derived from DNA polymerases δ, η, ι and κ (Bruning & Shamoo, 2004 ▶; Hishiki et al., 2009 ▶), the DNA-replication and cell-cycle inhibitor p21Cip1 (Gulbis et al., 1996 ▶) and the nuclease Fen1 (Bruning & Shamoo, 2004 ▶; Sakurai et al., 2005 ▶).
The archaeal chromosomal DNA replication machinery resembles a simplified form of that found in eukaryotic cells and has attracted attention as a model system for studying the processes of DNA replication at the molecular level (Grabowski & Kelman, 2003 ▶). Archaeal PCNA complexes have also been identified and subjected to extensive biochemical and structural analysis. Crystal structures have been obtained for the homotrimeric PCNA complexes of the euryarchaeal species Archaeoglobus fulgidus (Chapados et al., 2004 ▶) and Pyrococcus furiosus (Matsumiya et al., 2001 ▶, 2002 ▶) as well as for the heterotrimeric PCNA from the crenarchaeal species Sulfolobus solfataricus (Williams et al., 2006 ▶). PIP peptide-bound structures are also available for A. fulgidus and P. furiosus PCNAs bound to PIP peptides derived from Fen1 and RFC, respectively (Chapados et al., 2004 ▶; Matsumiya et al., 2002 ▶), and for S. solfataricus PCNA bound to PIP peptides from Fen1 (Williams et al., 2006 ▶) and the translesion DNA polymerase Dpo4 (Xing et al., 2009 ▶).
Realising the full potential of archaea as a model for PCNA function requires a coupling of biochemical and genetic approaches, yet this is hindered by the fact that the majority of well studied archaeal organisms are refractive to genetic analysis (Allers & Mevarech, 2005 ▶). However, this is not the case for several of the halophilic euryarchaea, such as Haloferax volcanii. This organism, which was first isolated in the Dead Sea, is an obligate halophile that grows optimally under aerobic conditions at 318 K in a medium containing 20% NaCl (Mullakhanbhai & Larsen, 1975 ▶). Under these conditions, the internal KCl concentration of the cells has been measured as 3–4 M (reviewed in Muller & Oren, 2003 ▶). A wide range of materials and methods for genetic manipulation of H. volcanii have been described, including an efficient transformation system, plasmid vectors, selectable and counter-selectable markers, constitutive and regulatable promoters and reporter genes (reviewed in Allers & Mevarech, 2005 ▶).
Recently, we initiated a reverse genetic analysis of H. volcanii PCNA (HfxPCNA) in order to allow us to probe questions of protein structure and function in living archaeal cells (unpublished results). Rather than rely on the structures of PCNA complexes from the hyperthermophilic species P. furiosus and A. fulgidus to guide our mutational analysis, we subjected HfxPCNA to structure determination by X-ray crystallography. In the present report, we describe the three-dimensional structure of PCNA from H. volcanii at 3.5 Å resolution, compare it with orthologues from different organisms and consider how this protein has adapted to its natural high-salt environment.
2. Material and methods
2.1. Gene cloning, expression and purification of HfxPCNA
The PCNA ORF was amplified by PCR from genomic DNA prepared from H. volcanii strain DS70 (Wendoloski et al., 2001 ▶) using the following oligonucleotide primers: HFXPCNA-5SAN (5′-GGTTGGTTGGGGGACCCGGGATGTTCAAGGCCATC-3′) and HFXPCNA-3BAM (5′-GGTGGTGGTTGGATCCTCAGTCGCTCTGGATGCG-3′). The PCR product was digested with the restriction enzymes SanDI and BamHI (sites shown in bold in the oligonucleotide sequences) and cloned into plasmid pET47b (Novagen) that had previously been digested with the same two enzymes so that the 247-amino-acid PCNA protein could be expressed as a fusion protein with a 20-amino-acid N-terminal sequence that included a hexahistidine tag (MAHHHHHHSAAELVLFQGPG). Expression was carried out in Escherichia coli Rosetta 2 (DE3) [pLysS] cells (Novagen). 1 l cultures were grown to mid-exponential phase (OD = 0.8) at 310 K and chilled on ice for 10 min before expression was induced by the addition of IPTG to 1 mM. Growth was continued for 18 h at 293 K, after which the cells were harvested by centrifugation, resuspended in lysis buffer (20 mM NaH2PO4, 20 mM imidazole, 2.0 M KCl pH 8.0) and lysed by sonication. Cleared extracts were then subjected to affinity chromatography using Ni–NTA agarose (GE Healthcare) at room temperature. Following extensive washing with lysis buffer, bound HfxPCNA was eluted in 20 mM NaH2PO4, 500 mM imidazole, 2.0 M KCl pH 8.0. Peak fractions were pooled and dialysed against 20 mM NaH2PO4, 20 mM imidazole, 2.0 M KCl pH 8.0 (Fig. 1 ▶ a).
Figure 1.

(a) Purified recombinant HfxPCNA (5 µg) visualized by PAGE Blue 90 staining following SDS–PAGE. The positions of molecular-weight markers (in kDa) are shown to the right. (b) Crystals of HfxPCNA belonging to monoclinic space group C2, with unit-cell parameters a = 174.4, b = 123.2, c = 123.3 Å, α = 90, β = 135, γ = 90°.
2.2. Crystallization and data collection
The concentrated protein was crystallized by the sitting-drop vapour-diffusion method at 293 K. Crystallization conditions were searched for using Hampton Research screening kits. Each droplet was made by mixing 1 µl reservoir solution with 1 µl protein solution, which was at a concentration of 4 mg ml−1 in 20 mM NaH2PO4 pH 8 containing 2 M potassium chloride and 20 mM imidazole. Crystals of cubic shape suitable for X-ray analysis were obtained using two different crystallization conditions: 1.6 M sodium citrate and 4 M sodium malonate. Both salt solutions were saturated, which fitted with the halophilicity of the investigated protein. Thus, neither different buffers nor additives could be used in order to improve the quality of the crystals. Cubic-shaped crystals grew in one month to a size of 0.15 mm only (Fig. 1 ▶ b). Further attempts to find different crystallization conditions were not successful. Unfortunately, the slightly larger crystals obtained using sodium citrate were not suitable for deep freezing; thus, we were forced to use the smaller crystals grown in sodium malonate for data collection at 100 K. The crystals appeared to be single; however, X-ray exposure of these crystals and data processing revealed twinned lattices from their diffraction images.
A diffraction data set was collected from a single crystal at a temperature of around 100 K on beamline ID23-2 at the European Synchrotron Research Facility (ESRF; Grenoble, France) using the sodium malonate reservoir solution as a cryoprotectant. Statistics of data collection are presented in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Resolution limits (Å) | 30–3.5 (3.65–3.5) |
| No. of observed reflections | 1454357 |
| No. of unique reflections | 26380 |
| Completeness (%) | 99.3 (86.3) |
| Average redundancy | 3.9 (3.8) |
| I/σ(I) | 8.6 (2.6) |
| Rmerge† (%) | 9.8 (57.7) |
| Wilson B factor (Å2) | 125.1 |
| Refinement | |
| No. of non-H protein atoms | 11214 |
| No. of solvent molecules | 31 |
| Resolution range (Å) | 30.0–3.5 |
| Rcryst‡ (%) | 23.7 |
| Rfree§ (%) | 25.0 |
| Ramachandran plot | |
| Most favourable regions (%) | 80.5 |
| Additional allowed regions (%) | 18.6 |
| Generously allowed regions (%) | 0.0 |
| Disallowed regions (%) | 0.9 |
| R.m.s. standard deviation | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.49 |
| Average B factor/SD (Å2) | 139.4 |
R
merge = 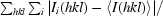
 , where I
i(hkl) is the scaled intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the mean intensity for that reflection.
, where I
i(hkl) is the scaled intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the mean intensity for that reflection.
R
cryst = 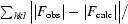
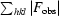 .
.
R free is the cross-validation R factor computed for a test set of 5% of the unique reflections.
2.3. Processing of the diffraction data and detection of twinning
The diffraction data were processed with the HKL-2000 package (Otwinowski & Minor, 1997 ▶). Auto-indexing of the diffraction data suggested six different lattices ranging from C-centred monoclinic to body-centred cubic, with a distortion index of between 0.89 and 2.98%. For comparison, all other lattices in the list showed a distortion index of greater than 14%. Initial processing of the data in space groups C2, F222, I222, I422, R32 and I23 showed a very similar R merge of around 11%. Examination of the distribution of the intensities 〈|I|2〉/(〈|I|〉)2 and the structure-factor amplitudes (〈|F|〉)2/〈|F|2〉 given by the program TRUNCATE from the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶) showed that the crystals were almost perfect pseudo-merohedral twins. This conclusion was confirmed by a twinning test run on the merohedral twinning server of the NIH MBI Laboratory (Yeates, 1997 ▶) and the phenix.xtriage routine from the program PHENIX (Adams et al., 2002 ▶). The twin law and twinning fraction were determined by phenix.xtriage to be h, −k, −h − l and 0.46, respectively. Close inspection of the systematic extinctions, the Laue symmetry of the diffraction pattern and a self-rotation function calculated in space group P1 suggested that the crystals can be characterized by unit-cell parameters a = 174.4, b = 123.2, c = 123.3 Å, α = γ = 90, β = 135° and the symmetry of the monoclinic space group C2, which is a subgroup of the higher symmetry space groups observed in the initial processing. The calculated Matthews coefficient in space group C2 had a value of 2.42 Å3 Da−1, which corresponds to the presence of six protein molecules with a molecular weight of 28 kDa in the asymmetric unit.
2.4. Structure solution and crystallographic refinement
Initial molecular-replacement trials were attempted in all suggested space groups using the program MOLREP as implemented in CCP4 (Collaborative Computational Project, Number 4, 1994 ▶), with the monomer structure of PCNA from A. fulgidus (PDB code 1rwz) as a Patterson search model. A sequence alignment performed with ClustalW (Thompson et al., 1994 ▶) revealed a sequence identity of 37%, which appeared to be the highest percentage of identical residues between HfxPCNA and all other known PCNA structures (Fig. 2 ▶). A correct molecular-replacement solution was only obtained in the monoclinic space group C2, in which six protein monomers in the asymmetric unit were found in individual searches one by one. Hence, the apparent monoclinic crystal lattice was confirmed.
Figure 2.

Sequence alignment of PCNAs from H. volcanii (PCNA_HALOV), P. furiosus (PCNA_PYRFU), A. fulgidus (PCNA_ARCFU), S. solfataricus (PCNA1_SULSO, PCNA2_SULSO, PCNA3_SULSO) and human (PCNA_HUMAN). The secondary-structure elements are shown above and labelled as they are found in the HfxPCNA structure; α-helices are shown in cyan, β-strands are shown in yellow and the loop connecting the N-terminal and C-terminal domains is highlighted in green. Identical residues are coloured green, homologous residues are coloured blue, the positively/negatively charged residues which are unique to HfxPCNA are coloured red and the residues that can be involved in PIP peptide interactions are highlighted in light grey.
2.4.1. Refinement
The crystallographic refinement was performed by phenix.refine (Adams et al., 2002 ▶), first using a rigid-body refinement protocol with six rigid groups and then using the maximum-likelihood target with respect to the twinning operator determined as described above. Bulk-solvent correction and noncrystallographic restraints between six protein subunits were applied throughout refinement. The procedure improved the initial electron-density map and allowed the building of almost all of the residues that had been replaced by alanine in the original model. The model was rebuilt with the graphics programs O (Jones et al., 1991 ▶) and Coot (Emsley & Cowtan, 2004 ▶). The progress of refinement was monitored by the free R factor, with 5% (1323 reflections) of the data put aside from the calculations. Several water molecules were built into the difference electron-density map at a late stage of refinement using Coot. The final model consisting of six protein subunits and 31 water molecules was refined at a resolution of 3.5 Å to an R cryst of 23.7% and an R free of 25%. The final refinement statistics are given in Table 1 ▶. The atomic coordinates and structure factors of PCNA from H. volcanii have been deposited in the Protein Data Bank (entry 3hi8).
3. Results and discussion
3.1. Overall structure of PCNA from H. volcanii
PCNA from H. volcanii, in common with all other known PCNA orthologues, is organized in a characteristic toroidal structure (Fig. 3 ▶ a) made up by three equivalent monomers. Every monomer in the ring interacts with two others in a head-to-tail manner. Thus, the six monomers found in the C2 asymmetric unit were organized into two separate rings. Each PCNA monomer contains 247 amino acids comprising two domains which are structurally duplicated and are connected to each other by long interdomain-connector loops (residues 116–128; Fig. 3 ▶ b). The association of three monomers in the ring has apparent threefold noncrystallographic symmetry and the presence of two symmetrical domains in every monomer gives the trimer a pseudo sixfold-symmetrical appearance.
Figure 3.

Structure of PCNA from H. volcanii (HfxPCNA). (a) Homotrimer; three subunits are shown in different colours. (b) Ribbon representation of monomeric HfxPCNA superimposed with PCNAs from A. fulgidus (brown; PDB code 1rwz; Chapados et al., 2004 ▶) and P. furiosus (green; PDB code 1ge8; Matsumiya et al., 2001 ▶). The N-terminal domain of HfxPCNA is shown in blue and the C-terminal domain is shown in cyan; secondary-structure elements are assigned and labelled with respect to the HfxPCNA structure. Figs. 3, 4 and 5 were generated with PyMOL (DeLano, 2002 ▶).
Each monomer domain consists of a nine-stranded β-sheet at one side and two α-helices at the other side. The middle β-strand (β4) interacts with the neighbouring β-strand (β18) from another domain by the formation of several hydrogen bonds. Thus, topologically the HfxPCNA monomer belongs to the family of α/β proteins and is composed of an 18-stranded antiparallel twisted β-sheet and four α-helices (Fig. 3 ▶ b). The helices of all three monomers are located inside the ring, whereas the extended β-sheet and connector loop form the outside surface of the ring.
The intersubunit contacts in the HfxPCNA trimer are formed by strand β9 and helix α2 of one subunit and strand β13 and helix α3 of the neighbouring subunit. Six main-chain amide-to-carbonyl hydrogen bonds are formed between antiparallel strands β9 (residues 103–109) and β13 (residues 171–177) and one hydrogen bond is found between the side chains of Tyr106 and Asp172. The α-helices provide hydrophobic interaction patches that are formed by Val76 and Met79 from one subunit and Met147 and Ala144 from another subunit. Despite the low sequence homology, this type of subunit interaction is well conserved among all known PCNA structures.
Many of the proteins that interact with PCNA in both archaeal and eukaryotic cells do so via an evolutionarily conserved peptide sequence known as the PIP peptide (Warbrick, 1998 ▶). In haloarchaeal cells, proteins containing PIP peptides include the DNA polymerases PolB and PolD, the large subunit of replication factor C and the nucleases Fen1 and Rnh2 (see Fig. 2 in MacNeill, 2009 ▶). In each of these, the PIP peptide is located at the extreme C-terminus of the protein. Structural analyses have shown that the PIP peptide binds tightly in a hydrophobic pocket on the surface of PCNA that is predominantly composed of residues from the PCNA interdomain-connector loop (IDCL) but which also involves the hydrophobic side chains of residues located towards the N- and C-termini of PCNA (Gulbis et al., 1996 ▶; Matsumiya et al., 2002 ▶; Chapados et al., 2004 ▶). Mutation of these residues results in impaired binding to the PIP peptide sequence. The residues that form this binding site in HfxPCNA are almost identical to those of other archaeal PCNA molecules. For example, eight of ten hydrophobic residues (Val39, Met41, Pro120, Pro123, Leu125, Leu127, Pro220 and Pro240) identified as being involved in A. fulgidus PCNA–FEN-1 interactions (Chapados et al., 2004 ▶) are identical in the HfxPCNA protein sequence (Fig. 2 ▶) and form a similar hydrophobic pocket in three dimensions (Fig. 4 ▶). The ninth and tenth residues implicated in formation of the A. fulgidus PCNA pocket, Tyr219 and Leu239, are replaced by Phe221 and Met239 in HfxPCNA, respectively (Figs. 2 ▶ and 4 ▶). Taken together, these observations indicate that the mode of PCNA–PIP peptide binding observed in mesohalic thermophilic archaeal organisms such as A. fulgidus is likely to be conserved in halophiles such as H. volcanii, but confirmation of this will require cocrystallization of a haloarchaeal PCNA with a cognate PIP peptide.
Figure 4.

Stereoview of a structural comparison of PIP peptide-binding sites in HfxPCNA (shown in cyan) and PCNA from A. fulgidus (shown in brown; PDB code 1rwz; Chapados et al., 2004 ▶) showing conserved hydrophobic side chains. The residues involved in the binding sites are represented by sticks.
3.2. Structural comparison of HfxPCNA and charge distribution on the surface
The structural comparison of HfxPCNA with archaeal orthologues from P. furiosus, A. fulgidus and three different PCNAs from S. solfataricus showed a very similar arrangement in homotrimers, with r.m.s.d. values of 1.6, 1.3, 2.6, 2.1 and 2.2 Å, respectively. The overlaid structures of monomers from the same organisms revealed a very similar arrangement in the central β-sheet; however, the flanking helices α2 and α3 are slightly shifted in the compared structures. The loops connecting secondary-structure elements are the most different parts in terms of length and conformation. Thus, in the N-terminal domain of HfxPCNA the loops 61–64, 80–83 and 101–103 are the shortest among the archaeal structures known so far, whereas the C-domain loops of HfxPCNA are the longest and have rather different conformations compared with the other orthologues. It is apparent from the r.m.s. deviation between different PCNAs that the protein from A. fulgidus is the closest structural homologue to HfxPCNA. The sequence alignment reveals a very low homology even among archaeal organisms; however, sequence analysis with the ProtParam tool (Gasteiger et al., 2005 ▶) showed a high level of positively and negatively charged surface residues, which is known to be a characteristic of extremophilic proteins (Karshikoff & Ladenstein, 2001 ▶). HfxPCNA, which is the first representative of a halophilic PCNA, showed many fewer positively charged residues (seven lysines and 11 arginines in HfxPCNA compared with 19 lysines and 12 arginines in A. fulgidus PCNA and 21 lysines and nine arginines in P. furiosus PCNA; Fig. 5 ▶ a) but a similar fraction of negatively charged aspartate and glutamate residues (28 aspartates and 21 glutamates in HfxPCNA compared with 19 aspartates and 25 glutamates in A. fulgidus PCNA and 17 aspartates and 33 glutamates in P. furiosus PCNA). A structural visualization of the charges on the surfaces of P. furiosus and A. fulgidus PCNA showed a rather typical distribution of positive and negative residues (Figs. 5 ▶ b and 5 ▶ c). The strong positive charge arising from several lysines and arginines is known to be responsible for the interaction with DNA and is located on the inner surface of the trimeric channel. Additional positively charged residues are equally distributed on the surface and are compensated by the negative charges from Asp and Glu residues. In contrast to the description given above, the structure of HfxPCNA showed a relatively weak positive charge inside the channel (only three lysines and two arginines compared with 11 lysines and one arginine in P. furiosus PCNA and seven lysines and one arginine in A. fulgidus PCNA). The negative charges formed by 22 Asp and 18 Glu residues are randomly distributed on the surface and cannot be compensated by the nine Arg and four Lys residues that are also found on the surface (Fig. 5 ▶ a). The most notable feature of protein halophilicity is the presence of clusters of non-interacting negatively charged surface residues. Thus, the distribution found in HfxPCNA is consistent with this feature. Such negatively charged clusters are associated with unfavourable electrostatic energy at low salt concentration and may account for the instability of the protein at a salt concentration lower than 2.5 M. The central channel in HfxPCNA contains only three lysine residues (Lys143, Lys201 and Lys205) and two arginine residues (Arg12 and Arg140) and thus is much less positively charged in comparison to all archaeal and previously determined PCNA structures. Notably, there are six aspartate and three glutamate residues in the central channel which are located near positively charged spots and seem to partially compensate for them. This local charge distribution is also consistent with adaptation to a high-salt environment.
Figure 5.

The distribution of negatively and positively charged residues on the surface of PCNA from (a) H. volcanii, (b) A. fulgidus (PDB code 1rwz; Chapados et al., 2004 ▶) and (c) P. furiosus (PDB code 1ge8; Matsumiya et al., 2001 ▶) and respective electrostatic potential maps in two different orientations. Residues are represented by sticks. Lysines/arginines are shown in blue and aspartates/glutamates are shown in red.
The charge distribution observed in the central channel of the halophilic HfxPCNA suggests an interaction mechanism with DNA which seems to be quite different from that at low-salt conditions. It is known that at high salt concentrations the negatively charged DNA phosphate groups as well as the negatively charged amino-acid side chains in proteins are surrounded by cations which can rapidly exchange their positions with other ligands (O’Brien et al., 1998 ▶). The increased number of negatively charged Asp and Glu side chains lining the central channel of HfxPCNA can serve as cation-binding sites. During HfxPCNA–DNA interaction the negatively charged DNA phosphate groups would be bound via intermediate cations forming bridges to the Asp and Glu side chains (O’Brien et al., 1998 ▶; Bergqvist et al., 2003 ▶). The central channel of HfxPCNA has a diameter of 36 Å, which is sufficient to accommodate double-stranded DNA (Krishna et al., 1994 ▶; Gulbis et al., 1996 ▶). In fact, room exists for one or two layers of water between the DNA and PCNA (Johnson & O’Donnell, 2005 ▶). Recent molecular-dynamics simulations suggest that the residues lining the PCNA channel interact with the phosphodiester backbone of DNA. However, these interactions are highly dynamic and seem to be frequently displaced by ions from the solution (Ivanov et al., 2006 ▶). We assume that in the case of HfxPCNA the bridging cations would favour a similarly dynamic manner of DNA binding. Recent measurements of diffusional properties as a function of viscosity and protein size suggested that PCNA moves along DNA using two different sliding mechanisms: (i) by rotationally tracking the helical pitch or (ii) by sliding uncoupled from the helical pitch (Kochaniak et al., 2009 ▶). The observation of only a very weak relation between ionic strength and diffusion properties suggested that PCNA maintains electrostatic contact with the DNA phosphate backbone while moving. In the high-salinity environment of HfxPCNA, screening of the repulsive interactions between Asp and Glu residues and the DNA phosphate backbone by cation bridges could allow a dynamic sliding interaction in a manner analogous to that described above.
The presence of mainly positive charges, as found in P. furiosus PCNA and A. fulgidus PCNA (Figs. 5 ▶ b and 5 ▶ c), in the DNA-interacting channel of a PCNA functioning at high salt concentration would be likely to lead to strong binding of anions which would unfavourably interfere with the DNA–PCNA interaction. Thus, the presence of a channel lined mainly by negative charges seems to represent an adaptive structural feature of halophilic PCNA molecules allowing binding interactions between two negatively charged binding partners via cation bridges.
4. Conclusion
The structure of PCNA from the extremely halophilic archaeon H. volcanii has revealed specific features of the protein that are likely to reflect adaptation to a high-salt environment. Our study suggests a different type of interaction with DNA in halophilic PCNAs that is mediated by cation bridges between the phosphodiester groups on DNA and abundant negative charges lining the central channel of PCNA. The ease with which H. volcanii can be cultured in the laboratory and the availability of tools for reverse genetic manipulation of this organism will allow these predictions to be tested by mutational analysis in vivo.
Supplementary Material
PDB reference: proliferating cell nuclear antigen, 3hi8, r3hi8sf
Acknowledgments
We thank Thorsten Allers (University of Nottingham, England) for supplying H. volcanii DS70, Xiaofeng Zhang for technical assistance during data collection and Malcolm White (University of St Andrews, Scotland) for advice. We gratefully acknowledge the access to synchrotron-radiation facilities at the ESRF (Grenoble, France) and thank the staff for the help at the beamline. FCG and SM were funded in Copenhagen by Forskningsrådet for Natur og Univers. SM is currently funded by the Scottish Universities Life Sciences Alliance (SULSA).
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Allers, T. & Mevarech, M. (2005). Nature Rev.6, 58–73. [DOI] [PubMed]
- Bergqvist, S., Williams, M. A., O’Brien, R. & Ladbury, J. E. (2003). Biochem. Soc. Trans.31, 677–680. [DOI] [PubMed]
- Bourenkov, G. P. & Popov, A. N. (2006). Acta Cryst. D62, 58–64. [DOI] [PubMed]
- Bowman, G. D., O’Donnell, M. & Kuriyan, J. (2004). Nature (London), 429, 724–730. [DOI] [PubMed]
- Bruning, J. B. & Shamoo, Y. (2004). Structure, 12, 2209–2219. [DOI] [PubMed]
- Chapados, B. R., Hosfield, D. J., Han, S., Qiu, J., Yelent, B., Shen, B. & Tainer, J. A. (2004). Cell, 116, 39–50. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. http://www.pymol.org.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D. & Bairoch, A. (2005). The Proteomics Protocols Handbook, edited by J. M. Walker, pp. 571–607. Totowa: Humana Press.
- Grabowski, B. & Kelman, Z. (2003). Annu. Rev. Microbiol.57, 487–516. [DOI] [PubMed]
- Gulbis, J. M., Kelman, Z., Hurwitz, J., O’Donnell, M. & Kuriyan, J. (1996). Cell, 87, 297–306. [DOI] [PubMed]
- Hishiki, A., Hashimoto, H., Hanafusa, T., Kamei, K., Ohashi, E., Shimizu, T., Ohmori, H. & Sato, M. (2009). J. Biol. Chem.284, 10552–10560. [DOI] [PMC free article] [PubMed]
- Ivanov, I., Chapados, B. R., McCammon, J. A. & Tainer, J. A. (2006). Nucleic Acids Res.34, 6023–6033. [DOI] [PMC free article] [PubMed]
- Johnson, A. & O’Donnell, M. (2005). Annu. Rev. Biochem.74, 283–315. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Karshikoff, A. & Ladenstein, R. (2001). Trends Biochem. Sci.26, 550–556. [DOI] [PubMed]
- Kochaniak, A. B., Habuchi, S., Loparo, J. J., Chang, D. J., Cimprich, K. A., Walter, J. C. & van Oijen, A. M. (2009). J. Biol. Chem.284, 17700–17710. [DOI] [PMC free article] [PubMed]
- Kontopidis, G., Wu, S. Y., Zheleva, D. I., Taylor, P., McInnes, C., Lane, D. P., Fischer, P. M. & Walkinshaw, M. D. (2005). Proc. Natl Acad. Sci. USA, 102, 1871–1876. [DOI] [PMC free article] [PubMed]
- Krishna, T. S., Kong, X. P., Gary, S., Burgers, P. M. & Kuriyan, J. (1994). Cell, 79, 1233–1243. [DOI] [PubMed]
- MacNeill, S. A. (2009). Biochem. Soc. Trans.37, 108–113. [DOI] [PubMed]
- Matsumiya, S., Ishino, S., Ishino, Y. & Morikawa, K. (2002). Genes Cells, 7, 911–922. [DOI] [PubMed]
- Matsumiya, S., Ishino, Y. & Morikawa, K. (2001). Protein Sci.10, 17–23. [DOI] [PMC free article] [PubMed]
- Mullakhanbhai, M. F. & Larsen, H. (1975). Arch. Microbiol.104, 207–214. [DOI] [PubMed]
- Muller, V. & Oren, A. (2003). Extremophiles, 7, 261–266. [DOI] [PubMed]
- O’Brien, R., DeDecker, B., Fleming, K. G., Sigler, P. B. & Ladbury, J. E. (1998). J. Mol. Biol.279, 117–125. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Sakurai, S., Kitano, K., Yamaguchi, H., Hamada, K., Okada, K., Fukuda, K., Uchida, M., Ohtsuka, E., Morioka, H. & Hakoshima, T. (2005). EMBO J.24, 683–693. [DOI] [PMC free article] [PubMed]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res.22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Vijayakumar, S., Chapados, B. R., Schmidt, K. H., Kolodner, R. D., Tainer, J. A. & Tomkinson, A. E. (2007). Nucleic Acids Res.35, 1624–1637. [DOI] [PMC free article] [PubMed]
- Warbrick, E. (1998). Bioessays, 20, 195–199. [DOI] [PubMed]
- Wendoloski, D., Ferrer, C. & Dyall-Smith, M. L. (2001). Microbiology, 147, 959–964. [DOI] [PubMed]
- Williams, G. J., Johnson, K., Rudolf, J., McMahon, S. A., Carter, L., Oke, M., Liu, H., Taylor, G. L., White, M. F. & Naismith, J. H. (2006). Acta Cryst. F62, 944–948. [DOI] [PMC free article] [PubMed]
- Xing, G., Kirouac, K., Shin, Y. J., Bell, S. D. & Ling, H. (2009). Mol. Microbiol.71, 678–691. [DOI] [PubMed]
- Yeates, T. O. (1997). Methods Enzymol.276, 344–358. [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: proliferating cell nuclear antigen, 3hi8, r3hi8sf
PDB reference: proliferating cell nuclear antigen, 3hi8, r3hi8sf