

Abstract
Accurate and complete measurement of single nucleotide (SNP) and copy number (CNV) variants, both common and rare, will be required to understand the role of genetic variation in disease. We present Birdsuite, a four-stage analytical framework instantiated in software for deriving integrated and mutually consistent copy number and SNP genotypes. The method sequentially assigns copy number across regions of common copy number polymorphisms (CNPs), calls genotypes of SNPs, identifies rare CNVs via a hidden Markov model (HMM), and generates an integrated sequence and copy number genotype at every locus (for example, including genotypes such as A-null, AAB and BBB in addition to AA, AB and BB calls). Such genotypes more accurately depict the underlying sequence of each individual, reducing the rate of apparent mendelian inconsistencies. The Birdsuite software is applied here to data from the Affymetrix SNP 6.0 array. Additionally, we describe a method, implemented in PLINK, to utilize these combined SNP and CNV genotypes for association testing with a phenotype.
Studies of SNPs and CNVs in human disease have to date been built on different analytical approaches, and somewhat based on conflicting assumptions. Specifically, SNP genotyping methods1,2 assume that every individual has two copies of each locus, whereas studies of copy number variation assume that individuals vary in their copy number across the genome. Because SNPs and CNVs coexist throughout the genome, they influence one another’s measurement, and may act both separately and in concert to influence human phenotypes. Ignoring CNVs during SNP genotyping results in failure to capture the true underlying sequence at many sites (genotypes like AAB and A), and can create the appearance of violations of mendelian inheritance or Hardy-Weinberg equilibrium where none in fact exists3,4. Ignoring SNPs in copy number analysis fails to incorporate allele-specific gains and losses, as well as the potential to exploit linkage disequilibrium between CNVs and nearby SNPs.
In addition, methods for copy number analysis have not previously separated the ideas of genotyping known copy number polymorphisms (CNPs) from discovery of rare (and thus previously unobserved) copy number variants (CNVs)5. In the former case, as in SNP genotyping, existing information about known polymorphisms can be used to design arrays, train clustering algorithms and assign a prior probability of aberrant copy number to guide interpretation of measurements. Discovery of rare variants, as in sequence analysis for rare mutations, is a much more difficult problem—both because it is more difficult to detect a single event than something seen many times, and because of the intrinsic low prior probability of there being such a variant at any particular location in the genome in any individual. Here, we develop separate methods for analysis of rare and common copy number variation. (For clarity, we use a nomenclature in which ‘copy number polymorphism’ (CNP) refers to the subset of ‘copy number variants’ (CNVs) that segregate at greater than 1% frequency in the population; this is parallel to the use of ‘single nucleotide polymorphism’ (SNP) to refer to sequence variants segregating at greater than 1% frequency in a population.)
Below we describe a new suite of algorithms instantiated in software for integrating these approaches. Our methods are informed on a small scale by the individual response of characteristics of each individual probe, and on a large scale by a high-resolution map of common CNPs. Briefly, the approach first assigns copy number across regions of known common CNPs. Second, at each SNP locus, samples expected to have two copies of the locus are assigned genotypes: AA, AB or BB. Third, informed by probe-specific mean and variance estimated in the second step, a hidden Markov model (HMM) is used to discover rare or de novo CNVs. Fourth, copy number and SNP allele information are combined to provide an integrated genotype at each locus (Fig. 1). Although initially developed for use with new, hybrid genotyping arrays designed to capture SNP and copy number information simultaneously6, these algorithmic approaches are general and can be applied to other genotyping platforms as well.
Figure 1.
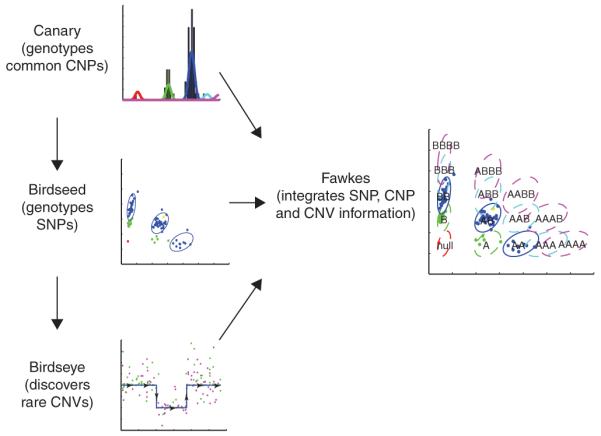
Overview of Birdsuite. In step 1, Canary estimates copy number across regions of known common copy number polymorphisms (‘CNP genotyping’). In step 2, Birdseed assigns canonical SNP genotypes (AA, AB or BB) to samples estimated by Canary to have two copies of a SNP locus (‘SNP genotyping’). Additionally, it calculates probe-specific mean and variances. In step 3, Birdseye estimates the likelihood of rare or de novo copy number variants, using probe-specific means and variances informed by Canary and Birdseed, and combining data across multiple probes in the region (‘CNV discovery’). In step 4, Fawkes combines copy number information for each sample at each locus with allele-specific information to assign a comprehensive SNP genotype, including noncanonical genotypes such as A-null or AAB.
RESULTS
Genotyping of common copy number polymorphisms
Previous approaches to copy number analysis7-10 involve searching a single individual’s genome for regions in which evidence of copy number deviation exceeds a genome-wide significance threshold—an approach that does not make use of prior knowledge. Yet the variation at more than 90% of the loci at which any two individuals differ in copy number across a region >10 kb in size seems due to a limited universe of common CNPs6. At such loci, a copy number variant unambiguously exists and segregates at an appreciable frequency, and the problem can be redefined not as a problem of ab initio discovery, but rather of accurate measurement (genotyping) of each individual’s integer copy number level5.
The first step in our methodology, Canary (copy number analysis routine), determines the copy number of each individual at each predefined CNP locus. A high-resolution map of common CNPs is needed to define these loci; we used the map of McCarroll et al.6, but improved maps can be substituted as they emerge. Although an individual probe inside a given CNP may not provide enough information to give an accurate integer measurement of copy number (a copy number ‘genotype’)11 (Fig. 2a), multiple probes that interrogate the same CNP segment typically show highly correlated and reproducible patterns of intensity6 (Fig. 2b,c). The measurements for the probes in the same CNP (or, in some cases, for a predefined high-performing subset of those probes; Fig. 2c) are combined into a single summarized intensity measurement, resulting in one summarized measurement per sample (Fig. 2d). The summarized measurements for a batch of samples are then clustered into discrete copy number classes using a one-dimensional Gaussian mixture model (GMM), where the expected location of copy number clusters is informed by the results of previous experiments (Fig. 2d-f). The resulting clusters are used to assign a CNP genotype to each sample at each CNP, as well as a score reflecting the confidence of each assignment (Supplementary Methods online).
Figure 2.

Schematic of how a CNP is processed through Canary illustrated with data from chromosome 4. (a) Histogram of raw data from a single copy number probe. (b) Cross-correlation matrix of neighboring probe intensity profiles across 88 HapMap samples (a depicts probe 13). For SNPs, the intensity used is the sum of the intensities for the A and B alleles. The high and consistent correlation in the center indicates copy number variation as opposed to random noise, demarcating the boundaries of the CNP. (c) Heat map depicting normalized intensities for adjacent probes across the 88 samples; red indicates low intensity, and yellow high intensity. Copy number probes are denoted by a pink dot below x axis. Probes used to summarize a CNP are notated with a black line at the bottom. (d) Summarized intensity measurements (across 88 samples) for this CNP, overlaid with the Gaussian clusters that Canary fit to the data, and colored by copy number. (e) Canary genotypes across six batches of HapMap data for a multiallelic CNP on the x axis is batch plus jitter, and on the y axis is sample intensity. Also along the y axis is represented the prior expectation of where the different copy number genotype classes should lie. The ‘mix’ batches are designed such that samples are not separated by ancestral group. (f) Same as e, except showing a YRI-specific simple deletion CNP for which batch effects are evident.
Validation of the CNP genotypes from such an approach is important, but currently hampered by the lack of a gold-standard set of reference genotypes (such as HapMap12 has provided for SNPs). The vast majority of CNPs have not been previously genotyped with accuracy demonstrated in a set of reference samples. We created one such reference dataset (on the basis of consistency across two independent studies of 263 HapMap samples) that has few mendelian inconsistencies, conforms to Hardy-Weinberg equilibrium and shows strong concordance to fosmid end-sequenced samples6. However, this specific dataset is inappropriate for validation of Canary, as it would be statistically overfit and therefore inflate measures of performance.
Instead, we assessed the quality of Canary-derived CNP genotypes by examining (i) inheritance in 91 independent parent-offspring trios and (ii) reproducibility across many laboratories. For the 1,177 diallelic CNPs tested (consisting of only a simple deletion or duplication, but not both), genotypes in 91 trios showed a mendelian inconsistency rate of approximately 0.005 per trio per CNP (Table 1). Copy number genotypes for 96 multiallelic CNPs6 were assessed for inheritance using Fisher’s h, which was distributed closely around 1.0, with only one CNP generating a P value <0.01. Canary genotypes were reproducible across the same HapMap samples run across seven independent labs, achieving an average sample call rate of 96.1% and a sample concordance with our reference dataset of 98.0%. Concordance with 783 independent copy number genotypes obtained by quantitative PCR (in 27 CNPs and 29 samples) averaged 97.6% across the seven labs. This is less complete and accurate than that for SNP genotypes, suggesting that further refinements are needed to either the algorithms or the underlying array data. Nonetheless, this performance across >1,000 CNPs far exceeds that of the small numbers (<100) of CNPs that have been genotyped in any previous study of appreciable sample size.
Table 1. Genotyping performance of Birdsuite judged by mendelian inheritance (MI) patterns.
| Diallelic CNPs | Autosomal SNPs | Autosomal SNPs within known6 CNPs | |
|---|---|---|---|
| Number in category | 1,177 | 872,276 | 11,256 |
| MI rate Birdseed | n.a. | 0.0868% | 0.1497% |
| MI rate Fawkes | 0.5200% | 0.0822% | 0.0926% |
These data were generated from a cohort of diverse ancestry which included 91 trios; we thus do not expect nor test for Hardy-Weinberg equilibria. The higher rate of MI observed for diallelic CNPs indicates that there is a substantial subclass of CNPs whose genotyping quality varies considerably from batch to batch. n.a., not applicable.
Genotyping of SNPs
We next turn to SNP genotyping (Fig. 3). For any given genomic segment containing a SNP, samples with two copies of the locus per diploid genome are expected to have one of the canonical SNP genotypes of AA, AB or BB. For most autosomal SNPs we expect all samples to have two copies, but for those overlapping a CNP other possibilities may be observed13 (Fig. 3a). For such SNPs, we use the information from Canary (above) to restrict initial SNP genotyping to those samples whose CNP genotype (integer copy number) is two (Fig. 3b). Such a partitioning allows for the model of diploid SNP clustering not to be misled by samples that have fewer or extra copies (as might happen if one clustered the raw SNP data shown in Figure 3a).
Figure 3.

Schematic of how a single SNP is processed through Birdsuite and evaluation of mendelian inconsistencies. (a) Raw data for the SNP, plotting allele-A intensity versus allele-B intensity. Unlike most SNPs on the array, this SNP does not form three discrete clusters because it lies in the common CNP depicted in Figure 2. (b) Same as a, colored by CNP genotype (as determined by Canary in Fig. 2d). (c) Birdseed uses those samples with two copies at this locus to determine allele-specific probe characteristics (‘clusters’) for the SNP. (d) Copy-variant clusters imputed on the basis of the two-copy models. These are in turn used to aid Birdseye in the search for undiscovered CNVs, and by Fawkes to assign allele-specific copy number genotypes such as A/null (lower right green region) or BBBB (upper left magenta region). (e) Plot showing the rate of mendelian inconsistency (MI) in SNPs that overlap a known CNP for 91 children using Birdseed alone (sans Canary) versus using the entire Birdsuite. Only copy-normal calls were used to test for a MI; rate of MI is the number of inconsistencies divided by the number of tests. (f) Histogram of MI rate using the Birdsuite divided by MI rate using Birdseed alone, calculated using all autosomal SNPs. MI rate decreases for all 91 samples, indicating that a considerable percentage of inconsistencies are due to either inherited, de novo or somatic copy number variation.
SNP genotyping is done using Birdseed (see Supplementary Methods and the BirdSuite web site), a specialized two-dimensional GMM, where the two dimensions are summarized probe intensities for each of the two alleles (A and B). Like Canary, Birdseed utilizes prior models representing the expected allele intensity information for each genotype class, built from previous data for samples of known genotype at each SNP (in this case, 270 HapMap samples and genotypes). Briefly, the algorithm utilizes expectation-maximization14 to determine the location of the AA, AB and BB clusters for each SNP (Fig. 3c and Supplementary Fig. 1 online). These clusters are used to assign a genotype (AA, AB or BB) to each sample along with a score reflecting the confidence of each call. Special procedures are used on the X, Y and mitochondrial chromosomes. Birdseed performance was validated on HapMap samples run on the Affymetrix SNP 6.0 array6 across seven different labs, not including any experiments used to generate the models or develop the algorithm. At the default confidence threshold, call rate on the HapMap samples was 99.47%, and these confident genotype calls were 99.74% concordant with HapMap genotypes, approaching the estimated error rate of HapMap itself. Like previous algorithms, Birdseed is not without bias: minor-allele homozygotes are less well genotyped when the minor allele frequency is low, affecting both call rate and concordance of this class of genotypes. However, Birdseed compares favorably to the BRLMM algorithm1 (comparison done on Affymetrix 500K data, as BRLMM does not work on the Affymetrix SNP 6.0 array; see Supplementary Fig. 2 online). Birdseed as a stand-alone program has been used to genotype over 50,000 samples at the Broad Institute with an average call rate >99% (S.B.G., unpublished observations).
Discovery and genotyping of rare CNVs
Although the first two steps in the framework focus on accurate typing of known, common polymorphisms, it is also possible using the same platforms to identify rare and de novo copy number variants for which there is no prior knowledge. Such problems of ab initio discovery are fundamentally more difficult because of the need to distinguish a relatively small number of real CNVs at unknown sites from the statistical fluctuations that arise in any genome-scale dataset. The heterogeneity of probe performances on array platforms further complicates this problem: different probes show different intrinsic measurement variance across samples (a fact seldom modeled by CNV discovery algorithms); furthermore, different SNP probe sets show different quantitative responses to having 0, 1 or 2 copies of each allele (Supplementary Fig. 1). We therefore sought to model the empirical properties of each probe in order to maximize the power to detect rare CNVs. As in most other algorithms10, we search for consistent evidence for copy number variation across multiple neighboring probes to reduce the effect of normal statistical fluctuations.
We consider first the task of accurately estimating copy number at a single location in the genome. Having previously run Canary and Birdseed aids in this task, in that they define copy number and allele-specific properties of each probe, as well as noise properties specific to each sample. The locations and variances of the Birdseed posterior clusters (corresponding to AA, AB and BB genotypes in intensity space) together represent an accurate estimate of the emission probability (the probability density function of intensity measurements given a particular underlying state) for each probe on the array in response to a sample with two copies at that locus. The expected intensity profile of ‘copy-variable’ genotypes (A-null, AAB and so on) can then be imputed from the locations of the Birdseed two-copy clusters (Fig. 3d). Combining these profiles across genotypes of equivalent copy number models the emission probability of a probe given a sample with 0, 1, 3 or 4 copies of a locus. For the copy number probes on the array, we model only a single cluster per locus representing two copies, which represents the emission probability of normal samples; alternative copy number emission probabilities are imputed analogous to the method for imputing additional SNP clusters (Supplementary Methods).
The assumption that copy number is an integer allows for predefined, strongly modeled states to increase sensitivity. For both SNP probe sets and copy number probes, these emission probabilities allow us to estimate the relative likelihood of each possible copy number level (0, 1, 2, 3 or 4) in a way that is informed by the specific performance of each probe. We note that although this considerably improves performance when assessing germline copy number, it may make Birdseye less suitable for applications where average copy number at a locus is noninteger (such as detection of mosaic copy number changes in heterogeneous tumor DNA).
Even after an empirically modeled interpretation of intensity measurements, the estimate of copy number from single probes can be noisy. The next step is therefore to integrate information across neighboring probes to find strong, consistent evidence for altered copy number states. Birdseye, an HMM-based algorithm, utilizes dynamic programming to perform this search quickly and efficiently across each chromosome15 (Fig. 4). Each segment of discrete copy number is assigned a lod score indicating the relative probability of the variant versus normal copy number in the region (which can be used in downstream analyses to prioritize discovered CNVs on the basis of confidence). Because the approach uses probe-specific variances, noisier probes are inherently downweighted with respect to more responsive probes, reducing the number of false positives one would find by assuming all probes are equal (Supplementary Methods).
Figure 4.

Discovery of unknown or de novo copy number variation using Birdseye. (a) Raw data from a copy number probe, with one sample (arrow) colored green (top left). Raw data from a neighboring SNP, with the same sample (arrow) colored green (top right). Although the sample is relatively low in intensity, one would not have confidence calling a deletion on the basis of these data alone (bottom). A view across a larger region surrounding these two probe locations. A point is placed at the estimated copy number for this sample at each queried locus (without taking into account neighboring probes). With enough probes to support the evidence of a deletion, the HMM transitions to call a heterozygous deletion in this sample across an 85-kb region (blue line). (b,c) In addition, calling the deletion in the sample shown in a, Birdseye determines the relative log-likelihood of the identical deletion in each parent of this sample. Owing to strong evidence against this deletion in the parents, the region represents a de novo event in the child. (d) Data from in silico gender-mixing experiment. Sensitivity and breakpoint accuracy to discover simulated deletions of varying size (left). A deletion was considered discovered only if the lod score for the deletion was above 2. Sensitivity to discover the simulated deletions plotted against expected number of false-positive discoveries per genome (right). Points are placed at lod thresholds of 5, 2, 1 and 0.
To assess the sensitivity and specificity of Birdseye (and lacking a gold-standard dataset), we simulated CNVs across a range of sizes via an in silico gender-mixing experiment. Intensity measurements from consecutive X-chromosome probes from a female sample were replaced with the intensities of the corresponding probes from a male sample, in order to create virtual samples with deletions at known locations (Methods).
Using this simulation framework, iterated thousands of times over dozens of independent female and male samples, we find that for deletions spanning 3, 5 and 10 probes (corresponding to mean sizes of 5 kb, 8 kb and 17 kb), Birdseye identified with lod of 2 or greater 10%, 51% and 97.5% of the events, respectively; as expected, mean reported lod score also increased with deletion size (Supplementary Table 1 online). Breakpoints were typically determined to within a single probe of the simulated CNV, and fewer than one false positive is expected per genome at a lod of 2 or greater (Fig. 4d). We note that because the simulation removes local autocorrelation of noise, this may overestimate performance on actual data; higher lod cutoffs may be appropriate in different datasets. Nonetheless, this simulation indicates that the combination of the Affymetrix SNP 6.0 array and the Birdsuite seems highly sensitive for deletions of 10 kb or larger.
Because the mutation rate to create de novo CNVs is exceedingly low6,16, when observed in individuals with a sporadic disease phenotype they are particularly good candidates to be causal factors5. However, in searching for de novo events it is critical not only to evaluate the evidence in favor of a CNV in a proband (or a tumor), but also to accurately estimate the likelihood against the presence of the CNV in the parents (or normal tissue). Birdseye is designed to address this need: in addition to reporting the evidence in favor of a CNV, it can be used to reassess a discovered region in other samples (such as the proband’s parents) using a framework that does not involve a stringent genome-wide discovery threshold (Fig. 4b,c). Thus one can filter a list of CNVs on the basis of strong evidence against variation in parents, as opposed to simply failing to achieve genome wide-significant evidence in favor of variation (which is frequently a false negative).
Combining copy number and SNP allele information
The CNV events identified by Birdseye, together with the Canary genotypes for common CNPs, yield an assessment of copy number for each sample across its genome; the SNP genotypes from Birdseed describe sequence variation for samples at SNPs with the expected two copies of each locus. The fourth component of Birdsuite, Fawkes (‘fast analysis with kopy-number et SNPs’), merges these results to yield an integrated picture of the genetic variation in each sample. Fawkes utilizes the imputed locations (in A/B intensity space) of copy-variable clusters to assign an allele-specific copy number genotype (such as AAB, ABBB, A or B) at each SNP (Fig. 1 and Supplementary Methods). Notably, the genotype assignment for a sample is constrained to the set of clusters corresponding to its integer copy number as determined by Canary and Birdseye; allelic copy number is thus informed by measurements not only from the SNP probe, but also from nearby SNP and copy number probes. This approach differs fundamentally from earlier attempts to estimate allele-specific copy number from intensity data at individual SNPs13,17.
We evaluated the genotypes provided by Fawkes of autosomal SNPs across a set of 790 ancestrally diverse samples. Across these samples (comprising 689,451,960 total genotypes) Fawkes changed 717,301 SNP genotypes in 267,070 unique SNPs as compared to running Birdseed alone. A small fraction of SNPs (5,600) had a copy-variable call in at least 1% of unrelated individuals. In the 91 parent-offspring trios available in this dataset, Birdsuite genotypes showed a lower rate of mendelian inconsistencies versus those from Birdseed alone (Table 1). In fact, every family analyzed showed a lower rate of mendelian inheritance errors, with an average decrease of 5% and a maximum decrease of 45% (Fig. 3f). This indicates that copy number variation is an infrequent but not insubstantial source of apparent mendelian inconsistency in all samples, and a major contributor in select samples (potentially owing to cell line artifacts that affect whole chromosomes). As expected, the rate of mendelian inconsistency was particularly reduced across the 11,256 SNPs that lie within common CNPs6, with an average reduction of 33% (Fig. 3e).
The removal of errors due to copy number variation, whether the CNVs originated in the germ line, somatically, or during cell culture, results in higher-quality data and increases the number of SNPs that pass typical filters applied to whole-genome association studies.
Comparison to other algorithms
We carried out a preliminary analysis to test the ability of Birdsuite as a whole to call CNVs. CGH- or sequence-confirmed CNVs discovered using fosmid end sequencing on eight HapMap samples were used as a reference dataset18. Combining Canary calls surpassing the default confidence threshold with Birdseye calls surpassing a lod cutoff of 5, we recovered 56% of the reference CNVs that overlap at least 2 probes on the array, and 94% of those that overlap at least 20 probes. We compared these results to those from two commercial copy number analysis platforms, Nexus and Partek, using the same set of CEL files as input and default thresholds. At these settings, Nexus recovers 26% and 73% of CNVs overlapping at least 2 probes and at least 20 probes, respectively, and Partek recovers 4% and 12%. Relaxing the Nexus parameters to allow significantly more total CNV calls per genome than Birdsuite calls boosts Nexus sensitivity to 36% and 74%, still well below that of Birdsuite (Supplementary Note online).
Association testing in regions of altered copy number
The methods above, in conjunction with a map of common polymorphisms (both single nucleotide and copy number) and hybrid arrays for detection6, allow characterization of the genetic variation in each sample with high accuracy and in a more comprehensive manner than previously possible. In addition to providing discrete calls, this framework provides a confidence of each call, which serves as a good guideline as to data and genotype quality. (In the downstream analyses that follow, we use these confidences only as a threshold for inclusion or exclusion of data; we note that such filtering has the potential to introduce bias, and methods that incorporate the uncertainty may perform better.)
The utility of genotypes from the Birdsuite, however, is not realizable unless analysis tools can accept and evaluate CNVs and noncanonical SNP genotypes, and test them for association with phenotype in a statistically robust manner. Specifically, in addition to performing the typical SNP test of association, one needs to assess the potential relationship of phenotypes to total copy number and allele-specific copy number (for example, AAB versus ABB). For example, a locus may be haploinsufficient when the remaining copy carries a low-expression allele, but not linked to the phenotype if the remaining copy is a high-expression allele. It is also important to assess association of phenotype with a collection of individually rare CNVs that overlie a common locus.
We have developed and implemented one such initial approach to test for such associations. For sites showing both allelic and copy number variation, we regress the phenotype (either a quantitative trait or disease status) on both the sum and the difference of the number of copies of each allele. A significant regression coefficient for the sum represents an association with overall copy number, whereas a significant coefficient for the difference represents an association with variation tagged by a SNP. If there is either no CNV or no SNP at a site, we fit a reduced model by removing the appropriate term (the sum or the difference, respectively), effectively giving a standard one-dimensional regression of phenotype.
Simulations indicate that this model is often considerably more powerful than the traditional approach that does not explicitly represent noncanonical genotypes (Supplementary Table 2 online). This joint approach is particularly powerful under certain scenarios, such as if a duplicated form of a ‘low-activity’ allele shows normal activity comparable with the wild-type allele (for example, when A and BB have similar phenotypes, different from B). In addition, the joint model can often disentangle SNP and CNP effects; for example, it can indicate whether a duplication or the allele that happens to be duplicated (or both) affects phenotype. The new release of the whole-genome association toolset PLINK19 now directly accepts Birdsuite output to perform these tests.
As has been observed in SNP genotyping20, results of association analyses using Canary CNP genotypes can be sensitive to differential bias that can arise when data from individual plates or batches are incorrectly clustered. It is prudent to search for any individual plates or batches for which the Canary clustering may be incorrect; this can be accomplished by visual inspection of the underlying intensity data, or by automated identification of individual plates for which the frequency of the observed genotype classes is statistically unusual given the distribution on the other plates.
For rare or de novo CNVs, one might expect a potentially stronger effect on phenotype—but in a smaller number of samples, as is the case with the 16p11.2 deletion recently discovered (using Birdseye) that seems to explain 1% of idiopathic autism21, or one of numerous deletions associated with schizophrenia22. In such a scenario, aggregation of events in cases at a particular locus, coupled with a lack of events in controls, can indicate that the region affects the assayed phenotype. The new release of PLINK includes a set of tools for manipulating, summarizing and analyzing rare and de novo CNVs output from Birdseye (see URLs section in Methods).
DISCUSSION
Birdsuite is qualitatively different from previous algorithms in that it approaches SNP genotyping and copy number analysis as a problem of joint estimation in which the sequence and copy number aspects of data analysis inform one another. Although we developed the Birdsuite to make use of data generated by a specific SNP and CNP genotyping array, the concepts and approach described here represent a general strategy that can be applied to any genotyping platform. The approach is model-based and empirically derived, offering a sensitive and mutually consistent description of sequence variation, and substantially reducing apparent errors (of mendelian inheritance and Hardy-Weinberg equilibrium) that actually reflect a true state of the individual’s genomic sequence.
Birdsuite is also the first algorithm that takes a central idea of SNP analysis—that an empirical catalog of polymorphisms can be used to disentangle the problem of ab initio discovery from that of highly accurate measurement—and applies it to copy number analysis. For SNPs, discovery and genotyping use separate technologies and algorithms (sequencing and genotyping technology, respectively), but in copy number analysis, these problems have been treated as one: CNV ‘calls’ have been based on the results of genome-wide discovery algorithms. This can lead to false negatives and positives that might be tolerated in the creation of initial CNV catalogs, but that create tremendous problems in association studies that rely on accurate genotyping across large cohorts.
The genotype-calling framework instantiated in Birdsuite and the new release of PLINK supports sensitive, high-resolution identification of CNVs, estimates of SNP and CNP allele frequencies, and tests of association with phenotype. When combined with higher-density hybrid arrays and maps of genome variation at lower frequencies and in more diverse samples (see URLs section in Methods) it should soon be possible to undertake a next generation of genome-wide association studies that provide unbiased, phenotype-driven genome screens for a deeper and more detailed examination of the role of DNA variation in human disease.
METHODS
Samples
DNA from the 270 HapMap individuals (Coriell) was independently prepared, labeled and hybridized to the SNP 6.0 arrays (in distinct plate layouts) at Affymetrix and the Broad Institute. Varying subsets of these samples were prepared and hybridized at seven different testing labs to test reproducibility of genotypes. DNA from 790 individuals (part of the collection of ‘HapMap Phase III’ samples (Coriell)) was prepared, labeled and hybridized at the Broad Institute.
Normalization and transformations of raw data
The set of 6.9 million probespecific measurements from each sample was normalized and summarized (using standard Affymetrix quantile normalization protocol) on a batch-by-batch basis. This yielded 2.7 million measurements per sample (from 932,915 copy number probes and 906,600 thousand SNPs with two measurements each (one per allele)).
in silico gender-mixing experiment
We created 4,000 simulated samples containing a known deletion-1,000 each of a 3-probe deletion, a 5-probe deletion, a 10-probe deletion and a 20-probe deletion. Each simulated sample with deletion size N was generated as follows: (i) choose a random female sample, (ii) choose a random male sample, (iii) randomly permute the order of both the SNP and copy number probes on chromosome X, excluding the pseudo-autosomal regions (this removes natural copy number variation that may occur in either sample), (iv) insert 200 probes from the female sample, (v) insert N probes from the male sample and (vi) insert 200 probes from the female sample. Each simulated deletion used a new random (female, male) pair.
Canary (‘CNP genotyping’)
Canary is a one-dimensional GMM to cluster samples into discrete copy number classes. The initial conditions for each cluster are specified in a prior-models file that contains CNP-specific estimates of cluster locations and variances; a series of models are tested consisting of different number and combination of genotype clusters. Cluster parameters are updated via expectation-maximization, iteratively estimating cluster membership (E step) and maximizing cluster parameters (M step).
A series of heuristics are used to determine which GMM model is best. Using this model, each sample i (with intensity xi) is genotyped as the copy number of the cluster j (with mean μj, s.d. σj and frequency wj), which maximizes the equation
Samples are furthermore assigned a confidence reflecting the relative likelihood of belonging to the next-best cluster (Supplementary Methods).
Birdseed (‘SNP genotyping’)
Birdseed is a two-dimensional GMM to cluster diploid samples into the canonical SNP genotype classes AA, AB and BB. The algorithm is analogous to that of Canary, extrapolated to two dimensions (Supplementary Methods).
Birdseye (‘CNV discovery’)
Birdseye is an HMM to find regions of variable copy number in a sample. The hidden state is the true copy number of the individual’s genome; the observed states are the normalized intensity measurements of each probe on the array.
For each copy number probe, emission probabilities are empirically estimated for an underlying hidden state of 2 copies as a normal distribution, with parameters determined by the intensities of all samples in the batch (excluding those already determined to be copy variable via Canary). Emission probabilities for a state of 0 or 1 copies are imputed using regression parameters learned from probes with known copy number variation (copy number probes on the X chromosome (males versus females), and single autosomal SNP probes (the A allele probe in AA, AB or BB samples)). Emission probabilities for extra copies are imputed assuming that the differences in like parameters between the model for each copy number state increase as a power law.
For each SNP, the emission probability for a state of 2 copies is the union of the three normal distributions specified by Birdseed (modeling the AA, AB and BB clusters). The natural copy number variation inherent to the individual alleles across these clusters allows for direct estimation of the null, A, B, AAB, ABB and AABB clusters. (Future iterations could model and compensate for crosstalk between the two alleles9.) Emission probabilities for increasing dosages of each allele are imputed similar to copy number probes.
The transition probabilities between underlying copy number states are asserted such that transitioning out of a state reflecting normal copy number (typically 2, but varying for the sex chromosomes) is low, whereas transitioning within the same state or returning to normal copy number is relatively high. Furthermore, the transition probability is dependent on the distance between neighboring probes8. (The algorithm is fairly robust to reasonable variations in these settings.)
The emission and transition probabilities are combined to find a path S = {s1, s2...sn} (representing the copy number state at each probe) that maximizes the probability of observing the data X
This maximization is carried out using the standard Viterbi algorithm14. Segments of continuous copy number C are assigned a lod score reflecting the log-likelihood of the path including the event (Si = {F1i, Ci, Ci, Ci, ... F2i} versus the log-likelihood of the path excluding the event Si = {F1i, F1i, F1i, F1i, ... F2i} or Si = {F1i, F2i, F2i, F2i, ... F2i}), where F represents the copy number of the flanking segment (Supplementary Methods).
Association testing of CNVs and embedded SNPs
Our model regresses the phenotype Y on both the sum and the difference of the allelic dosage (0,1,2,3 or 4) for the two alleles, A and B:
A 2-degree-of-freedom test of the null hypothesis H0: b1 = b2 = 0 provides a combined test of CNV and allelic variation; the null H0: b1 = 0 gives a test of copy number variation; the null H0: b2 = 0 gives a test of allelic SNP effects. If there is either no copy number variation or no SNP variation at a site, we fit a reduced model by removing the appropriate term (A + B or A - B, respectively), equivalent to a standard regression of phenotype on allele dosage (assuming constant copy number) or copy number alone (assume a constant allelic background). Moderate correlation between CNV and allelic variation can impact the interpretation of specific tests of either b1 or b2, although the joint 2-d.f. test will be valid.
Supplementary Material
ACKNOWLEDGMENTS
We wish to thank G. Getz for discussions on algorithms and comments regarding the supplemental methods. We also thank E. Lander and J. Hirschhorn for their readings and feedback. Finally, we are indebted to the testing labs that provided us with many replicates of HapMap samples run on the Affymetrix SNP 6.0 array. S.A.M. was supported by a Lilly Life Sciences Research Fellowship.
Footnotes
URLs. Birdsuite, http://www.broad.mit.edu/mpg/birdsuite/; PLINK CNV analysis tools, http://pngu.mgh.harvard.edu/purcell/plink/cnv; 1000 Genomes, http://www.1000genomes.org.
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturegenetics/.
References
- 1.Rabbee N, Speed TP. A genotype calling algorithm for affymetrix SNP arrays. Bioinformatics. 2006;22:7–12. doi: 10.1093/bioinformatics/bti741. [DOI] [PubMed] [Google Scholar]
- 2.Nicolae DL, Wu X, Miyake K, Cox NJ. GEL: a novel genotype calling algorithm using empirical likelihood. Bioinformatics. 2006;22:1942–1947. doi: 10.1093/bioinformatics/btl341. [DOI] [PubMed] [Google Scholar]
- 3.McCarroll SA, et al. Common deletion polymorphisms in the human genome. Nat. Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- 4.Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- 5.McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat. Genet. 2007;39(Suppl):S37–S42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- 6.McCarroll SA, et al. Integrated detection and population-genetic analysis of SNPs and copy-number variation. Nat. Genet. 2008 September 7; doi: 10.1038/ng.238. advance online publication, doi:10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 7.Komura D, et al. Genome-wide detection of human copy number variations using high-density DNA oligonucleotide arrays. Genome Res. 2006;16:1575–1584. doi: 10.1101/gr.5629106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fiegler H, et al. Accurate and reliable high-throughput detection of copy number variation in the human genome. Genome Res. 2006;16:1566–1574. doi: 10.1101/gr.5630906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 10.Wang K, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bengtsson H, Irizarry R, Carvalho B, Speed TP. Estimation and assessment of raw copy numbers at the single locus level. Bioinformatics. 2008;24:759–767. doi: 10.1093/bioinformatics/btn016. published online 19 January 2008. [DOI] [PubMed] [Google Scholar]
- 12.The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macconaill LE, Aldred MA, Lu X, Laframboise T. Toward accurate high-throughput SNP genotyping in the presence of inherited copy number variation. BMC Genomics. 2007;8:211. doi: 10.1186/1471-2164-8-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. J. Roy. Statist. Soc. B. 1977;39:1–38. [Google Scholar]
- 15.Viterbi AJ. Error bounds for convolutional codes and an asymptotically optimum decoding algorithm. IEEE Trans Info Theory. 1967;IT-13:260–269. [Google Scholar]
- 16.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laframboise T, Harrington D, Weir BA. PLASQ: a generalized linear model-based procedure to determine allelic dosage in cancer cells from SNP array data. Biostatistics. 2007;8:323–336. doi: 10.1093/biostatistics/kxl012. [DOI] [PubMed] [Google Scholar]
- 18.Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clayton DG, et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat. Genet. 2005;37:1243–1246. doi: 10.1038/ng1653. [DOI] [PubMed] [Google Scholar]
- 21.Weiss LA, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. published online 9 January 2008. [DOI] [PubMed] [Google Scholar]
- 22.The International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008 July 30; doi: 10.1038/nature07239. advance online publication, doi:10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.