

Abstract
The topological control hypothesis presented by Bostick and Brooks [Proc. Natl. Acad. Sci. USA 104:9260, 2007] has sought to explain binding selectivity in potassium channels on the basis of universal environment-independent features related to ion coordination. This leads to the view that ion selectivity is predominantly controlled by the number of ligands coordinating the ion, and that the chemical type of those ligands plays a minor role. The significance of the topological control hypothesis and its ability to predict ion selectivity in protein binding sites are examined. It is shown that the framework encounters increasing difficulties when different protein binding sites with similar coordination numbers are considered.
Introduction
Understanding the microscopic mechanisms by which proteins can bind specific ions is a question that has fascinated scientists for decades. Ion binding to specific protein sites occurs for purposes of stabilization as well as playing central roles in enzyme catalysis [1, 2, 3, 4]. Ion movements across the cell membrane is tightly controlled by specialized proteins such as pumps, transporters, and ion channels [5]. The high discrimination displayed by membrane channels and transporters between K+ and Na+ is particularly remarkable because these two monovalent cations are very similar, differing only slightly in their atomic radii. For example, K+ channels can conduct K+ ions at a rate near the diffusion limit and remain highly selective over Na+.
While permeation is a non-equilibrium process, several aspects of selectivity in membrane channels may be understood on the basis of equilibrium thermodynamic of ion binding [6, 7, 8]. In this context, selectivity is governed by the relative free energy of ion i and j,
| (1) |
where is the free energy difference between ion i and j in the bulk solvent, and is the free energy difference between ion i and j in the binding site. Because small ions are strongly bound to water molecules in bulk solutions, the protein site must provide coordinating groups that help compensate the loss of hydration. Binding selectivity arises when this energetic compensation is more favorable for one type of ion than for another, relative to the hydration free energy.
A particularly important system where those concepts may be applied is the K+-selective KcsA channel. The determination of its structure to 2.0 Å resolution by X-ray crystallography revealed the chemistry of K+ coordination in the narrowest part of the pore to atomic details [9]. Selectivity was explained by pointing out that the coordinating carbonyl ligands were positioned to precisely fit K+ but not Na+ [10]. Molecular dynamics (MD) simulations, however, indicated that the structure was too flexible to be consistent with this explanation [11]. Results from free energy perturbation (FEP/MD) simulations showed that the pore was nevertheless highly selective for K+ over Na+ in spite of the fluctuations; the most selective location in the selectivity filter being the site S2 with a ΔΔGNa,K of about 5.5 kcal/mol [11, 12]. This value is in good agreement with the most accurate measurements of binding site selectivity in K+ channels, which have been obtained with the large conductance MaxiK channel using barium block assays [6, 7, 8]. The ΔΔGNa,K was measured as 5.5 kcal/mol = kBT ln(27mM/2.7μM), the ratio of the dissociation constants of K+ and Na+ to the external “lock-in” site; the latter corresponds to the binding site S2 [13]. Further analysis shed light on the mechanism by which selectivity for K+ over Na+ can be robustly maintained, in spite of the thermal fluctuations of a flexible binding site [12, 14, 15].
Considering additional molecular systems helps further illuminate the microscopic basis of ion binding selectivity. Generallly, the results from FEP/MD simulations based on molecular mechanical force fields are consistent with experimental observations: KcsA [11, 12, 16, 17] and valinomycin [12, 18, 19] are highly selective for K+, the NaK channel is permissive for Na+ and K+ [20, 21, 22], and the two binding sites of the leucine transporter LeuT are highly selective for Na+ [23, 24]. The large difference in hydration free energy between Na+ and K+, about 17.2 kcal/mol [25], clearly sets a fundamental “baseline” for the selectivity of cation binding sites in proteins. Since is the same regardless of the protein binding site, a reasonable view is that the microscopic causes of selectivity are to be understood from the detailed interactions controlling the quantity itself. This is indeed the case: in the site S2 of the KcsA channel, the K+ is coordinated by eight carbonyl oxygen from the protein backbone; in the most selective site of the leucine transporter LeuT, the Na+ is coordinated by six ligands including one negatively charged carboxylate oxygen [23, 24]; in the non-selective sites of the NaK channel, the cation is coordinated by 6-8 carbonyls and water molecules [21, 22]. These considerations lead to the view that selectivity in flexible protein binding sites is controlled both by the number and the chemical type of ion-coordinating ligands [12, 21, 24].
Alternatively, some theoretical studies have sought to explain binding selectivity on the basis of universal environment-independent features associated with ion coordination. This perspective leads to the view that ion selectivity is predominantly controlled by the number of ligands coordinating the ion and that their chemical type plays a minor role [26, 27, 28]. Such a seductively simple idea, based on an assumed equivalence among the ion-coordinating ligands, goes back to classic work by Mullins [29]. In the “topological control” hypothesis presented by Bostick and Brooks [28], this proposition was put on a quantitative footing by proposing that differences in the probabilities Pi(n) and Pj(n) of the n-coordinated states of ions i and j occurring spontaneously in the bulk phase can be translated into an intrinsic “topological” free energy, ΔJij(n) = kBT ln[Pj(n)/Pi(n)], that directly reflects the relative free energy ΔΔGij governing ion selectivity in a protein binding site. A two-dimensional topological control free energy surface ΔJNa,K(n,m) = kBT ln[PK(n)/PNa(m)] was constructed from MD simulations of Na+ and K+ in bulk water to predict selectivity in protein binding sites [28]. Indeed, this construct predicts that the S2 binding site of KcsA with its n = 8 backbone carbonyl ligands is selective for K+ over Na+, in agreement with FEP/MD simulations [11, 12]. According to this view, the ion bound in the site S2 of KcsA is “over-coordinated”, leading to a less favorable free energy for Na+ relative to K+ [26, 27, 28].
However, the topological control hypothesis encounters increasing difficulties when additional systems are considered. For example, valinomycin binds K+ with six ligands, but is highly selective for K+ over Na+ [12, 18, 19]. The NaK channel presents a coordination structure that is similar to that of KcsA but is non-selective [21]. The amino acid transport LeuT possesses two Na+-selective binding sites comprising five (3.2 kcal/mol) and six (6.1 kcal/mol) ligands [24]; mapping those coordination states onto ΔJNa,K would actually predict that those sites would be non-selective. Moreover, a survey of bound K+ and Na+ in the protein database indicates a large overlap of coordination numbers, suggesting that this is not a strong discriminating factor for Na+ or K+ binding [24]. In spite of these difficulties, the notion that selectivity in protein binding sites can be understood solely from the permeant ion's coordinated state remains very attractive. In a recent study, the topological free energy map has been used to assess the selectivity of the cationic NaK channel [30]. In other words, the ΔJNa,K(n, m) reported in Ref. [28] is starting to be treated as a universal “free energy meter” that can be utilized as a substitute for genuine FEP/MD simulations for estimating ΔΔGNa,K. The purpose of this communication is to clarify the significance of the topological control hypothesis and examine the validity of the topological free energy surface ΔJNa,K to evaluate the selectivity of ion binding sites.
Methods
Theoretical Developments
To clarify the role of hydration number and the significance of the topological control hypothesis, let us re-express the relative free energy of ion i and j in the bulk solvent in terms of constrained n-coordinated states. This is achieved by introducing a Kroenecker discrete delta function δnn′, equal to 1 only when the instantaneous coordination number of the ion n′ is equal to n [31]. By definition, Pi(n) is equal to 〈δnn′〉(i). It follows that,
| (2) |
where is the free energy difference between ion i and j in the bulk for a solvation shell constrained to have a coordination number equal to n and ΔJij(n) = kBT ln[Pj(n)/Pi(n)]. Accordingly, the total free energy difference from Eq. (1) may be expressed as,
| (3) |
The step-by-step thermodynamic procedure underlying the topological hypothesis stated by Eq. 3) is illustrated schematically in Fig. 1.
Figure 1.
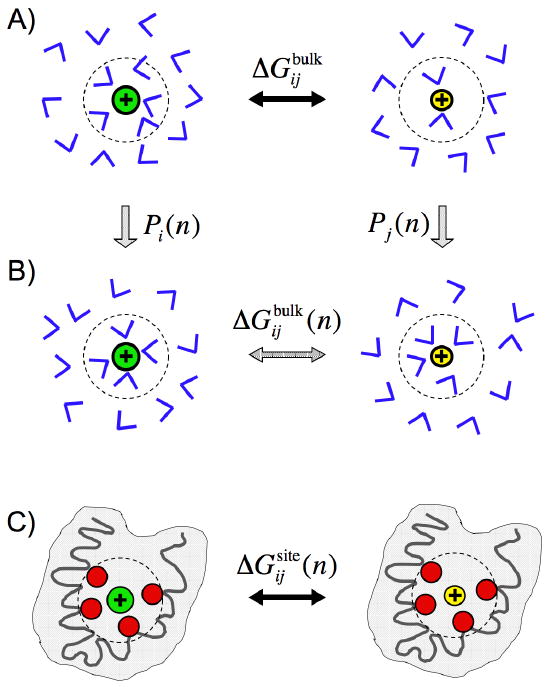
Schematic representation of the role of coordination states in bulk solvent in ion selectivity. In (A), the ions i and j are free in solution; the free energy difference is . In (B), the ions i and j are immersed in the bulk phase but constrained to be coordinated by n solvent molecules; the free energy difference is . The probabilities for ion i and j to spontaneously realize a n-coordinated state in solution are Pi(n) and Pj(n), respectively. In (C), the ions i and j are in the protein binding site surrounded by n ligands; the free energy difference is . Ion binding sites in proteins are typically embedded in a nonpolar low dielectric shell (colored grey) [45]. The notation is meant to emphasize that the binding site is effectively constrained to provide n coordinating ligands.
It is of practical interest to further reduce the complexity of the multi-dimensional configurational integrals in Eq. (2) by integrating out the degrees of freedom that do not participate directly in ion coordination. For any instantaneous configuration R, we can write n′(R) = Σi hi, where hi is a step-function equal to 1 when the i-th molecule is inside the ion's coordination shell region and zero otherwise. It follows that,
| (4) |
where X represents the coordinates of the ion and the n water molecules in the coordination shell, Hbulk(n) = h1h2 … hn is a step function enforcing the confinement of the n water molecules, and ΔWbulk is the PMF representing the influence of the surrounding bulk phase onto the ion and the n water molecules in the coordination shell [31, 32]. Although a unique Hbulk(n) was assumed for ion i and j in Eq. (4), in principle the step function enforcing the n-coordination state could depend on the ion type.
Computational Details
The simulation methodology and PARAM27 force field parameters have been described elsewhere [11, 12, 21]. The simulations with PARAM27 [33] and Drude polarizable [34, 35, 36] force fields were performed with CHARMM c35a1 [37]; those with the Amoeba polarizable force field [38, 39] were performed with Tinker [40]. For the bulk solvation properties of Table 1, a box of 216 water molecules under periodic boundary conditions was simulated at 298 K and 1 atm. The ΔJNa,K(n) was calculated by directly counting the probability of the n-coordinated structure (Pi(n)) in unbiased 1 ns MD simulations. The coordination number of of Na+ and K+ in bulk water were calculated by counting the frequency of having exactly n water within a distance of 3.5 Å. The distance of 3.5 Å closely corresponds to the the minimum in the radial distribution function of K+ (at 3.6 Å), and the minimum in the radial distribution function of Na+ (at 3.2 Å) with both the non-polarizable and polarizable force fields. The coordination number distributions are very similar with both force fields. The unconstrained and the constrained were calculated using FEP/MD simulations. The 8-coordinated state was enforced by a steep flat-bottom harmonic restraint with a force constant of 100 kcal/mol/Å2 during the FEP/MD simulation. The relative free energy of Na+ and K+ in a droplet of eight water confined by a half-harmonic potential at 3.5 Å with a force constant of 100 kcal/mol/Å2 was calculated using FEP/MD simulations [12, 21]. Trajectories of 1 ns were generated and combined using the weighted histogram analysis method (WHAM) [41, 42]. The results are shown in Figs. 2 and 3. The probabilities of the 8-coordinate states of K+ and Na+ in the KcsA channel were calculated from a trajectory of 2 ns. The results are shown in Fig. 4.
Table 1. Free energy and Na+/K+ 8-coordination state (kcal/mol).
| Model | ΔΔG | ΔGsite(n) | ΔGbulk | ΔGbulk(n) | ΔJ(n) |
|---|---|---|---|---|---|
| Param27 | 5.93 | -12.37 | -18.30 | -16.96 | 1.61 |
| Drude | 6.24 | -11.06 | -17.30 | -15.80 | 1.12 |
Figure 2.

Relative K+/Na+ free energy of hydration in the gas phase as a function of the number of water molecules in the cluster from simulations with different potential models and experimental data [25, 43]. For a direct comparison, the results taken from Figure 2A of Ref. [28] are included. The relative hydration free energy of K+ and Na+ at 298.15 K with SWM4-NDP, TIP3P and Amoeba are -17.3, -18.3 and -17.3 kcal/mol, respectively. The confinement is imposed as a flat-bottom half-harmonic potential of 100 kcal/mol/Å2 at 3.5 Å.
Figure 3.

Free energy in reduced models of eight ligands. Relative K+/Na+ free energy as a function of the well-depth (∈min) of the Lennard-Jones WALL confining potential in Tinker.
Figure 4.

Probability to find exactly eight oxygen ligands (backbone carbonyls, water, hydroxyl or carboxylate) around K+ or Na+ in the binding site S2 of the KcsA channel (solid lines). At the larger distances, the average number of ligands is 10 when the eight carbonyls and the 2 nearest water molecules are included (dashed lines).
Results and Discussion
It is important to delineate the conditions under which the the topological free energy contribution ΔJij(n) can be used to ascertain ion selectivity in protein binding sites. According to Eq. (3), the contribution from ΔJij(n) dominates when its magnitude is much larger than the difference between and . The implication is that the relative free energy of ion i and j in a n-coordinated state in two different environments, bulk water and protein binding site, must nearly cancel out. This is illustrated schematically in Fig. 1. For the n-coordinated state in the bulk phase, the n solvent molecules fluctuate within the confined sub-volume of the coordination shell while they are surrounded by a high dielectric fluid (Fig. 1B). In contrast, the n ligands in the binding site are tethered to the protein structure and are held in place via a wide range of architectural forces, including covalent bonding, hydrogen bonding, and van der Waals packing (Fig. 1C). For example, a bound ion in the site S2 of the KcsA K+ channel finds itself in an anhydrous environment where it is primarily coordinated by eight carbonyl backbone oxygens. Despite the considerable differences of the binding site S2 with bulk water, the topological control hypothesis postulates that ΔJNa,K(n) calculated from K+ and Na+ in bulk water can serve to explain the high selectivity of KcsA [28].
To ascertain the applicability of the topological control hypothesis expressed by Eq. (3), we first examine the quantitative equivalence between and by characterizing the free energy contributions corresponding to Fig. 1 for ions binding to the site S2 of the KcsA channel. The results are given in Table 1 for the standard PARAM27 force field [33] and a more accurate model accounting for induced polarization [34, 35, 36]. The quantities , , and ΔJNa,K(n = 8), calculated independently, satisfy the constraint imposed by the thermodynamic cycle of Fig. 1 and Eq. 3): is indeed equal to . The 8-coordinate state constraint yields a topological free energy ΔJNa,K of about 1-2 kcal/mol. This value is considerably smaller than that indicated in Ref. [28]. The discrepancy is partly explained by noting that the topological free energy map ΔJNa,K(n,m) in Ref. [28] was produced by fitting Gaussian functions to the distribution of coordination number PK(n) and PNa(m). The topological free energy ΔJNa,K tends to be overestimated because the the true distributions are not gaussian-like (the problem arises from the tail of PNa(n) near n = 8 where its value is close to zero). While the topological constraint ΔJNa,K displays a slight preference in favor of K+, it differs quantitatively from ΔΔGNa,K calculated by FEP/MD [12, 21]. Based on the present analysis and the results given in Table 1, the free energies and are not equivalent.
To shed additional light on the selectivity displayed by a n-coordinated state, it is interesting to consider a simple reduced (“toy”) model comprising only the ion surrounded by n water molecules restricted to remain within a maximum distance. The free energy difference in such a reduced model is realized by setting the influence of the surrounding bulk ΔWbulk to zero in Eq. (4),
| (5) |
The relative free energy of K+ and Na+ in the confined droplet was calculated using FEP/MD simulations. Fig. 2 shows that a confined droplet comprising eight water molecules displays a small preference of 1-2 kcal/mol for K+ over Na+. This result is obtained from a variety of models, polarizable and nonpolarizable alike, and is consistent with the topological contribution ΔJNa,K calculated from the probability of 8-coordinate states in liquid water (Table 1). It is noteworthy that the calculated relative hydration free energy of K+ and Na+ in small confined droplets of water molecules approaches bulk-like values for n equal to 4-6, consistent with experimental gas phase data [25, 43]. Hence, the lack of prominent selectivity in the small water droplet is an unavoidable trend, as long as the confinement imposed by Hbulk does not interfere too much with the ion coordination.
In apparent contradiction with the above results, Bostick and Brooks [28] presented results indicating that a confined droplet of eight water molecules was selective for K+ over Na+ by about 4 kcal/mol. This value, which is almost equal to the ΔΔGNa,K obtained from all-atom FEP/MD simulations in the S2 binding site of the KcsA channel as well as a “toy model” of eight confined carbonyl CO [11, 12], provided a strong support for the topological control hypothesis [28]. But as seen in Fig. 2, the free energies reported in Ref. [28] using Amoeba are outliers with respect to all other results, including a new set of calculations with Amoeba. Such quantitative discrepancy between well-converged FEP/MD simulations generated with reduced systems comprising only a small number of particles must be explained. The origin of the difference can be traced back to the details of the simulations of the reduced binding site model Eq. (5) generated in Ref. [28]. The spatial confinement Hbulk on the eight Amoeba water molecules was enforced using the WALL option of the program Tinker [40]. This involves applying a radial Lennard-Jones (LJ) 6-12 potential to all the particles contained inside a spherical region. By default, the WALL option of Tinker includes an attractive energy well applied to each of the three atoms forming a water molecule. According to a simple perturbative analysis, the attractive LJ well uwell is akin to an extra “structural” potential, which yields an additional free energy contribution, 〈uwell〉(Na) − 〈uwell〉(K). The latter enhances selectivity for the larger cation: the attractive LJ well “pulls” the water molecules away from the central cation, effectively favoring K+ over Na+. The WALL confinement was used only for the calculations involving Amoeba, whereas all the other systems shown in Figure 2A of Ref. [28] were taken directly from previously published values generated using a steep flat bottom half-harmonic potential [21]. For this reason, the comparison of 8-coordinated systems, water or carbonyl CO, shown in Figure 2A of Ref. [28] is invalid, and the statement “water molecules and carbonyl groups can both provide K+ selective environments if equivalent constraints are imposed on the coordination number of the complex” is incorrect. As shown in Fig. 3 the system become increasing more selective for K+ when the LJ well deepens. When the well depth is negligible, the binding site model of eight water molecules displays only a marginal selectivity for K+ over Na+. All the results are consistent once this is taken into account. Under identical conditions, the flexible binding site comprising eight carbonyl CO is considerably more selective than the corresponding system with eight water molecules (Fig. 3). A recent study confirms this observation and clarifies its origin [15].
It is important to realize that the magnitude of ΔJNa,K is directly affected by the criteria used to define the n-coordinated state in Eq. (2). In a fluid, it is customary to define the “coordination shell” of the ion based on the first minimum in the ion-solvent radial distribution function. However, such a simple definition becomes problematical in the case of an ion bound to a protein. A protein is not a fluid, and the concept of a minimum in a radial distribution function is not applicable. Here, we used a unique maximum distance of 3.5 Å for all states based on the dominant 8-coordinated state observed in the KcsA binding site (Fig. 4). Fundamentally, this choice is motivated by Fig. 1, which seeks to establish the equivalence of and that is implicitly assumed in the topological control hypothesis [28]. The present choice is also consistent with previous studies [12, 21] and a recent analysis based on the topological hypothesis [30]. In principle, one could choose different distances to define the first coordination shell of K+ and Na+ in the bulk phase and in the binding site. However, the four independent distances required to decompose the free energies in Eq. (1) would undermines the transparency and clarity of a simple argument drawn from the concept of a constrained coordination state aimed at explaining ion selectivity. As shown in Fig. 4, the probability of the 8-coordinate states for a cation in the binding site S2 of the KcsA channel reaches a maximum around 3.5 Å; the maximum is at 3.25 Å for Na+ and 3.75 Å for K+. On average there are roughly eight ligands within a distance of 3.5 Å from the ion, but there are frequent excursion to various coordination states (from 6 to 9). It is therefore an approximation to assume that n = 8 represents the coordination state of a cation in the S2 site of KcsA.
More generally, a strict decomposition of the free energy based on the coordination states of the ion in a protein binding site would require consideration of the probabilities and of the spontaneous occurrence of n-coordinated states for ions i and j in the protein binding site. The latter would then have to be translated into a second “topological” free energy for the protein binding site, leading to the relation . While a complete decomposition of and can certainly be carried out, the advantages of pursuing this formal route are unclear. Partly to address such issues, Bostick and Brooks proposed to consider the coordinated states of an ion immersed in a “soup” of freely moving protein ligands [28]. The quantity obtained by counting the number of ligands around the ion would then be used as a yardstick to assess the role played by topological constraints in the protein site. However, the concept of such a “soup” of ligands is problematical for two reasons. First, the probabilities of n-coordinate states depend on the ion-ligand interactions as well as the density and excess chemical potential of the ligands forming the “soup” [31], but there is no obvious prescription for setting the value of those parameters. Second, there is no reason to expect that should mirror the values of in the protein.
One may argue that the relative free energy of K+ and Na+ in the cluster of n = 8 water molecules observed in Fig. 2 is nevertheless revelatory of a fundamental mechanism that is pertinent to explain the occurrence of low selectivity binding sites [28]. There is indeed a slight preference of about 1-2 kcal/mol for K+ over Na+ according to the computations. However, because the calculated free energy differences are small and there is considerable variability variations between the different models, such results should be interpreted with great caution. For example, the ΔΔG(n = 8) is predicted to be 2.1 kcal/mol for the Drude polarizable model, but only 1.4 kcal/mol for Amoeba. In fact, the limitations of current computational models are uncovered by considering the enthalpy of small K+ hydrates in the gas phase. For clusters of n=1-6 water molecules, the values from the Drude model are 17.5, 33.0, 46.2, 57.7, 67.0, and 76.0 kcal/mol, respectively [35]. For Amoeba, the values are 16.9, 31.9, 44.3, 55.1, 64.8, and 72.2 kcal/mol, respectively [38, 39]. By comparison, the gas phase experimental values are 17.9, 34.0, 47.2, 59.0, 69.7, and 79.7 kcal/mol, respectively [25, 43]. The discrepancies between the different values are on the order of 0.5-1.0 kcal/mol for n = 1, but become considerably larger as n grows.
There are good reasons to expect that relative free energies are more accurate than indicated by this comparison, and to some extent, meaningful computational studies are made possible by relying on cancellation of errors [44]. Conclusions drawn from an analysis of the the most selective sites in KcsA [11, 12] and LeuT [24] exhibiting large free energy differences (4-5 kcal/mol) are presumably less sensitive to such inaccuracies. But ultimately, the comparison with the experimental gas phase data shows that all potential functions have limitations. Quantum mechanical (QM) calculations might, in principle, provide information based on first principles. However, utilization of QM approaches is not without problems. First, it is important to realize that there are underlying approximations to the potential energy surface obtained from current QM and density functional methods [35]. Furthermore, configurational sampling at finite temperature is also an important issue. Because they are computationally expensive, QM studies of ion selectivity are commonly based on a comparison of energy-minimized configurations rather than Boltzmann averages [26, 27]. Accordingly, the free energy difference between K+ and Na+ is approximated by taking the difference of two separate minimum energies for the ion and its n coordinating ligands. One can utilize FEP/MD simulations based on a polarizable potential function to illustrate and assess the magnitude of the error arising from an energy minimization procedure. If straight energy minimization is performed until the gradient is small, tests calculations show that the system will unavoidably get trapped in one of the multiple local energy minima. This gives rise to uncorrelated errors that are on the order of 2 kcal/mol with n = 8 compared to the free energy computed by FEP/MD. Test computations show that there exist an increasingly large number of local minima for ions bound to small cluster for n > 4. Alternatively, if efforts are made to search extensively to discover the absolute energy minimum, systematic errors on the order of 2.5 kcal/mol ensue for n = 8 compared to the relative free energy computed by FEP/MD on the same system. These considerations suggests that estimating quantitatively the slight free energy preference for K+ over Na+ in the confined water cluster relative to bulk water is at the limit of current computational methods.
In summary, whether the topological free energy ΔJNa,K(n = 8) is taken from bulk solution (Table 1) or estimated from reduced models (Fig. 2), its value is close to 1-2 kcal/mol according to the present calculations. This is clearly too small to account for the binding free energy at the site S2 of the K+ channels based on the most reliable estimate from experiments [6, 7, 8] as well as all-atoms FEP/MD simulations [11, 12]. The chemical type of ligands in the site S2 has an important impact on the magnitude of relative free energies and cannot be neglected [21]. A binding site of eight carbonyls or eight water molecules differ substantially. Lastly, the present analysis shows that the topological map ΔJNa,K generated from MD simulations of ions in bulk water does not provide a universal measure of transfer free energy to estimate the selectivity of protein binding sites and that it cannot be used as a substitute for genuine FEP/MD simulations.
Acknowledgments
The authors are grateful to Drs. Alan Grossfield, and Jay Ponder for their help with the program Tinker. Clarifying discussions with Dilip Asthagiri, Troy Whitfield and Albert C. Pan are acknowledged. The work was funded by grant GM-62342 from the National Institute of Health (NIH) for H.Y. and B.R.. S.Y.N. is supported by a Discovery Grant RGPIN-315019 from the Natural Sciences and Engineering Research Council of Canada (NSERC) and by the Alberta Heritage Foundation for Medical Research (AHFMR) Scholar.
References
- 1.Harris ME, Christian EL. Recent insights into the structure and function of the ribonucleoprotein enzyme ribonuclease P. Curr Opin Struct Biol. 2003;13:325–333. doi: 10.1016/s0959-440x(03)00069-1. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett GJ, Borkakoti N, Thornton JM. Catalysing new reactions during evolution: economy of residues and mechanism. J Mol Biol. 2003;331:829–860. doi: 10.1016/s0022-2836(03)00734-4. [DOI] [PubMed] [Google Scholar]
- 3.Di Cera E. Thrombin: a paradigm for enzymes allosterically activated by monovalent cations. C R Biol. 2004;327:1065–1076. doi: 10.1016/j.crvi.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 4.Sigel RK, Pyle AM. Alternative roles for metal ions in enzyme catalysis and the implications for ribozyme chemistry. Chem Rev. 2007;107:97–113. doi: 10.1021/cr0502605. [DOI] [PubMed] [Google Scholar]
- 5.Hille B. Ion channels of excitable membranes. 3rd. Sinauer Associates; Sunderland, MA: 2001. [Google Scholar]
- 6.Neyton J, Miller C. Potassium blocks barium permeation through a calcium-activated potassium channel. J Gen Physiol. 1988;92:549–567. doi: 10.1085/jgp.92.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neyton J, Miller C. Discrete Ba2+ block as a probe of ion occupancy and pore structure in the high-conductance Ca2+ -activated K+ channel. J Gen Physiol. 1988;92:569–586. doi: 10.1085/jgp.92.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vergara C, Alvarez O, Latorre R. Localization of the K+ lock-In and the Ba2+ binding sites in a voltage-gated calcium-modulated channel. Implications for survival of K+ permeability. J Gen Physiol. 1999;114:365–376. doi: 10.1085/jgp.114.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 10.Gouaux E, Mackinnon R. Principles of selective ion transport in channels and pumps. Science. 2005;310:1461–1465. doi: 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- 11.Bernèche S, Roux B. Energetics of ion conduction through the K+ channel. Nature. 2001;414:73–77. doi: 10.1038/35102067. [DOI] [PubMed] [Google Scholar]
- 12.Noskov SY, Bernèche S, Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–834. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Y, MacKinnon R. The Barium Site in a Potassium Channel by X-Ray Crystallography. J Gen Physiol. 2000;115:269–272. doi: 10.1085/jgp.115.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asthagiri D, Pratt L, Paulaitis M. Role of fluctuations in a snug-fit mechanism of KcsA channel selectivity. J Chem Phys. 2006;125:24701. doi: 10.1063/1.2205853. [DOI] [PubMed] [Google Scholar]
- 15.Dixit PD, Merchant S, Asthagiri D. Ion selectivity in the KcsA potassium channel from the perspective of the ion binding site? Biophys J. 2009;96:2138–2145. doi: 10.1016/j.bpj.2008.12.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen TW, Bliznyuk A, Rendell A, Kuyucak S, Chung SH. The potassium channel: Structure, selectivity and diffusion. J Chem Phys. 2000;112:8191–8204. [Google Scholar]
- 17.Luzhkov V, Åqvist J. K(+)/Na(+) selectivity of the KcsA potassium channel from microscopic free energy perturbation calculations. Biochim Biophys Acta. 2001;1548:194–202. doi: 10.1016/s0167-4838(01)00213-8. [DOI] [PubMed] [Google Scholar]
- 18.Åqvist J, Alvarez O, Eisenman G. Ion-selective properties of a small ionophore in methanol studied by free energy perturbation simulations. J Phys Chem. 1992;96:10019–10025. [Google Scholar]
- 19.Marrone T, Merz K., Jr Molecular recognition of K+ and Na+ by valinomycin in methanol. J Am Chem Soc. 1995;117:779–791. [Google Scholar]
- 20.Shi N, Ye S, Alam A, Chen L, Jiang Y. Atomic structure of a Na+- and K+-conducting channel. Nature. 2006;440:570–574. doi: 10.1038/nature04508. [DOI] [PubMed] [Google Scholar]
- 21.Noskov SY, Roux B. Importance of hydration and dynamics on the selectivity of the KcsA and NaK channels. J Gen Physiol. 2007;129:135–143. doi: 10.1085/jgp.200609633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alam A, Jiang Y. Structural analysis of ion selectivity in the NaK channel. Nat Struct Mol Biol. 2009;16:35–41. doi: 10.1038/nsmb.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamashita A, Singh S, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 24.Noskov SY, Roux B. Control of ion selectivity in LeuT: Two Na+ binding sites with two different mechanisms. J Mol Biol. 2008;377:804–818. doi: 10.1016/j.jmb.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tissandier MD, Cowen KA, Feng WY, Gundlach E, Cohen MH, Earhart AD, Coe JV, Tuttle TR. The proton's absolute aqueous enthalpy and Gibbs free energy of solvation from cluster-ion solvation data. J Phys Chem A. 1998;102:7787–7794. [Google Scholar]
- 26.Varma S, Rempe S. Tuning ion coordination architectures to enable selective partitioning. Biophys J. 2007;93:1093–1099. doi: 10.1529/biophysj.107.107482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas M, Jayatilaka D, Corry B. The predominant role of coordination number in potassium channel selectivity. Biophys J. 2007;93:2635–2643. doi: 10.1529/biophysj.107.108167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bostick DL, Brooks CL. Selectivity in K+ channels is due to topological control of the permeant ion's coordinated state. Proc Natl Acad Sci USA. 2007;104:9260–9265. doi: 10.1073/pnas.0700554104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mullins L. The penetration of some cations into muscle. J Gen Physiol. 1959;42:817–829. doi: 10.1085/jgp.42.4.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fowler PW, Tai K, Sansom MS. The selectivity of K+ ion channels: testing the hypotheses. Biophys J. 2008;95:5062–5072. doi: 10.1529/biophysj.108.132035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng Y, Roux B. Computation of binding free energy with molecular dynamics and grand canonical Monte Carlo simulations. J Chem Phys. 2008;128:115103. doi: 10.1063/1.2842080. [DOI] [PubMed] [Google Scholar]
- 32.Rogers DM, Beck TL. Modeling molecular and ionic absolute solvation free energies with quasichemical theory bounds. J Chem Phys. 2008;129:134505. doi: 10.1063/1.2985613. [DOI] [PubMed] [Google Scholar]
- 33.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 34.Lamoureux G, Harder E, Vorobyov IV, Roux B, MacKerell AD. A polarizable model of water for molecular dynamics simulations of biomolecules. Chem Phys Lett. 2006;418:245–249. [Google Scholar]
- 35.Whitfield TW, Varma S, Harder E, Lamoureux G, Rempe SB, Roux B. Theoretical study of aqueous solvation of K+: Comparing ab initio, polarizable, and fixed-charge models. J Chem Theo Comput. 2007;3:2068–2082. doi: 10.1021/ct700172b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu H, Whitfield TW, Mazzanti CT, Koeppe RE, II, Andersen OS, Roux B. Solvation of KCl and NaCl in N-methylacetamide: A combined experimental and modeling study. 2008 doi: 10.1021/ja103270w. To be submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM - A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 38.Ren PY, Ponder JW. Polarizable atomic multipole water model for molecular mechanics simulation. J Phys Chem B. 2003;107:5933–5947. [Google Scholar]
- 39.Grossfield A, Ren PY, Ponder JW. Ion solvation thermodynamics from simulation with a polarizable force field. J Amer Chem Soc. 2003;125:15671–15682. doi: 10.1021/ja037005r. [DOI] [PubMed] [Google Scholar]
- 40.Ponder JW. TINKER: Software Tools for Molecular Design. 4th. Washington University School of Medicine; Saint Louis, MO: 2004. [Google Scholar]
- 41.Kumar S, Bouzida D, Swendsen R, Kollman P, Rosenberg J. The Weighted Histogram Analysis Method for free-energy calculations on biomolecules. I. The method. J Comp Chem. 1992;13:1011–1021. [Google Scholar]
- 42.Souaille M, Roux B. Extension to the Weighted Histogram Analysis Method: Combining umbrella sampling with free energy calculations. Comp Phys Comm. 2001;135:40–57. [Google Scholar]
- 43.Dzidic I, Kebarle P. Hydration of alkali ions in gas phase. Enthalpies and entropies of reactions M+(H2O)n-1+H2O = M+(H2O)n. J Phys Chem. 1970;74:1466–1474. [Google Scholar]
- 44.Roux B, Berneche S. On the potential functions used in molecular dynamics simulations of ion channels. Biophys J. 2002;82:1681–1684. doi: 10.1016/S0006-3495(02)75520-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamashita M, Wesson L, Eisenman G, Eisenberg D. Where metal ions bind in proteins. Proc Natl Acad Sci USA. 1990;87:5648–5652. doi: 10.1073/pnas.87.15.5648. [DOI] [PMC free article] [PubMed] [Google Scholar]