

Abstract
Class I phosphoinositide 3-kinase (PI3K) is a dimeric enzyme, consisting of a catalytic and a regulatory subunit. The catalytic subunit occurs in four isoforms designated as p110α, p110β, p110γ and p110δ. These combine with several regulatory subunits; for p110α, β and δ the standard regulatory subunit is p85, for p110γ it is p101. PI3Ks play important roles in human cancer. PIK3CA, the gene encoding p110α, is mutated frequently in common cancers, including carcinoma of the breast, prostate, colon and endometrium. Eighty percent of these mutations are represented by one of three amino acid substitutions in the helical or kinase domains of the enzyme. The mutant p110α shows a gain of function in enzymatic and signaling activity and is oncogenic in cell culture and in animal model systems. Structural and genetic data suggest that the mutations affect regulatory inter- and intramolecular interactions and support the conclusion that there are at least two molecular mechanisms for the gain-of-function in p110α. One of these mechanisms operates largely independently of binding to p85, the other abolishes the requirement for an interaction with Ras. The non-alpha isoforms of p110 do not show cancer-specific mutations. However, they are often differentially expressed in cancer and, in contrast to p110α, wild-type non-alpha isoforms of p110 are oncogenic when overexpressed in cell culture. The isoforms of p110 have become promising drug targets. Isoform-selective inhibitors have been identified. Inhibitors that target exclusively the cancer-specific mutants of p110α constitute an important goal and challenge for current drug development.
Keywords: PI3K, PTEN, Akt, Ras, p85
Introduction
This contribution will present a brief review of Class I phosphatidylinositol 3-kinases (PI3Ks) and their oncogenic activities, focusing on cancer-specific mutations and on differential expression of the four catalytic subunits of this enzyme family.
Class I PI3Ks phosphorylate phosphatidylinositol 4,5 bisphosphate (PIP2) at the 3 position of the inositol ring. The product, phosphatidylinositol 3,4,5-trisphosphate (PIP3), functions as a second cellular messenger that controls cell growth, survival, proliferation, motility and morphology (Bader et al., 2005; Cantley, 2002; Deane and Fruman, 2004; Engelman et al., 2006; Hawkins et al., 2006; Katso et al., 2001; Okkenhaug and Vanhaesebroeck, 2003; Vanhaesebroeck et al., 2001; Vanhaesebroeck and Waterfield, 1999; Vivanco and Sawyers, 2002). The phosphatase PTEN (phosphatase and TENsin homolog deleted on chromosome 10) hydrolyzes PIP3 to PIP2, thus acting as the catalytic antagonist of PI3K (Maehama and Dixon, 1998; Myers et al., 1998; Stambolic et al., 1998). Mutational activation and overexpression of class I PI3K and genetic or epigenetic inactivation of PTEN result in enhanced PI3K signaling which is associated with oncogenic cellular transformation and cancer (Ali et al., 1999; Bachman et al., 2004; Bader et al., 2005; Broderick et al., 2004; Campbell et al., 2004; Cantley, 2002; Cully et al., 2006; Eng, 2003; Fruman, 2004; Hartmann et al., 2005; Kang et al., 2005b; Lee et al., 2005; Leslie and Downes, 2004; Levine et al., 2005; Li et al., 2005; Maehama et al., 2001; Saal et al., 2005; Salmena et al., 2008; Samuels et al., 2004; Simpson and Parsons, 2001; Vogt et al., 2007; Wang et al., 2005; Wishart and Dixon, 2002). Because of the enzymatic antagonism of PI3K and PTEN, it is tempting to equate loss of PTEN with gain in PI3K function. However, there is mounting evidence that a loss of PTEN results in cellular changes that are quite different from those induced by a gain of function in PI3K (Blanco-Aparicio et al., 2007). The enzymatic antagonism is not the only determining factor that characterizes the balance of PTEN and PI3K in the cell. The cellular distribution of the two proteins is different, and these differences can be enhanced by external and internal stimuli. Interaction with other proteins could also gravely affect the balance between PTEN and PI3K. Tumors that have lost PTEN often show drug sensitivities that are different from those that have a direct gain of PI3K function (Salmena et al., 2008).
Only Class I PI3Ks are involved in cancer; there are no data linking Class II PI3Ks or Class III PI3K (Vsp34p) to oncogenesis. This fact probably reflects the different product and substrate specificities of the three classes of PI3K. Only Class I PI3Ks can use PIP2 to generate PIP3, Class II PI3Ks produce the 3,4-bisphosphate and the 3-monophosphate of inositol lipids, and Class III can only make the 3-monophosphate. PIP3 is a critical component in the control of cell growth and replication, and the ability to produce this important second messenger molecule confers oncogenic potential to the lipid kinase. Class I PI3Ks have both lipid and protein kinase activities (Dhand et al., 1994; Foukas et al., 2004; Foukas and Shepherd, 2004). Genetic experiments have shown that lipid kinase is essential for oncogenic activity; p110 engineered to have only protein kinase activity is non-oncogenic (Kang et al., 2006). Whether protein kinase plays a role in conjunction with lipid kinase is not known.
The canonical PI3K signaling pathway
In normal cells, the activity of class I PI3Ks is tightly controlled. Upstream signals recruit the cytosolic PI3Ks to the plasma membrane. This relocation is mediated by interactions with receptor tyrosine kinases (RTK) (Skolnik et al., 1991) or G-protein-coupled receptors (GPCR) (Stephens et al., 1994). Interaction with Ras also contributes to the activation of PI3K (Chan et al., 2002; Rodriguez-Viciana et al., 1994; Rodriguez-Viciana et al., 1996) (Fig. 1). The product of class I PI3K, PIP3, recruits proteins that contain a pleckstrin homology (PH) domain to cellular membranes (Corvera and Czech, 1998). Among these are the serine-threonine kinase Akt (cellular homolog of murine thymoma virus Akt8 oncoprotein), as well as its activating kinase PDK1 (3-phosphoinositide-dependent kinase 1). PDK1 phosphorylates and thereby activates Akt at threonine 308 (Alessi et al., 1997). Signals originating from Akt control the initiation of protein synthesis through a cascade of interactions that proceeds through the tuberous sclerosis complex (TSC), Rheb (Ras homolog enriched in brain) and TOR (target of rapamycin) to two critical downstream targets, S6K (p70 S6 kinase) and 4EBP (eukaryotic initiation factor 4E-binding protein) (Bader and Vogt, 2004; Garami et al., 2003; Inoki et al., 2003; Inoki et al., 2002; Tee et al., 2003; Zhang et al., 2003). Akt signals also regulate transcription, inducing phosphorylation-dependent degradation of FOXO1 (forkhead box O transcription factor) (Biggs et al., 1999; Brunet et al., 1999; Kops et al., 1999; Takaishi et al., 1999; Tang et al., 1999) and inactivation of GSK3β (glycogen synthase kinase-3β) (Cross et al., 1995). Important targets of FOXO1 are the growth-attenuating p27(Kip1) (Medema et al., 2000) and p21(Cip1) (Seoane et al., 2004), and pro-apoptotic BIM (Bcl-2 interacting mediator of cell death) proteins (Arden, 2004; Gilley et al., 2003; Stahl et al., 2002). GSK3β regulates the potentially oncogenic transcription factors Jun (cellular homolog of the Jun oncoprotein of avian retrovirus ASV17) and Myc (cellular homolog of the avian myelocytoma retroviral oncogene) (de Groot et al., 1993; Gregory et al., 2003; Nikolakaki et al., 1993; Sears et al., 2000; Wei et al., 2005). In a positive feedback loop, TOR, in complex with the Rictor (Rapamycin-insensitive companion of TOR) protein phosphorylates and thereby additionally activates Akt at serine 473 (Sarbassov et al., 2005). S6K can introduce an inhibitory phosphorylation on IRS1 (insulin receptor substrate 1), mediating a negative feedback loop (Harrington et al., 2004). Ras is also linked to the PI3K pathway. Activated Ras enhances the activities of PI3K; in turn, the product of PI3K, PIP3, stimulates Ras activation (Chan et al., 2002; Rodriguez-Viciana et al., 1994; Rodriguez-Viciana et al., 1996). The overall effect of the combined PI3K signals is to enhance the stimulation of cellular replication and survival and to reduce growth inhibition and apoptosis.
Figure 1.
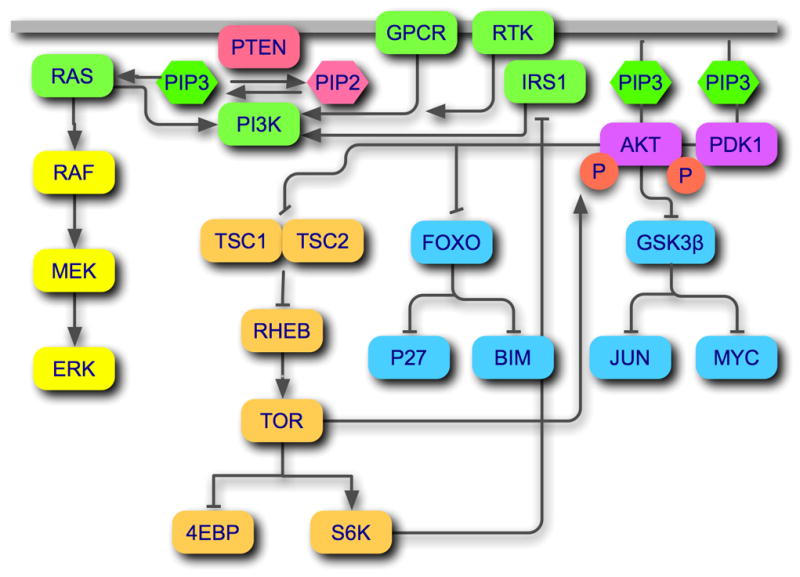
The canonical PI3K signaling pathway. PI3Ks can be activated by RTKs (with or without adaptors such as IRS1) or GPCRs. Ras is an additional positive regulator of PI3K, probably by facilitating membrane localization. The phosphatase PTEN dephosphorylates the product of PI3K, PIP3 at the 3-positiion and thus acts as the exact enzymatic antagonist of PI3K. PIP3 initiates downstream signaling by recruiting the serine-threonine kinases AKT and PDK1. PDK1 phosphorylates and thereby activates Akt. Three major signaling branches originate from Akt. Akt-mediated phosphorylation of GSK3β and of FOXO directly and indirectly controls transcriptional activities and cellular growth and survival (blue icons). The signal proceeding through the TSC complex, RHEB, and TOR affects primarily protein synthesis (beige icons). A positive feed-back loop extends from the TOR-RICTOR complex to Akt, resulting in additional activating phosphorylation of Akt. A negative feed-back loop consists of the S6K-mediated phosphorylation of IRS1.
Class I PI3Ks are heterodimeric proteins that consist of a catalytic subunit and a regulatory subunit, also referred to as an adaptor (Fig. 2). There are four isoforms of the catalytic subunit: p110α, p110β, p110δ and p110γ. They share the same domain composition: an amino-terminal adaptor-binding domain (ABD) that provides the principal interaction surface with the regulatory subunit, a Ras-binding domain (RBD) that mediates the interaction between p110 and Ras-GTP and contributes to the stimulation of PI3K and to the Ras-driven signaling pathway, a C2 (protein-kinase-C homology-2) domain with affinity for lipid membranes, a helical domain acting as a scaffold for other domains of p110, and a carboxyl-terminal kinase domain (Walker et al., 1999). Class I PI3Ks are further subdivided according to their regulatory subunits (Vanhaesebroeck et al., 1997a). Class IA, encompassing p110α, p110β and p110δ, associates with the regulatory subunits p85α, p85β, p55α, p55γ and p50α. Class IB, consisting only of p110γ, binds the regulatory subunits p101, p84 and p87PIKAP. The representative regulatory subunit, p85α, contains several modular protein-protein interaction domains: a Src-homology 3 (SH3) domain, a breakpoint clustered homology (BH) domain, two Src-homology 2 (SH2) domains, and an inter-SH2 (iSH2) domain. The iSH2 domain is the primary p110-binding domain (Dhand et al., 1994; Klippel et al., 1994). Regulatory subunits link p110 to upstream signals, interacting with RTKs and GPCRs. In the absence of upstream signals, the regulatory subunits stabilize p110 and suppress its catalytic activities (Luo et al., 2005; Yu et al., 1998).
Figure 2.
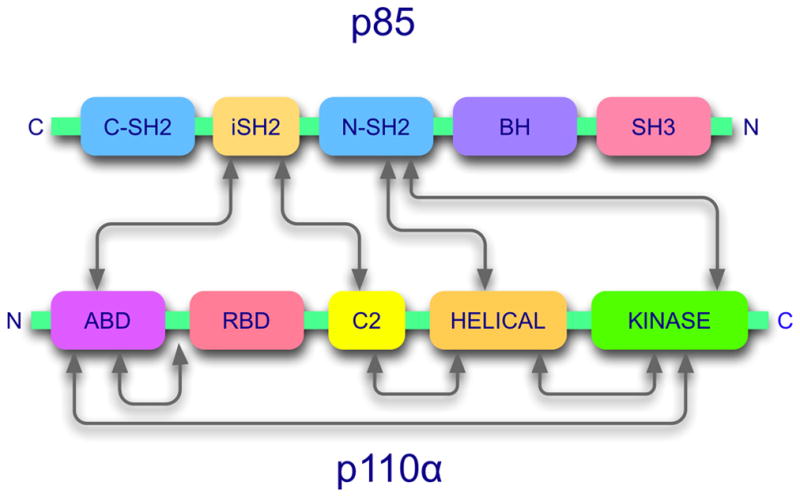
PI3K is a dimeric enzyme. The figure shows the domain structure and domain interaction map of the standard regulatory subunit, p85 and the catalytic subunit, p110 (Huang et al., 2007; Miled et al., 2007; Pacold et al., 2000; Shekar et al., 2005; Walker et al., 1999).
Cancer-specific mutations in PIK3CA
The gene coding for p110α, PIK3CA, is mutated in various human cancers (Samuels and Ericson, 2006; Samuels et al., 2004). The mutations are non-synonymous, arising from single-nucleotide substitutions. They occur in around 30% of several common cancers, including carcinoma of the breast, the colon, endometrium and prostate (Catalogue of Somatic Mutations in Cancer, http://www.sanger.ac.uk/genetics/CGP/cosmic). These cancers carry a single p110α mutation, and 80 % of the mutated proteins contain one of three “hot spot” mutations. Two of these hot spot mutations map to the helical domain of p110α, and the third resides in the kinase domain. The hot spot mutations induce a gain of function in p110α. The lipid kinase activity of the mutant protein is significantly upregulated (Ikenoue et al., 2005; Kang et al., 2005a; Samuels et al., 2004). Mutant-expressing cells show constitutive downstream signaling detectable by el“evated phosphorylation of Akt, S6K, 4EBP and GSK3β. p110α carrying one of the hot spot mutations shows oncogenic activity. It can transform primary fibroblasts in culture, induce anchorage-independent growth and cause tumors in animal model systems (Bader et al., 2006; Ikenoue et al., 2005; Isakoff et al., 2005; Kang et al., 2005a; Zhao et al., 2005). This oncogenic potential probably contributes to the neoplastic phenotype of the human cancer cells carrying mutant p110α; the mutations can therefore be regarded as driver” mutations. The clustering of p110α mutations in hot spots suggests that the mutations provide a selective growth advantage to the cell. In addition to the three hot spot mutations which account for four fifths of the p110α mutations, numerous different rare cancer-specific mutations have been identified (for selected examples see Fig. 3). They are widely distributed over the coding sequence and occur in all domains of p110α except the RBD. Most of these rare mutations also show a gain of function (Gymnopoulos et al., 2007; Ikenoue et al., 2005). However, quantitative measurements of oncogenic activity show that these rare mutants are far less potent than the hot spot mutants. This lower level of oncogenic activity may translate into a weaker selective advantage for the mutant-carrying cell and may explain the rarity of these marginally oncogenic mutations. A recurring theme in the gain-of-function mutations of p110α is substitution of an acidic or neutral residue with a basic residue and location of the substituted residue on the surface of the protein. Indeed, some of the engineered point mutations of p110α that meet these criteria show a gain of function (Gymnopoulos et al., 2007)
Figure 3.
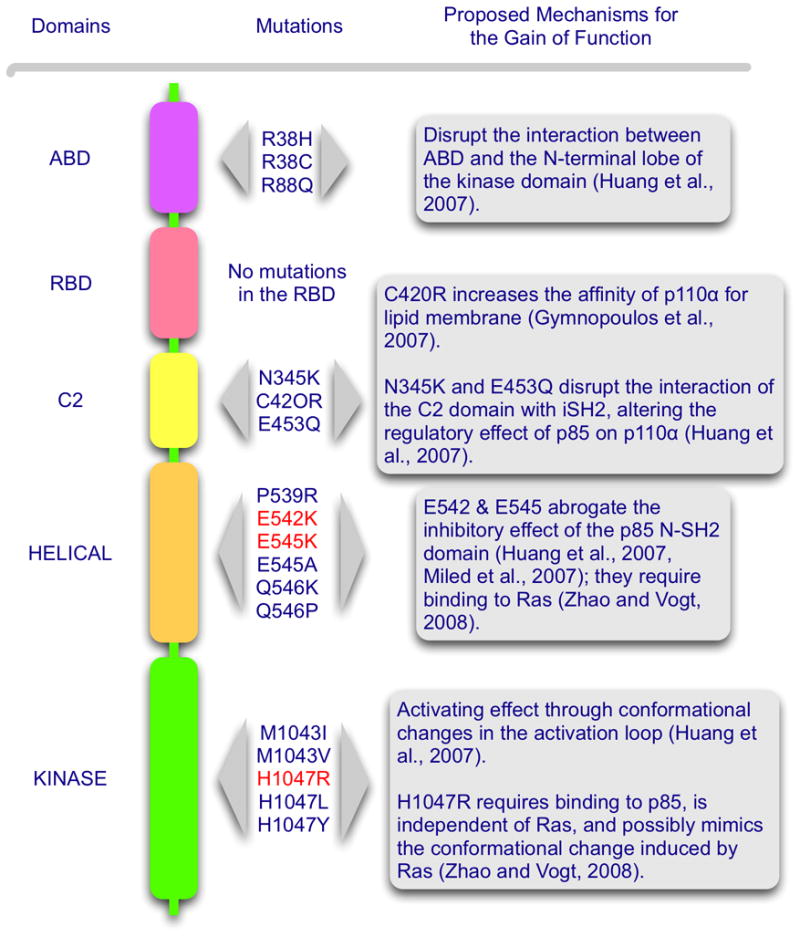
A map of selected cancer-specific gain-of-function mutations in p110α. Suggested mechanisms for the gain of function are listed at the right. The three hot-spot mutations are in red.
Structural data
The X-ray crystal structure of p110α in complex with a portion of p85α has been solved (Huang et al., 2008; Huang et al., 2007). The data show that the ABD of p110α not only binds to the iSH2 domain of p85 (the region responsible for high affinity binding between p85 and p110), but also interacts with the kinase domain and a linker region between ABD and RBD. R38 and R88 of p110α form hydrogen-bonds with Q738, D743 and D746 of the N-terminal lobe of the kinase domain. The rare cancer-specific mutations, R38H, R38C and R88Q possibly disrupt these interactions, resulting in a conformational change of the kinase domain. An ABD deletion mutant of wild-type p110α shows enhanced lipid kinase activity compared to its full-length counterpart (Zhao et al., 2005; Zhao and Vogt, 2008). This increase in activity may be due to the loss of an inhibitory interaction between ABD and the kinase domain of p110α, or it may reflect a relief of the inhibition that is caused by the binding of the N-SH2 (N-terminal SH2) domain of p85 to the helical domain of p110 (see below). Deletion of part of the p85-binding domain in wild-type p110α also reveals a low level of oncogenic transforming activity that is readily demonstrable in cell culture (Zhao and Vogt, 2008).
The C2 domain has been postulated to facilitate recruitment of p110 to the plasma membrane (Nalefski and Falke, 1996; Newton and Johnson, 1998; Rizo and Sudhof, 1998). This function is evident in the crystal structure of p110γ (Walker et al., 1999). Positively charged amino acids are critically involved in membrane binding (Heo et al., 2006). Cancer-specific mutations (N345K and C420R) in the C2 domain of p110α increase the positive surface charge of the domain and were thought to mediate improved binding to the cell membrane, making lipid kinase activity independent of signals transmitted through the regulatory subunit (Gymnopoulos et al., 2007). However in the co-crystal structure of p110α and p85, N345 of p110α forms a hydrogen bond with N564 and D560 in the iSH2 domain of p85. Therefore, N345K is likely to disrupt this interaction of the C2 and iSH2 domains and thus alter the regulatory effect of p85 on p110α (Huang et al., 2007). The electron density of the C420 residue in the C2 domain of p110α is not seen in the co-crystal structure. The C420R mutation may function to increase the affinity of p110α for lipid membranes as previously proposed (Gymnopoulos et al., 2007). Another C2 domain mutation, E453Q, also disrupts the interaction of the C2 domain with iSH2, similar to the N345K mutation (Huang et al., 2007).
Biochemical and structural modeling studies provide evidence for an interaction between the helical domain of p110α and the N-SH2 domain of p85 (Miled et al., 2007; Shekar et al., 2005; Wu et al., 2007). In the co-crystal structure of p110α/p85, the N-SH2 domain is not highly ordered. However, a structural model can be generated if biochemically identified interactions are taken into account. In this model, the N-SH2 domain of p85 binds to the interface between the kinase and the helical domains of p110α (Huang et al., 2007). These interactions may be responsible for the p85-induced inhibition of p110α. The helical domain mutations (E542K and E545K) could interfere with this p85-p110α interaction and thus could relieve the inhibition. The phosphorylated insulin receptor substrate activates the lipid kinase activity of wild-type p110α, presumably by engaging the N-SH2 domain of p85 and thus lifting the inhibitory hold of N-SH2 on the helical domain. This activation is not seen with the helical domain mutants suggesting that the inhibitory interaction with the N-SH2 domain of p85 has been weakened or interrupted by the mutations (Carson et al., 2007). However, helical domain mutations that carry an ABD truncation show significantly higher oncogenic activity than the truncated wild-type p110α (Zhao and Vogt, 2008). Since both have lost p85 binding, the difference in oncogenic potency suggests an effect of the helical domain mutations that goes beyond interference with p85 binding. The relevant intra- and intermolecular interactions are schematically summarized in Fig. 2.
The genetics of cancer-specific mutations in PIK3CA
Genetic experiments provide insight into the molecular mechanisms of mutant-induced gain of function in p110α (Kang et al., 2005a; Liu and Roberts, 2006; Zhao et al., 2005; Zhao and Vogt, 2008). The location of the hot spot mutations in two different domains of the protein, E542K and E545K in the helical and H1047R in the kinase domain, suggests that they operate by different mechanisms. This proposal is supported by the observation that combining helical and kinase domain hot spot mutations in the same molecule has a strongly synergistic effect on downstream signaling and on oncogenic potency. The double mutant, E545K/H1047R, has also been found in human cancer (Lee et al., 2005). The case for mechanistic differences between helical and kinase domain mutations is further strengthened by the interactions with p85 and Ras (Zhao and Vogt, 2008). A truncation of the p85-binding domain that eliminates the interaction with p85 does not silence oncogenic and signaling activities of the helical domain mutants, but completely abolishes oncogenicity in the kinase domain mutant. Curiously, the thus incapacitated kinase-domain mutant still signals through Akt and TOR, albeit at lower levels. Disabling the Ras-p110α interaction by the K227E mutation in the RBD has the opposite effect on the hot spot mutants of p110α. Interaction of GTP-bound Ras with wild-type p110α is known to augment the activity of p110α (Chan et al., 2002), possibly by inducing a conformational change in the substrate-binding site (Pacold et al., 2000). In turn, PI3K is an important Ras effector, mediating the proliferative and survival functions of Ras (Rodriguez-Viciana et al., 1994; Rodriguez-Viciana et al., 1996). Introducing the Ras-binding mutation into the hot spot mutants causes a complete loss of oncogenic potency in the helical domain mutations together with a cessation of signaling, whereas the kinase domain mutant is unaffected by the absence of Ras-binding (Figs. 3 and 4). The kinase domain mutant is even able to rescue helical domain mutants that were incapacitated by the absence of Ras-binding, restoring oncogenic and signaling activities to the synergistic levels seen with helical-kinase domain double mutants. The kinase domain mutation maps close to the activation loop and may affect the conformation of the loop, altering the interaction with the substrate (Huang et al., 2008; Huang et al., 2007). Previous structural studies on the p110γ-Ras complex have demonstrated a change in the conformation of the substrate-binding site as a result of the interaction with Ras (Pacold et al., 2000). The kinase domain mutation H1047R may induce a similar conformational change in the absence of Ras and thus gain Ras-independence. The data on p85 and Ras interaction strongly support the existence of two distinct molecular mechanisms for the mutation-induced gain of function in p110α. The data are compatible with the suggestion that the helical domain mutations lift the inhibitory interaction between N-SH2 of p85 and the helical domain of hot spot mutants of p110α and that the kinase domain mutation mimics the conformational change that is triggered by the interaction with Ras (Zhao and Vogt, 2008). These straight-forward interpretations ascribe the effect of the mutations in the p85 and Ras interacting domains to the specific elimination of p85 and Ras-binding respectively. However, the possibility that these mutant effects are caused by some conformational change that is independent of the targeted protein-protein interactions has not been ruled out. For instance, the co-crystal structure of p110α and p85 also reveals an unexpected interaction between p85-binding domain and kinase domain, which would be affected by the truncation of the p85-binding region (Huang et al., 2007). The ultimate test of these ideas will be the co-crystal structure of mutant p110α bound to the full-length p85 regulatory subunit.
Figure 4.

The interactions with p85 and with Ras define two distinct molecular mechanisms for the gain of function seen in the hot spot mutations in p110α. The helical domain mutations are largely but not completely independent of binding to p85 but require the interaction with Ras. The kinase domain mutation completely depends on the interaction with p85 but is not affected by a loss of Ras-binding. However, the kinase domain mutation still shows residual signaling activity in the absence of p85-binding.
The non-alpha isoforms of class I PI3K
Although the four isoforms of class I PI3K have identical enzymatic activities, they have different, non-redundant cellular functions. Their patterns of expression are distinct, ubiquitous for the p110α and p110β isoforms and largely leukocyte-specific for p110γ and p110δ (Sawyer et al., 2003; Vanhaesebroeck et al., 1997b). Genetic inactivation of p110α and p110β in mice leads to early embryonic lethality (Bi et al., 2002; Bi et al., 1999); p110γ and p110δ knock-out mice are viable but show defective immune responses (Ali et al., 2004; Clayton et al., 2002; Hirsch et al., 2000; Jou et al., 2002; Laffargue et al., 2002; Li et al., 2000; Okkenhaug et al., 2002; Rodriguez-Borlado et al., 2003; Sasaki et al., 2000). Conditional and tissue-specific mutations of the p110 isoforms and experiments with isoform-specific antibodies have generated a steadily increasing catalog of diverse isoform-specific activities (Ali et al., 2008; Bony et al., 2001; Foukas et al., 2006; Graupera et al., 2008; Hooshmand-Rad et al., 2000; Ji et al., 2007; Leverrier et al., 2003; Suire et al., 2006; Vanhaesebroeck et al., 2005; Vanhaesebroeck et al., 1999; Yip et al., 2004). The general conclusions emerging from this work place p110γ and δ firmly in the realm of the immune system, assign p110α to cell growth and reveal an interesting connection between p110β and blood clotting (Ono et al., 2007; van der Meijden et al., 2008). Class I p110α has attracted much attention because of its involvement in cancer, documented by the frequent occurrence of gain-of-function, cancer-specific mutations. No such cancer-specific mutations have been identified in the non-alpha isoforms. Yet there is evidence that non-alpha isoforms of p110 are involved in the development and progression of malignancies. Consistent overexpression of p110δ is seen in acute myeloblastic leukemia (Sujobert et al., 2005). Inhibitors of p110δ specifically interfere with the growth of the leukemic cells, suggesting that p110δ can function as an oncoprotein (Sadhu et al., 2003). Elevated expression of p110γ is observed in chronic myeloid leukemia (Hickey and Cotter, 2006; Skorski et al., 1997). Further data suggest a role of non-alpha isoforms in cancers of the bladder, brain and colon (Benistant et al., 2000; Knobbe et al., 2005; Mizoguchi et al., 2004). Overexpression of non-alpha isoforms in cancer is significant in view of observations in cell culture. Unlike wild-type p110α which lacks oncogenic activity when expressed in primary fibroblasts, the wild-type non-alpha isoforms are oncogenic. This surprising activity of p110β, γ and δ was first documented in avian cells (Kang et al., 2006), but has now been observed in rodent cells as well (Ueno and Vogt, 2007, unpublished). The absence of cancer-specific mutations in the non-alpha isoforms may therefore reflect an inherent oncogenic potential that can be activated by differential expression in the absence of mutation.
A study of cells transformed by p110 isoforms has revealed striking isoform-specific activities that group p110β and p110γ together, placing them apart from p110α and p110δ (Fig. 5) (Denley et al., 2007). In PI3K-transformed cells, wild-type p110δ signals constitutively as does the H1047R mutant of p110α. Transformation by p110β and p110γ does not result in constitutive downstream signaling. This deficiency can be remedied by the addition of a myristylation signal to p110β and p110γ which also results in an enhancement of oncogenic activity (Fig. 4). Additional criteria also show similarities between p110β and p110γ: oncogenicity and signaling of these isoforms require interactions with Ras. The disabling mutation of the RBD K227E eliminates transforming and signaling activities of p110β and p110γ. In contrast, the activities of p110α H1047R and of wild-type p110δ are not affected by this mutation in the RBD. Again, myristylation of p110β and p110γ can substitute for Ras-binding, restoring oncogenic and signaling potential (Fig. 6A). This observation suggests that an essential function of Ras in PI3K signaling is the recruitment of p110β and p110γ to the cell membrane. This recruitment function of Ras may also explain the Ras-independence of p110δ. p110δ has a unique accumulation of basic residues in its C2 domain which probably mediate a direct interaction with the cell membrane. Mutating these basic residues results in an inactive protein. The dependence of p110β and p110γ on Ras is also seen in their sensitivity to inhibitors of the MAP kinase pathway. The Raf inhibitor BAY43-9006 and the MEK1/2 inhibitor U0126 interfere with oncogenicity and signaling of p110β and p110γ (Fig. 6B).
Figure 5.
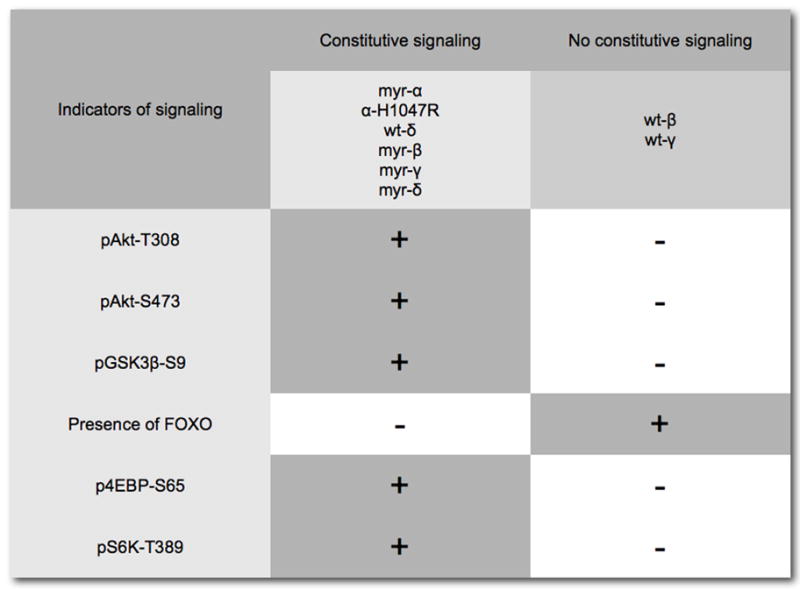
Cells transformed by the four isoforms of Class I p110 show distinct patterns of constitutive downstream signaling that group p110α–H1047R together with p110δ, as both constitutively activate Akt and downstream components of the pathway. In contrast, p110β–and p110γ–transformed cells do not show this constitutive activation of Akt, but this deficiency can be remedied by linking a myristylation signal to the N-terminus of p110.
Figure 6.
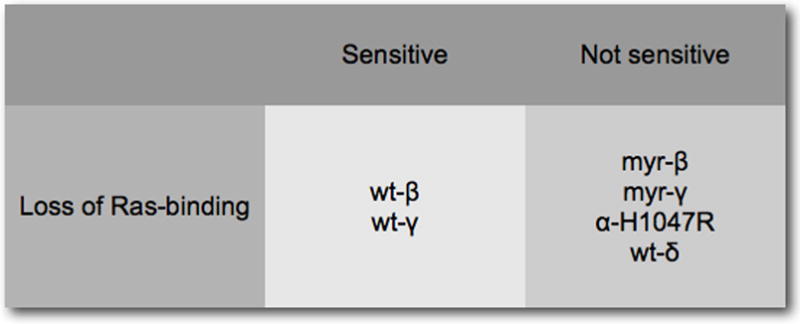
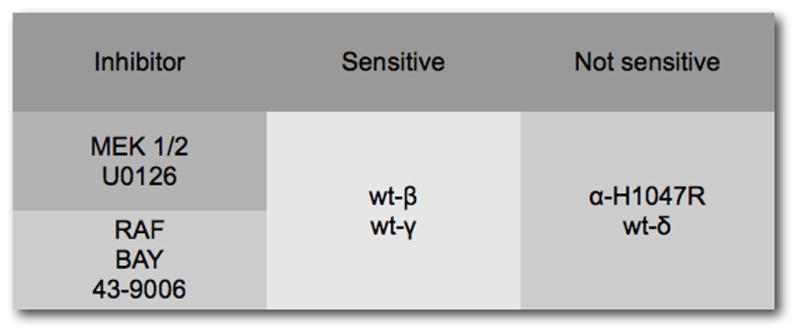
(A) Loss of Ras-binding inactivates wild-type p110β and p110γ, but not p110α–H1047R and p110δ. A myristylation signal can substitute for Ras-binding in p110β and p110γ, suggesting that Ras functions as membrane anchor. (B) The dependence on Ras is also reflected by the sensitivity of p110β and p110γ to inhibitors of the MAP kinase pathway.
Evolving small molecule inhibitors of p110
The distinctive properties of the p110 isoforms lead to the question of small molecule inhibitors and their specificity. The standard PI3K inhibitors for experimental work, Wortmannin and LY294002, are not isoform-specific. However, there are several inhibitors that show significant selectivity for one of the isoforms (Denley et al., 2007) (Fig. 7) However, they are ATP-competitive inhibitors, and because ATP-binding pockets of different kinases are structurally similar, such inhibitors usually show activities against several kinases. Of the ones shown in the figure, PI 103 also inhibits TOR, and TGX221 is effective against class III PI3K. All four p110 isoforms are promising targets for small molecule inhibitors, each isoform is linked to a specific set of clinical indications. The identification and development of such inhibitors with drug-like properties and therapeutic potential has proceeded at a rapid pace in recent years (for example, see (Aftab et al., 2008; Bruce et al., 2007; Folkes et al., 2007; Knight et al., 2006; Knight and Shokat, 2007; Mutton and Pass, 2007; Quattropani et al., 2007; Raynaud et al., 2007)). The success of these efforts and the initiation of clinical trials will require a careful examination of side effects, important especially in long-term use. For p110α, the cancer-specific mutations offer a solution to this potential problem: the identification and development of mutant-specific inhibitors. These could interfere with the oncogenic versions of p110α and leave the important normal functions of wild-type p110α untouched. The genetic and biochemical data on the p110α mutants suggest that mutant-specificity could be attainable. A crystal structure of the mutant proteins would provide decisive guidance in this effort.
Figure 7.
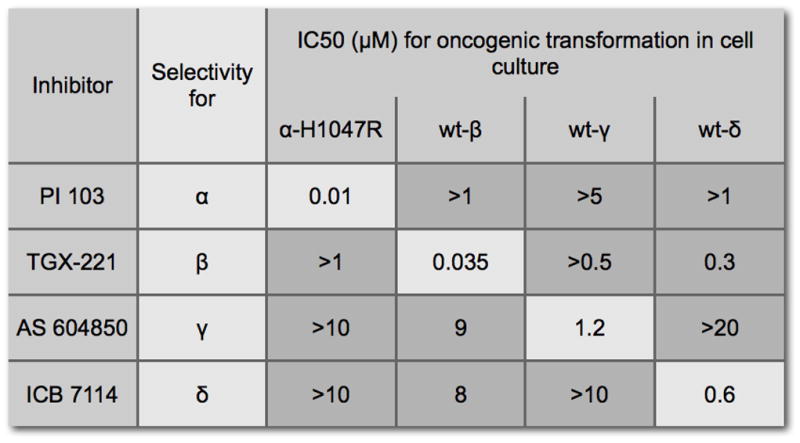
Isoform-selective inhibitors of PI3K. The IC50 values were determined by measuring oncogenic activity in cell culture (Denley et al., 2007).
Acknowledgments
This work was supported by grants from the National Cancer Institute and by The Stein Foundation. Dr. Li Zhao is the recipient of a postdoctoral fellowship from the National Cancer Institute (F32CA130304). This is manuscript no. 19561-MEM from The Scripps Research Institute. We thank Dr. Jonathan Hart and Dr. Marco Gymnopoulos for thoughtful discussions and critical information.
References
- Aftab DT, Laird DA, Lamb P, Martini J-FA. Exelixis, Inc; USA WO: 2008. p. 368. [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Ali IU, Schriml LM, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst. 1999;91:1922–32. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- Ali K, Bilancio A, Thomas M, Pearce W, Gilfillan AM, Tkaczyk C, et al. Essential role for the p110delta phosphoinositide 3-kinase in the allergic response. Nature. 2004;431:1007–11. doi: 10.1038/nature02991. [DOI] [PubMed] [Google Scholar]
- Ali K, Camps M, Pearce WP, Ji H, Ruckle T, Kuehn N, et al. Isoform-specific functions of phosphoinositide 3-kinases: p110 delta but not p110 gamma promotes optimal allergic responses in vivo. J Immunol. 2008;180:2538–44. doi: 10.4049/jimmunol.180.4.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden KC. FoxO: linking new signaling pathways. Mol Cell. 2004;14:416–8. doi: 10.1016/s1097-2765(04)00213-8. [DOI] [PubMed] [Google Scholar]
- Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3:772–5. doi: 10.4161/cbt.3.8.994. [DOI] [PubMed] [Google Scholar]
- Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A. 2006;103:1475–9. doi: 10.1073/pnas.0510857103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5:921–9. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- Bader AG, Vogt PK. An essential role for protein synthesis in oncogenic cellular transformation. Oncogene. 2004;23:3145–50. doi: 10.1038/sj.onc.1207550. [DOI] [PubMed] [Google Scholar]
- Benistant C, Chapuis H, Roche S. A specific function for phosphatidylinositol 3-kinase alpha (p85alpha-p110alpha) in cell survival and for phosphatidylinositol 3-kinase beta (p85alpha-p110beta) in de novo DNA synthesis of human colon carcinoma cells. Oncogene. 2000;19:5083–90. doi: 10.1038/sj.onc.1203871. [DOI] [PubMed] [Google Scholar]
- Bi L, Okabe I, Bernard DJ, Nussbaum RL. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm Genome. 2002;13:169–72. doi: 10.1007/BF02684023. [DOI] [PubMed] [Google Scholar]
- Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J Biol Chem. 1999;274:10963–8. doi: 10.1074/jbc.274.16.10963. [DOI] [PubMed] [Google Scholar]
- Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–6. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28:1379–86. doi: 10.1093/carcin/bgm052. [DOI] [PubMed] [Google Scholar]
- Bony C, Roche S, Shuichi U, Sasaki T, Crackower MA, Penninger J, et al. A specific role of phosphatidylinositol 3-kinase gamma. A regulation of autonomic Ca(2)+ oscillations in cardiac cells. J Cell Biol. 2001;152:717–28. doi: 10.1083/jcb.152.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick DK, Di C, Parrett TJ, Samuels YR, Cummins JM, McLendon RE, et al. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004;64:5048–50. doi: 10.1158/0008-5472.CAN-04-1170. [DOI] [PubMed] [Google Scholar]
- Bruce I, Hayler JF, Bloomfield GC, Edwards L, Cox B, Howsham C. Novartis A.-G., Switz.; Novartis Pharma G.m.b.H.; WO: 2007. p. 82. [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64:7678–81. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Carson JD, Van Aller G, Lehr R, Sinnamon RH, Kirpatrick RB, Auger KR, et al. Effects of oncogenic p110alpha subunit mutations on the lipid kinase activity of phosphatidylinositol 3-kinase. Biochem J. 2007 doi: 10.1042/BJ20070681. [DOI] [PubMed] [Google Scholar]
- Chan TO, Rodeck U, Chan AM, Kimmelman AC, Rittenhouse SE, Panayotou G, et al. Small GTPases and tyrosine kinases coregulate a molecular switch in the phosphoinositide 3-kinase regulatory subunit. Cancer Cell. 2002;1:181–91. doi: 10.1016/s1535-6108(02)00033-8. [DOI] [PubMed] [Google Scholar]
- Clayton E, Bardi G, Bell SE, Chantry D, Downes CP, Gray A, et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med. 2002;196:753–63. doi: 10.1084/jem.20020805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvera S, Czech MP. Direct targets of phosphoinositide 3-kinase products in membrane traffic and signal transduction. Trends Cell Biol. 1998;8:442–6. doi: 10.1016/s0962-8924(98)01366-x. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–92. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- de Groot RP, Auwerx J, Bourouis M, Sassone-Corsi P. Negative regulation of Jun/AP-1: conserved function of glycogen synthase kinase 3 and the Drosophila kinase shaggy. Oncogene. 1993;8:841–7. [PubMed] [Google Scholar]
- Deane JA, Fruman DA. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu Rev Immunol. 2004;22:563–98. doi: 10.1146/annurev.immunol.22.012703.104721. [DOI] [PubMed] [Google Scholar]
- Denley A, Kang S, Karst U, Vogt PK. Oncogenic signaling of class I PI3K isoforms. Oncogene. 2007 doi: 10.1038/sj.onc.1210918. [DOI] [PubMed] [Google Scholar]
- Dhand R, Hiles I, Panayotou G, Roche S, Fry MJ, Gout I, et al. PI 3-kinase is a dual specificity enzyme: autoregulation by an intrinsic protein-serine kinase activity. Embo J. 1994;13:522–33. doi: 10.1002/j.1460-2075.1994.tb06290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–98. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Folkes A, Shuttleworth S, Chuckowree I, Oxenford S, Wan NC, Castanedo G, et al. Piramed Limited, UK; Genentech, Inc.; Goldsmith, Richard; WO: 2007. p. 206. [Google Scholar]
- Foukas LC, Beeton CA, Jensen J, Phillips WA, Shepherd PR. Regulation of phosphoinositide 3-kinase by its intrinsic serine kinase activity in vivo. Mol Cell Biol. 2004;24:966–75. doi: 10.1128/MCB.24.3.966-975.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, et al. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature. 2006;441:366–70. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- Foukas LC, Shepherd PR. Phosphoinositide 3-kinase: the protein kinase that time forgot. Biochem Soc Trans. 2004;32:330–1. doi: 10.1042/bst0320330. [DOI] [PubMed] [Google Scholar]
- Fruman DA. Towards an understanding of isoform specificity in phosphoinositide 3-kinase signalling in lymphocytes. Biochem Soc Trans. 2004;32:315–9. doi: 10.1042/bst0320315. [DOI] [PubMed] [Google Scholar]
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–66. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162:613–22. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008 doi: 10.1038/nature06892. [DOI] [PubMed] [Google Scholar]
- Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem. 2003;278:51606–12. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104:5569–74. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–23. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann C, Bartels G, Gehlhaar C, Holtkamp N, von Deimling A. PIK3CA mutations in glioblastoma multiforme. Acta Neuropathol (Berl) 2005;109:639–42. doi: 10.1007/s00401-005-1000-1. [DOI] [PubMed] [Google Scholar]
- Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans. 2006;34:647–62. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, et al. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314:1458–61. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey FB, Cotter TG. BCR-ABL regulates phosphatidylinositol 3-kinase-p110gamma transcription and activation and is required for proliferation and drug resistance. J Biol Chem. 2006;281:2441–50. doi: 10.1074/jbc.M511173200. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–53. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- Hooshmand-Rad R, Hajkova L, Klint P, Karlsson R, Vanhaesebroeck B, Claesson-Welsh L, et al. The PI 3-kinase isoforms p110(alpha) and p110(beta) have differential roles in PDGF- and insulin-mediated signaling. J Cell Sci. 2000;113:207–14. doi: 10.1242/jcs.113.2.207. [DOI] [PubMed] [Google Scholar]
- Huang CH, Mandelker D, Gabelli SB, Amzel LM. Insights into the oncogenic effects of /PIK3CA/ mutations from the structure of p110alpha/p85alpha. Cell Cycle. 2008;7 doi: 10.4161/cc.7.9.5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–8. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65:4562–7. doi: 10.1158/0008-5472.CAN-04-4114. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–1000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- Ji H, Rintelen F, Waltzinger C, Bertschy Meier D, Bilancio A, Pearce W, et al. Inactivation of PI3Kgamma and PI3Kdelta distorts T-cell development and causes multiple organ inflammation. Blood. 2007;110:2940–7. doi: 10.1182/blood-2007-04-086751. [DOI] [PubMed] [Google Scholar]
- Jou ST, Carpino N, Takahashi Y, Piekorz R, Chao JR, Wang D, et al. Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol Cell Biol. 2002;22:8580–91. doi: 10.1128/MCB.22.24.8580-8591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005a;102:802–7. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Bader AG, Zhao L, Vogt PK. Mutated PI 3-kinases: cancer targets on a silver platter. Cell Cycle. 2005b;4:578–81. doi: 10.4161/cc.4.4.1586. [DOI] [PubMed] [Google Scholar]
- Kang S, Denley A, Vanhaesebroeck B, Vogt PK. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2006;103:1289–94. doi: 10.1073/pnas.0510772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annual Review of Cell & Developmental Biology. 2001;17:615–75. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- Klippel A, Escobedo JA, Hirano M, Williams LT. The interaction of small domains between the subunits of phosphatidylinositol 3-kinase determines enzyme activity. Mol Cell Biol. 1994;14:2675–85. doi: 10.1128/mcb.14.4.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–47. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Shokat KM. Chemically targeting the PI3K family. Biochem Soc Trans. 2007;35:245–9. doi: 10.1042/BST0350245. [DOI] [PubMed] [Google Scholar]
- Knobbe CB, Trampe-Kieslich A, Reifenberger G. Genetic alteration and expression of the phosphoinositol-3-kinase/Akt pathway genes PIK3CA and PIKE in human glioblastomas. Neuropathol Appl Neurobiol. 2005;31:486–90. doi: 10.1111/j.1365-2990.2005.00660.x. [DOI] [PubMed] [Google Scholar]
- Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398:630–4. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- Laffargue M, Calvez R, Finan P, Trifilieff A, Barbier M, Altruda F, et al. Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity. 2002;16:441–51. doi: 10.1016/s1074-7613(02)00282-0. [DOI] [PubMed] [Google Scholar]
- Lee JW, Soung YH, Kim SY, Lee HW, Park WS, Nam SW, et al. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24:1477–80. doi: 10.1038/sj.onc.1208304. [DOI] [PubMed] [Google Scholar]
- Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J. 2004;382:1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverrier Y, Okkenhaug K, Sawyer C, Bilancio A, Vanhaesebroeck B, Ridley AJ. Class I phosphoinositide 3-kinase p110beta is required for apoptotic cell and Fcgamma receptor-mediated phagocytosis by macrophages. J Biol Chem. 2003;278:38437–42. doi: 10.1074/jbc.M306649200. Epub 2003 Jul 16. [DOI] [PubMed] [Google Scholar]
- Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 2005;11:2875–8. doi: 10.1158/1078-0432.CCR-04-2142. [DOI] [PubMed] [Google Scholar]
- Li VS, Wong CW, Chan TL, Chan AS, Zhao W, Chu KM, et al. Mutations of PIK3CA in gastric adenocarcinoma. BMC Cancer. 2005;5:29. doi: 10.1186/1471-2407-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 2000;287:1046–9. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- Liu Z, Roberts TM. Human tumor mutants in the p110alpha subunit of PI3K. Cell Cycle. 2006;5:675–7. doi: 10.4161/cc.5.7.2605. [DOI] [PubMed] [Google Scholar]
- Luo J, Field SJ, Lee JY, Engelman JA, Cantley LC. The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J Cell Biol. 2005;170:455–64. doi: 10.1083/jcb.200503088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Maehama T, Taylor GS, Dixon JE. PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem. 2001;70:247–79. doi: 10.1146/annurev.biochem.70.1.247. [DOI] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–7. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317:239–42. doi: 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- Mizoguchi M, Nutt CL, Mohapatra G, Louis DN. Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol. 2004;14:372–7. doi: 10.1111/j.1750-3639.2004.tb00080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutton SP, Pass M. Astrazeneca AB, Swed.; Astrazeneca UK Limited; WO: 2007. p. 109. [Google Scholar]
- Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, et al. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998;95:13513–8. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 1996;5:2375–90. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AC, Johnson JE. Protein kinase C: a paradigm for regulation of protein function by two membrane-targeting modules. Biochim Biophys Acta. 1998;1376:155–72. doi: 10.1016/s0304-4157(98)00003-3. [DOI] [PubMed] [Google Scholar]
- Nikolakaki E, Coffer PJ, Hemelsoet R, Woodgett JR, Defize LH. Glycogen synthase kinase 3 phosphorylates Jun family members in vitro and negatively regulates their transactivating potential in intact cells. Oncogene. 1993;8:833–40. [PubMed] [Google Scholar]
- Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297:1031–4. doi: 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3:317–30. doi: 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- Ono A, Lim J, Hamilton JR, Jackson SP, Schoenwaelder SM. SELECTIVE SIGNALLING ROLE FOR PI 3-KINASE P110BETA IN THROMBIN-INDUCED CLOT RETRACTION. Journal of Thrombosis and Haemostasis. 2007;5:W-238. [Google Scholar]
- Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103:931–43. doi: 10.1016/s0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- Quattropani A, Pomel V, Rueckle T, Grippi-Vallotton T. Laboratoires Serono S.A., Switz; WO: 2007. p. 76. [Google Scholar]
- Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–50. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- Rizo J, Sudhof TC. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem. 1998;273:15879–82. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Borlado L, Barber DF, Hernandez C, Rodriguez-Marcos MA, Sanchez A, Hirsch E, et al. Phosphatidylinositol 3-kinase regulates the CD4/CD8 T cell differentiation ratio. J Immunol. 2003;170:4475–82. doi: 10.4049/jimmunol.170.9.4475. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–32. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. Embo J. 1996;15:2442–51. [PMC free article] [PubMed] [Google Scholar]
- Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65:2554–9. doi: 10.1158/0008-5472-CAN-04-3913. [DOI] [PubMed] [Google Scholar]
- Sadhu C, Dick K, Tino WT, Staunton DE. Selective role of PI3K delta in neutrophil inflammatory responses. Biochem Biophys Res Commun. 2003;308:764–9. doi: 10.1016/s0006-291x(03)01480-3. [DOI] [PubMed] [Google Scholar]
- Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–14. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–6. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- Sawyer C, Sturge J, Bennett DC, O’Hare MJ, Allen WE, Bain J, et al. Regulation of breast cancer cell chemotaxis by the phosphoinositide 3-kinase p110delta. Cancer Res. 2003;63:1667–75. [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–14. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–23. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- Shekar SC, Wu H, Fu Z, Yip SC, Nagajyothi, Cahill SM, et al. Mechanism of constitutive phosphoinositide 3-kinase activation by oncogenic mutants of the p85 regulatory subunit. J Biol Chem. 2005;280:27850–5. doi: 10.1074/jbc.M506005200. [DOI] [PubMed] [Google Scholar]
- Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264:29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- Skolnik EY, Margolis B, Mohammadi M, Lowenstein E, Fischer R, Drepps A, et al. Cloning of PI3 kinase-associated p85 utilizing a novel method for expression/cloning of target proteins for receptor tyrosine kinases. Cell. 1991;65:83–90. doi: 10.1016/0092-8674(91)90410-z. [DOI] [PubMed] [Google Scholar]
- Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16:6151–61. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, et al. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–31. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Stephens L, Smrcka A, Cooke FT, Jackson TR, Sternweis PC, Hawkins PT. A novel phosphoinositide 3 kinase activity in myeloid-derived cells is activated by G protein beta gamma subunits. Cell. 1994;77:83–93. doi: 10.1016/0092-8674(94)90237-2. [DOI] [PubMed] [Google Scholar]
- Suire S, Condliffe AM, Ferguson GJ, Ellson CD, Guillou H, Davidson K, et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol. 2006;8:1303–9. doi: 10.1038/ncb1494. [DOI] [PubMed] [Google Scholar]
- Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, Verdier F, et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood. 2005;106:1063–6. doi: 10.1182/blood-2004-08-3225. [DOI] [PubMed] [Google Scholar]
- Takaishi H, Konishi H, Matsuzaki H, Ono Y, Shirai Y, Saito N, et al. Regulation of nuclear translocation of forkhead transcription factor AFX by protein kinase B. Proc Natl Acad Sci U S A. 1999;96:11836–41. doi: 10.1073/pnas.96.21.11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741–6. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–68. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- van der Meijden PE, Schoenwaelder SM, Feijge MA, Cosemans JM, Munnix IC, Wetzker R, et al. Dual P2Y 12 receptor signaling in thrombin-stimulated platelets--involvement of phosphoinositide 3-kinase beta but not gamma isoform in Ca2+ mobilization and procoagulant activity. FEBS J. 2008;275:371–85. doi: 10.1111/j.1742-4658.2007.06207.x. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci. 2005;30:194–204. doi: 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Jones GE, Allen WE, Zicha D, Hooshmand-Rad R, Sawyer C, et al. Distinct PI(3)Ks mediate mitogenic signalling and cell migration in macrophages. Nature Cell Biology. 1999;1:69–71. doi: 10.1038/9045. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends in Biochemical Sciences. 1997a;22:267–72. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Experimental Cell Research. 1999;253:239–54. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U S A. 1997b;94:4330–5. doi: 10.1073/pnas.94.9.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Vogt PK, Kang S, Elsliger MA, Gymnopoulos M. Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem Sci. 2007;32:342–9. doi: 10.1016/j.tibs.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–20. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- Wang Y, Helland A, Holm R, Kristensen GB, Borresen-Dale AL. PIK3CA mutations in advanced ovarian carcinomas. Hum Mutat. 2005;25:322. doi: 10.1002/humu.9316. [DOI] [PubMed] [Google Scholar]
- Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Wishart MJ, Dixon JE. PTEN and myotubularin phosphatases: from 3-phosphoinositide dephosphorylation to disease. Trends Cell Biol. 2002;12:579–85. doi: 10.1016/s0962-8924(02)02412-1. [DOI] [PubMed] [Google Scholar]
- Wu H, Yan Y, Backer JM. Regulation of class IA PI3Ks. Biochem Soc Trans. 2007;35:242–4. doi: 10.1042/BST0350242. [DOI] [PubMed] [Google Scholar]
- Yip SC, El-Sibai M, Hill KM, Wu H, Fu Z, Condeelis JS, et al. Over-expression of the p110beta but not p110alpha isoform of PI 3-kinase inhibits motility in breast cancer cells. Cell Motil Cytoskeleton. 2004;59:180–8. doi: 10.1002/cm.20032. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18:1379–87. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5:578–81. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102:18443–8. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008;105:2652–7. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]