

Abstract
Over 100 mutations in the gene encoding human copper-zinc superoxide dismutase (SOD1) cause an inherited form of the fatal neurodegenerative disease amyotrophic lateral sclerosis (ALS). Two pathogenic SOD1 mutations, His46Arg (H46R) and His48Gln (H48Q), affect residues that act as copper ligands in the wild type enzyme. Transgenic mice expressing a human SOD1 variant containing both mutations develop paralytic disease akin to ALS. Here we show that H46R/H48Q SOD1 possesses multiple characteristics that distinguish it from the wild type. These properties include: 1) an ablated copper-binding site; 2) a substantially weakened affinity for zinc; 3) a binding site for calcium ion; 4) the ability to form stable heterocomplexes with the Copper Chaperone for SOD1 (CCS); and 5) compromised CCS-mediated oxidation of the intrasubunit disulfide bond in vivo. The results presented here, together with data on pathogenic SOD1 proteins coming from cell culture and transgenic mice, suggest that incomplete posttranslational modification of nascent SOD1 polypeptides via CCS may be a characteristic shared by fALS SOD1 mutants, leading to a population of destabilized, off-pathway folding intermediates that are toxic to motor neurons.
The homodimeric antioxidant enzyme copper-zinc superoxide dismutase (SOD1) has been studied for nearly four decades. In 1993, interest in the molecule intensified when mutations in the gene encoding SOD1 were linked to the lethal neurodegenerative disease amyotrophic lateral sclerosis (ALS) (1, 2). Since then, ∼100 distinct pathogenic mutations have been documented [reviewed in (3)], with most resulting in single amino acid substitutions and a few in truncations in the C-terminal portion of the poypeptide.
Landmark studies in transgenic mice established that pathogenic SOD1 proteins elicit motor neuron dysfunction through the acquisition of a deleterious property and not a loss of enzymatic function (4-6). SOD1-enriched inclusions are observed in cell culture model systems, ALS-SOD1 transgenic mice, and in fALS patients, suggesting that SOD1- linked ALS pathology is related to misfolding or aggregation [reviewed in (7-9)]. However, the precise molecular mechanism(s) underlying SOD1 toxicity to motor neurons is unknown, and it remains to be clarified whether the observed inclusions are causal or symptomatic of motor neuron dysfunction.
Given the observations described above, ALS-mutant SOD1 proteins likely possess properties distinct from the wild type enzyme that facilitate their aggregation in vivo. Initial structural studies suggested that metal-deficient, pathogenic SOD1 dimers might be ideal candidates for self-assembly because they have been observed to engage in non-native SOD1-SOD1 interactions that propagate bidirectionally to form linear and helical filamentous arrays (10). Subsequent biochemical studies suggested that dissociated, monomeric pathogenic SOD1 may aggregate (11, 12), and this suggestion focused attention on the structural elements responsible for stabilization of the homodimer (13). More recent studies highlighted the critical roles of both metal binding and the presence of an oxidized intrasubunit disulfide bond to SOD1 dimer stability (14-16). These findings turned attention to a helper protein, the Copper Chaperone for SOD1 (CCS), which specifically recognizes newly translated SOD1 and activates it by inserting the catalytic copper ion and catalyzing the oxidation of the SOD1 intrasubunit disulfide bond (17-19). Because these two posttranslational modifications are integral to SOD1 stability, pathogenic SOD1 mutations that might (directly or indirectly) interfere with metal-binding, disulfide bond oxidation, and/or interaction with CCS are increasingly under scrutiny.
Of the ∼100 distinct disease-causing mutations that have been identified, two (H46R and H48Q) map to copper ligands in the active site. In rodent models of the disease, rats expressing H46R SOD1 (20), mice expressing the H46R/H48Q double mutant (21), and mice expressing a quadruple SOD1 mutant that disrupts all four copper ligands (22), all develop motor neuron disease characterized by the appearance of fibrillar SOD1-containing aggregates. Because the overexpressed human SOD1 proteins in these models all harbor a compromised copper-binding site, these studies together strongly suggest that copper-binding is not a requirement in SOD1-linked ALS etiology.
Here we report crystal structures of human H46R/H48Q SOD1 in two distinct crystal systems and characterize the biophysical properties of this pathogenic SOD1 variant and its complexes with CCS. The structural and biochemical data presented here, together with results from previous studies in cell culture and in transgenic mice, suggest a characteristic common to all fALS mutant SOD1 proteins may be that a fraction of the newly translated mutants “fail to mature”. That is, for each pathogenic mutant, a subset of the newly translated SOD1 proteins are not stabilized via posttranslational modification by CCS, leading to what are in essence off pathway folding intermediates that are toxic to motor neurons.
Experimental Procedures
Materials
Monobasic and dibasic potassium phosphate, acetonitrile (Optima grade), formic acid, sodium hydroxide, yeast extract, peptone, dextrose (glucose), EDTA, sodium chloride, and sodium acetate were obtained from Fischer Scientific. Ammonium sulfate was purchased from US Biological. Polyethelyne glycol 1000 came from Fluka. Tris was purchased from Research Products International. Primers came from Invitrogen. Agarose glycerol, DEAE Sephadex, and PMSF were obtained from Sigma. Pfu DNA polymerase and deoxyribonucleotides were purchased from Stratagene. Glass beads were obtained from Biospec. Tris(2-carboxyethyl)phosphine (TCEP), crystallization screening kits, and crystal growth trays were purchased from Hampton Research. Phenyl Sepharose and Sephadex G-75 came from Pharmacia. All solutions unless otherwise noted in the text were prepared using de-ionized water passed through a Millipore ultra purification system. In experiments using reducing agent, TCEP was added as a buffered solution with the pH adjusted to the values indicated.
SOD1 Cloning, Expression and Purification
DNA fragments encoding the human H46R/H48Q SOD1 double mutant were a amplified by PCR and ligated into the YEP351-hSOD plasmid, where expression of the SOD1 protein is directed under the control of its own promoter. The protein was expressed, purified, and characterized as previously described (15) with the addition of a DEAE-Sephadex chromatography step between the hydrophobic interaction chromatography and gel filtration column steps. Metal content of purified SOD1 proteins was determined using inductively-coupled plasma mass spectrometry (ICP-MS).
CCS Cloning, Expression and Purification
DNA fragments encoding wild type yeast and human CCS were generated by polymerase chain reaction (PCR) from plasmids generously supplied by J.S. Valentine (UCLA). CCS constructs were cloned into a pkA6H vector, which contains an inducible LacZ promoter, a 6×-N-terminal His-tag, and a tobacco etch virus (TEV) cleavage site. CCS proteins were expressed in Escherichia coli BL21 (DE3). Cells containing these expression plasmids were grown in LB media at 37 °C to an OD600 of 0.6 to 0.8. The hCCS expression media was supplemented with ZnSO4 to final concentration of 200 μM. After induction with IPTG, the cells were transferred to 30 °C for an additional 4 hours before being harvested. CCS proteins were purified using a HisTrap HP nickel column purchased from Amersham. After purification, the hexa-His-tag was removed from the CCS proteins using TEV protease produced in-house and engineered to contain its own non-cleavable hexa-His tag. After digestion, TEV protease was removed from the CCS sample by a final pass through the nickel column. This procedure leaves a two residue (Gly-His) extension on the CCS N-termini. Metal content of purified CCS proteins was determined using inductively-coupled plasma mass spectrometry (ICP-MS) at the Chemical Analysis Laboratory at the University of Athens, Georgia.
Crystallization, Structure Determination, and Refinement
Purified H46R/H48Q SOD1 was concentrated to 15 mg/ml in 2.25 mM potassium phosphate, pH 7.0 (protein solution). Crystals were grown by the hanging drop vapor diffusion method. For crystals grown in space group P21, protein solution was mixed with an equal volume of precipitating solution containing 25 % (v/v) polyethylene glycol (PEG) 550 MME, 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) at pH 5.9, and 0.01 M ZnSO4. Rectangular plates appeared after about a week of incubation at 22 °C. For crystals grown in space group C2221, protein solution was mixed with an equal volume of precipitating solution containing 20 % (v/v) polyethylene glycol (PEG) 1000, 0.1 M imidazole at pH 8.0, and 0.2 M calcium acetate. Rectangular prisms grew after about a week of incubation at 22 °C. Both crystal forms were cryoprotected in a solution of the mother liquor plus 10 % (v/v) PEG 200 and flash frozen in liquid nitrogen prior to data collection.
X-ray diffraction data were acquired on a Rigaku FR-D rotating copper anode X-ray generator equipped with RAXIS-HTC image plate detectors. The oscillation range was 1° and 0.5° for crystals in space groups P21 and C2221, respectively, and the crystal-to-detector distance was 150 mm for both crystal forms. Crystals were cooled during data acquisition using an X-STREAM low-temperature system. Diffraction data were processed using HKL2000 (23). Five percent of the reflections were selected at random as a function of resolution shell and set aside during refinement for cross-validation (24). The structure of the human G37R variant of SOD1 (pdb code 1AZV (25) was used as the search model in molecular replacement performed by the program MOLREP (26), which positioned the SOD1 dimers in the unit cells of both crystal forms. Rigid-body refinement using monomers as individual rigid elements was followed by iterative cycles of positional and atomic displacement parameter (B-factor) refinement in REFMAC (27) and manual model building using the program COOT (28). Translational, librational, and screw (TLS) parameters (29) were implemented in the final refinement cycles.
Native Gel-Shift
10-15 μg each of wild type and H46R/H48Q SOD1 and y/hCCS in 50 mM MES pH 6.0, 150 mM NaCl with or without 10 mM TCEP (reducing agent) were run on 12 % non-denaturing polyacrylamide gels. SOD1/CCS proteins were mixed in a 1:1 stoichiometric ratio and immediately loaded onto the native gel and run at 120 mV. The running buffer was 25 mM Tris pH 8.0, 200 mM glycine.
Zinc Release Assay
The relative affinities of H46R/H48Q and wild type SOD1 proteins for zinc ion were compared using a slightly modified 2-pyridylazo-resorcinol (PAR) assay (30). All measurements were acquired in triplicate in assay buffer containing 100 μM PAR, 50 mM Tris pH 8.1, 150 mM NaCl, with and without 10 mM TCEP on a Beckman DU7400 spectrophotometer. The extinction coefficient for the zinc-PAR2 complex at 500 nm (ε500 nm) was 84,000 M-1 cm-1, as determined experimentally from standard curves derived from known concentrations of ZnSO4 in assay buffer. 50-60 μg of human wild type and H46R/H48Q SOD1, and equal amounts of yeast and human CCS were added to a quartz cuvette containing assay buffer with and without TCEP and absorbance readings were taken at one minute intervals for one hour at room temperature. After 60 minutes, 0.5 mM EDTA was added to strip the zinc from PAR, quenching the absorbance at 500 nm.
Analytical Ultracentrifugation
Sedimentation velocity experiments were performed with a Beckman XL-A analytical ultracentrifuge using absorbance optics. Sedimentation velocity experiments were performed at 60,000 rpm over four hours at 4 °C. The concentration of the protein samples corresponded to OD280nm readings of 0.8 (SOD1 = ∼2.4 mg/mL, yCCS = ∼0.8 mg/mL, and hCCS = ∼1.9 mg/mL). Sedimentation velocity data were analyzed using the method of van Holde and Weischet (31) to determine the sedimentation coefficient distribution G(s), followed by 2-D Spectrum and genetic algorithm analyses to acquire estimates of the molecular weights of the protein components as implemented in the ULTRASCAN software package (32).
Gel Filtration Chromatography
A Superdex 200 column (Amersham) was equilibrated with a solution containing 50 mM MES pH 6.0 and 150 mM NaCL with or without 10 mM TCEP and calibrated with purified human SOD1 (32 kDa) and human CCS (58 kDa) proteins plus proteins from High and Low Molecular Weight gel filtration calibration kits (Amersham). These proteins included thyroglobulin (669 kDa), Ferritin (440 kDa), albumin (65 kDa), ovalbumin (43 kDa), and ribonuclease A (13.7 kDa). H46R/H48Q SOD1 and y/hCCS proteins were mixed in a 1:1 stoichiometric ratio and immediately loaded onto and run through a Superdex 200 column at constant rate of 0.5 mL/min.
Determination of SOD1 Disulfide Status in Cells
4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS), which covalently modifies cysteine thiolates, was employed to monitor the status of the SOD1 intrasubunit disulfide bond in yeast (33) and in human embryonic kidney (HEK 293) cells (34) as described previously.
Results
Structures of the H46R/H48Q SOD1 Double Mutant
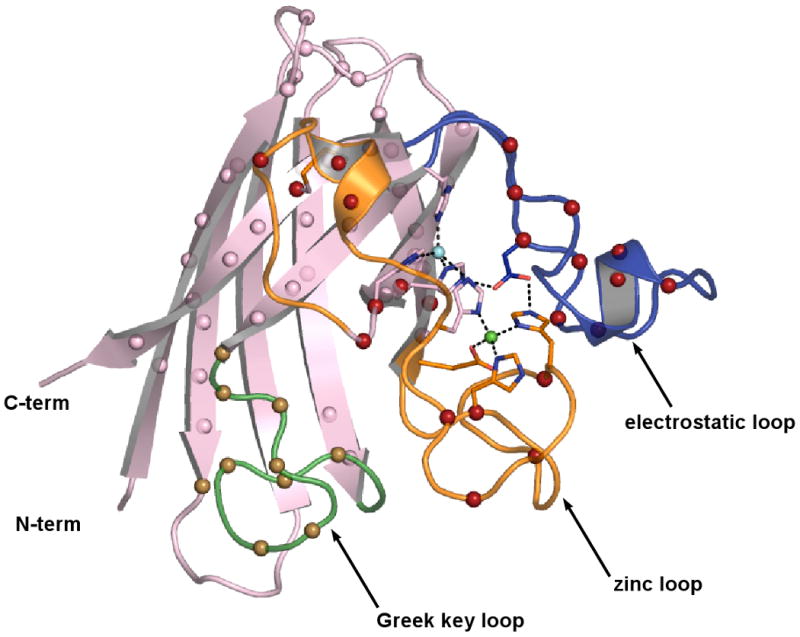
Crystal structures of H46R/H48Q SOD1 were determined and refined in space groups P21 and C2221 to resolutions of 2.3 and 2.2 Ǻ, respectively. X-ray diffraction data collection and protein structure refinement statistics are shown in Table 1. The H46R/H48Q SOD1 subunits in space group P21 are designated A, B, C, and D with AB and CD representing distinct SOD1 homodimers. The H46R/H48Q SOD1 subunits in space group C2221 are designated E, F, G, H, I, and J, with EF, GH, and IJ representing homodimers. In the wild-type enzyme, the metal-binding sites are formed by H46, H48, and H120 coming from the exterior of the β-barrel and by H63, H71, H80, and D83 coming from loop IV. Loop IV (residues 49-83), also known as the “zinc loop”, contains a substructure we designate the “disulfide loop” (residues 49-60). Loop VII (residues 121-142), also known as the “electrostatic loop”, forms a large portion of the active site channel and contains charged amino acids that help to guide the negatively charged superoxide substrate to the catalytic copper ion (35). The electrostatic loop also contains D124, the side chain of which helps to stabilize both the copper- and zinc-binding sites (see Figure 2B). Figure 1A shows the active site region from chain A of H46R/H48Q SOD1 in space group P21, which is representative of the active sites in all chains in both crystal systems. None of the subunits in either space group contain a metal ion in what is clearly an ablated copper-binding site. In contrast, the zinc-binding sites of all subunits contain a zinc ion coordinated in the same fashion as the wild-type enzyme.
Table 1.
X-ray diffraction data and refinement statistics for H46R/H48Q crystals used in this study
| PDB accession code | 2NNX | xxxx |
| Space Group | P21 | C2221 |
| Diffraction Data | ||
| Wavelength (Å) | 1.5418 | 1.5418 |
| Space Group | P21 | C2221 |
| Unit Cell dimensions (Å) | a = 76.5, b = 63.5, c = 85.1 | a = 112.6, b = 194.3, c = 143.2 |
| a = 90.0°, β = 116°, g = 90.0° | a = β = g = 90.0° | |
| Diffraction resolution (Å) | 50 – 2.30 | 50 – 2.20 |
| No. of Observations | 139, 672 | 361, 783 |
| No. of Unique Reflections | 32, 846 | 79, 657 |
| Completeness (%)a | 100.0 (100.0) | 99.8 (100.0) |
| Mean I/σI | 15.8 (2.6) | 14.7 (2.9) |
| Rsym | 0.092 (0.531) | 0.088 (0.503) |
| Structure Refinement | ||
| No. protein atoms | 4440 | 6660 |
| No. water molecules | 308 | 504 |
| No. Zn ions | 8 | 6 |
| No. sulfate ions | 1 | 0 |
| No. Ca2+ ions | 0 | 5 |
| Rcryst | 0.186 (0.217) | 0.204 (0.239) |
| Rfreeb | 0.241 (0.313) | 0.241 (0.264) |
| RMSD Bond Lengths (Å) | 0.011 | 0.011 |
| RMSD bond angles (deg.) | 1.4 | 1.3 |
| Ramachandran Plotd | ||
| Favored (%) | 88.0 | 90.2 |
| Allowed (%) | 11.8 | 9.4 |
| Generous (%) | 0.2 | 0.4 |
| Disallowed (%) | 0.0 | 0.0 |
| Average B-factors (Å2) | ||
| Protein | 28 | 31 |
| Water | 33 | 38 |
| Zn | 43 | 28 |
| Sulfate | 24 | NA |
| Ca2+ | NA | 56 |
Values in parentheses are for the highest resolution shell.
5 % of reflections were chosen randomly in each resolution shell for cross-validation.
Generated using the program PROCHECK (58)
Figure 2. H46R/H48Q SOD1 has weakened affinity for zinc.
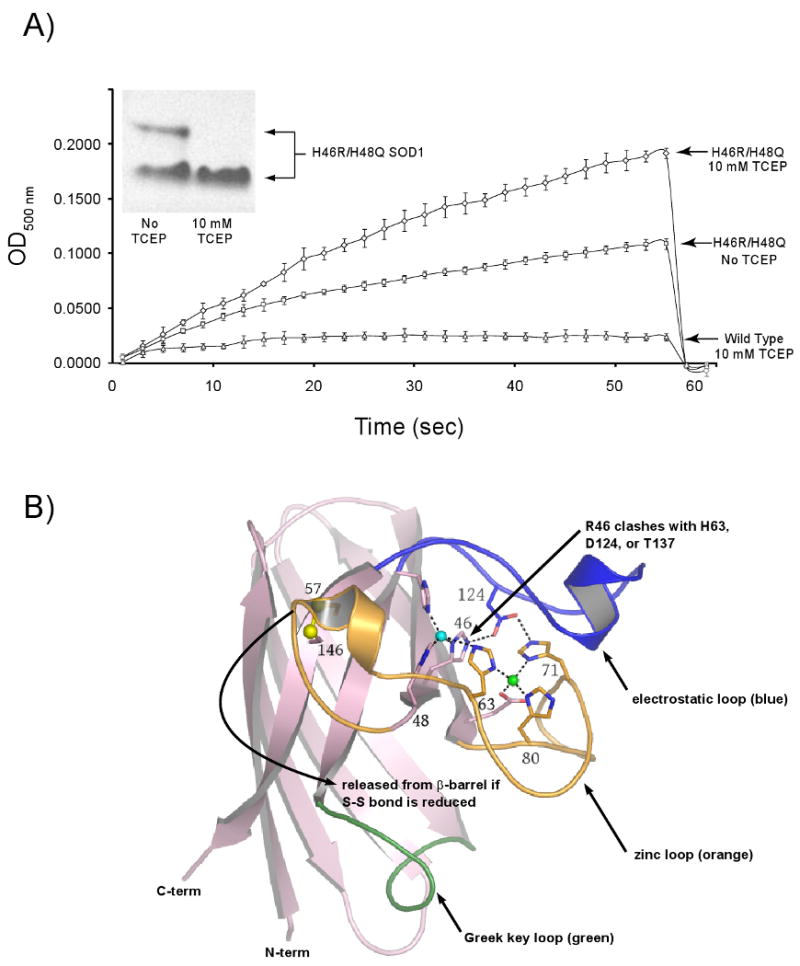
A) Zinc Release Assay. In an effort to understand the molecular basis for the native gel-shift shown by the H46R/H48Q SOD1 double mutant in the presence of reducing agent (inset), the chelator 4-(2-Pyridylazo)-resorcinol (PAR) was used to monitor zinc release by H46R/H48Q SOD1 as shown by increased absorbance due to a newly formed PAR2-Zinc complex. In the absence of reducing agent (middle trace), PAR slowly leaches zinc from the enzyme. In the presence of TCEP (top trace) zinc is released relatively rapidly from H46R/H48Q SOD1. Wild type SOD1 (lower trace) retains its copper and zinc ions under all conditions and even in the presence of 2 M GdnHCl on this timescale (30) (see text). B) Putative release of the zinc loop (loop IV, orange) from the β-barrel (pink) upon reduction of the intrasubunit disulfide bond as depicted in the wild type enzyme. The Sγ atoms of C57 and C146 are shown as yellow spheres, the copper ion is shown as a cyan sphere, and the zinc ion is shown as a green sphere. The electrostatic loop (loop VII) is shown in blue and is linked to the active site by hydrogen bonds between D124 and the nonliganding imidazole nitrogen atoms of copper ligand H46 and Zn ligand H71. H46R substitution is expected to impact H63, D124, and T137 weakening the interaction between the zinc and electrostatic loop elements as described in the text.
Figure 1. The structure of H46R/H48Q SOD1.
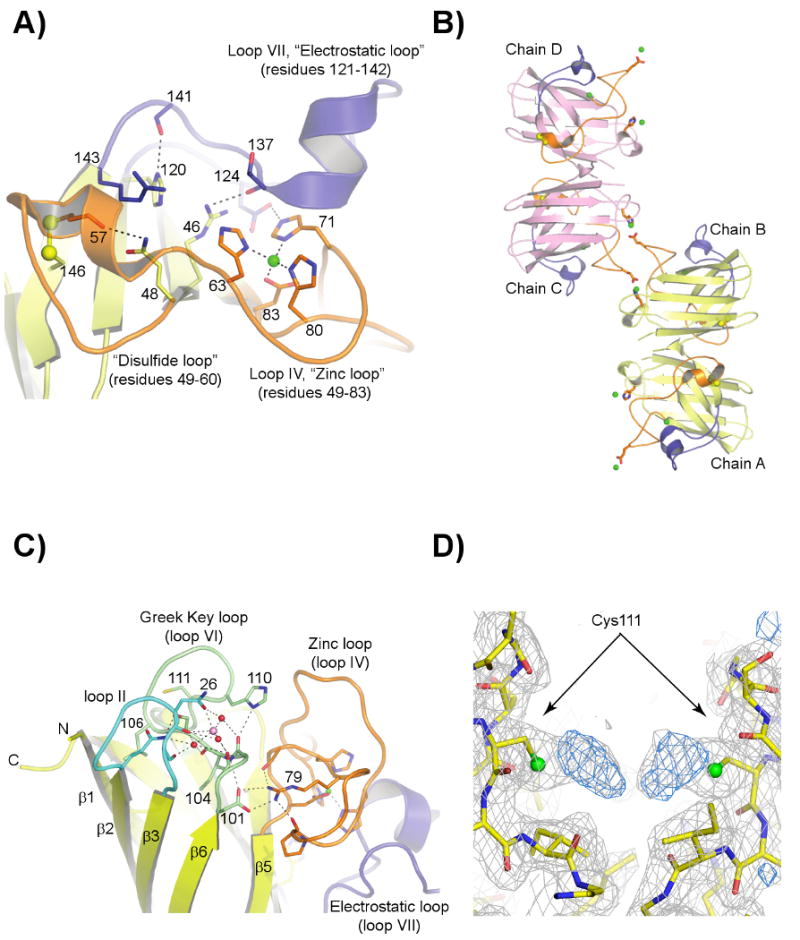
A) The H46R/H48Q active site region in subunit A, which is similar to all subunits in both crystal forms. The zinc and electrostatic loop elements are colored orange and blue, respectively. The disulfide bond between C57 of the disulfide loop and C146 of the β-barrel is shown in yellow ball-and-stick. The zinc ion is shown as a green sphere. The R46 sidechain spans the active site channel and donates a hydrogen bond to a sidechain oxygen atom of D124 and the carbonyl oxygen of T137, both of the electrostatic loop. The Q48 sidechain displaces the guanidinium moiety of R143, preventing it from forming its normal hydrogen bonds to the carbonyl oxygen of C57. B) External zinc-binding site. The zinc and electrostatic loop elements are colored as in panel A). The sidechains of E77 and H110 from symmetry-related H46R/H48Q subunits coordinate zinc ions (green spheres) that bridge SOD1 dimers along the b-axis of the P21 crystal form. A zinc ion also occupies the zinc-binding site, capping the C-terminus of the small α-helix in the electrostatic loop. C) The calcium-binding site within each SOD1 monomer in the C2221 structure (see text). The zinc and electrostatic loop elements are colored as in panels A) and B). The calcium ion (pink sphere) is held in a cleft between loop II (cyan) and the Greek key loop (loop VI, pale green) by the sidechain and carbonyl oxygen atoms of N26 and the carbonyl oxygen atoms of G27 and S102 in all but one subunit in the C2221 crystal form. Water molecules acting as ligands to the calcium ion are shown as red spheres. D) 2FoFc (gray) and FoFc (blue) SIGMAA electron density for what appear to be diatomic covalent adducts on the Sγ atoms of C111 in the P21 structure. The 2FoFc and FoFc maps are contoured at 1 and 3 σ, respectively.
Figure 1B shows that each H46R/H48Q subunit is bridged to another subunit along the crystallographic b axis by two external zinc ions such that chain A of one dimer is linked to chain D of a second dimer, and chain B of this second dimer is linked to chain C of a third SOD1 dimer. Together with a water molecule from the solvent, each subunit contributes the sidechains of E77 and H110 to act as liganding residues to the external bridging zinc ions.
Figure 1C shows a representative calcium binding site formed by the sidechain and carbonyl oxygen atoms of N26 and the carbonyl oxygen atoms of G27 and S102 as observed in chain H of the C2221 crystal form. Nearly identical calcium binding sites are observed in chains E, F, G, H, and J in the C2221 crystal system, and two modes through which calcium ions mediate crystal packing are shown in panels B) and C) of Supplementary Figure 1.
Figure 1D shows 2Fo-Fc and Fo-Fc SIGMAA electron density superimposed on a region of the dimer interface of chains A and B in the P21 crystal form, revealing what appear to be diatomic adducts bound to the Sγ atoms of Cys111 residues in both subunits. Adducts very similar in appearance are also observed on chains C and D.
Zinc Release/Affinity Assay
To probe whether the gel-shift demonstrated by H46R/H48Q SOD1 shown in the inset to Figure 2A upon addition of reducing agent might be due to the loss of zinc, the absorbance of H46R/H48Q SOD1 at 500 nm was monitored in the presence of PAR and in the presence and absence of TCEP. ICP-MS (Table 2) and direct observation of the crystal structure confirm that H46R/H48Q SOD1 does not bind metal ions in the copper-binding site - thus the increase in absorbance in these experiments comes from the release of zinc from the enzyme. The rapid rise in absorbance at 500 nm for H46R/H48Q SOD1 in the presence of reducing agent reveals a substantial decrease in affinity for zinc as compared with wt-SOD1 under the same conditions. The individual curves were fit to a hyperbolic equation to estimate the total zinc release and this value was in close agreement to the total metal content as determined by ICP-MS (data not shown). Because either dissociation to monomers or loss of zinc ion would enhance the rate of migration in the native gel due to the decrease in molecular mass and the loss of two positive charges, respectively, analytical gel filtration analyses were performed in an effort to differentiate between the two possibilities (see two sections below).
Table 2.
Inductively-Coupled Plasma Mass Spectrometry (ICP-MS) analyses of the proteins used in this study.
| wt-hSOD1 | wt-ySOD1 | H46R/H48Q | yCCS | hCCS | |
|---|---|---|---|---|---|
| as-isolated proteins | 1.8 Cu | 1.6 Cu | 0.1 Cu | 0.0 Cu | 0.15 Cu |
| (eq./dimer) | 2.1 Zn | 2.0 Zn | 1.5 -2.0 Zn | 0.0 Zn | 2.2 Zn |
| after dialysis vs. TCEP | 1.8 Cu | 1.6 Cu | 0.06 Cu | 0.0 Cu | 0.0 Cu |
| (eq./dimer) | 1.9 Zn | 1.9 Zn | 0.16 Zn | 0.0 Zn | 1.6 Zn |
| after metal stripping | 0.0 Cu | 0.0 Cu | 0.0 Cu | N/A | N/A |
| (eq./dimer) | 0.0 Zn | 0.0 Zn | 0.0 Zn |
Nondenaturing Gel Electrophoresis
To monitor the properties of wild type SOD1 and H46R/H48Q SOD1 in the presence of CCS, stoichiometeric ratios of SOD1 and CCS polypeptides (calculated on a per monomer basis) were used in each assay shown in Figure 3. Equimolar ratios of SOD1-to-CCS were chosen to avoid mass action effects that might tend to drive complex formation. Figure 3A shows the results of the nondenaturing gel electrophoresis experiments, which reveal that wild-type human and yeast SOD1 do not form stable complexes with CCS under any conditions tested. In contrast, and as shown in Figure 3B, a nearly quantitative gel-shift arising from formation of a H46R/H48Q SOD1/CCS complex is observed, but only in lanes in which the reducing agent TCEP is present.
Figure 3. Non-denaturing gel electrophoresis of long-lived H46R/H48Q SOD1–CCS complexes.
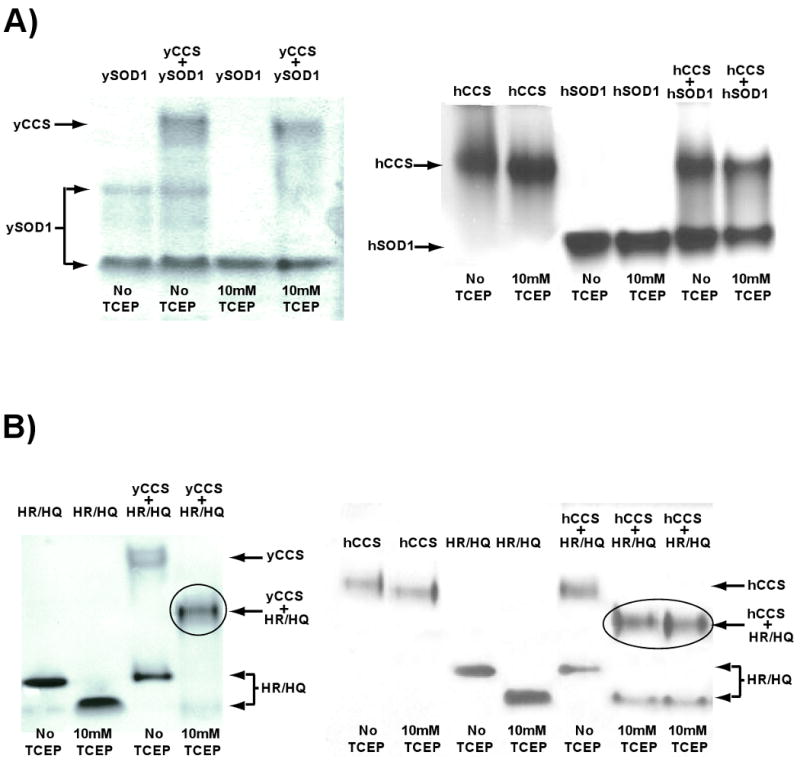
A) Wild-type yeast (left) and human (right) SOD1 proteins do not stably interact with their cognate copper chaperone proteins in non-denaturing PAGE of gel filtration chromatography (see Figure 4). B) A gel-shift characteristic of a 1:1 complex (circled) between H46R/H48Q SOD1 and yeast CCS (left) and human CCS (right) is observed, but only in the presence of reducing agent.
Analytical Gel Filtration
In analytical gel filtration experiments using a Superdex 200 column, an equimolar mixture of H46R/H48Q SOD1 + yCCS or hCCS produces two major peaks in the absence of reducing agents. As shown in Figure 4A, when H46R/H48Q SOD1 is mixed with yCCS, the first peak emerges at a volume consistent with a molecular mass of ∼55 kDa, which SDS-PAGE analyses confirm contain yCCS alone and the second peak emerges at a volume consistent with a molecular mass of ∼32 kDa, and SDS-PAGE analyses demonstrate that this peak contains only the SOD1 double mutant (data not shown). Thus, Peak 1 contains a yCCS homodimer and Peak 2 contains a H46R/H48Q SOD1 homodimer. Interestingly, H46R/H48Q SOD1 alone in the presence of 10 mM TCEP elutes at a volume identical to that for the H46R/H48Q SOD1 dimer under nonreducing conditions (compare Supplementary Figure 2A to gray elution profiles in Figure 4), suggesting that the native gel shift described in the preceding section is due to zinc ion loss and not monomerization.
Figure 4. Isolation and characterization of the H46R/H48Q SOD1/CCS complex using analytical gel-filtration chromatography.

A) The elution profile for a mixture of H46R/H48Q SOD1 and yeast CCS (gray trace) consists of two distinct peaks that emerge at a volume characteristic of a 54 kDa yCCS dimer and a slower eluting 32 kDa SOD1 dimer. The same 1:1 mixture of H46R/H48Q SOD1 in the presence of 10 mM TCEP (black trace) elutes from the column as a single peak characteristic of a 42 kDa SOD1/CCS heterodimer. B) The human form of CCS produces a similar elution profile to the yeast version when mixed with the SOD1 double mutant. Reducing agent must be present to induce complex formation.
The eluates of the H46R/H48Q SOD1 + CCS mixtures shown in Figure 4A were collected, combined, and run again over the same column, but this time in the presence of 10 mM TCEP. As shown in Figure 4B, a single distinct peak appeared at a volume consistent with a molecular mass of ∼43 kDa, corresponding to the calculated molecular mass of a SOD1/yCCS heterodimer. SDS-PAGE confirmed that both SOD1 and yCCS were present within the sample peak (data not shown).
When run alone, hCCS eluted from the Superdex 200 column as two peaks, whether 10 mM TCEP is present or not. The major peak elutes at a volume consistent with a ∼58 kDa homodimer and the faster eluting minor peak at a volume consistent with a ∼116 kDa homotetramer. As observed with yCCS, hCCS does not interact with the SOD1 double mutant under non-reducing conditions. However, inclusion of 10 mM TCEP induces complex formation between hCCS and H46R/H48Q SOD1, and this complex elutes from the column as two peaks. The major peak corresponds to a volume consistent with a hCCS/SOD1 heterodimer, while the minor peak elutes at a volume consistent with a molecular weight of ∼92 kDa. SDS-PAGE analyses revealed hCCS and H46R/H48Q SOD1 in both peaks suggesting heterodimeric and heterotetrameric H46R/H48Q/hCCS complexes, respectively.
Analytical Ultracentrifugation
As shown in Supplementary Figure 2B, the results of sedimentation velocity experiments on SOD1 proteins alone and when mixed with CCS proteins mirrored the analytical gel filtration results described above.
Disulfide Bond Status of H46R/H48Q SOD1 in Cells
Figure 5 shows the results of thiol modification experiments designed to probe the status of the Cys57-Cys146 intrasubunit disulfide bond in H46R/H48Q SOD1 expressed in yeast and human embryonic kidney (HEK) cells. In fresh yeast lysates, the disulfide bond is mostly reduced, although there is a small fraction of disulfide-oxidized protein in CCS-replete cells not present in CCS knockout cells. Co-transfection of the genes encoding human CCS and H46R/H48Q in HEK cells gives a result similar to that observed in CCS-replete yeast.
Figure 5. Disulfide status of H46R/H48Q SOD1.
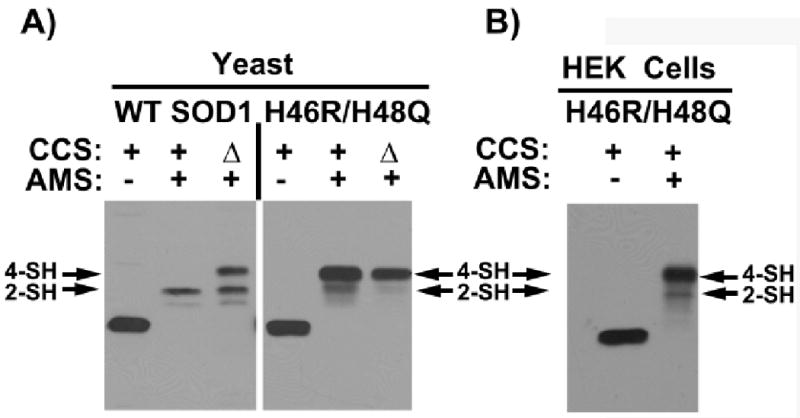
The status of the intrasubunit disulfide bond in H46R/H48Q SOD1 coming from freshly lysed yeast and human embryonic kidney (HEK) cells probed using of 4-acetamide-4′-meleimidylstilbene-2, 2′-disulfunic acid (AMS), which modifies free thiols. The SOD1 monomer contains four cysteine residues, two of which make up a conserved disulfide bond. A) Yeast and B) HEK cells expressing H46R/H48Q SOD1 are lysed in the presence of AMS and the lysates are run on SDS-PAGE and probed with antibody specific for SOD1 as described (33, 34). In yeast, H46RH48Q SOD1 is expressed in ccs1-replete and ccs1Δ cells. In HEK cells, H46R/H48Q and hCCS are co-transfected as described previously (33). As indicated, AMS-modified thiols produce gel shifts to higher molecular weight such that two are modified in the disulfide oxidized enzyme and four are modified in the disulfide reduced enzyme. The in vivo disulfide status of the SOD1 double mutant whether from yeast cells or HEK cells remains predominantly reduced and the presence of CCS has no effect.
Discussion
The H46R/H48Q SOD1 Metal Binding Sites
Transgenic mice expressing H46R/H48Q SOD1 developed paralytic symptoms concomitant with the appearance of SOD1-enriched proteinaceous inclusions in their motor neurons (21). The pathology of the H46R/H48Q mice is dominated by fibrillar (Thioflavin-S-positive) inclusions and similar inclusions are evident in mouse models that express the G37R and G93A variants of human SOD1 (21). However, unlike the G37R and G93A “wild type-like” SOD1 variants, H46R/H48Q SOD1 proteins from mammalian cells grown in the presence of 64Cu were found to be devoid of copper in audioradiograms of cell lysates run on non-denaturing and non-reducing gels (36). In the same study, we provided a snapshot of the active site, confirming that the H46R/H48Q SOD1 protein coming from the P21 structure (pdb code 2NNX) was incapable of binding metal ions, but we did not describe the structural and biophysical properties of the H46R/H48Q SOD1 molecule further. Taken together, however, the results of these studies strongly suggest that copper-binding is not a prerequisite for toxicity in SOD1-linked ALS (36).
The catastrophic effects of the dual H46R/H48Q substitutions for the copper-binding site are shown in Figure 1A, which reveals that the R46 sidechain physically occludes the active site channel as it forms a hydrogen bond to the carbonyl oxygen of T137. The Q48 sidechain also partially fills the copper-binding site and disrupts the hydrogen bonds in the wild type enzyme donated by the epsilon and guanidinium nitrogen atoms of R143 to the carbonyl oxygen of C57, interactions critical for the positioning of the positively-charged guanidinium group of R143 for optimal guidance of substrate into the active site (35). As described below, we suspect that, in addition to preventing CCS-mediated posttranslational modification of nascent H46R/H48Q SOD1, these substitutions exert effects on the zinc and electrostatic loop elements, contributing to enhanced recognition of the protein by CCS and facilitating the ability of the H46R/H48Q SOD1 protein to form stable SOD1/CCS heterocomplexes.
Surface Binding Site for Zinc
As shown in Figure 1B, E77 and H110 in each subunit of H46R/H48Q SOD1 in the P21 structure coordinate zinc such that each dimer is bridged to two other dimers by a total of four zinc ions to form tightly-linked layers that run the entire length of the crystal along the crystallographic b axis. A similar zinc-mediated linking of pathogenic SOD1 dimers was recently observed in the structure of the G85R SOD1 variant, except in that case, E24 and H110 participated as ligands to the bridging zinc ions (37). C111 sits adjacent to H110, providing another potential metal-binding ligand in the vicinity. It is currently unknown whether the external zinc-binding sites observed in these studies are relevant to pathogenesis, although metal ions such as copper and zinc are abundant in brain and spinal tissue and have been postulated to play a role as bridging entities in aggregates found in a range of proteins that cause human neurodegenerative diseases, including Alzheimer peptides (38) and prion proteins [(39) - for a review, see (40)].
Calcium Binding Site
Five of the six subunits in the C2221 H46R/H48Q SOD1 structure contain electron density for what appears to be a calcium ion bound in the cleft between loop II (residues 23-28) and the Greek key loop (loop VI, residues 101-113). Calcium is suspected because the crystallization experiment contained 0.2 M calcium acetate and the ligands are all oxygen atoms at distances ranging from 2.3-2.7 Ǻ. As shown in Figure 1C, each calcium ion is 6-coordinate, with the sidechain and carbonyl oxygen atoms of N26, the carbonyl oxygen atom of S102, and three water molecules acting as calcium ligands. In addition, each liganding water molecule donates a minimum of two hydrogen bonds to backbone oxygen atoms, resulting in an extensive network of calcium- and water-mediated interactions linking these two loops.
The observation of a calcium binding site in SOD1 is intriguing because in recently, SOD1 was found to activate calcineurin in brain and spinal cord via direct CN-SOD1 protein-protein interactions (41). Calcineurin is an iron- and zinc-containing metallophosphatase that is activated following calcium entry through ion channels (42), although it is currently unknown whether the SOD1-calcineurin interactions are mediated by calcium. SOD1 and calcineurin were found to be co-localized in the cytoplasm and membranes of neurons, and SOD1 co-immunoprecipitated with calcineurin from homogenates of brain hippocampus. SOD1 is thought to exert its stabilizing/activating effects on calcineurin by preventing iron and zinc loss from the calcineurin active site. Interestingly, SOD1-mediated activation of calcineurin appears to be dependent on the conformation of the zinc and electrostatic loop elements, as loss of zinc from SOD1 abolishes activation of calcineurin (41). As mentioned above, alteration of the conformation of the zinc loop is predicted to disrupt the calcium-binding site in SOD1, and it is tempting to speculate that calcium-binding by SOD1 may be linked to regulation of calcineurin activity. It should be noted, however, that the C2221 crystals were grown in the presence of 200 mM calcium acetate, and given this high concentration, the significance of the calcium binding site observed in this study remains to be determined.
Modification of Cys111
The identity of the adducts on the Sγ atoms of Cys111 residues shown in Figure 1D is currently unknown. They are intriguing, however because the electron density appears to correspond to a diatomic species and is not consistent with oxidation of Cys111 sulfur atoms to the sulfenic, sulfinic, and sulfonate forms. These adducts are observed clearly on the H46R/H48Q SOD1 variant in the P21 crystal form but not in the C2221 crystal form, even though they were grown from the same protein sample. However, we have previously observed the crystallization process “select” for certain SOD1 species over others in the same crystallization solution for recruitment into distinct crystal forms. An excellent example of this selectively is observed in the crystal structure of G85R SOD1, which crystallizes in four distinct crystal forms in which each crystal form contains a metal-bound species distinct from those found in the other crystal forms (37). To our knowledge, however, the Cys111 adducts observed here have not been reported in other SOD1 structures (wild type or mutant) determined to date. Additional work is necessary to pursue these observations and to ascertain the relevance (if any) to SOD1-linked ALS.
Weakened Affinity for Zinc
As shown in Table 2, ICP-MS analyses indicate that, depending on the preparation, the purified SOD1 double mutant contains between 1.5-2.0 zinc ions per dimer (approximately one per subunit), consistent with the crystallographic observation of a zinc ion in each subunit in both the P21 and C2221 crystal systems and with the total amount of zinc released from the H46R/H48Q SOD1 protein as calculated in the PAR assay under reducing conditions. However, the PAR chelation assay also reveals that the H46R/H48Q double mutant holds zinc ion only tenuously relative to the wild type enzyme. As shown in Figure 2A, under non-reducing conditions, zinc is slowly leached from this SOD1 variant by PAR. Upon reduction of the sample with TCEP, however, release of zinc is accelerated. In contrast, release of zinc ion from the wild type enzyme is negligible over days whether in the presence of reducing agent or not. In fact, the wild type enzyme loses zinc with a half-life of 11 hours, but only in the presence of chaotrope [2 M GdHCl (30)]. Thus, the H46R/H48Q mutations somehow dramatically reduce the affinity of the enzyme for zinc.
Structural Basis for Decreased Affinity for Zinc in H46R/H48Q SOD1
Figure 2B shows that the sidechain of D124 of the electrostatic loop (loop VII, residues 121-142) stabilizes the metal-binding sites in wild type SOD1 by forming hydrogen bonds simultaneously with the nonliganding imidazole nitrogen atoms of copper ligand H46 and zinc ligand H71. The importance of the proper positioning of the D124 sidechain for zinc-binding was demonstrated in previous studies where D124N and D124G mutants were found to be severely zinc-deficient, even after dialysis against 0.5 M ZnCl2 at neutral pH for extended periods (43). In subsequent work on crystal structures of H46R SOD1 (without the H48Q mutation), the side chain of the R46 pathogenic mutation was observed to clash with and displace the sidechain of D124 (10, 44), zinc was not bound to the zinc-binding site, and the electrostatic and zinc loops were both highly disordered, suggesting that the observed displacement of D124 by R46 was responsible for these phenomena (10, 44). In addition, a recent structure of the pathogenic SOD1 mutant D124V reveals that the protein contains no metal in the zinc-binding site [SVS and PJH in preparation].
As shown in Figure 1A, although we observe van der Waals contact between the guanidinium nitrogen of R46 and a sidechain oxygen of D124 in H46R/H48Q SOD1, we do not observe a substantial displacement of the D124 sidechain. In addition, zinc is observed bound to the zinc-binding site and the disulfide bond between C57 and C146 is oxidized in the crystal. Taking these observations and those of the previous studies together, we speculate that the weakened affinity for zinc demonstrated by H46R/H48Q SOD1 in Figure 2A arises from a combination of perturbation of the D124 sidechain by the sidechain of R46 SOD1 together with the reduction of the intrasubunit disulfide bond by TCEP, which would permit enhanced mobility of metal-liganding residues of the zinc loop (loop IV, residues 49-83), including H63, H71, H80, and D83.
Long-lived H46R/H48Q SOD1/CCS Complexes
The stable complexes between human H46R/H48Q SOD1 and both yeast and human CCS in non-denaturing gel-shift, gel filtration chromatography, and analytical ultracentrifugation experiments are reminiscent of those observed previously for the H48F mutant of yeast SOD1 with yeast CCS (45). In that study, however, the ySOD1/yCCS proteins were loaded onto the gel filtration column in a 2:1 molar ratio under reducing conditions and the addition of exogenous zinc ion greatly facilitated complex formation (45). We find that even when the H46R/H48Q SOD1 protein is completely demetallated, it quantitatively forms heterocomplexes with both yeast and human CCS, just as long as reducing agent is present (data not shown). We speculate that the perturbations in the electrostatic and zinc loop elements described above in reduced H46R/H48Q SOD1 (which loses its zinc) prolong the presentation of binding determinants coming from these elements to CCS that are not as accessible in the wild type enzyme. For example, in the crystal structure of metal-free, wild type human SOD1, the electrostatic and zinc loop elements are disordered in one-half of the molecules in the asymmetric unit and ordered in the other half (46), suggesting an equilibrium in the metal-free enzyme between the disordered and ordered, mature conformations of these loop elements. We suggest that the H46R/H48Q substitutions in essence shift this equilibrium toward the disordered state, and that it may be the amino acid residues of these mobile loop elements that are recognized by CCS. In this model, CCS has a lower affinity for the mature conformation because it represents the conformation that is the product of the CCS-mediated posttranslational modification events.
The notion that a partially folded nascent SOD1 structure is the substrate recognized by CCS is supported in recent synchronized cell-free SOD1 translation and protein folding assays in reticulocyte extracts replete with the full complement of molecular chaperones and folding facilitators that are normally required to fold this metalloenzyme. Kopito and colleagues detected the appearance of a partially protease resistant intermediate after about 4 minutes that persisted before being converted to a fully resistant form over a longer timeframe (t1/2 = ∼8 min) (47). They also observed that the fully resistant form was metal-replete and disulfide oxidized, while the partially resistant form was metal-deficient and disulfide reduced. These data seem consistent with the suggestion that CCS is recognizing mobile elements of the zinc and electrostatic loop elements that become ordered upon metal ion binding and disulfide bond formation.
However, as shown in Figures 2 and 5, despite the fact that the H46R/H48Q SOD1 variant forms stable complexes with both yeast and human CCS in the presence of reducing agent, these CCS molecules are apparently unable to catalyze the oxidation of the SOD1 intrasubunit disulfide bond in this SOD1 variant. CCS-mediated copper delivery and disulfide bond oxidation may in fact be linked events because CCS itself must be loaded with copper in order to catalyze the thioldisulfide oxidation. Blackburn and colleagues have shown that CCS forms multinuclear clusters with copper at the domain 3 CXC binding site, and it has been suggested that a similar cluster may form within the CCS-SOD heterocomplex involving the CCS CXC motif and the SOD1 C57 or C146 thiols (48, 49). It is possible that the H48Q mutation might interfere with the mechanics of disulfide bond oxidation by copper-loaded CCS, as the displacement of Arg143 prevents the formation of two hydrogen bonds with the carbonyl oxygen of Cys57, which is one-half of the disulfide bonding pair.
Hindered CCS-Mediated Posttranslational Modification: a Common Element in SOD1-Linked ALS?
The inability of H46R/H48Q SOD1 to attain the fully mature and enormously stable holo state raises the possibility that ALL pathogenic SOD1 mutations give rise to off-pathway folding intermediates. How might this occur? At steady-state, the SOD1/CCS protein ratio is estimated to be between 15-30:1 (50) and CCS must therefore cycle through the nascent SOD1 pool to activate these molecules (51). In order to activate SOD1, CCS must itself be copper-loaded, and this likely requires cycling back to the plasma membrane to acquire copper from the copper transporter CTR1 at least once for each SOD1-CCS maturation event. Thus the ability of CCS to activate the newly translated pathogenic SOD1 proteins is likely dependent on the kinetics of copper loading into CCS and the biophysical properties of the pathogenic SOD1 proteins including their overall stability and the 3D location of the mutations, which vary widely. In addition, different pathogenic mutations are likely to hinder CCS action at different points in the SOD1 maturation cycle [reviewed in (3)].
At one end of the spectrum are those pathogenic substitutions that severely destabilize nascent SOD1. For example, the nascent L126 truncation variant is so destabilized relative to the nascent wild type enzyme (52, 53) that CCS likely encounters and posttranslationally modifies only a small fraction of these molecules before they are degraded, oligomerize, or enter the insoluble fraction. Indeed, CCS would likely fail to stabilize the nascent L126 molecules it does encounter because the latter molecule is completely lacking a β-strand believed necessary for wild type SOD1 heterodimerization with CCS domain II.
Another class of pathogenic SOD1 mutations is defined by those that fall in the β-barrel, such as A4V, G37R, and G93A (among ∼55 others). These SOD1 variants are not as radically destabilized as L126Z in their nascent forms, but they are still enormously destabilized relative to the nascent wild type enzyme as evidence by their significantly decreased melting points (Tm) in differential scanning calorimetry experiments (54). We propose that this instability in the metal-free and disulfide-reduced state prevents CCS from cycling through the entire pool of these “wild type-like” nascent SOD1 pathogenic mutants before a significant number are degraded, enter the insoluble fraction, or assemble into soluble oligomeric species. Indeed, it has been shown that A4V and G41D SOD1 mutants are rapidly degraded in ccs1Δ yeast and that insolubility of G37R SOD1 is enhanced in CCSΔ- fibroblasts (33, 34). We consider pathogenic SOD1 molecules not acted upon by CCS to be essentially off-pathway folding intermediates prone to aggregate or interfere with other cellular processes.
It is true that pathogenic A4V. G37R, G93A and other β-barrel mutants have been isolated in metal-bound and disulfide-oxidized WT-like forms from yeast and insect cells, but this may be deceiving to a certain degree because experimentalists tend to focus on the proteins that can be purified and not on the fraction of molecules that cannot. For example, the literature is replete with discussions of pathogenic SOD1 proteins “losing” their metal ions and/or disulfide bond. Our view is that these metal-deficient, disulfide-reduced proteins actually quite likely never acquired these posttranslational modifications in the first place. It is important to note that in contrast to the pathogenic SOD1 variants, newly translated wild type SOD1 has been under evolutionary pressure to remain stable and soluble until acted upon by CCS and NOT to aggregate or interact adversely with cellular machinery, which would obviously be detrimental to the cell. Thus, in our model, nascent wild type SOD1 does not represent an off-pathway folding intermediate.
At the other end of the spectrum, and in contrast to L126Z and the “wild type-like” β-barrel mutants described above, the “metal binding mutants” such as H46R, H48Q, H46R/H48Q, H80R, and D124V (among many others), are not destabilized relative to the wild type enzyme when in the nascent state. However, CCS can never fully stabilize these metal-free molecules via posttranslational modification because the nature of these mutations is such that they directly prevent metal-binding and/or interfere with SOD1-CCS protein-protein interactions. CCS can also never stabilize the pathogenic SOD1 mutants C57R and C146R, which cannot make the intrasubunit disulfide bond. Thus, these pathogenic SOD1 variants are arrested in their maturation and exist as off-pathway folding intermediates. Figure 6 shows the positions of the pathogenic SOD1 mutations color-coded to reflect at which point in the SOD1 maturation pathway each might be predicted to exert their inhibitory effect in the context of CCS action.
Figure 6. Positions of fALS associated SOD1 mutations and their postulated inhibitory effects on the SOD1 maturation pathway.
The pink spheres represent mutations located within the β-barrel of SOD1. These mutations destabilize the metal-free disulfide-reduced (nascent) protein decreasing the fraction of molecules that are available for posttranslational modified by CCS. Unmodified nascent SOD1 proteins are either turned over or associate into soluble oligomeric complexes on the pathway to toxicity. The red spheres correspond to ALS mutations that adversely affect metal binding, the conformation of the zinc and electrostatic loop elements, and disulfide status. Although nascent pathogenic SOD1 mutants of this class are not generally destabilized relative to the nascent wild-type enzyme, full maturation cannot be achieved due to abrogated metal binding/coordination and/or disulfide oxidation, and these molecules remain off-pathway folding intermediates. The gold spheres correspond to ALS mutations that fall in the Greek key loop and might impact calcium ion binding and the stability of the nascent SOD1 protein.
Finally, the hypothesis that immature pathogenic SOD1 molecules may represent the noxious species in SOD1-linked fALS is gathering momentum as evidenced by recent studies in cell culture and in transgenic mice in which it is observed that much of the pathogenic SOD1 protein in the soluble fraction is metal-deficient, disulfide-reduced, and/or monomeric (11, 55, 56). Interestingly, in a pair of recent studies, it was found that over-expression of CCS greatly accelerated disease in a G93A SOD1 mouse model in the absence of visible proteinaceous inclusions (34, 57). Unexpectedly, however, CCS over-expression failed to enhance oxidation of the G93A SOD1 disulfide bond, and in fact, elevated the population of disulfide-reduced G93A SOD1 in the soluble fraction of brain and spinal cord of these animals (34). These data suggest that CCS may be interacting with novel “non-productive” conformation of nascent G93A (which is at a stoichiometric ratio of approximately 1:1 in these animals) hindering its maturation as well as its turnover and/or aggregation, and that the observed elevated levels of soluble disulfide-reduced G93A SOD1 augments the mitochondrial pathology resulting in significantly earlier onset of paralytic symptoms. However, it remains unclear why CCS over-expression in these animals results in elevated levels of disulfide reduced G93A SOD1, but it suggests that like H46R/H48Q SOD1, CCS binding alone is not sufficient to convert nascent G93A SOD1 into its mature holo form.
In summary, the results presented here on the structure and biophysical properties of H46R/H48Q SOD1, together with data on pathogenic SOD1 proteins coming from cell culture and in transgenic mice, suggest that incomplete posttranslational modification of nascent SOD1 polypeptides via CCS may be a characteristic shared by fALS SOD1 mutants, resulting in a population of destabilized, off-pathway folding intermediates that are toxic to motor neurons.
Supplementary Material
Acknowledgments
We thank Borries Demeler and Virgil Schirf for help with analytical ultracentrifugation, and Rodney Levine, Edith Gralla, and Lawrence Hayward for helpful discussions.
This work was supported by grants NIH-NINDS R01-NS39112 (PJH), P01-NS04913 (Joan S. Valentine, DRB, and PJH), NIH-GMS GM50016 (VCC) and the JHU NIEHS center (VCC). DDW was supported in part by the American Foundation for Aging Research (AFAR). JPS was supported by NIH T32AG021890-03. JBP was supported by NIEHS training grant ES 07141. XC was supported in part by the William and Ella Owens Medical Research Foundation and the Judith and Jean Pape Adams Charitable Foundation. Support for the X-ray Crystallography Core Laboratory and the Center for Analytical Ultracentrifugation of Macromolecular Assemblies by the UTHSCSA Executive Research Committee and the San Antonio Cancer Institute is also gratefully acknowledged.
Abbreviations and Textual Footnotes
- SOD1
copper-zinc superoxide dismutase
- hSOD1
human copper-zinc superoxide dismutase
- ySOD1
yeast copper-zinc superoxide dismutase
- ALS
amyotrophic lateral sclerosis
- fALS
familial amyotrophic lateral sclerosis
- H46R/H48Q
copper-zinc superoxide dismutase with His46 and His48 substituted with Arg and Gln, respectively
- CCS
copper chaperone for SOD1
- hCCS
human copper chaperone for SOD1
- yCCS
yeast copper chaperone for SOD1
- EDTA
ethylenediaminetetraacetic acid
- PMSF
phenylmethylsulphonyl fluoride
- TCEP
tris(2-carboxyethyl)phosphine
- PCR
polymerase chain reaction
- TEV
tobacco etch virus
- MES
2-(N-morpholino)ethanesulfonic acid
- PEG
polyethylene glycol
- PAR
2-pyridylazo-resorcinol
- ICP-MS
inductively coupled plasma mass spectrometry
- MME
monomethylether
Footnotes
Supporting Information Available: Supplementary Figure 1 shows additional details of the calcium binding site and its role in SOD1 crystal packing interactions. Supplementary Figure 2 shows additional details of H46R/H48Q SOD1 interactions with CCS in solution using analytical ultracentrifugation and analytical gel filtration. This material is available free of charge via the Internet at http://pubs.acs.org”.
References
- 1.Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, Warner C, Deng G, Soriano E, Smyth C, Parge HE, Ahmed A, Roses AD, Hallewell RA, Pericak-Vance MA, Siddique T. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 2.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH., Jr Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 3.Winkler DD, Prudencio M, Karch CM, Borchelt DR, Hart PJ. Copper-Zinc Superoxide Dismutase, its Copper Chaperone, and Familial Amyotrophic Lateral Sclerosis. In: Dobson CM, Kelly JW, Ramirez-Alvarado M, editors. Protein Misfolding Diseases: Current and Emerging Principles and Therapies. John Wiley & Sons, Inc.; Hoboken: 2009. p (in press) [Google Scholar]
- 4.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 5.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 6.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 7.Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 8.Hart PJ. Pathogenic superoxide dismutase structure, folding, aggregation and turnover. Curr Opin Chem Biol. 2006;10:131–138. doi: 10.1016/j.cbpa.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 9.Valentine JS, Doucette PA, Potter SZ. Copper-Zinc Superoxide Dismutase and Amyotrophic Lateral Sclerosis. Annu Rev Biochem. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- 10.Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, Hart PJ. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol. 2003;10:461–467. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]
- 11.Rakhit R, Crow JP, Lepock JR, Kondejewski LH, Cashman NR, Chakrabartty A. Monomeric Cu, Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J Biol Chem. 2004;279:15499–15504. doi: 10.1074/jbc.M313295200. [DOI] [PubMed] [Google Scholar]
- 12.Ray SS, Nowak RJ, Strokovich K, Brown RH, Jr, Walz T, Lansbury PT., Jr An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry. 2004;43:4899–4905. doi: 10.1021/bi030246r. [DOI] [PubMed] [Google Scholar]
- 13.Rakhit R, Robertson J, Van de Velde C, Horne P, Ruth DM, Griffin J, Cleveland DW, Cashman NR, Chakrabartty A. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med. 2007;13:754–759. doi: 10.1038/nm1559. [DOI] [PubMed] [Google Scholar]
- 14.Arnesano F, Banci L, Bertini I, Martinelli M, Furukawa Y, O'Halloran TV. The unusually stable quaternary structure of human Cu, Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J Biol Chem. 2004;279:47998–48003. doi: 10.1074/jbc.M406021200. [DOI] [PubMed] [Google Scholar]
- 15.Doucette PA, Whitson LJ, Cao X, Schirf V, Demeler B, Valentine JS, Hansen JC, Hart PJ. Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant. Insights into the molecular basis for dimer stability. J Biol Chem. 2004;279:54558–54566. doi: 10.1074/jbc.M409744200. [DOI] [PubMed] [Google Scholar]
- 16.Lindberg MJ, Normark J, Holmgren A, Oliveberg M. Folding of human superoxide dismutase: disulfide reduction prevents dimerization and produces marginally stable monomers. Proc Natl Acad Sci U S A. 2004;101:15893–15898. doi: 10.1073/pnas.0403979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Culotta VC, Klomp LW, Strain J, Casareno RL, Krems B, Gitlin JD. The copper chaperone for superoxide dismutase. J Biol Chem. 1997;272:23469–23472. doi: 10.1074/jbc.272.38.23469. [DOI] [PubMed] [Google Scholar]
- 18.Brown NM, Torres AS, Doan PE, O'Halloran TV. Oxygen and the copper chaperone CCS regulate posttranslational activation of Cu, Zn superoxide dismutase. Proc Natl Acad Sci U S A. 2004;101:5518–5523. doi: 10.1073/pnas.0401175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furukawa Y, Torres AS, O'Halloran TV. Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. Embo J. 2004;23:2872–2881. doi: 10.1038/sj.emboj.7600276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, Brown RH, Jr, Itoyama Y. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis. 2002;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR. Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet. 2003;12:2753–2764. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- 23.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW, Sweet RM, editors. Methods Enzymol. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 24.Brünger AT. Free R value: cross-validation in crystallography. In: Carter CW, Sweet RM, editors. Methods Enymol. Academic Press; New York: 1997. pp. 366–396. [DOI] [PubMed] [Google Scholar]
- 25.Hart PJ, Liu H, Pellegrini M, Nersissian AM, Gralla EB, Valentine JS, Eisenberg D. Subunit asymmetry in the three-dimensional structure of a human CuZnSOD mutant found in familial amyotrophic lateral sclerosis. Protein Sci. 1998;7:545–555. doi: 10.1002/pro.5560070302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vagin AA, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 27.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 29.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 30.Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- 31.van Holde KE, Weischet WO. Biopolymers. 1978;17:1387–1403. [Google Scholar]
- 32.Demeler B. UltraScan 6.2 - An integrated data analysis software package for sedimentation experiments. University of Texas Health Science Center at San Antonio, Department of Biochemistry 2004 [Google Scholar]
- 33.Carroll MC, Outten CE, Proescher JB, Rosenfeld L, Watson WH, Whitson LJ, Hart PJ, Jensen LT, Cizewski Culotta V. The effects of glutaredoxin and copper activation pathways on the disulfide and stability of Cu, Zn superoxide dismutase. J Biol Chem. 2006;281:28648–28656. doi: 10.1074/jbc.M600138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Proescher JB, Son M, Elliott JL, Culotta VC. Biological effects of CCS in the absence of SOD1 enzyme activation: implications for disease in a mouse model for ALS. Hum Mol Genet. 2008;17:1728–1737. doi: 10.1093/hmg/ddn063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Getzoff ED, Tainer JA, Weiner PK, Kollman PA, Richardson JS, Richardson DC. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature. 1983;306:287–290. doi: 10.1038/306287a0. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Martin E, Gonzales V, Borchelt DR, Lee MK. Differential regulation of small heat shock proteins in transgenic mouse models of neurodegenerative diseases. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao X, Antonyuk SV, Seetharaman SV, Whitson LJ, Taylor AB, Holloway SP, Strange RW, Doucette PA, Valentine JS, Tiwari A, Hayward LJ, Padua S, Cohlberg JA, Hasnain SS, Hart PJ. Structures of the G85R variant of SOD1 in familial amyotrophic lateral sclerosis. J Biol Chem. 2008;283:16169–16177. doi: 10.1074/jbc.M801522200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curtain CC, Ali F, Volitakis I, Cherny RA, Norton RS, Beyreuther K, Barrow CJ, Masters CL, Bush AI, Barnham KJ. Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem. 2001;276:20466–20473. doi: 10.1074/jbc.M100175200. [DOI] [PubMed] [Google Scholar]
- 39.Jobling MF, Huang X, Stewart LR, Barnham KJ, Curtain C, Volitakis I, Perugini M, White AR, Cherny RA, Masters CL, Barrow CJ, Collins SJ, Bush AI, Cappai R. Copper and zinc binding modulates the aggregation and neurotoxic properties of the prion peptide PrP106-126. Biochemistry. 2001;40:8073–8084. doi: 10.1021/bi0029088. [DOI] [PubMed] [Google Scholar]
- 40.Bush AI. Metals and neuroscience. Curr Opin Chem Biol. 2000;4:184–191. doi: 10.1016/s1367-5931(99)00073-3. [DOI] [PubMed] [Google Scholar]
- 41.Agbas A, Hui D, Wang X, Tek V, Zaidi A, Michaelis EK. Activation of brain calcineurin (Cn) by Cu-Zn superoxide dismutase (SOD1) depends on direct SOD1-Cn protein interactions occurring in vitro and in vivo. Biochem J. 2007;405:51–59. doi: 10.1042/BJ20061202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mansuy IM, Mayford M, Jacob B, Kandel ER, Bach ME. Restricted and regulated overexpression reveals calcineurin as a key component in the transition from short-term to long-term memory. Cell. 1998;92:39–49. doi: 10.1016/s0092-8674(00)80897-1. [DOI] [PubMed] [Google Scholar]
- 43.Banci L, Bertini I, Cabelli DE, Hallewell RA, Tung JW, Viezzoli MS. A characterization of copper/zinc superoxide dismutase mutants at position 124. Zinc-deficient proteins, Eur J Biochem. 1991;196:123–128. doi: 10.1111/j.1432-1033.1991.tb15794.x. [DOI] [PubMed] [Google Scholar]
- 44.Antonyuk S, Elam JS, Hough MA, Strange RW, Doucette PA, Rodriguez JA, Hayward LJ, Valentine JS, Hart PJ, Hasnain SS. Structural consequences of the familial amyotrophic lateral sclerosis SOD1 mutant His46Arg. Protein Sci. 2005;14:1201–1213. doi: 10.1110/ps.041256705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lamb AL, Torres AS, O'Halloran TV, Rosenzweig AC. Heterodimer formation between superoxide dismutase and its copper chaperone. Biochemistry. 2000;39:14720–14727. doi: 10.1021/bi002207a. [DOI] [PubMed] [Google Scholar]
- 46.Strange RW, Antonyuk S, Hough MA, Doucette PA, Rodriguez JA, Hart PJ, Hayward LJ, Valentine JS, Hasnain SS. The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J Mol Biol. 2003;328:877–891. doi: 10.1016/s0022-2836(03)00355-3. [DOI] [PubMed] [Google Scholar]
- 47.Bruns CK, Kopito RR. Impaired post-translational folding of familial ALS-linked Cu, Zn superoxide dismutase mutants. Embo J. 2007;26:855–866. doi: 10.1038/sj.emboj.7601528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barry AN, Clark KM, Otoikhian A, van der Donk WA, Blackburn NJ. Selenocysteine Positional Variants Reveal Contributions to Copper Binding from Cysteine Residues in Domains 2 and 3 of Human Copper Chaperone for Superoxide Dismutase. Biochemistry. 2008;47:13074–13083. doi: 10.1021/bi801438g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barry AN, Blackburn NJ. A selenocysteine variant of the human copper chaperone for superoxide dismutase. A Se-XAS probe of cluster composition at the domain 3-domain 3 dimer interface. Biochemistry. 2008;47:4916–4928. doi: 10.1021/bi8001049. [DOI] [PubMed] [Google Scholar]
- 50.Rothstein JD, Dykes-Hoberg M, Corson LB, Becker M, Cleveland DW, Price DL, Culotta VC, Wong PC. The copper chaperone CCS is abundant in neurons and astrocytes in human and rodent brain. J Neurochem. 1999;72:422–429. doi: 10.1046/j.1471-4159.1999.0720422.x. [DOI] [PubMed] [Google Scholar]
- 51.Furukawa Y, O'Halloran TV. Posttranslational modifications in Cu, Zn-superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid Redox Signal. 2006;8:847–867. doi: 10.1089/ars.2006.8.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jonsson PA, Ernhill K, Andersen PM, Bergemalm D, Brannstrom T, Gredal O, Nilsson P, Marklund SL. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain. 2004;127:73–88. doi: 10.1093/brain/awh005. [DOI] [PubMed] [Google Scholar]
- 53.Wang J, Xu G, Li H, Gonzales V, Fromholt D, Karch C, Copeland NG, Jenkins NA, Borchelt DR. Somatodendritic accumulation of misfolded SOD1-L126Z in motor neurons mediates degeneration: alphaB-crystallin modulates aggregation. Hum Mol Genet. 2005;14:2335–2347. doi: 10.1093/hmg/ddi236. [DOI] [PubMed] [Google Scholar]
- 54.Rodriguez JA, Shaw BF, Durazo A, Sohn SH, Doucette PA, Nersissian AM, Faull KF, Eggers DK, Tiwari A, Hayward LJ, Valentine JS. Destabilization of apoprotein is insufficient to explain Cu, Zn-superoxide dismutase-linked ALS pathogenesis. Proc Natl Acad Sci U S A. 2005;102:10516–10521. doi: 10.1073/pnas.0502515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Lindberg M, Oliveberg M, Marklund SL. Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain. 2006;129:451–464. doi: 10.1093/brain/awh704. [DOI] [PubMed] [Google Scholar]
- 56.Zetterstrom P, Stewart HG, Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Oliveberg M, Marklund SL. Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc Natl Acad Sci U S A. 2007;104:14157–14162. doi: 10.1073/pnas.0700477104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Son M, Puttaparthi K, Kawamata H, Rajendran B, Boyer PJ, Manfredi G, Elliott JL. Overexpression of CCS in G93A-SOD1 mice leads to accelerated neurological deficits with severe mitochondrial pathology. Proc Natl Acad Sci U S A. 2007;104:6072–6077. doi: 10.1073/pnas.0610923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laskowski RA, McArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallog. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.