

Abstract
Interferon regulatory factors (IRFs) are essential in the innate immune response and other physiological processes. Activation of these proteins in the cytoplasm is triggered by phosphorylation of Ser/Thr residues in a C-terminal autoinhibitory region, which stimulates dimerization, transport into the nucleus, assembly with the coactivator CBP/p300 and initiation of transcription. The novel crystal structure of the transactivation domain of pseudophosphorylated human IRF5 reveals a striking dimer in which the bulk of intersubunit interactions involve a highly extended C-terminal region. The corresponding region has previously been shown to block CBP/p300 binding to unphosphorylated IRF3. Mutation of key interface residues supports the observed dimer as the physiologically activated state of IRF5 and IRF3. Thus phosphorylation likely activates IRF5 and other family members by triggering remarkable conformational rearrangements that switch the C-terminal segment from an autoinihibitory to a dimerization role.
Our survival depends on molecular defense mechanisms that include both tumor suppression and immunogenic responses to pathogens. Breakdown of these mechanisms can lead to cancer and infection, whereas hyperactivity can result in autoimmune disease. The interferon (IFN) regulatory factors (IRFs) are key transcription factors involved in such defense mechanisms, having been directly implicated in the innate immune response and other physiological roles1-3.
IRF5 is a particularly interesting IRF family member with a variety of activities, including activation of type I IFN genes4,5, inflammatory cytokines6 and tumor suppressors7,8. Expression of IRF5 has been detected primarily in B cells and dendritic cells and is stimulated by type I IFN, viral expression and the tumor suppressor p539-12. While type I IFN responses are beneficial to the host, inflammatory cytokines that are stimulated by IRF5 can be a major contributor to the morbidity and mortality associated with viral pathogenesis. In particular, mutants in IRF5 are associated with pathogenesis of autoimmune diseases including systemic lupus erythematosis13,14, Sjogren’s syndrome15, multiple sclerosis16 and inflammatory bowel disease17. Understanding how IRF5 activity is controlled at the molecular level, therefore, is potentially important clinically, as therapeutic agents that enhance activity could combat virus infection or tumor growth18, whereas agents that attenuate activity could be used to minimize harmful inflammatory responses. The physiological importance of IRF5 makes it an important prototype for investigating regulation of IRF proteins.
IRF proteins, including IRF5, comprise two major domains, an N-terminal DNA binding domain (DBD) and a C-terminal domain largely responsible for activation. The well conserved DBD (~120 amino acids) recognizes positive regulatory domain DNA sequences in the IFN-β enhancer region as well as IFN stimulated response elements (ISRE) in IFN induced genes. Details of the interaction of various IRF DBDs with DNA and their participation with other factors in transcriptional activation of the IFN-β gene have been elucidated by a number of crystallographic analyses19-22. The C-terminal portions regulate the transcriptional activity of the IRFs. IRF3 through IRF9 possess a C-terminal IRF association domain (IAD) that is responsible for homo- and heteromeric interactions among the IRFs as well as interaction with transcriptional co-modulators. The transactivation functions of the IAD of IRF3, IRF4, IRF5 and IRF7 have been shown to be suppressed by autoinhibitory structures flanking the IAD10,23,24.
The most extensively studied member of this family is IRF3. IRF3 is constitutively expressed in all cell types, remaining in the cytosol in a latent, autoinhibited form until activated. Upon viral infection, IRF3 is phosphorylated by two serine/threonine kinases, TBK1 and IKKε25,26. Phosphorylation induces IRF3 to homo or heterodimerize, translocate into the nucleus and assemble with CREB binding protein (CBP), or the closely related p300, allowing promoter binding and activation of the IFN-β gene27-29. Crystal structures of the transactivation domain of IRF330,31 revealed the conformation of an autoinhibitory region near the C-terminus that contains the key phosphorylation sites. A subsequent crystal structure of CBP in complex with IRF3 lacking the C-terminal phosphorylation region demonstrated that CBP binds to the surface masked by the autoinhibitory region in the monomeric structure32. Thus, these structures strongly suggest that phosphorylation triggers an unfolding of a C-terminal autoinhibitory region, but do not explain how such structural rearrangements are coupled to dimerization or why dimerization favors binding of CBP/p300.
To provide a structural basis for IRF5 function and to understand how the phosphorylation of IRF proteins activates dimerization and binding to CBP/p300, we have determined the crystal structure of the transactivation domain of human IRF5 with a phosphomimetic mutation that enhances dimer formation. The C-terminal autoinhibitory region, including key phosphorylation sites, adopts a highly extended conformation that mediates homodimerization. In the process, the CBP/p300 binding surface is largely unmasked in the dimer, providing a clear structural link between phosphorylation, dimerization and CBP/p300 binding.
RESULTS
IRF5 phosphomimetic mutants
We investigated the self-association and CBP interactions of purified IRF5 (residues 222-467) and potential phosphomimetic mutants (S421D, S425D, S427D, S430D, S436D and S457D) by size-exclusion chromatography, analytical sedimentation velocity and isothermal titration calorimetry (ITC). (At least nine different isoforms of IRF5 have been shown to result from differential splicing11, leading to variations in residue numbering. Our studies are on variant 4.) ITC experiments with CBP (residues 2067-2112) showed that S421D and S457D weakened interactions with CBP, but the other four mutations increased affinity by factors of 1.7 to 2.9 fold relative to wild-type (Table 1, Supplementary Fig. 1 online). These results are consistent with previous experiments that showed mutation of these four serines to aspartate rendered IRF5 constitutively active33. Of these mutants, S430D exhibited the strongest affinity with CBP. Size-exclusion chromatography showed that mixing of CBP with both IRF5 (222-467) and IRF5 (222-467) S430D resulted in a shift in elution volume that is consistent with oligomerization (Supplementary Fig. 2). Sedimentation velocity experiments confirm the conclusion from gel exclusion chromatography that IRF5 (222-467) and IRF5 (222-467) S430D form IRF52CBP2 assemblies in the presence of CBP (Supplementary Fig. 2). These results strongly suggest that CBP binding and IRF5 dimerization are coupled events. Furthermore, sedimentation experiments show that IRF5 (222-467) S430D assembles into stable homodimers in the absence of CBP (Supplementary Fig. 2b and 3). Since IRF5 (222-467) S430D forms dimers, representative of activated IRF5, structural studies were initiated on this form.
Table 1.
Summary of thermodynamic results for interactions of CBP interaction with IRF5 (222-467) and its mutants
| Complex | n | ΔH | -TΔS | ΔG | Kd | -fold |
|---|---|---|---|---|---|---|
| kcal/mol | kcal/mol | kcal/mol | μM | |||
| CBP-IRF5 | 1.05 ± 0.09 | -14.4 ± 1.1 | 6.5 ± 3.0 | -7.9 ± 0.2 | 1.64 ± 0.34 | 1.0 |
| CBP-IRF5 (S457D) | 1.08 ± 0.12 | -10.3 ± 0.9 | 2.6 ± 0.4 | -7.7 ± 0.1 | 2.41 ± 0.05 | 0.7 |
| CBP-IRF5 (S421D) | 1.03 ± 0.15 | -6.4 ± 0.3 | 1.3 ± 0.5 | -7.7 ± 0.5 | 2.28 ± 0.35 | 0.7 |
| CBP-IRF5 (S427D) | 0.98 ± 0.06 | -12.9 ± 0.4 | 4.8 ± 0.6 | -8.2 ± 0.1 | 0.96 ± 0.34 | 1.7 |
| CBP-IRF5 (S425D) | 1.04 ± 0.06 | -13.3 ± 1.3 | -4.9 ± 1.6 | -8.4 ± 0.2 | 0.71 ± 0.71 | 2.3 |
| CBP-IRF5 (S436D) | 1.07 ± 0.21 | -6.8 ± 1.5 | -1.6 ± 1.7 | -8.4 ± 0.8 | 0.67 ± 0.09 | 2.4 |
| CBP-IRF5 (S430D) | 1.02 ± 0.09 | -15.2 ± 1.2 | 6.7 ± 1.4 | -8.5 ± 0.1 | 0.56 ± 0.04 | 2.9 |
Crystallographic analysis of IRF5 S430D
We determined the crystal structure of the transactivation domain of IRF5 (residues 222-467) S430D (hereafter referred to as IRF5) to 2.0Å resolution using single heavy atom isomorphous replacement to solve the phase problem. The crystals contain one polypeptide chain per asymmetric unit. An atomic model has been refined to a conventional R-factor of 0.209 and a free R of 0.244.
Tertiary structure of IRF5
The tertiary structure of the IRF association domain (IAD) of IRF5 is very similar to that of IRF3 (Fig. 1). Alpha carbon coordinates for core structures of IRF5 and IRF3 can be aligned with a root-mean-square (rms) deviation of 1.4Å for 138 (out of 185) IAD residues. (Residues used in this alignment are shown in cyan in Fig. 1). However, the C-terminal regions (IRF5 residues 423-457, magenta) exhibit dramatically different conformations. The C-terminal region of the IRF3 monomer forms an autoinhibitory structure whose position masks a hydrophobic surface for binding CBP/p30031,32. In IRF5, this region is in an extended conformation, which strongly contributes to dimerization (see below) and largely exposes the CBP/p300 binding surface. A short N-terminal helix remains packed against the CBP/p300 binding surface; in the co-crystal structure of truncated IRF3 with CBP 32, a similar short N-terminal helix becomes displaced upon binding CBP (Fig. 1c). Other structural differences are observed in the lengths of helix 4, helix 5 and the conformation of the connecting polypeptide. Given the structural similarities of the IAD of IRF5 and IRF3 many of these differences likely represent structural changes that occur upon activation and dimerization. Phosphorylation sites (yellow balls) evidently trigger switching between these dramatically different conformations.
Figure 1.
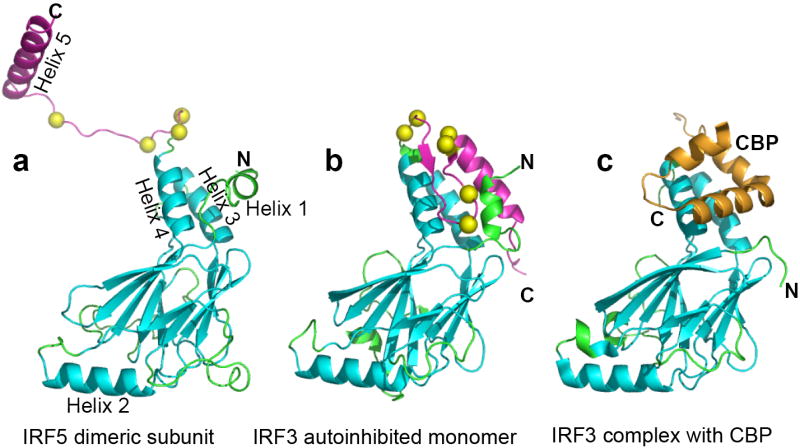
Ribbon diagrams of the C-terminal transactivation domain of IRF5 and IRF3. Monomers from three crystal structures have been aligned using 138 residues of the IAD (cyan). The rest of the IAD is in green, the C-terminal autoinhibitory region is in magenta and putative phosphorylation sites are shown as yellow balls. (a) IRF5 (222-267) S430D has a very extended C-terminal region that participates in dimer formation. This leaves most of the CBP binding site, formed by helices 3 and 4, exposed to solvent except for that covered by helix 1. (b) In the autoinhibited IRF3 (173-427) monomer, the autoinhibitory C-terminal region binds to and masks the CBP binding site 31. (c) In the complex of IRF3 (173-394) with CBP (2067-2112), CBP (gold) binds to helices 3 and 4, which were exposed by the removal of residues 395-427, and displaces the N-terminal helix 1 32.
IRF5 S430D Dimer
The homodimer of IRF5 likely represents the activated form and exhibits an extensive intermolecular surface with very favorable interactions (Fig. 2a). The dimer assembles from two IRF5 monomers that are related by a crystallographic two-fold axis. Intradimer contacts involve helix 4, helix 5 and the extended connecting polypeptide from one subunit packing against a second subunit at the surface formed by helix 2 and a number of interstrand loops. A total surface area of 5800Å2 is buried in the dimeric interface, with the C-terminal autoinhibitory/dimerization region (residues 423-457) participating in 74% of this buried surface. The buried surface area in the IRF5 dimer is substantially larger than the 2200Å2 surface area buried by the autoinhibitory region of monomeric IRF3 and the 1800Å2 buried in the IRF3/CBP complex 32. These observations strongly implicate the C-terminal region as having a key role in activation.
Figure 2.
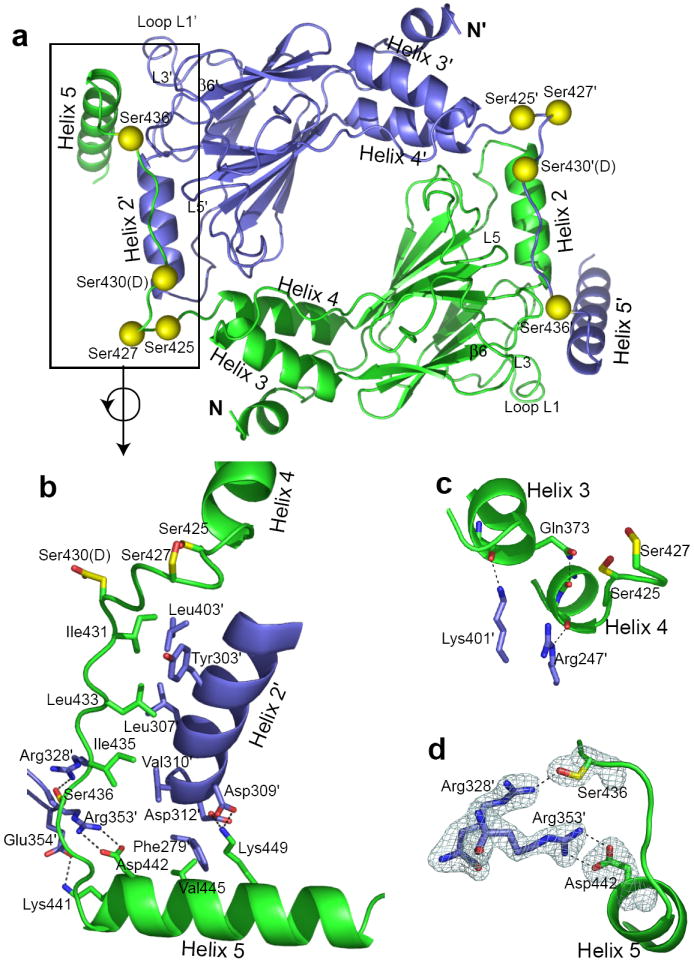
IRF5 dimer. (a) A ribbon diagram of the crystallographic IRF5 dimer is shown with one subunit in blue and one in green. Putative phosphorylation sites are shown as yellow balls. The C-terminal region lays across the surface of the second subunit making extensive contacts particularly involving helix 4, helix 5 and the interhelical region interacting with helix 2′ and loops L1′, L3′ and L5′. (Primes designate the second subunit.) (b) An expanded view of the major interacting region between subunits, rotated from a. Color code is the same as in a with key side chains shown, including those of putative phosphorylation sites with yellow carbon atoms. A hydrophobic patch on Helix 2′, formed from residues Y303′, L307′, V310′ and L403′ interacts with residues I431, L433 and I435 from the extended peptide between helices 4 and 5. Ionic interactions are contributed by residues in helix 5, including D442 with R353′, K441 with E354′ and K449 with D309′ and D312′; likely hydrogen bonds are shown as dashed lines. (c) Interface helix capping of helices 3 and 4. K401′ forms a likely hydrogen bond with the F372 carbonyl at the C-terminus of helix 3. Q373 forms a likely intrasubunit hydrogen bond with the F420 carbonyl at the C-terminus of helix 4, whereas R247′ forms a likely hydrogen bond with the main-chain carbonyl oxygen of S421. These interactions may stabilize the shorter length of helix 4 in dimeric IRF5 compared with monomeric IRF3. (d) Fo-Fc simulated annealing omit map showing the dimeric interactions of S436 with R328’ and D442 with R353′, whose atoms were removed from the atomic model for the simulated annealing process. Electron density is shown at the 3σ level. The interaction between R328′ and S436 is likely to be strengthened by phosphorylation of S436.
The dimeric interface involves a mixture of hydrophobic and hydrophilic contacts (Fig. 2b). Contributing centrally to the hydrophobic dimeric interactions are Ile431, Leu433 and Ile435, in the extended region of polypeptide between helix 4 and helix 5. These residues pack against a hydrophobic surface formed by residues Tyr303′, Leu307′ and Val310′ in helix 2 and Leu403′ from loop L5. (Primes designate partner subunit.) IRF5 residues Ile431, Leu433 and Ile435 are homologous to IRF3 residues Val391, Leu393 and Ile395, respectively, which pack against the CBP/p300 binding surface in monomeric IRF3 31. These three positions are largely occupied by Ile, Leu and Val residues among the IRF family members, as are many of the acceptor residues (Supplementary Fig. 4). Ile, Leu and Val residues have been shown in folding studies to be particularly suitable for forming stable hydrophobic clusters34. Thus, packing of these branched hydrophobic amino acids likely contributes to the overall stability of the dimeric IRF5 interface.
Hydrophilic interactions provide specificity to the dimer and dominate the contact involving the region near the beginning of helix 5 (Fig. 2b). Key hydrophilic interactions involve Asp442 (helix5) with Arg353′ (loop3), Lys449 (helix 5) with Asp309′ and Asp312′ (in helix 2), Lys441 (helix 5) with Glu354′ (loop L3) and Ser436 with Arg328′ (strand β6) (Fig. 2b and Supplementary Fig. 5). This constellation of hydrophilic interactions contributes to the complementarity of the dimeric interface.
One exception to the hydrophilic interface interactions of helix 5 is Val445 (Fig. 2b). Val445 is within 4.5Å of three hydrophobic residues in the partner subunit: Leu269, Leu278 and Phe279. These residues are in loop L1, which is substantially longer in IRF5 than in several other IRF family members (Supplementary Fig. 4). Although these interactions are not tight, the exclusion of water may contribute to greater stability of the IRF5 dimer relative to those of other IRF family members.
Helix capping hydrogen bonds35 involving carbonyl oxygen atoms at the C-termini of helices 3 and 4 (Fig. 2c) also contribute to the dimeric interface. Helix 3 is capped by a likely hydrogen bond with Lys401′. The last turn of helix 4 in dimeric IRF5 is unwound relative to that of monomeric IRF3 and appears to be stabilized by two helix capping hydrogen bonds, involving Arg247′ and Gln373 (Fig. 2c). These hydrogen bonds may play a central role in the formation of the dimer by favoring unwinding of the last turn of helix 4, thus contributing to the extended polypeptide between helices 4 and 5 that participates in other key dimeric interactions.
Role of potential phosphorylated Serine residues
ITC experiments (Table 1) implicate Ser425, Ser427, Ser430 and Ser436 as important phosphorylation sites. The IRF5 crystal structure shows that phosphorylated Ser425, Ser427 and Ser430 will not participate in dimeric contacts; rather phosphorylation of these residues likely contributes to activation by triggering the unfolding of the autoinhibitory structure in the monomer. In contrast, Ser436 is within hydrogen bonding distance of Arg328′ (Fig. 2d); Arg328 is one of the few absolutely conserved residues in human IRF3 through IRF9 (Supplementary Fig. 4). This interaction is likely to be strengthened upon phosphorylation of Ser436. The homologous residue in IRF3, Ser396, is very important for phosphorylation induced IRF3 activation28,36,37, with several studies suggesting that this site 2 residue is important for releasing autoinhibition but that phosphorylation of a site 1 residue (S385/S386) is required for dimerization38,39. Our structure suggests a more direct role for Ser436 in dimerization of IRF5. S436D showed a similar increase in affinity of IRF5 for binding CBP as did S430D (Table 1), despite a potential role of phosphorylated S436 in stabilization of the activated dimer. That S436D is not more stabilizing for CBP binding suggests that the phosphomimetic may not accurately mimic the stabilizing interactions of phosphoserine at 436 in the dimeric contact due to stereochemical and charge differences between aspartate and phosphoserine. Thus, phosphorylation of Ser425, Ser427 and Ser430 are likely to contribute to activation primarily by destabilizing the autoinhibitory conformation of C-terminal region, but the phosphorylation of Ser436 may additionally contribute directly to stabilization of the activated dimer.
IRF5 mutants of key dimeric interface residues
We investigated the relevance of the crystallographically observed dimer by site-directed mutagenesis of key interface residues. Sites selected for mutation were based on their involvement in the dimeric interface, but also their likelihood of being exposed to solvent in the monomeric state. Mutants chosen to disrupt the hydrophobic packing interactions were L307D, V310D and L403D and those designed to disrupt ionic/hydrophilic interactions included D442R, R353D and R328E (Fig. 2b).
Dimerization of IRF5 mutants was tested using size-exclusion chromatography with CBP. Each mutant was constructed in IRF5 (222-467) S430D to investigate if mutations were capable of disrupting dimer formation even in the presence of this dimer-favoring phosphomimetic. The double mutant R328E/S436D mutant was also constructed. Mutants V310D, R353D, D442R, and R338E (both in the context of S430D and S436D) were investigated by size-exclusion chromatography. (L307D S430D and L403D S430D did not express well in E. coli.) None of these mutants forms a dimer even in the presence of CBP (Fig. 3a). SDS-PAGE on concentrated fractions of the IRF5 V310D S430D peak run with CBP showed no evidence of CBP (data not shown) suggesting that mutants that are unable to dimerize also are unable to bind CBP. These results indicate that the dimer observed in the crystal structure is formed in solution.
Figure 3.

Structure guided mutagenesis of IRF5 and IRF3. (a) Gel exclusion chromatography on IRF5 dimeric interface mutants in the presence of CBP, all of which run as monomers in contrast to IRF5 S430D. (b) Gel exclusion chromatography on IRF3 (173-427) mutants in the background of the IRF3 S386D/S396D phosphomimetic, showing R285E and L362E disrupt dimer formation. (c) IFNβ luciferase activity measured from HEK293 cells transfected with the IFNβ reporter gene, TK-renilla, WT or mutants of IRF5 and increasing concentrations of MyD88 (20, 40, 80ng). (d) For IRF3 HEK assays, cells were transfected with the IFNβ reporter, TK-renilla and IRF3 as indicated and either uninfected (mock) or infected with NDV (8HAU/ml) for 16hrs prior to measuring luciferase activity. Western blot analysis of transfected proteins is shown in panel insets for both IRF5 (c) and IRF3 (d).(e) IRF3-deficient fibroblasts were transfected with WT or mutant versions of IRF3 and IFNβ reporter gene activity measured in uninfected cells (mock) or after NDV infection. In all cases, data are expressed as fold induction relative to the reporter-only control and are the mean ± SD.
To test the physiological relevance of this dimer, we engineered dimer-destabilizing mutations in a full length IRF5 vector and tested using an IRF5 reporter assay in HEK293 cells. Upon activation by the TLR adapter MyD88, IRF5 translocates to the nucleus, where it binds to and activates transcription of the IFNβ reporter gene. Activation of IRF5 was disrupted with mutants of both hydrophobic and hydrophilic interface residues. Single mutants R338D, R353D, L307D and L403D, as well as double mutants L307D V310D and D442R R353D and a triple mutant L307D V310D L403D showed severely compromised activation of IRF5 that was comparable to use of an empty vector (Fig. 3b). Although still defective, the single site mutants V310D and D442R showed only a partial signal reduction. We verified equivalent expression of all mutants by western blot analysis of the same lysates as those analyzed for luciferase activity (Fig. 3b). Thus, the crystallographically observed dimer is physiologically relevant as its disruption inhibits activation of IRF5.
IRF3 dimeric activation
Comparison of the autoinhibited IRF3 and dimeric IRF5 structures suggests that, for the most part, IRF3 should be able to attain a similar dimer to that found for IRF5. The only region where such dimer formation would induce unfavorable contacts is in Loop L5. The observed L5 structural differences could reflect distinctions between IRF3 and IRF5, or dimerization induced conformational transitions. Comparison of crystal structures of IRF3 in the autoinhibited monomer with the IRF3/CBP complex show conformational changes of L5 as a result of crystal contacts31,32. Interestingly, loop L5 in the IRF3/CBP crystal structure is more similar with that of the IRF5 dimer than it is with that in the autoinhibited monomer (Supplementary Fig. 6). Apparently, loop L5 has intrinsic flexibility that allows its rearrangement upon dimerization. A conserved glycine, Gly390 in IRF5, near the beginning of loop L5 (Supplementary Fig. 4) may contribute to this plasticity and allow IRF3 to assemble into a similar dimer to that observed here for IRF5.
To directly test whether IRF3 forms an activated dimer similar to that of IRF5, we mutated the two interface residues that are absolutely conserved in family members IRF3 through IRF9. In the IRF5 dimer, Arg328′ contacts Ser436 and Leu403 contributes to hydrophobic interactions. Both R285E (homologous with Arg328 in IRF5) and L362D (homologous with Leu403 in IRF5) were constructed in the background of the S386D S396D double mutant that dimerizes in the presence of CBP37. In both cases, size exclusion chromatography indicates that these triple mutants elute as monomers in the presence of CBP (Fig. 3c), strongly supporting the hypothesis that IRF3 forms a very similar dimer to that observed in the IRF5 crystal structure.
To test the physiological relevance of this IRF3 dimer, we engineered the same mutations in a full length IRF3 vector for use with assays in HEK293 cells and fibroblasts from IRF3-deficient mice. Transfection of HEK293 cells with wild type IRF3 alone dose dependently increased IFNβ reporter gene activity, which was further enhanced upon NDV (Newcastle Disease Virus) infection, as expected. In contrast, the R285E mutant was much less active either when transfected alone or in the presence of NDV infection (Fig. 3d). The L362D mutant was completely defective in driving the IFNβ reporter alone and did not enhance the NDV response above baseline levels. Western blot analysis of the highest dose of transfected IRF3 in each case revealed equivalent expression levels (panel inset). Since HEK293 cells express IRF3 endogenously, we also tested the ability of these mutants to reconstitute NDV induced IFNβ reporter gene activation in fibroblasts lacking functional IRF3. These fibroblasts fail to induce IFNβ reporter gene activity in response to NDV infection, but when transfected with WT IRF3, NDV induced IFNβ reporter gene activation. However, neither the R285D nor the L362D mutants could reconstitute signaling in these cells (Fig. 3e). Taken together, these data support the idea that activation of IRF3 involves formation of a similar dimer to that of IRF5.
DISCUSSION
The first direct high resolution structural information of an activated IRF reveals a dramatic phosphorylation-dependent structural rearrangement of the C-terminal region that triggers dimerization of IRF5. Thus, this work provides a structural basis for understanding how phosphorylation activates IRF family members.
Our mutagenesis studies support the observed dimer as representing the activated state of both IRF5 and IRF3. Published data on IRF3 mutants further supports the conclusion that IRF3 dimerizes similarly to IRF5. Qin et al.31 reported a triple mutation of loop L6 residues in which Val391, Leu393 and Ile395 (homologous with IRF5 residues Ile431, Leu433 and Ile435, respectively) were simultaneously mutated to Arg to disrupt the autoinhibitory contacts. As hypothesized, this triple mutant abolished the virus-dependent activation, but, unexpectedly, also interfered with IRF3 oligomerization. Such an interference with oligomerization of IRF3 is precisely the expected effect if the IRF3 dimer is similar to the IRF5 dimer, with these three residues in the interface. Additional published IRF3 mutants are also consistent with similar dimer formation to that of IRF5. These include double mutations, R211A R213A and K360A R361A, and triple mutations, R255A R262A H263A and R285A H288A H290A, that have been shown to inhibit viral-induced dimerization of IRF3 30,31. In each mutant protein, one or more of the mutated residues is homologous with a residue in the IRF5 interface. Taken together, both the published data and the results presented here strongly support the crystallographically observed IRF5 dimer as representing the activated state of both IRF5 and IRF3.
The dramatic structural transitions observed for IRF5 appear to be a signature of the IRF family that is not shared with the evolutionarily related30,31 Smad proteins involved in TGF-β signaling. In both IRF and Smad proteins, phosphorylation of Ser/Thr residues in the C-terminal region triggers oligomerization. In Smad proteins, phosphorylation of serine residues in the C-terminal SSXS motif drives homo and hetero trimerization due to the direct binding of these phosphorylated serines in the trimer interface40. In contrast, the phosphorylation sites in IRFs are more than 20 residues from the C-terminus. As a result, phosphorylation triggers much larger structural changes. Such structural changes can expose binding sites for other proteins, including CBP/p300, that contribute to signaling. In addition, the structural complexity of phosphorylation mediated signaling in IRFs suggests differential roles for individual sites: Ser425, Ser427 and Ser430 act primarily to destabilize the autoinhibited form, whereas Ser436 contributes to dimeric stability. These considerations suggest that conformational changes in IRF signaling may permit a complex differentiated biological response due to particular site specific phosphorylations.
Following phosphorylation IRF5 is translocated into the nucleus. Two potential nuclear localization signals (NLS) have been identified 10. In contrast to IRF3 and IRF7, IRF5 can be detected in the nucleus of uninfected cells, which could result from the presence of two NLSs9,10. Also unlike other IRFs, one putative NLS has been identified in the C-terminal transactivation domain. NLS signals are normally on unstructured surface loops that permit binding to importin-α (karyopherin-α)41-43. The residues identified in the C-terminal domain as an NLS (398PREKKLI404) are in loop L5 that is intimately involved in the dimeric interface, which may mask them from binding importin-α. Thus the mutagenesis results on the putative NLS residues10 may need to be reevaluated, as such mutants likely disrupt dimerization which, in and of itself, could alter nuclear localization.
Dimerization of the IRF5 transactivation domain initiates the process leading to binding of full-length IRF5 to DNA. While no crystal structure of the N-terminal DNA binding domain (DBD) of IRF5 with DNA is available, structures of the well-conserved DBDs from several other IRF family members with DNA are available19-22. The human IFN-β enhancer comprises binding sites for up to four IRF DBDs as well as activators NFκB and ATF-2/Jun. Pairs of DBDs that could reasonably be part of an IRF dimer have C-terminal α-carbon atoms separated by 42-49Å21,22. These distances are consistent with the subunit arrangement for the IRF5 transactivation domain observed here, in which the N-terminal α-carbon atoms of the two IRF5 subunits are 59Å apart. Thus, the ~75 residue linker region between the N-terminal and C-terminal domains is not required to substantially alter the spacing between subunits for optimal DNA binding. However, this region must permit differences in the orientation of the two DBDs upon DNA binding since the two-fold symmetry relating the two C-terminal domains in the dimer is not present in the tandem bound DBDs (Fig. 4). As a result, dimerization of the transactivation domain brings the DBDs into only approximate position for binding DNA.
Figure 4.

Schematic diagram for the activation of IRF family members, in which the interferon association domain (IAD) is shown as a crescent and the DNA Binding Domain (DBD) is shown as an oval behind the IAD. Unphosphorylated IRF proteins generally remain in the cytoplasm in an autoinhibited state, with the CBP/p300 binding site masked. Phosphorylation induces an unfolding of the C-terminal region that triggers its assembly with another IRF to form a dimer and unmask the CBP binding site. Phosphorylated IRF dimers are translocated into the nucleus, where they interact with CBP/p300 and other transcription factors to form complexes that bind to the IFN-β enhancer region as well as other promoters. The DBDs bind to DNA in a tandem, rather than a two-fold, arrangement requiring that the two-fold symmetry of the IAD does not extend to the DBDs when bound to DNA.
IRF5 and IRF3 show substantially different responses to phosphorylation mimetics, with changes in affinity between CBP and IRF3 that are much more dramatic than those reported here for IRF5. In the case of IRF3, single phosphomimetic mutations increased affinity for CBP up to 19-fold and double mutations increased affinity up to 120-fold37. In contrast, the largest change in affinity for binding CBP with IRF5 was only 2.9-fold (Table 1). These differences are consistent with earlier studies indicating that, unlike IRF3 and IRF7, an unphosphorylated autoinhibitory domain is not able to completely inhibit activation of IRF5 10. Thus the phosphorylation dependent switch between autoinhibition and activation is more finely tuned in IRF5 than in IRF3.
The striking differences in sensitivity to activation between IRF3 and IRF5 likely represent different functional requirements that reflect distinct physiological roles of different IRFs. For instance, IRF3 is constitutively expressed in all cells, acting as a molecular sentry for viral infection. As such, it must be strongly autoinhibited, activated only in response to a clear signal. The C-terminal autoinhibitory/dimerization region plays a central role in activation. This region shows low sequence similarity (Supplementary Fig. 4) among IRFs, including no absolutely conserved residues among IRF3 through IRF8. For IRF3, a large number of hydrophobic residues in the IAD likely contribute to its lower basal activity compared with IRF7 and IRF5 31. An additional factor is the stability of the activated dimer. The IRF5 structure highlights a number of hydrophobic and ionic interactions that stabilize the dimer and may contribute to its greater basal activity. For instance, the ionic interaction between Arg353′ and Asp442 appears likely to be absent in IRF3 and IRF7 as the equivalent residues are Lys313 and Ser402 in IRF3 and Arg398 and Tyr488 in IRF7. In IRF3, K313A does not show a defect in virus-induced of dimer formation31 (the only reported non-defective IRF3 mutation of a residue homologous to an IRF5 dimeric interface residue). The absence of dimer stabilization by Lys313 may contribute to the tight autoinhibitory control of IRF3. Additionally, the hydrophobic surface formed by residues in the L1 loop that interacts with a portion of helix 5′ in IRF5 is absent in IRF3 and likely absent in IRF7 (Supplementary Fig. 4), which could also weaken IRF3 and IRF7 dimers relative to the IRF5 dimer. Therefore the sensitivity of a given IRF family member is likely controlled through the balance between intramolecular autoinhibitory interactions and intermolecular dimeric interactions.
In conclusion, the structure of dimeric IRF5 reveals that phosphorylation-induced activation of IRF5 and other IRF family members involves a dramatic structural transition of the C-terminal autoinhibitory/dimerization region. Phosphorylation triggers unfolding of this region, allowing an extended conformation that binds to another IRF5 monomer. In doing so, the CBP binding site is exposed, providing a clear structural link between phosphorylation, dimerization and CBP/p300 binding (Fig. 4), key steps in transcriptional activation of IFN-β and associated genes.
METHODS
Protein expression
The cDNA fragment of human IRF5 (residues 222-467) (variant 4 ref. 11) was generated by PCR and subcloned into the pGEX-6p-1 vector (Amersham). The IRF5 protein was expressed in E. coli strain HB101 with a glutathione S-transferase (GST) tag, extracted by glutathione sepharose, and released by PreScission Protease (Phamacia Biotech). Eluted protein was dialyzed in DEAE buffer (20 mM Tris, pH 7.3, 10 mM NaCl, 0.1 mM EDTA, 1 mM DTT and 1 mM PMSF). Protein was purified using a DEAE-sepharose column and eluted by a gradient of 10 to 300 mM NaCl. Site-directed mutants of IRF5 (215-477) were generated using the Quik-Change kit (Strategene) and confirmed by sequencing.
The cDNA fragment of CBP (residues 2067-2112) was generated by PCR and subcloned into the pGEX-6p-1 vector (Amersham). The GST-CBP fusion protein was expressed with a GST tag and extracted by glutathione sepharose. Eluted proteins were dialyzed in DEAE buffer. Protein was purified using an SP-sepharose column equilibrated with DEAE buffer and eluted by a gradient of 10 to 300 mM NaCl.
Size exclusion chromatography and ITC measurements
To assess the level of assembly and CBP binding to IRF5, complexes were analyzed by size-exclusion chromatography on a Superdex 200 HR column using the Akta Explorer 10 FPLC system (Pharmacia Biotech, Superdex ™ 200, 10/30) at room temperature in FPLC buffer (20 mM HEPES pH 7.3, 0.1 mM EDTA, 100 mM NaCl, and 1 mM DTT). IRF5 (or mutants) and mixtures of IRF5 (or mutants) with a 3-fold molar excess of CBP were incubated overnight at 4 °C. Prior to loading onto the column, the samples were incubated in 1 mM TCEP for 60 min at room temperature. FPLC was operated and data were analyzed with UNICORN software. The column was calibrated with blue dextran (to determine void volume) and molecular weight standards ranging from 43 kDa to 440 kDa and run at a flow rate of 0.5 ml min-1.
To measure the binding of purified CBP (2067-2112) with IRF5 and its mutants, isothermal titration calorimetry (ITC) was used44 with a VP-ITC calorimeter (MicroCal, Northampton, MA). All samples were dialyzed against ITC buffer (20 mM HEPES, pH 7.3, 100 mM NaCl, 0.1 mM EDTA, 1 mM TCEP) and degassed prior to titration. Experiments were performed at 25°C. Isothermal interactions between proteins were measured by titrating over 55 injections using 1.4 ml of 40 μM protein against 300 μl of 400 μM CBP. The heats of dilution were measured by titrating CBP into ITC buffer; these values were subtracted for data analysis. Data were analyzed with Origin 7.0 software (MicroCal), using a single-site binding model. ΔH, ΔS, and Ka values were experimentally determined, and ΔG was calculated from the equation, ΔG = −RT ln Ka.
Crystallization and crystal structure determination
Crystallization was carried out with hanging drops of 2 μl protein mixed with an equal volume of precipitating solution that were allowed to equilibrate with the precipitating solution at 23°C by vapor diffusion. The crystals used for structure determination were grown using a 16 mg/ml protein solution that was equilibrated with 100mM imidazole (pH 7.0) and 0.5-2.0% (w/v) PEG 6000.
Diffraction data were collected to 2.0Å resolution from a native crystal at the National Synchrotron Light Source (NSLS, Brookhaven NY) beamline X29. The crystal was flash frozen in the cold nitrogen stream following a brief soak in a cryoprotectent solution of 100 mM imidazole (pH 7.0), 1% PEG 6000, 15% ethylene glycol, and 15% glycerol. A heavy atom derivative was obtained by soaking these crystals in 2 mM mercury-[(o-carboxyphenyl) thio] ethy sodium salt for five days. Diffraction data from this derivative were collected on our R-AXIS IV image plate system (Rigaku, USA). Diffraction data were processed using HKL2000, DENZO and SCALEPACK45.
Heavy atom analysis, phase improvement and initial model building were carried out with the SOLVE/RESOLVE package46. Four heavy atom sites were found and refined, resulting in a figure of merit of 0.48 to 3.2Å resolution. Density modification was applied with RESOLVE incorporating all the data to 2.0Å resolution, resulting in a figure of merit of 0.51. (This analysis clearly showed space group P3121 rather than its enantiamorph P3221 was correct.) Automatic model building by RESOLVE succeeded in building 179 residues into the structure. This represented 76% of the 236 residues in the final refined structure. Iterative cycles of refinement, with CNS 47 and REFMAC548 (including TLS refinement49), and model building in O50 and COOT51 were carried out to complete the structure (Table 2). Residues 458-467 were not visible in the electron density maps and are not included in the refined model. All residues in the model have conformations that are either in the most favored region (91.2%) or additional allowed region (8.8%) of a Ramachandran plot. Figures 2 & 3 and Supplementary Figures 3 & 4 were generated with PyMOL52.
Table 2.
Data collection and refinement statistics on IRF5
| Native | Hg derivative | |
|---|---|---|
| Data collection | ||
| Space group | P3121 | P3121 |
| Cell dimensions a, b, c (Å) | 66.16, 66.16, 156.25 | 66.22, 66.22, 156.09 |
| Resolution (Å) | 100-2.0 (2.07-2.0)* | 100-3.2 (3.31-3.2) |
| Rsym (%) | 5.7 (22.9) | 6.4 (19.7) |
| I / σI | 13.8 (5.1) | 14.0 (5.2) |
| Completeness (%) | 96.9 (78.8) | 99.4 (99.9) |
| Redundancy | 9.9 (7.2) | 6.9 (7.1) |
| Refinement | ||
| Resolution (Å) | 60.0 – 2.0 | |
| No. reflections | 25,440 | |
| Rwork / Rfree (%) | 20.5 / 24.2 | |
| No. atoms | ||
| Protein | 1935 | |
| Water | 132 | |
| B-factors (Å2) | ||
| Protein | 44.4 | |
| Water | 47.8 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.013 | |
| Bond angles (°) | 1.36 |
Values in parentheses are for highest-resolution shell. One crystal was used for each data set.
Lucerferase reporter assays
The IFNβ luciferase, full length IRF5-v4-Flag, IRF3-Flag and MyD88 were as previously described4. pGL4-TK renilla luciferase was from Promega (Madison, WI). For HEK293 assays, 2.104 cells/well in 96-well plates were transfected with 40ng of the IFNβ luciferase reporter gene and 40ng of the thymidine kinase Renilla-luciferase reporter gene together with the indicated doses of either wild type or mutant IRFs using Genejuice (Novagen, Madison, WI). Where indicated cells were also transfected with MyD88 expression plasmids. Transfected cells were either untreated or where indicated were infected with NDV (LaSota Strain, 8HAU/ml) for 16 hours. IRF3-deficient fibroblasts were prepared from IRF3-deficient mice originally obtained from T. Taniguchi (Tokyo, Japan). IRF3-deficient fibroblasts, 104 cells/well in 24-well plates were transfected with 250ng of IFNβ-luciferase, 250ng TK-renilla reporter and 20, 40 or 80ng of the IRF constructs. 8 hours later cells were either uninfected (mock) or infected with NDV for 16 hours. In all cases, luciferase activity was measured as previously described4 and data were normalized according to the manufacturer recommendations (Promega).
Supplementary Material
Acknowledgments
This paper is dedicated to the memory of Dr. Kai Lin, whose guidance initiated our investigations. We thank Brendan Hilbert for technical assistance and Drs. Reid Gilmore, Mary Munson and Melissa Moore for helpful comments on the manuscript. This work was supported by NIH grants AI056080 (WER & CAS) and AI067497 (KAF). We thank the staff of National Synchrotron Light Source beamline X29. Financial support for this facility comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources of the National Institutes of Health. All Analytical Ultracentrifugation runs were done in the UMMC AUC Facility.
Footnotes
Accession code. Protein Data Bank: The IRF5 atomic coordinates and structure factors have been deposited under accession code 3DSH.
AUTHOR CONTRIBUTIONS: K.L., W.C., W.E.R., C.A.S., J.J.C. and K.A.F. designed the experiments; W.C., S.S.L. and H.S. prepared, characterized and crystallized the protein; W.C. and C.A.S. carried out and analyzed the ITC experiments, W.C. and W.E.R. performed the crystallographic analysis; Z.J. and K.A.F. performed and analyzed the cell-based assays; J.J.C. carried out and analyzed the sedimentation experiments, W.E.R., W.C., K.A.F., C.A.S. and J.J.C. prepared the manuscript.
References
- 1.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–58. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 2.Paun A, Pitha PM. The IRF family, revisited. Biochimie. 2007;89:744–53. doi: 10.1016/j.biochi.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF Family Transcription Factors in Immunity and Oncogenesis. Annu Rev Immunol. 2008;26:535–84. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 4.Schoenemeyer A, et al. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem. 2005;280:17005–12. doi: 10.1074/jbc.M412584200. [DOI] [PubMed] [Google Scholar]
- 5.Yasuda K, et al. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. J Immunol. 2007;178:6876–85. doi: 10.4049/jimmunol.178.11.6876. [DOI] [PubMed] [Google Scholar]
- 6.Takaoka A, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–9. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 7.Hu G, Mancl ME, Barnes BJ. Signaling through IFN regulatory factor-5 sensitizes p53-deficient tumors to DNA damage-induced apoptosis and cell death. Cancer Res. 2005;65:7403–12. doi: 10.1158/0008-5472.CAN-05-0583. [DOI] [PubMed] [Google Scholar]
- 8.Yanai H, et al. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc Natl Acad Sci U S A. 2007;104:3402–7. doi: 10.1073/pnas.0611559104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes BJ, Moore PA, Pitha PM. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J Biol Chem. 2001;276:23382–90. doi: 10.1074/jbc.M101216200. [DOI] [PubMed] [Google Scholar]
- 10.Barnes BJ, Kellum MJ, Field AE, Pitha PM. Multiple regulatory domains of IRF-5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Mol Cell Biol. 2002;22:5721–40. doi: 10.1128/MCB.22.16.5721-5740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mancl ME, et al. Two discrete promoters regulate the alternatively spliced human interferon regulatory factor-5 isoforms. Multiple isoforms with distinct cell type-specific expression, localization, regulation, and function. J Biol Chem. 2005;280:21078–90. doi: 10.1074/jbc.M500543200. [DOI] [PubMed] [Google Scholar]
- 12.Mori T, et al. Identification of the interferon regulatory factor 5 gene (IRF-5) as a direct target for p53. Oncogene. 2002;21:2914–8. doi: 10.1038/sj.onc.1205459. [DOI] [PubMed] [Google Scholar]
- 13.Graham RR, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38:550–5. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 14.Sigurdsson S, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76:528–37. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miceli-Richard C, et al. Association of an IRF5 gene functional polymorphism with Sjogren’s syndrome. Arthritis Rheum. 2007;56:3989–94. doi: 10.1002/art.23142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kristjansdottir G, et al. Interferon Regulatory Factor 5 (IRF5) Gene Variants are Associated with Multiple Sclerosis in Three Distinct Populations. J Med Genet. 2008 doi: 10.1136/jmg.2007.055012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dideberg V, et al. An insertion-deletion polymorphism in the Interferon Regulatory Factor 5 (IRF5) gene confers risk of inflammatory bowel diseases. Hum Mol Genet. 2007 doi: 10.1093/hmg/ddm259. [DOI] [PubMed] [Google Scholar]
- 18.Hu G, Barnes BJ. Interferon regulatory factor-5-regulated pathways as a target for colorectal cancer therapeutics. Expert Rev Anticancer Ther. 2006;6:775–84. doi: 10.1586/14737140.6.5.775. [DOI] [PubMed] [Google Scholar]
- 19.Escalante CR, Yie J, Thanos D, Aggarwal AK. Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature. 1998;391:103–6. doi: 10.1038/34224. [DOI] [PubMed] [Google Scholar]
- 20.Panne D, Maniatis T, Harrison SC. Crystal structure of ATF-2/c-Jun and IRF-3 bound to the interferon-beta enhancer. Embo J. 2004;23:4384–93. doi: 10.1038/sj.emboj.7600453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Escalante CR, Nistal-Villan E, Shen L, Garcia-Sastre A, Aggarwal AK. Structure of IRF-3 bound to the PRDIII-I regulatory element of the human interferon-beta enhancer. Mol Cell. 2007;26:703–16. doi: 10.1016/j.molcel.2007.04.022. [DOI] [PubMed] [Google Scholar]
- 22.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell. 2007;129:1111–23. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin R, Mamane Y, Hiscott J. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol. 1999;19:2465–74. doi: 10.1128/mcb.19.4.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin R, Mamane Y, Hiscott J. Multiple regulatory domains control IRF-7 activity in response to virus infection. J Biol Chem. 2000;275:34320–7. doi: 10.1074/jbc.M002814200. [DOI] [PubMed] [Google Scholar]
- 25.Fitzgerald KA, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–6. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 26.Sharma S, et al. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–51. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 27.Sato M, Tanaka N, Hata N, Oda E, Taniguchi T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett. 1998;425:112–6. doi: 10.1016/s0014-5793(98)00210-5. [DOI] [PubMed] [Google Scholar]
- 28.Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol. 1998;18:2986–96. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weaver BK, Kumar KP, Reich NC. Interferon regulatory factor 3 and CREB-binding protein/p300 are subunits of double-stranded RNA-activated transcription factor DRAF1. Mol Cell Biol. 1998;18:1359–68. doi: 10.1128/mcb.18.3.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahasi K, et al. X-ray crystal structure of IRF-3 and its functional implications. Nat Struct Biol. 2003;10:922–7. doi: 10.1038/nsb1001. [DOI] [PubMed] [Google Scholar]
- 31.Qin BY, et al. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol. 2003;10:913–21. doi: 10.1038/nsb1002. [DOI] [PubMed] [Google Scholar]
- 32.Qin BY, et al. Crystal structure of IRF-3 in complex with CBP. Structure. 2005;13:1269–77. doi: 10.1016/j.str.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 33.Cheng TF, et al. Differential activation of IFN regulatory factor (IRF)-3 and IRF-5 transcription factors during viral infection. J Immunol. 2006;176:7462–70. doi: 10.4049/jimmunol.176.12.7462. [DOI] [PubMed] [Google Scholar]
- 34.Wu Y, Vadrevu R, Kathuria S, Yang X, Matthews CR. A tightly packed hydrophobic cluster directs the formation of an off-pathway sub-millisecond folding intermediate in the alpha subunit of tryptophan synthase, a TIM barrel protein. J Mol Biol. 2007;366:1624–38. doi: 10.1016/j.jmb.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aurora R, Rose GD. Helix capping. Protein Sci. 1998;7:21–38. doi: 10.1002/pro.5560070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Servant MJ, et al. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J Biol Chem. 2003;278:9441–7. doi: 10.1074/jbc.M209851200. [DOI] [PubMed] [Google Scholar]
- 37.Chen W, et al. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J Mol Biol. 2008;379:251–60. doi: 10.1016/j.jmb.2008.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panne D, McWhirter SM, Maniatis T, Harrison SC. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J Biol Chem. 2007;282:22816–22. doi: 10.1074/jbc.M703019200. [DOI] [PubMed] [Google Scholar]
- 39.Mori M, et al. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J Biol Chem. 2004;279:9698–702. doi: 10.1074/jbc.M310616200. [DOI] [PubMed] [Google Scholar]
- 40.Wu JW, et al. Crystal structure of a phosphorylated Smad2. Recognition of phosphoserine by the MH2 domain and insights on Smad function in TGF-beta signaling. Mol Cell. 2001;8:1277–89. doi: 10.1016/s1097-2765(01)00421-x. [DOI] [PubMed] [Google Scholar]
- 41.Conti E, Uy M, Leighton L, Blobel G, Kuriyan J. Crystallographic analysis of the recognition of a nuclear localization signal by the nuclear import factor karyopherin alpha. Cell. 1998;94:193–204. doi: 10.1016/s0092-8674(00)81419-1. [DOI] [PubMed] [Google Scholar]
- 42.Cutress ML, Whitaker HC, Mills IG, Stewart M, Neal DE. Structural basis for the nuclear import of the human androgen receptor. J Cell Sci. 2008;121:957–68. doi: 10.1242/jcs.022103. [DOI] [PubMed] [Google Scholar]
- 43.Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol. 2007;8:195–208. doi: 10.1038/nrm2114. [DOI] [PubMed] [Google Scholar]
- 44.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–7. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 46.Terwilliger TC. SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 47.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 48.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 49.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–33. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 50.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 51.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 52.DeLano WL. The PyMOL Molecular Graphics System. 2002 http://www.pymol.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.