

Summary
Decades ago it was proposed that exocytosis involves invagination of the target membrane, resulting in a highly localized site of contact between the bilayers destined to fuse. The vesicle protein synaptotagmin-I (syt) bends membranes in response to Ca2+, but whether this drives localized invagination of the target membrane to accelerate fusion has not been determined; previous studies relied on reconstituted vesicles that were already highly curved and used mutations in syt that were not selective for membrane-bending activity. Here, we directly address this question by utilizing vesicles with different degrees of curvature. A tubulation-defective syt mutant was able to promote fusion between highly curved SNARE-bearing liposomes, but exhibited a marked loss of activity when the membranes were relatively flat. Moreover, bending of flat membranes by adding an N-BAR domain rescued the function of the tubulation-deficient syt mutant. Hence, syt-mediated membrane bending is a critical step in membrane fusion.
Introduction
Chernomordik and co-workers have envisioned membrane fusion and fission as similar processes that pass through analogous intermediate membrane structures, but in opposite directions as dictated by distinct proteins (Kozlov and Chernomordik, 2002). A key step in endocytosis involves localized invagination of the plasma membrane, allowing a nascent vesicle to form. This process is mediated by a set of proteins that are able to deform membranes (Farsad and De Camilli, 2003). Interestingly, invagination of the plasma membrane has also been observed during exocytosis in some secretory cells (Monck and Fernandez, 1994). This remodeling would result in a curved dimple that points toward secretory vesicles, bringing the two membranes into close proximity at a small point of contact to reduce the energy barrier for fusion. Therefore, ‘dimpling’ of the plasma membrane might constitute an essential step in regulated secretion (Monck and Fernandez, 1994). However, proteins that mediate this putative invagination step during exocytosis have yet to be identified.
Recent studies have shown that synaptotagmin-I (syt), a Ca2+ sensor that triggers rapid neuronal exocytosis, is able to tubulate membranes in response to Ca2+ (Arac et al., 2006; Martens et al., 2007). Hence, syt might operate by buckling the plasma membrane to lower the energy barrier for vesicle fusion.
Attempts have been made to correlate syt's ability to tubulate membranes with its ability to promote fusion of small unilamellar vesicles (SUVs) (Martens et al., 2007). However, the SUV membrane is already highly curved; as reported here, using more physiologically relevant lipid mixtures, syt-induced membrane tubules have diameters that are comparable to SUVs. These findings indicate that SUV-SUV fusion assays are not dependent on the ability of syt to bend membranes, since SUVs are already fully ‘bent’. In addition, previous work on this problem relied on mutant forms of syt, which, as shown in the present study, also affect the interaction of syt with SNARE proteins. Because syt functions in part by engaging SNAREs (Chapman, 2008), it could not be determined whether the differences in fusion activity observed for these syt mutants were due to changes in membrane-bending or SNARE-binding activity.
Here, we directly test the hypothesis that syt must bend membranes in order to promote fusion. This was achieved by analyzing SNARE-mediated fusion of giant unilamellar vesicles (GUVs, diameter > 1 μm) in parallel with SUVs (diameter ∼ 65 nm). We found that a syt mutant with compromised membrane-bending activity failed to stimulate fusion when the membrane was relatively flat (i.e., when using GUVs), but functioned effectively when the membrane was already highly curved (i.e., when using SUVs). Addition of the N-BAR domain of endophilin, which plays a critical role in endocytosis - presumably by bending the plasma membrane to facilitate vesicle budding (Farsad et al., 2001) - rescued the function of a membrane-bending deficient syt mutant during regulated GUV-GUV fusion. These findings indicate that exo- and endocytosis proceed via common intermediate membrane structures. Finally, we demonstrate that the cytoplasmic domain of syt - when targeted to either synaptic vesicles or the pre-synaptic plasma membrane - can rescue rapid exocytosis in syt knock-out neurons, validating the use of this protein fragment in reconstituted fusion assays.
Results
Syt senses membrane-curvature: steady state and time-resolved quantitative analysis
To set the stage for assessing the functional significance of syt's membrane bending activity, we quantitatively analyzed the ability of syt to sense membrane curvature. The cytoplasmic domain of syt consists of tandem C2 domains - C2A and C2B - that bind Ca2+ via two flexible loops that protrude from one end of each domain (Figure 1A) (Fernandez et al., 2001; Sutton et al., 1995). Upon binding Ca2+, these loops partially insert into the hydrophobic core of the lipid bilayer (Bai et al., 2004; Chapman and Davis, 1998). Penetration-loops, with substantial molecular volume, should prefer membranes with relatively loose lipid packing, as in the outer leaflet of positively curved bilayers. Indeed, a previous study revealed that C2A-C2B co-sediments more avidly with small as opposed to larger liposomes (Martens et al., 2007). Here, we applied three independent assays to gain quantitative insights into the curvature sensing ability of C2A-C2B.
Figure 1. Biophysical analyses of membrane curvature sensing ability of syt.

(A) Model depicting the interaction of the cytoplasmic domain of syt (C2A-C2B) with membranes. The solution structure of C2A and C2B were rendered from (Shao et al., 1998) and (Fernandez et al., 2001). Ca2+ ions = red spheres. Two Ca2+ binding loops (L1 and L3) in each C2 domain partially penetrate the lipid bilayer in response to Ca2+. (B) Co-sedimentation experiments were used to assess the membrane-curvature preference of C2A-C2B. Shown is a representative gel of the remaining protein in the supernatant of each sample. With increasing [lipid], C2A-C2B was depleted from the supernatant fraction. (C) Quantification of the gels in panel B; error bars represent SEM from triplicate determinations. (D) Representative calorimetric heat flow signals obtained when vesicles are injected into a solution containing C2A-C2B. V105nm suspension (20 mM [lipid]) was titrated against C2A-C2B (20 μM) in the presence of 1 mM Ca2+ (top panel), 0.2 mM EGTA (middle panel), or V252nm in 1 mM Ca2+ (lower panel). (E) Integrated heat plotted as a function of [lipid]/[C2A-C2B]; thermodynamic parameters are summarized in Table S2. The binding isotherm of V252nm is right-shifted as compared to that of V105nm. (F) Reaction scheme for the stopped-flow rapid mixing experiments; vesicle = blue sphere. (G) Representative traces of the time course of Ca2+•C2A-C2B assembly with V105nm and V252nm, at 1 mM [lipid]. (H) kobs was plotted as a function of [lipid]. Kinetic parameters are summarized in Table S3. (I) Fold-increase in C2A-C2B•membrane complex Kd values as a function of liposome diameter.
We first compared the binding affinities of C2A-C2B with liposomes of different diameters: 105 nm (V105nm), 138 nm (V138nm), 189 nm (V189nm), and 252 nm (V252nm), by assaying C2A-C2B•liposome cosedimentation as a function of lipid concentration (Figure 1B); depletion of protein from the supernatant was used to monitor binding. The resulting titration curves confirmed that C2A-C2B prefers highly curved membranes and resolved a ∼1.9-fold difference in the apparent dissociation constants (Kd) between V105nm and V252nm (Figure 1C, Table S1). However, C2A-C2B exhibited similar binding affinities for V252nm and giant unilamellar vesicles (diameters of a few microns), suggesting that C2A-C2B is relatively insensitive to curvature when the vesicle diameter is above 252 nm (Figure S2). A preference for highly curved membranes was exhibited by a range of syt mutants that were analyzed in parallel (Figure S3 and S4; Table S1).
We then carried-out isothermal titration calorimetry (ITC) experiments to determine the thermodynamics of C2A-C2B•membrane interactions. Aliquots of V105nm or V252nm were injected into a sample chamber that contained C2A-C2B; titrations using V252nm exhibited a shallow, right-shifted binding isotherm (Figure 1D, E), confirming the idea that syt preferentially binds to highly curved membranes. The isotherm for Ca2+•C2A-C2B•V105nm complex formation exhibited two phases, which are likely due to a binding step followed by lipid rearrangements. This notion is supported by the distinct broadening of peaks in the exothermic phase compared to the earlier endothermic phase, indicating slow lipid rearrangements (Heimburg and Biltonen, 1994). We fitted the biphasic binding isotherm using a two-site model (Table S2). Here, we only discuss the first site, which is likely to correspond to the lipid binding step. The stoichiometry (N) was determined to be 81 ± 4. Given that liposomes contained 25% phosphatidylserine (PS) and C2A-C2B only inserts into one leaflet, we estimate that each C2A-C2B binds ∼10 PS molecules.
The experiments described thus far were carried out under equilibrium conditions. However, syt might drive slow yet dramatic shape changes in membranes (shown later in Figure 2 and Figure S5-7), which would affect measurements of the apparent binding strength. To test this, we utilized a stopped-flow rapid mixing approach to examine C2A-C2B•liposome binding at the initial instant of assembly. Reactions were monitored via fluorescence resonance energy transfer (FRET) between the native aromatic residues in C2A-C2B and dansyl-PE in the membrane of vesicles with different diameters (Figure 1F). Rapid mixing of C2A-C2B with liposomes resulted in a rapid increase in fluorescence (Figure 1G) and these data were used to determine kinetic parameters, as summarized in Table S3. These measurements revealed that a reduction in vesicle diameter - from 252 nm to 105 nm - enhances the affinity of the C2A-C2B•membrane interaction 9.3-fold; this is a more dramatic increase than the 1.9-fold difference resolved in the cosedimentation assays (Figure 1C). The fold enhancement in Kd as a function of liposome diameter is plotted in Figure 1I.
Figure 2. Syt utilizes C2B to bend membranes in a response to Ca2+.
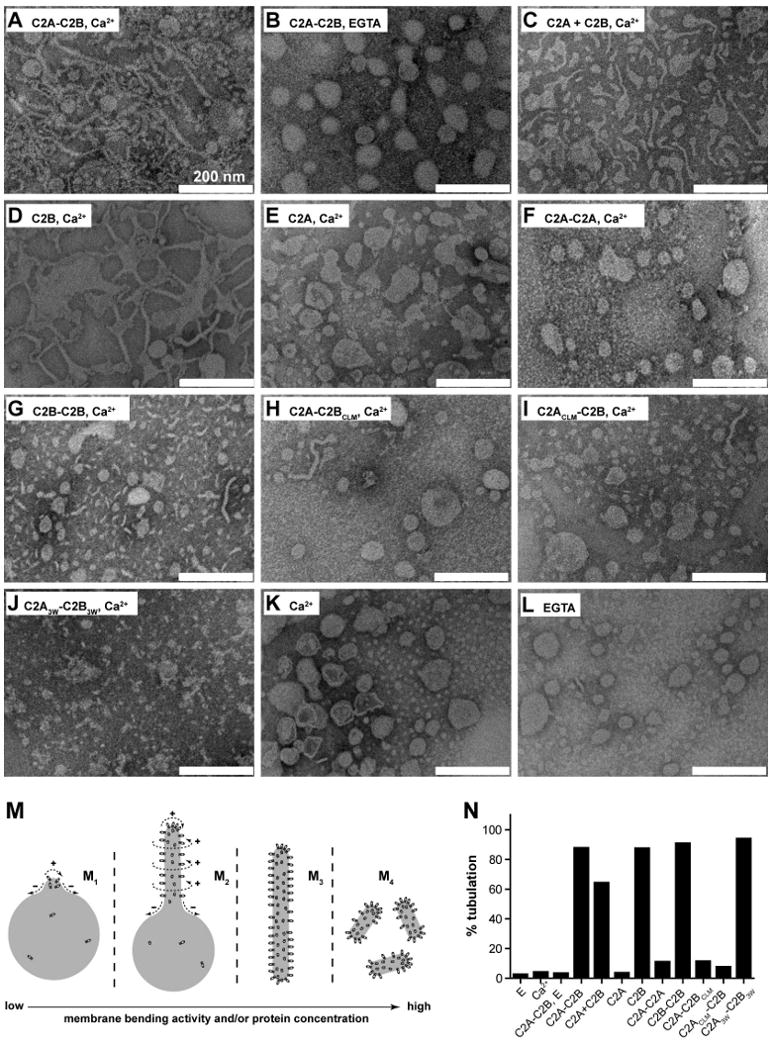
(A-L) Electron micrographs of Folch liposomes incubated with the indicated syt fragments (10 μM) for 10 hr. Scale bars correspond to 200 nm. The diameters of the lipid tubules were (mean ± SD, n = 30): 16 ± 4 nm for C2A-C2B; 11 ± 2 nm for C2B and 10 ± 3 nm for C2A3W-C2B3W. (M) Model showing that different degrees of membrane-bending may result in different morphologies of membranes. Membranes are shown in gray, syt fragments are shown as small ovals. Positive and negative curvatures are indicated as ‘+’ and ‘−’, respectively. Penetration of syt into the membrane drives deformation of the bilayer. The resulting morphologies can be classified into four categories, as follows: (M1), formation of a ‘nipple’, which has positive curvature at the tip, but negative curvature at the base, as indicated by dashed arrows. Protein prefers positively curved membranes and tends to cluster at the nipple tip. This in turn helps to maintain the shape of the nipple. (M2), more extensive membrane bending causes elongation of the nipple, forming a lipid tubule protruding from the parental liposome. New areas with a high degree of positive curvature emerge on the radial plane resulting in increased membrane area that is favorable for protein binding. (M3), even more extensive membrane bending would transform a whole liposome into a long tubule, removing any negative curvature. (M4), in extreme cases, the long lipid tubules are broken into short tubules or even tiny vesicles, further increasing the area with positive curvature. (N) Quantification of the fraction of tubulated liposomes obtained in the presence of the indicated syt construct.
Clearly, kinetic and steady state measurements of the curvature-dependence of syt•membrane interactions differ. These differences are likely to be the result of ‘work’ that syt carries out to bend membranes after the initial binding step.
Syt utilizes C2B to bend membranes in a Ca2+ dependent manner
We explored the ability of syt to induce membrane curvature using liposomes made from Folch fraction I brain lipid extract; structures were visualized via negative stain electron microscopy. In the presence of Ca2+, C2A-C2B converted Folch liposomes into long thin lipid tubules (Figure 2A, M3). Tubulation was not observed in the absence of Ca2+ (Figure 2B) or in the absence of protein (Figure 2K, L), consistent with previous studies (Arac et al., 2006; Martens et al., 2007). However, in contrast to Martens et al. (2007), membrane tubulation activity did not require the tethering of C2A with C2B, as C2A + C2B also drove tubulation (Figure 2C, M2), albeit to a lesser degree than the intact cytoplasmic domain of syt (C2A-C2B). A striking result was obtained when we tested each isolated C2-domain; the C2B domain tubulated membranes as well as C2A-C2B (Figure 2D, N) while C2A failed to tubulate membranes (Figure 2E). Even C2A-C2A, in which two C2A domains are tethered together, failed to tubulate membranes (Figure 2F). In contrast, C2B-C2B caused fragmentation of the lipid membranes (Figure 2G, M4), indicative of stronger tubulation activity. Together, these experiments demonstrate that the C2B domain of syt is both necessary and sufficient to drive tubulation. The fact that isolated C2A avidly penetrates bilayers (Chapman and Davis, 1998; Davis et al., 1999) yet fails to drive tubulation indicates that penetration is not sufficient for membrane bending.
Having established that C2B is the membrane-bending domain of syt, we next determined the tubulation activity of C2A-C2BCLM, in which the C2B domain does not bind membranes due to neutralization of its Ca2+ ligands (CLM; Ca2+ ligand mutations) (Bai et al., 2002). This mutant exhibited only trace levels of membrane-tubulation activity (Figure 2H, N). Surprisingly, C2ACLM-C2B, which carries analogous Ca2+ ligand mutations only within C2A, also exhibited a large defect in membrane tubulation (Figure 2I, N). This indicates that in the context of C2A-C2B, a ‘dead’ C2A domain can inhibit the membrane-bending activity of an adjacent C2B domain.
Because nonpolar side chains in the Ca2+-binding loops of syt insert into the hydrophobic core of the bilayer, increasing the hydrophobic volume of these residues should strengthen C2A-C2B•bilayer interactions, which in turn should translate to greater binding energy for deforming membranes. We therefore tested the tubulation activity of C2A3W-C2B3W, in which six nonpolar residues within the membrane-penetration loops were replaced with tryptophans. This mutant caused the formation of short membrane tubules and tiny vesicles (Figure 2J, M4), suggesting enhanced membrane-bending activity. At relatively low concentrations (1 μM), C2A3W-C2B3W induced the formation of long tubules from Folch liposomes (Figure S6).
The tubulation assay described above utilized liposomes made from Folch lipid extract, which is commonly used to study membrane-bending activity. However, this lipid mixture contains much higher levels of PS (∼50%) than occur in cells (7.5%-15%) (Steenbergen et al., 2006); such high levels of PS are likely to exaggerate the membrane-bending activity of syt by enhancing the affinity of Ca2+•syt•membrane complexes. To determine whether syt can tubulate more physiologically relevant membranes, we incubated syt fragments with liposomes containing 15% PS + 30% phosphatidylethanolamine (PE) + 55% phosphatidylcholine (PC); i.e. the lipid mix that is used in the reconstituted fusion assays detailed further below, and which contains all of the major phospholipids found in cells in proper ratios. In the presence of Ca2+, C2A-C2B and isolated C2B were both able to tubulate membranes (Figure S7A, C), although to a lesser extent than was observed using Folch liposomes (Figure 2). C2A failed to induce any detectable tubulation (Figure S7B), as observed with Folch liposomes (Figure 2E). C2A3W-C2B3W induced dramatic tubulation (Figure S7D).
For membranes that harbored 15% PS, C2A-C2B induced the formation of tubules that were 54 ± 20 nm (Figure S7A), which is markedly greater than what we observed using Folch liposomes that contain ∼50% PS (∼16 nm, Figure 2B; an inverse relationship between fraction of PS and tubule diameter is shown in Figure S8. Thus, the diameter of C2A-C2B induced membrane tubules (54 ± 20 nm) is comparable to the diameter of the SUVs (65 ± 19 nm) used in standard reconstituted fusion assays. These results indicate that C2A-C2B is unlikely to further bend membranes during reconstituted SUV-SUV fusion (Martens et al., 2007), and the function of the membrane-bending activity of syt has yet to be directly determined.
Mutations that affect the membrane-bending activity of syt also affect interactions with t-SNAREs
The membrane tubulation activities of a handful of mutant syts were previously reported to correlate with their abilities to stimulate fusion of SNARE-bearing SUVs (Martens et al., 2007); e.g., C2A2W-C2B2W, a Trp mutant with enhanced tubulation activity exhibited a greater ability to stimulate fusion. An analogous Ala mutant, C2A2A-C2B2A, which fails to tubulate membranes, was defective in the in vitro fusion assay. These correlations led to the suggestion that membrane bending is essential for syt function. However, such correlations do not hold for other mutants, e.g., C2A-C2BCLM does not tubulate membranes to any significant degree, but stimulates fusion almost as well as wild-type (WT) C2A-C2B (Bhalla et al., 2005; Stein et al., 2007). Moreover, although the membrane-bending activity of C2B is similar to that of C2A-C2B (Figure 3D compared to Figure 3A), the ability of isolated C2B to stimulate fusion is much lower (Gaffaney et al., 2008).
Figure 3. Mutations that alter membrane-bending also affect the ability of syt to bind t-SNAREs.

(A) Co-immunoprecipitation (IP) of syt fragments with t-SNARE heterodimers was carried out using an anti-syntaxin (syx) antibody. Equal fractions of total input (T) and immunoprecipitated material (E: 0.2 mM EGTA; Ca: 1 mM Ca2+) were subjected to SDS-PAGE. The 31 samples were resolved in two separate gels, which were juxtaposed at the position indicated by the arrow. (B) t-SNARE binding activity of each syt protein was quantified by densitometry. Data are reported as mean ± SD, n = 3. (C) Trp/Ala mutations within the membrane-penetration loops of syt strongly affect syt's ability to engage t-SNAREs in response to Ca2+, as assessed using co-flotation assays with t-SNARE-bearing, PS-free vesicles. Trp mutants (C2A2W-C2B2W, C2A3W -C2B3W) co-floated more avidly than WT C2A-C2B, while Ala mutant (C2A2A -C2B2A) exhibited diminished binding. (D) Gels in panel C were quantified by densitometry, and molar ratios of syts and syx were plotted against log[Ca2+]. Error bars represent the SEM (n ≥ 3).
Syt must engage both membranes and t-SNAREs to regulate SNARE catalyzed fusion (Chapman, 2008), yet the t-SNARE binding activities of the syt tubulation mutants have not been compared in a systematic way; it is possible that the differential activities of these mutants in the SUV-SUV fusion assay are due to changes in their abilities to engage t-SNAREs. We therefore compared the t-SNARE binding activities of the aforementioned syt mutants. Immunoprecipitation (IP) showed that all mutants tested exhibited significantly altered t-SNARE binding activity (Figure 3A, B). Two Trp mutants, C2A2W-C2B2W and C2A3W-C2B3W exhibited enhanced t-SNARE binding activity, while all of the other mutants tested showed reductions in binding.
In contrast to our findings, another group did not detect significant differences in SNARE binding activity of the Trp and Ala mutant forms of syt (Lynch et al., 2008). In order to resolve this issue, we carried out coflotation assays as an independent approach to compare the t-SNARE binding of the syt mutants (Figure 3C). The extent of t-SNARE binding was markedly enhanced for the two Trp mutants, and was clearly reduced for the Ala mutant (Figure 3D). These experiments demonstrate that the Trp and Ala mutations affect not only the membrane-tubulation activity of syt but also affect interactions with t-SNAREs. Hence, it remained unclear whether syt's ability to bend membranes has any functional role during fusion.
Using vesicles with different curvatures to determine directly whether syt must bend membranes to regulate fusion
To directly assess the function of the membrane-bending activity of syt during membrane fusion, we utilized two sets of SNARE-bearing vesicles with distinct intrinsic membrane curvatures: the commonly used SUVs (Chicka et al., 2008; Martens et al., 2007; Stein et al., 2007; Tucker et al., 2004; Weber et al., 1998), whose membranes are already highly curved; and GUVs, whose membranes are relatively flat (Figure 4A, B). Fusion between SNARE-bearing vescles was monitored using FRET (Tucker et al., 2004; Weber et al., 1998). The rationale was as follows: mutants that are tubulation-deficient but are still able to engage t-SNAREs should be able to stimulate SUV-SUV fusion, but should exhibit loss-of-function in the GUV-GUV fusion assay. By contrast, syt mutants that efficiently bend membranes should be relatively insensitive to membrane curvature. We selected WT, a gain-of-function mutant (C2A3W-C2B3W), a loss-of-function mutant (C2A-C2BCLM), and isolated C2B for these experiments. All of these syt proteins retain some degree of t-SNARE binding activity (Figure 3A), but have distinct membrane-bending abilities (Figure 2).
Figure 4. A tubulation-deficient syt mutant regulates the fusion of small, but not large, liposomes.

(A) Syt-stimulated fusion of v-SNARE vesicles, containing donor and acceptor FRET pairs, with unlabeled t-SNARE vesicles leads to an increase in FRET-donor fluorescence. Fusion assays were carried out with either GUVs or SUVs. (B) Representative fluorescence microscopy (left) and electron microscopy (right) images of vesicles used in this study. Scale bars: 8 μm (left) and 200 nm (right). (C) Absolute NBD de-quenching signals for C2A-C2B/C2A-C2BCLM-stimulated fusion of GUVs (left) and SUVs (right). Addition of detergent produced a maximum fluorescence signal for normalization. (D) Comparison of the % syt stimulated fusion for WT C2A-C2B and the tubulation deficient mutant, C2A-C2BCLM, for GUV (left) and SUV (right) fusion assays. Relative to WT, the extent of C2A-C2BCLM stimulated fusion shows a clear loss-of-function in the GUV system (left) as compared to the SUV system. 1 mM Ca2+ was added at t = 2 min to activate syt and trigger fusion. (E) Extent of stimulated fusion for GUV (left) and SUV (right) systems plotted for four syt constructs. (F) Left: GUV-GUV fusion was blocked by pretreatment with botulinum neurotoxin B or by the use of a truncated form of SNAP-25, which mimicked the cleavage product of botulinum neurotoxin E, or by addition of the cytoplasmic domain of synaptobrevin (cd-syb) or the cytoplasmic domain of the t-SNARE heterodimers (cd-dimer). Right An SDS-PAGE gel showing that botulinum neurotoxin B efficiently cleaved GUV embedded syb.
To measure the effect of membrane curvature on syt-stimulated SNARE-mediated fusion, syt proteins were added to assays containing either SNARE-bearing SUVs or GUVs. For GUV-GUV fusion, C2A-C2BCLM could only stimulate fusion to 19% of the levels of WT (Figure 4D, left). By contrast, in the SUV-SUV system, the extent of C2A-C2BCLM stimulated fusion reached 70% of the WT level (Figure 4D, right) (Bhalla et al., 2005). Thus, mutations that disrupt the ability of the C2B domain to sense Ca2+ significantly decrease syt's ability to stimulate fusion of vesicles with a low degree of membrane curvature. These findings explain why Ca2+-ligand mutations in C2B completely disrupt the function of syt in neurons (Mackler et al., 2002; Nishiki and Augustine, 2004) but have little effect on syt-regulated fusion in in vitro fusion assays utilizing highly curved SUVs (Bhalla et al., 2005; Stein et al., 2007). By mimicking the low-curvature of the plasma membrane through the use of GUVs, a greater loss-of-function can be detected for C2A-C2BCLM, more closely matching the loss-of-function synaptic transmission phenotype. Notably, the functional deficiency exhibited by C2A-C2BCLM was insensitive to changes in membrane fluidity and rigidity (Figure S11).
Isolated C2B possesses similar membrane-bending activity but lower t-SNARE binding activity as compared to C2A-C2B (Figure 2D, N and Figure 3A, B). As expected, isolated C2B exhibited a reduced ability to stimulate SUV-SUV fusion than C2A-C2B, in accord with a previous report (Gaffaney et al., 2008). This loss-of-function is most likely due to compromised t-SNARE binding activity, since the ability of C2B to promote membrane fusion is insensitive to membrane curvature: C2B is equally efficient in GUV-GUV and SUV-SUV fusion assays (55 ± 13% versus 65 ± 9%, p > 0.05).
C2A3W-C2B3W is endowed with enhanced membrane bending and t-SNARE binding activity (Figure 2J, N and Figure 3A-D). As expected, this mutant was even more effective than WT C2A-C2B in both SUV-SUV and GUV-GUV fusion assays (132 ± 10% and 151 ± 11%, respectively). Notably, the enhancement in activity of the Trp mutant was more prominent in the GUV-GUV fusion assay, presumably due to stronger membrane-bending activity.
We next sought to determine which membrane -t- or v-SNARE - must be bent in order for syt to regulate membrane fusion. This was accomplished using asymmetric systems composed of either t-GUV + v-SUV or t-SUV + v-GUV vesicles (Figure S12). While decreases in curvature of either the donor or acceptor membrane resulted in lower extents of fusion, a more significant loss in C2A-C2BClm stimulated fusion relative to WT C2A-C2B occurred using the t-GUV/v-SUV system. These data suggest that the curvature of the t-SNARE membrane has a greater effect on syt function and supports the idea that syt regulates fusion by doing work on the plasma membrane (Bai et al., 2004; Chicka et al., 2008; Stein et al., 2007).
Finally, GUV-GUV fusion was efficiently blocked by agents that cleave SNAREs or prevent trans-SNARE pairing, demonstrating that these fusion reactions were mediated by functional SNARE complexes (Figure 4F).
The function of a membrane-bending-defective syt mutant can be rescued in trans by the N-BAR domain from endophilin
The data presented above demonstrate that Ca2+•syt-mediated membrane bending comprises an essential step during Ca2+-triggered fusion. Thus, syt might act, at least in part, by buckling the plasma membrane towards the vesicle membrane. Based on our tubulation data (Figure S7), we predict that syt-driven plasma membrane dimples have a curvature that is similar to synaptic vesicles (∼ 50 nm diameter). Such dimples have curvatures that are similar to buds that form during endocytosis. Therefore, we next determined if an endocytic protein could compensate for the membrane-bending defects of the syt mutant, C2A-C2BCLM.
The N-BAR domain of endophilin, a protein involved in clathrin-mediated endocytosis, was utilized for these experiments. N-BAR domains are highly conserved protein motifs that occur in a number of endocytic proteins. These domains assemble into crescent-shaped dimers that avidly bend membranes (Casal et al., 2006). Here, we show that the N-BAR of endophilin interacts with PS in a Ca2+-independent manner (Figure 5A) and does not exhibit significant interactions with SNAREs (Figure 5B, C). N-BAR of endophilin was able to deform PS-containing vesicles into tubules with diameters of 30-90 nm (Figure 5D); addition of N-BAR to GUVs resulted in tubulation of the membranes within a few minutes (Figure S13).
Figure 5. N-BAR domain induced membrane-bending rescues C2A-C2BCLM-regulated membrane fusion.

(A) Representative cosedimentation gels showing that N-BAR binds PS harboring liposomes in a Ca2+-independent manner. With increasing levels of PS, N-BAR shifted from the supernatant (s) to the pellet (p) fraction. At the same %PS, N-BAR binds more avidly to V105nm than to V252nm. (B) N-BAR does not exhibit significant t- or v-SNARE binding activity, while C2A-C2BClm and C2A-C2B bind t-SNAREs in a Ca2+-dependent manner, as assessed using coflotation assays. Protein free, t-SNARE- and v-SNARE-bearing vesicles are indicated as pf, Tr and Vr, respectively. (C) Quantification of coflotation assays. (D) Electron micrographs showing that 1 μM N-BAR tubulated liposomes composed of 100% Folch lipids or 15% PS/30% PE/55% PC, after 15 min incubation; the diameters of the tubules ranged from 30 nm to 90 nm. (E) In vitro membrane fusion assay setup. v-SNARE and t-SNARE GUVs were mixed with 10 μM C2A-C2BClm and 1 μM N-BAR. (F) Addition of N-BAR greatly enhanced both the rate and extent of Ca2+-triggered C2A-C2BCLM-stimulated GUV-GUV fusion, but had modest effects on WT C2A-C2B-stimulated fusion; in the absence of N-BAR, C2A-C2BCLM-stimulated GUV-GUV fusion was negligible (green trace). 1 mM Ca2+ was added at t = 20 min. (G) Sequential addition of 10 μM C2A-C2BCLM (t = 0), 1 mM CaCl2 (t = 15 min), and 1 μM N-BAR (t = 25 min) to GUV t-SNARE and v-SNARE vesicles, as indicated by arrows. (H) Sequential addition experiment as carried out in C, with N-BAR added at t = 0 and C2A-C2BCLM added at t = 25 min, as indicated by arrows. (I) N-BAR-rescued fusion was inhibited by the same agents used in Figure 4F.
As discussed in detail above, C2A-C2BCLM alone stimulates very low levels of SNARE-mediated GUV-GUV fusion (Figure 5F, green). Notably, addition of N-BAR to the GUV-GUV + C2A-C2BClm system resulted in a marked increase in membrane fusion relative to C2A-C2BCLM alone (Figure 5F, red). This result clearly demonstrates that the membrane-curvature generating ability of N-BAR can rescue - in trans - the membrane-bending defects of C2A-C2BCLM, resulting in the reconstitution of regulated membrane fusion. The extent of N-BAR + C2A-C2BClm regulated fusion was only slightly less than that for WT C2A-C2B alone (Figure 5F, cyan), suggesting N-BAR efficiently rescued the function of C2A-C2BCLM. We also found that N-BAR (Figure 5F, blue), as well as hypertonic sucrose, which was used to buckle GUV membranes through osmotic shrinkage (Figure S14), yielded only modest increases in fusion regulated by WT C2A-C2B as compared to C2A-C2BCLM. Together, these results confirm the idea that N-BAR rescues C2A-C2BCLM by restoring membrane-bending activity to fusion reactions.
In the absence of C2A-C2BCLM, N-BAR affected neither the extent nor the rate of SNARE-mediated GUV fusion (Figure 5F, orange), further demonstrating that membrane bending alone is not sufficient for regulating fusion. The fact that N-BAR stimulates fusion only in the presence of C2A-C2BCLM provides strong evidence that the t-SNARE binding activity of syt plays a critical role in the regulation of fusion. N-BAR did not enhance the ability of C2A-C2BCLM to stimulate SUV-SUV fusion (data not shown), further supporting the idea that SUVs are not an appropriate system to assess the significance of protein-mediated membrane bending. Notably, N-BAR-rescued GUV-GUV fusion was blocked by the same agents used in Figure 4E, demonstrating that ‘rescued fusion’ also proceeds through trans-SNARE pairing (Figure 5I).
The experiments above were carried out with C2A-C2BCLM and N-BAR added concurrently to GUVs 20 min before addition of Ca2+. We next added the C2A-C2BCLM and N-BAR proteins sequentially and in different orders to monitor N-BAR mediated membrane bending and the action of C2A-C2BCLM on SNAREs in a step-wise fashion. In either case, sequential addition of C2A-C2BCLM followed by N-BAR (Figure 5G) or of N-BAR followed by C2A-C2BCLM (Figure 5H) resulted in fast membrane fusion only after the addition of the second component. These experiments further confirm that the presence of both C2A-C2BCLM and N-BAR are required for the rapid and robust Ca2+-triggered fusion of GUVs.
These experiments demonstrate that syt operates by both bending membranes and by engaging SNARE proteins, and that these two functions can be dissociated from one another.
Finally, although both N-BAR and syt engage PS-harboring membranes, N-BAR does not significantly affect the interaction of C2A-C2B or C2A-C2BCLM with liposomes (Figure S15). Thus, the marked rescue of regulated fusion by N-BAR is likely to be a direct consequence of N-BAR-mediated membrane bending.
Targeting of the cytoplasmic domain (C2A-C2B) of syt to pre-synaptic boutons rescues rapid synaptic transmission in syt I KO neurons
While it is well established that C2A-C2B is able to accelerate reconstituted SNARE-catalyzed fusion in a Ca2+-dependent manner (Chicka et al., 2008; Martens et al., 2007; Stein et al., 2007; Tucker et al., 2004), studies with reconstituted, membrane-embedded syt have not resulted in a clear picture. Namely, membrane-embedded syt was reported to facilitate Ca2+-independent fusion in one study (Mahal et al., 2002), and to inhibit fusion in response to Ca2+ in another study (Stein et al., 2007). These reports raise questions regarding the analysis of syt in reduced, reconstituted fusion assays such as those used in the current study. In our laboratory, full-length syt aggregates and causes vesicles that harbor the protein to become unstable (J.D.G. and E.R.C. unpublished observations), so we have relied largely on C2A-C2B. It has been reported that splice variants of some syt isoforms lack a transmembrane domain (TMD) (Craxton, 2004), suggesting that a membrane anchor is not essential for some aspects of function, but this might not be the case for syt I, which is the topic of study here. So, we carried out a series of experiments to determine whether C2A-C2B can regulate membrane fusion in syt I KO neurons.
First, to determine whether the TMD of syt is essential for function, we fused C2A-C2B to another vesicle protein, synaptophysin (Figure 6A, upper). This fusion protein (physin-C2A-C2B), when expressed in syt I KO cortical neurons via lentiviral infection, was targeted to secretory vesicles (Figure S16). We monitored the release of inhibitory neurotransmitter from infected neurons via post-synaptic recordings. Syt I KO neurons produced only slow asynchronous GABA release in response to action potentials; rapid and robust release was restored by expressing WT syt in these neurons. Strikingly, rapid evoked release of GABA was also observed for physin-C2A-C2B expressing neurons. The IPSCs exhibited similar kinetics and peak amplitudes as compared to neurons rescued by full length WT protein (Figure 6B-D). Furthermore, both WT syt and physin-C2A-C2B expressing neurons released more GABA than syt I KO neurons (Figure 6E). These results suggest that the TMD is not essential for syt to accelerate synaptic vesicle exocytosis.
Figure 6. Synaptic vesicle and plasma membrane localized C2A-C2B both rescue rapid exocytosis in syt knock-out neurons.

(A) Diagram showing the C2A-C2B fusion proteins that were expressed in syt I KO neurons. C2A-C2B was targeted to synaptic vesicles by fusing it to the C-terminus of full-length synaptophysin, or was targeted to the presynaptic plasma membrane by fusing it to the first twenty residues of GAP43. (B) Typical traces of evoked IPSCs recorded from syt I KO neurons (KO), and lentivirus infected KO neurons expressing WT syt (syt rescue), GAP43-C2A-C2B, physin-C2A-C2B, GAP43-C2A-C2BCLM or physin-C2A-C2BCLM. (C) Average normalized cumulative IPSC charge transfer over 1.5 s for KO neurons expressing the indicated constructs (D & E) Summaries of the peak amplitude (D) and total charge transfer over 1.5 s (E) of evoked IPSCs.
We next determined whether the localization of syt to vesicles is required for its function. C2A-C2B was fused to the N-terminal membrane targeting motif of GAP43 (Figure 6A, lower), a protein that is anchored to the inner leaflet of the plasma membrane via two palmitoylated cysteines (Liu et al., 1991). This fusion construct (GAP43-C2A-C2B) was indeed targeted to the plasma membrane (Figure 6A and Figure S16), and rescued rapid synaptic transmission in syt I KO neurons (Figure 1B). The peak IPSC amplitude was similar to cortical neurons expressing WT syt or physin-C2A-C2B. The total charge transfer was slightly lower than neurons rescued by WT syt, yet significantly larger than the syt I KO control.
We next determined whether the ability of membrane targeted C2A-C2B to rescue rapid synaptic transmission depended on its Ca2+-activated membrane bending activity. The tubulation deficient mutant, C2A-C2BCLM, was fused to synaptophysin or GAP43 and expressed in syt I KO cortical neurons. The fusion proteins - physin-C2A-C2BCLM and GAP43-C2A-C2BCLM - were correctly targeted to presynaptic boutons (Figure S17), but failed to rescue rapid synaptic transmission (Figure 6B, inset). These data further confirm the idea that membrane bending comprises a critical step during syt-regulated membrane fusion.
From these data, we conclude that neither the TMD nor vesicle localization is required for syt function during exocytosis; C2A-C2B is capable of promoting rapid release of neurotransmitters when attached to either the vesicle or plasma membrane.
Discussion
Dual activities of syt: sensing and inducing membrane curvature
Curvature-sensing activity can be considered as a ‘passive’ property of syt, which is likely to be a direct result of its membrane penetration activity. In contrast, the curvature-inducing activity can be envisioned as an ‘active’ property of syt, by which syt can create favorable curvature for binding to membranes. Because syt both senses and induces positive curvature, its cytoplasmic domain is likely to associate with membranes in a cooperative manner, i.e., the binding of syt bends the underlying membrane and favors the subsequent binding of additional copies of syt. This mechanism would enhance the ability of syt to engage relatively flat membranes under equilibrium conditions, thus ‘blurring’ the observed curvature sensitivity to some extent in steady-state experiments.
While all of the syt constructs tested in this study sensed membrane curvature (Figure S3-S4), some lacked the ability to induce membrane curvature (Figure 2). These data suggest that the abilities of syt to sense and induce curvature are not intrinsically coupled properties. Notably, in contrast to isolated C2A, isolated C2B was able to mediate bending of membranes, contradicting a previous report (Martens et al., 2007), but this earlier study used a truncated C2B domain (residues 273 - 408) that lacked a α helix in the C-terminal portion of the domain (residues 409 - 418) (Figure 1A). Combined with our finding that the tandem C2-domain construct, C2A-C2A, is unable to efficiently tubulate liposomes, we conclude that tethering of C2 domains is neither sufficient nor necessary for tubulation activity.
It is somewhat surprising that isolated C2B avidly tubulates membranes, while the homologous C2A domain does not; thus, membrane insertion alone is not sufficient to drive membrane-bending. Membrane deformation has been suggested to result from the coordinated effort of two or more interactions from one protein (Gallop et al., 2006), or a collective effort from multiple copies of a protein (Ayton et al., 2007). We speculate that the unique structural features of C2B, e.g., two α helices at the ‘bottom’ of the domain, in conjugation with its membrane penetration loops at the ‘top’, somehow endow C2B with membrane-bending activity. As noted above, a truncated form of C2B that lacked the C-terminal α helix was unable to tubulate membranes, consistent with the notion that this helix plays a critical role in the membrane-bending process. It has been shown that C2A-C2B forms ring-like and filamentous structures in a Ca2+ and PS-dependent manner and C2B appears to be crucial for this activity (Wu et al., 2003). Thus, it is tempting to consider the possibility that the C terminal α helices mediate dimerization or oligomerization of C2B to facilitate membrane bending.
Functional analysis of the membrane-bending activity of syt
The use of GUVs in the reconstituted fusion system provides the first direct evidence for the idea that in addition to critical interactions with t-SNAREs, syt regulates fusion by bending membranes. Because the standard in vitro fusion assays utilize SUVs whose membranes are already highly curved, defects in the ability of C2A-C2BCLM to stimulate fusion via loss of membrane-bending activity could not be discerned. GUVs better mimic the lack of curvature of the plasma membrane in cells, and reveal a striking loss-of-function for this mutant (Figure 4) that agrees with the inability of C2B Ca2+-ligand mutants to support synaptic transmission (Mackler et al., 2002; Nishiki and Augustine, 2004). Hence, improvements in reconstituted fusion assays continue to result in the convergence of this system with findings obtained using cell based assays (Chicka et al., 2008). A model for the function of syt during regulated membrane fusion is shown in Figure 7.
Figure 7. Model depicting the role of syt during Ca2+-triggered exocytosis of synaptic vesicles.

(A) Before Ca2+ influx, partially assembled SNARE complexes form between the vesicle and target membrane. The two membranes are held close to each other but cannot fuse due, in part, to the clamping activity of syt, which serves to arrest trans-SNARE pairs (Chicka et al., 2008). Ca2+ independent interactions with t-SNARE and plasma membrane lipid PIP2 (Bai et al., 2004; Chicka et al., 2008) steer C2B toward the plasma membrane. (B) Upon Ca2+ influx, the Ca2+-binding loops of the C2B domain rapidly insert into the target membrane (Chicka et al., 2008). Insertion might be accompanied by a rotation (indicated by the curved arrow) and/or oligomerization of this domain. (C) The concerted actions of multiple C2B domains serve to locally bend the plasma membrane toward the vesicle membrane (only two C2B domains are shown in the model for simplicity). This invagination brings the two membranes into closer proximity and significantly lowers the energy barrier for bilayer merger (Martens et al., 2007; Monck and Fernandez, 1994). (D) Ca2+-syt simultaneously triggers structural transitions t-SNAREs, perhaps driving complete zippering of SNARE complexes, to initiate fusion.
How does the membrane-bending activity of syt accelerate fusion? First, bending of the plasma membrane serves to bring the two curved bilayers into closer proximity, making it possible for lipid molecules to transfer from one membrane to the other (Marrink and Mark, 2003). Second, a plasma membrane dimple would greatly reduce the contact area of the apposing membranes, thereby minimizing the repulsive hydration force (Chernomordik and Kozlov, 2003). Third, curvature may increase tension in the plasma membrane to partially expose the nonpolar interior of the bilayer, allowing hydrophobic attractions between the interiors of the two bilayers (Chanturiya et al., 2002).
Given the extremely rapid kinetics of synaptic vesicle exocytosis, syt-mediated membrane deformations must occur on even faster time-scales. However, due to technical limitations, ms or sub-ms remodeling of membranes cannot be captured by standard electron microscopy methods. Current tubulation assays indicate that lipid tubules formed within 30 min at 25 °C (Figure S5). The relatively slow kinetics of tubulation do not imply that membrane-bending is a slow process, since tubulation represents extensive membrane remodeling and is the collective outcome of potentially rapid local membrane-bending events that accumulate over time. Indeed, the rate at which syt penetrates membranes approaches the collision-limit for this reaction (Bai et al., 2004), thus local membrane deformations are likely to occur on rapid time scales.
An endocytic protein motif rescues the activity of a tubulation-deficient syt mutant during fusion
Syt must engage both membranes and t-SNAREs to operate as a Ca2+ sensor for exocytosis (Chapman, 2008). By using the N-BAR domain of endophilin, we dissociated these two activities at a functional level. Separately, C2A-C2BCLM or the N-BAR domain of endophilin had little effect on the fusion of relatively flat membranes (GUVs). Strikingly, when presented simultaneously in the GUV fusion assay, these proteins drove rapid and robust membrane fusion. These data clearly demonstrate that the ability of syt to carry out work on membranes and SNARE proteins (Bhalla et al., 2006) can be separated from one-another. The N-BAR rescue experiment suggests that it is possible that different copies of syt could have distinct roles in exocytosis, e.g., some might only bind/bend membranes (mimicked by N-BAR), while other copies might only engage t-SNAREs (mimicked by C2A-C2BCLM).
It is rather remarkable that a motif from an endocytic protein can drive membrane fusion (in the presence of a mutant form of syt that can interact with and activate t-SNAREs), underscoring the conclusion that membrane fusion and fission proceed through similar transient membrane structures.
The transmembrane domain is not required for syt to accelerate membrane fusion in neurons
There is good agreement among numerous groups that the cytoplasmic domain of syt renders SNARE-catalyzed fusion dependent upon Ca2+. However, it has previously remained unclear as to whether the TMD of syt plays a significant role during synaptic vesicle exocytosis. In the current study, we found that targeting of C2A-C2B to either synaptic vesicles or the presynaptic plasma membrane is sufficient to restore rapid synaptic transmission in syt I KO neurons. In particular, the finding that GAP43-C2A-C2B rescues the syt I KO phenotype argues against a model in which syt works by physically pulling the vesicle and plasma membranes together. Clearly, C2A-C2B must be targeted to presynaptic boutons in order to function, and such targeting normally requires the amino terminal portion of the molecule (including the TMD), but this is not required in reconstituted fusions assays where the concentration of C2A-C2B can be optimized.
Concluding remarks
In closing, we reiterate that since the interaction of syt with membranes and SNAREs appear to be intrinsically coupled binding reactions (Bhalla et al., 2006), mutations that specifically affect syt's membrane-bending activity might not be forthcoming, and, as we show here, all current membrane-bending mutants also exhibit altered SNARE binding activity. Here, we devised a different strategy to approach this problem; by utilizing membranes with different degrees of curvature we were able to directly determine whether or not the membrane-bending activity of syt plays a role in regulated fusion. This approach underscores the utility of reconstituted systems, where starting parameters - including membrane curvature - can be readily controlled, making it possible to elucidate the nano-mechanics of membrane fusion reactions. The data reported here demonstrate that the interaction of syt with lipid bilayers serves to drive localized invagination of the target membrane to facilitate fusion (Monck and Fernandez, 1994), and suggest that the fusion pore might be composed of both lipids as well as SNARE TMDs (Han et al., 2004). Because similar membrane bending steps occur during early steps in vesicle retrieval (Kozlov and Chernomordik, 2002), exo- and endocytosis appear to share common intermediate membrane structures.
Experimental Procedures
Liposomes
Protein-free liposomes were prepared by extrusion through polycarbonate filters with pore sizes of 50, 100, 200 and 400 nm (Avanti Polar Lipids). The hydrodynamic diameters of the extruded liposomes were 105 ± 37, 138 ± 32, 189 ± 74 and 252 ± 103 nm, respectively, as determined using a PCS Submicron Particle Size Analyzer (Beckman Coulter). These liposome suspensions - when made from identical amounts of lipid - possess similar binding capacities for C2A-C2B and exhibited ∼100% sedimentation efficiency under the conditions used in our assays as shown in Figure S1 and Table S4. Lipid composition was 25% PS/75% PC for cosedimentation and ITC experiments, and 5% dansyl-PE/25% PS/70% PC for stopped-flow experiments. Rapid injections of liposomes during ITC and stopped-flow experiments do not significantly alter the size of the liposomes, as shown in Table S5.
For tubulation assays, brain lipid extract (Folch fraction I) was dried and then resuspended at 1 mg/ml in HEPES buffered saline (50 mM HEPES-NaOH, 100 mM NaCl, pH 7.4). Folch liposomes were produced by sonication for 10 min. Liposomes used in Figure 5D, S7 and S8 -composed of 15% PS/30% PE/55% PC - were prepared by extrusion through a polycarbonate filter with a pore size of 400 nm.
For in vitro fusion assays, SNARE-bearing SUVs were prepared as described previously (Tucker et al., 2004) using 15% PS/30% PE/55% PC for t-SNARE vesicles and 15% PS/27% PE/ 55% PC/1.5% NBD-PE/1.5% rhodamine-PE for v-SNARE vesicles. SNARE-bearing GUVs were formed via the electroformation method using SNARE-bearing SUVs by centrifuging the SUVs for 90 min at 85,000 rpm. The pelleted vesicles were then resuspended in a low salt HEPES buffer (5 mM HEPES-NaOH, 5 mM NaCl, pH 7.5), dispensed onto an indium tin oxide coated glass slide and dried under vacuum. The dried film was re-hydrated with 200 mM sucrose while a 1V-10Hz sinusoidal electric field was applied for 3 hr. T- and v-SNARE bearing GUVs were analyzed under a microscope and found to have diameters of 4.7 ± 1.6 μm. The SNARE bearing SUVs were analyzed using electron microscopy and their diameters were 65 ± 19 nm (mean ± SD, n = 31). The drying process during GUV preparation appears to have little, if any, effect on the activity/conformation of SNARE proteins (Figure S9). During preparation of GUVs from SUVs, equal percentages of SNAREs and lipids are lost (40% - 60%); hence, the SNARE/lipid ratios of GUVs are similar to SUVs (Figure S10).
Electron microscopy
0.3 mg/ml Folch liposomes or artificial liposomes (15% PS/30% PE/ 55% PC) were incubated with the indicated concentrations of N-BAR or syt proteins, in the presence of 1 mM CaCl2 or 0.2 mM EGTA at RT for 15 min to 10 hr, as indicated. Samples were loaded on a pioloform coated (Ted Pella Inc) 300-mesh grid (glow discharged), and stained with nano-W (methylamine tungstate, Nanoprobes Inc). Negatively stained samples were viewed on a Philips CM120 scanning transmission electron microscope.
Fusion assays
Fusion assays were carried out as described previously (Tucker et al., 2004). All reactions contained a v-SNARE to t-SNARE vesicle ratio of 1:2 and 0.1mM EGTA. Fusion was triggered by addition of 1 mM CaCl2. For all experiments, t-SNARE vesicles were first mixed with HEPES buffer (50 mM HEPES-NaOH, 100 mM NaCl, pH 7.4), EGTA and syt/N-BAR and allowed to warm to 37°C before addition of v-SNARE vesicles. The total reaction volume was 100 μl; data were normalized to the initial time point and the maximum fluorescence in n-dodecyl β-D-maltoside.
Statistical methods
Statistical significance was evaluated by two-tailed unpaired Student's t test: *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary Material
Acknowledgments
We thank J. Gaffaney and Z. Wang for providing materials and technical suggestions for experiments in Figure 3, R. Massey and M. Stowell for assistance and suggestions regarding tubulation assays, and D. McCaslin for ITC training. We also thank M. Jackson, J. Audhya, J. Weisshaar and members of Chapman lab for helpful discussions and critical comments. This study was supported by grants from the US Institutes of Health (MH 61876) and the American Heart Association (0440168N) to E.R.C.. E.H. was supported by an American Heart Association predoctoral fellowship. E.R.C. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arac D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Sudhof TC, Rizo J. Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- Ayton GS, Blood PD, Voth GA. Membrane remodeling from N-BAR domain interactions: insights from multi-scale simulation. Biophys J. 2007;92:3595–3602. doi: 10.1529/biophysj.106.101709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nature structural & molecular biology. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- Bai J, Wang P, Chapman ER. C2A activates a cryptic Ca(2+)-triggered membrane penetration activity within the C2B domain of synaptotagmin I. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:1665–1670. doi: 10.1073/pnas.032541099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla A, Chicka MC, Tucker WC, Chapman ER. Ca(2+)-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nature structural & molecular biology. 2006;13:323–330. doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- Bhalla A, Tucker WC, Chapman ER. Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Molecular biology of the cell. 2005;16:4755–4764. doi: 10.1091/mbc.E05-04-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casal E, Federici L, Zhang W, Fernandez-Recio J, Priego EM, Miguel RN, DuHadaway JB, Prendergast GC, Luisi BF, Laue ED. The crystal structure of the BAR domain from human Bin1/amphiphysin II and its implications for molecular recognition. Biochemistry. 2006;45:12917–12928. doi: 10.1021/bi060717k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanturiya A, Scaria P, Kuksenok O, Woodle MC. Probing the mechanism of fusion in a two-dimensional computer simulation. Biophys J. 2002;82:3072–3080. doi: 10.1016/S0006-3495(02)75648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER. How Does Synaptotagmin Trigger Neurotransmitter Release? Annual Review of Biochemistry. 2008 doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- Chapman ER, Davis AF. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. The Journal of biological chemistry. 1998;273:13995–14001. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- Chernomordik L, Kozlov M. PROTEIN-LIPID INTERPLAY IN FUSION AND FISSION OF BIOLOGICAL MEMBRANES *. Annual Review of Biochemistry. 2003;72:175–207. doi: 10.1146/annurev.biochem.72.121801.161504. [DOI] [PubMed] [Google Scholar]
- Chicka MC, Hui E, Liu H, Chapman ER. Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca(2+) Nat Struct Mol Biol. 2008;15:827–835. doi: 10.1038/nsmb.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craxton M. Synaptotagmin gene content of the sequenced genomes. BMC Genomics. 2004;5:43. doi: 10.1186/1471-2164-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AF, Bai J, Fasshauer D, Wolowick MJ, Lewis JL, Chapman ER. Kinetics of synaptotagmin responses to Ca2+ and assembly with the core SNARE complex onto membranes. Neuron. 1999;24:363–376. doi: 10.1016/s0896-6273(00)80850-8. [DOI] [PubMed] [Google Scholar]
- Farsad K, De Camilli P. Mechanisms of membrane deformation. Curr Opin Cell Biol. 2003;15:372–381. doi: 10.1016/s0955-0674(03)00073-5. [DOI] [PubMed] [Google Scholar]
- Farsad K, Ringstad N, Takei K, Floyd SR, Rose K, De Camilli P. Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J Cell Biol. 2001;155:193–200. doi: 10.1083/jcb.200107075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez I, Arac D, Ubach J, Gerber SH, Shin O, Gao Y, Anderson RG, Sudhof TC, Rizo J. Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron. 2001;32:1057–1069. doi: 10.1016/s0896-6273(01)00548-7. [DOI] [PubMed] [Google Scholar]
- Gaffaney JD, Dunning FM, Wang Z, Hui E, Chapman ER. Synaptotagmin C2B domain regulates Ca2+-triggered fusion in vitro: Critical residues revealed by scanning alanine mutagenesis. J Biol Chem. 2008 doi: 10.1074/jbc.M803355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallop JL, Jao CC, Kent HM, Butler PJ, Evans PR, Langen R, McMahon HT. Mechanism of endophilin N-BAR domain-mediated membrane curvature. Embo J. 2006;25:2898–2910. doi: 10.1038/sj.emboj.7601174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Wang CT, Bai J, Chapman ER, Jackson MB. Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science (New York, NY) 2004;304:289–292. doi: 10.1126/science.1095801. [DOI] [PubMed] [Google Scholar]
- Heimburg T, Biltonen RL. Thermotropic behavior of dimyristoylphosphatidylglycerol and its interaction with cytochrome c. Biochemistry. 1994;33:9477–9488. doi: 10.1021/bi00198a013. [DOI] [PubMed] [Google Scholar]
- Kozlov MM, Chernomordik LV. The protein coat in membrane fusion: lessons from fission. Traffic. 2002;3:256–267. doi: 10.1034/j.1600-0854.2002.030403.x. [DOI] [PubMed] [Google Scholar]
- Liu YC, Chapman ER, Storm DR. Targeting of neuromodulin (GAP-43) fusion proteins to growth cones in cultured rat embryonic neurons. Neuron. 1991;6:411–420. doi: 10.1016/0896-6273(91)90249-y. [DOI] [PubMed] [Google Scholar]
- Lynch KL, Gerona RR, Kielar DM, Martens S, McMahon HT, Martin TF. Synaptotagmin-1 Utilizes Membrane Bending and SNARE Binding to Drive Fusion Pore Expansion. Mol Biol Cell. 2008 doi: 10.1091/mbc.E08-03-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE. The C(2)B Ca(2+)-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature. 2002;418:340–344. doi: 10.1038/nature00846. [DOI] [PubMed] [Google Scholar]
- Mahal LK, Sequeira SM, Gureasko JM, Sollner TH. Calcium-independent stimulation of membrane fusion and SNAREpin formation by synaptotagmin I. J Cell Biol. 2002;158:273–282. doi: 10.1083/jcb.200203135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink SJ, Mark AE. Molecular dynamics simulation of the formation, structure, and dynamics of small phospholipid vesicles. J Am Chem Soc. 2003;125:15233–15242. doi: 10.1021/ja0352092. [DOI] [PubMed] [Google Scholar]
- Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- Monck JR, Fernandez JM. The exocytotic fusion pore and neurotransmitter release. Neuron. 1994;12:707–716. doi: 10.1016/0896-6273(94)90325-5. [DOI] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ. Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J Neurosci. 2004;24:8542–8550. doi: 10.1523/JNEUROSCI.2545-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X, Fernandez I, Sudhof TC, Rizo J. Solution structures of the Ca2+-free and Ca2+-bound C2A domain of synaptotagmin I: does Ca2+ induce a conformational change? Biochemistry. 1998;37:16106–16115. doi: 10.1021/bi981789h. [DOI] [PubMed] [Google Scholar]
- Steenbergen R, Nanowski TS, Nelson R, Young SG, Vance JE. Phospholipid homeostasis in phosphatidylserine synthase-2-deficient mice. Biochimica et biophysica acta. 2006;1761:313–323. doi: 10.1016/j.bbalip.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Stein A, Radhakrishnan A, Riedel D, Fasshauer D, Jahn R. Synaptotagmin activates membrane fusion through a Ca2+-dependent trans interaction with phospholipids. Nature structural & molecular biology. 2007;14:904–911. doi: 10.1038/nsmb1305. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Davletov BA, Berghuis AM, Sudhof TC, Sprang SR. Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell. 1995;80:929–938. doi: 10.1016/0092-8674(95)90296-1. [DOI] [PubMed] [Google Scholar]
- Tucker WC, Weber T, Chapman ER. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science (New York, NY) 2004;304:435–438. doi: 10.1126/science.1097196. [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Wu Y, He Y, Bai J, Ji SR, Tucker WC, Chapman ER, Sui SF. Visualization of synaptotagmin I oligomers assembled onto lipid monolayers. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2082–2087. doi: 10.1073/pnas.0435872100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.