

Abstract
It has been suggested that the neonatal Fc receptor (FcRn) is a primary determinant of the distribution of IgG to the brain. In the present report, 125I-labeled 7E3, a monoclonal IgG1 antibody, was injected intravenously to groups of FcRn-deficient mice and C57BL/6J control mice. Sub-groups of three mice were sacrificed at several time points. Blood and brain tissue were harvested and radioactivity was assessed. Antibody concentrations in brain were corrected for residual blood using 51Cr-labeled red blood cells. Data were analyzed via WinNonlin, and areas under plasma and tissue concentration vs. time curves (AUCs) were assessed via the Bailer method. The apparent plasma elimination half-life and clearance of 7E3 were 13.61 ± 0.61 days and 6.5 ± 0.10 ml/day/kg in control mice and 0.70 ± 0.05 days and 63.5 ± 2.7 ml/day/kg in the knockout mice. Plasma and brain AUCs (0–10 days) were found to be 3,338.7 ± 50.4 and 7.46 ± 0.5 nM day in control animals and 781.2 ± 16.6 and 1.65 ± 0.1 nM day in FcRn-deficient animals. There was no significant difference between brain-to-plasma AUC ratios in control and FcRn-deficient mice (0.0022 ± 0.00015 vs. 0.0021 ± 0.00011, p = 0.3347). Two-way analysis of variance showed no significant differences, at any time point, between brain-to-plasma concentration ratios determined from control and knockout animals. The results suggest that FcRn does not contribute significantly to the “blood–brain barrier” for IgG in mice, and the data suggest that FcRn is not responsible for the low exposure of IgG in the brain relative to plasma.
Key words: antibody, blood brain barrier, FcRn, IgG, pharmacokinetics
INTRODUCTION
In adult animals and humans, the neonatal Fc receptor (FcRn) is expressed within endothelial cells (1), epithelial cells (2,3), hepatocytes (4), intestinal macrophages, peripheral blood monocytes, dendritic cells (5), and at the maternal–fetal barrier (6–8). FcRn expression has been demonstrated in vascular endothelial cells in the brain (9), and Pardridge and coworkers have hypothesized that FcRn may play a role in the efflux of IgG from brain tissue, contributing to the “blood–brain barrier” (BBB) for IgG antibodies (9,10). Recent work published by Boado et al. described the use of a fusion protein that crosses the BBB, binds to amyloid beta (Aβ) fibrils, and is then transported out of the brain (11). Their results are consistent with Fc receptor-mediated transport, and the authors concluded that FcRn is responsible for the efflux of the fusion protein. However, no data were provided to demonstrate convincingly that efflux was mediated by FcRn. In a second study, Deane et al. also suggested that FcRn effluxes Aβ–IgG complexes from the brain (12). However, some of their data are inconsistent with the hypothesis that FcRn is the only, or primary, Fc receptor mediating brain efflux of IgG. For example, using FcRn knockout mice, the authors found that the administration of excess anti-Aβ IgG (4G8) resulted in a complete inhibition of 125I-4G8 efflux. Additionally, the authors also reported that in the FcRn knockout mice, serum 125I-4G8 levels were significantly reduced (>90%) upon central administration of excess unlabeled 4G8. These data suggest that additional Fc receptors mediate some, or all, of the observed efflux of IgG from the brain, thus raising questions regarding the role played by FcRn. In the present study, we applied a simple straightforward strategy to test the hypothesis that FcRn is a primary determinant of IgG exposure in the brain.
MATERIALS AND METHODS
Materials and Animals
β-2-Microglobulin knockout mice, lacking functional expression of FcRn, and C57BL/6J control mice, 19–22 g, were purchased from Jackson Laboratory (Bar Harbor, ME, USA). 7E3, a monoclonal murine anti-human anti-platelet IgG1 antibody, was produced and purified in our laboratory (13). The 7E3 antibody demonstrates high affinity for human glycoprotein IIb/IIIa; however, the antibody does not bind to murine glycoprotein IIb/IIIa (14). Water and food were provided ad libitum. 125Iodine and 51chromium were obtained from GE Healthcare (Piscataway, NJ, USA). All animal experiments were approved by the Institutional Animal Care and Use Committee of the State University of New York at Buffalo.
Radiolabeling of 7E3 with 125Iodine
7E3 was radiolabeled with 125I using a modified chloramine T method (15). This method of iodination is relatively mild and has previously been shown not to affect the disposition of 7E3 (16). We found that there was no significant difference (p > 0.05) in the half-life (15.6 ± 1.8 vs. 14.6 ± 3.1 days) and clearance (CL; 6.2 ± 1.4 vs. 5.7 ± 1.0 ml/day/kg) of labeled 7E3 compared to unlabeled 7E3 (16). Briefly, 10 µl of 125I (100 mCi/ml) was added to 40 µl of IgG (~2 mg/ml in phosphate buffered saline (PBS)), followed by addition of 20 µl of 1 mg/ml chloramine T in phosphate buffer. The reaction was stopped after 90 s by the addition of 25 µl of 2 mg/ml sodium metabisulfite in phosphate buffer and 40 µl of 10 mg/ml potassium iodide in double distilled water. The mixture was immediately loaded on a Sephadex G-25 M pre-packed column (GE Healthcare, Piscataway, NJ, USA). The mixture was eluted with PBS and 0.5 ml fractions were collected. In order to locate the labeled antibody, 2 μl samples from each fraction were analyzed for radioactivity. Total concentrations of IgG were estimated by UV absorbance, assuming 1.35 AU = 1 mg/ml. The specific activity of the labeled preparation was approximately 10–15 mCi/mg. The radiochemical purity of the labeled preparation was >99% as confirmed by instant thin layer chromatography and immunoprecipitation. Labeled antibody was stored at 4°C until needed.
Radiolabeling of Red Blood Cells with 51Chromium
To allow accurate determination of the quantity of residual blood in samples of brain tissue, 51Cr-labeled red blood cells (RBCs) were prepared and co-injected with 125I-7E3. Red cell labeling was based on the method proposed by the International Committee for Standardization in Hematology (ICSH) (17). The 51Cr method for estimating the volume of RBC (or the residual blood) has been designated as the ICSH-selected method “on the basis of reliability, reproducibility, and ease of operation in routine clinical use” (18). The accuracy of this method has also been confirmed by a recent meta-analysis (19). Briefly, 1 ml blood was collected from untreated anaesthetized mice. Blood was centrifuged at 150 g for 5 min. The supernatant was discarded and the pellet was washed three times with isotonic sodium chloride. The cells were reconstituted with 0.5 ml normal saline. Na512CrO4 was added to the cell suspension and the mixture was incubated within a fume hood for 45 min at room temperature. The cell suspension was centrifuged at 150 g for 5 min and supernatant was discarded. Excess 51Cr was removed by successive washing of the pellet with saline. The labeled cells were suspended in saline and radioactivity was measured by gamma counting.
Pharmacokinetics of 125I-7E3 in Mice
Two days before the experiment, mice were given potassium iodide (0.2 g/l) in drinking water to block thyroid uptake of 125I. 125I-labeled 7E3, at a dose of 8 mg/kg and 400 µCi/kg 125I activity (~10 µCi/mouse), was mixed with 400 µCi/kg 51Cr-labeled RBCs (~10 µCi/mouse). Mice were anesthetized with 3% isoflurane and oxygen for induction and maintained at 2% isoflurane and oxygen, with the use of a nose cone, to facilitate the administration of the antibody–RBC mixture. The duration of anesthesia was less than 5 min. The mixture was injected via the penile vein to groups of FcRn-deficient mice and C57BL/6J control mice. At various time points (i.e., 1, 2, 6, and 12 h, 1, 2, 3, and 4 days for the FcRn knockout mice and 1, 2, 6, and 12 h, 1, 2, 4, 7, and 10 days for the FcRn control mice), sub-groups of three mice were sacrificed via exsanguination. Blood and brain tissue were collected and weighed in borosilicate glass culture tubes. 125I and 51Cr activities were measured in the samples via gamma counting (LKB Wallac 1272, Wallac, Turku, Finland). The blood-to-plasma concentration ratio was calculated to be 0.55 ± 0.045 and was used to convert blood concentrations to plasma concentrations. Previous work conducted in our laboratory has shown that following 125I-labeled 7E3 administration to mice, >90% of the radioactivity associated with blood and with various tissues is trichloroacetic acid precipitable, at all time points, up to 10 days (16). Radioactive counts were corrected for background and decay. The amount of residual blood within each brain sample, expressed as grams of blood per gram of brain, was calculated as the ratio between 51Cr counts per minute per gram of brain and counts per minute per gram of blood (20). These values were converted into milliliters of blood per gram of tissue using 1 g/ml as the density of blood. Brain samples were corrected for residual blood content using the following equation:
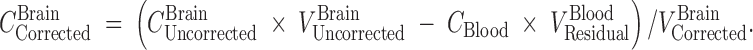 |
In this equation, 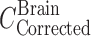 and
and 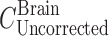 are the residual blood-corrected and residual blood-uncorrected brain concentrations, respectively.
are the residual blood-corrected and residual blood-uncorrected brain concentrations, respectively. 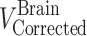 , which is the volume of the residual blood-corrected brain sample, was calculated by subtracting the volume of trapped blood (
, which is the volume of the residual blood-corrected brain sample, was calculated by subtracting the volume of trapped blood (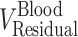 ) in the brain from the total volume of the brain sample (
) in the brain from the total volume of the brain sample (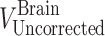 ). CBlood is the 7E3 concentration in the blood. Residual blood volume was estimated from the quotient of total 51Cr activity in brain tissue samples and the measured concentration of 51Cr activity in blood. That is,
). CBlood is the 7E3 concentration in the blood. Residual blood volume was estimated from the quotient of total 51Cr activity in brain tissue samples and the measured concentration of 51Cr activity in blood. That is, 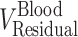 (milliliter) = 51Cr activity in brain sample/51Cr activity per milliliter of blood. The density of brain was assumed to be 1 g/ml (21).
(milliliter) = 51Cr activity in brain sample/51Cr activity per milliliter of blood. The density of brain was assumed to be 1 g/ml (21).
Pharmacokinetic Analysis
Pharmacokinetic parameters for the plasma data were assessed using the standard non-compartmental techniques in WinNonlin version 5.0 (Pharsight Corporation, Palo Alto, CA, USA). Plasma and brain AUCs were calculated using the Bailer method (22,23).
RESULTS
7E3 concentration–time profiles in plasma and in the brain of control and FcRn-deficient mice are shown in Fig. 1. Brain-to-plasma AUC ratio values in control and knockout mice are presented in Table I and Fig. 2. Consistent with results obtained by other investigators, we found that control animals showed little distribution of IgG to the brain, as indicated by the low brain/plasma AUC ratio (0.0022 ± 0.00015). Nearly identical results were obtained from FcRn-deficient mice, where the brain/plasma AUC ratio was 0.0021 ± 0.00011 (p = 0.3347 for the comparison of control vs. knockout AUC ratios). Furthermore, two-way analysis of variance showed that there were no significant differences between the control and the knockout animals in the brain-to-plasma concentration ratios at all time points (p > 0.05).
Fig. 1.

Plasma and brain disposition of 7E3. Plasma concentration vs. time profiles and brain concentration vs. time profiles 7E3 are shown for control and FcRn-deficient mice. Brain concentrations were corrected for residual blood content, as described in the text. Error bars indicate the standard deviation associated with each mean concentration
Table I.
Plasma and Brain AUC and AUC Ratios for Control and FcRn-Deficient Mice
| AUC (nM day) | Brain/plasma AUC ratio | ||
|---|---|---|---|
| Plasma | Brain | ||
| Control mice | 3,338.7 ± 50.4 | 7.46 ± 0.5 | 0.0022 ± 0.00015 |
| Knockout mice | 781.2 ± 16.6 | 1.65 ± 0.1 | 0.0021 ± 0.00011 |
Values are listed as mean ± standard deviation (n = 3). Standard deviation of the AUC was calculated by the Bailer method
AUC area under plasma and tissue concentration vs. time curve
Fig. 2.

Brain to plasma AUC ratios in control and in FcRn-deficient mice. Areas under the plasma and brain concentration vs. time curves were assessed with the linear trapezoidal method, and variability estimates were determined by the Bailer method. Error bars indicate the standard deviation of the mean AUC ratios
The plasma CL, as calculated by non-compartmental techniques in WinNonlin, for control and knockout animals were 6.5 ± 0.10 and 63.5 ± 2.7 ml/day/kg, respectively. The plasma half-lives for control and knockout animals were 13.6 ± 0.6 and 0.70 ± 0.05 days, respectively. Similarly, brain half-lives for control and knockout animals were found to be 6.5 ± 2.1 and 0.8 ± 0.1 days, respectively. The plasma CL and half-life values in control and knockout animals are comparable to values reported in the literature for both iodinated (16) and unlabeled (24) IgG1 monoclonal antibody 7E3. The residual blood content in brain, expressed as percentage of residual blood (milliliters blood per 100 g tissue), was found to be 0.96 ± 0.19% for the control animals and 0.97 ± 0.44% for the knockout animals.
DISCUSSION
In 1964, Brambell and coworkers proposed the existence of a transport protein that functions to protect IgG antibodies from catabolism (25). The transporter was first isolated from neonatal rat intestinal epithelium and was thus named as the Fc receptor of the neonate (26–28). The receptor has been shown to be a heterodimer of β2-microglobulin (β2m) and a major histocompatibility complex class 1-like α-chain (29), and it is now known that FcRn is expressed in a wide variety of adult tissues, including epithelial cells (2,3), hepatocytes (4), intestinal macrophages, peripheral blood monocytes, dendritic cells (5), and vascular endothelial cells (1). Studies conducted in vitro have shown that FcRn facilitates IgG transport across monolayers in a bidirectional manner (i.e., in the apical-to-basolateral and in basolateral-to-apical direction) (30), suggesting that FcRn may play an important role in the extravasation and distribution of IgG. Zhang and Pardridge proposed an Fc receptor-mediated “reverse transcytosis” of IgG from the brain to blood direction (10). Later, using 1G3 (an anti-rat FcRn antibody), Schlachetzki et al. found expression of FcRn at the BBB (9). These results, in combination, have led to the hypothesis that FcRn actively effluxes IgG out of the brain, thereby contributing to the BBB for IgG antibodies.
The role of FcRn in the efflux of IgG from the brain may have significant implications for the development of monoclonal antibodies (mAb) for several conditions, including Alzheimer’s disease and brain cancers. For example, if FcRn is shown to contribute significantly to the low brain exposure associated with IgG antibodies, this finding may lead to efforts to engineer mAb for reduced affinity for FcRn. Such engineered mAb might be expected to demonstrate improved exposure in brain tissues and reduced systemic exposure, potentially allowing for greater selectivity and safety.
To test the hypothesis that FcRn is a primary determinant of brain exposure to IgG, we studied the plasma and brain pharmacokinetics of systemically administered 7E3 in control and FcRn-light chain (β2m) knockout mice. If FcRn-mediated efflux is a significant determinant of brain exposure, this would be indicated by increased brain exposure in the β-2-microglobulin knockouts, which do not express functional FcRn. Our simple experimental design is similar to that used in a wide array of studies that have investigated the role of specific transporters (e.g., p-glycoprotein) as determinants of the brain exposure of specific substrates (31–36). Although it may have been preferable to use FcRn-heavy chain knockouts, these animals were not commercially available at the time of our study. β2m knockouts have been used for the vast majority of experiments assessing role of FcRn in IgG disposition (37–39), and prior work has shown that there are only slight differences in IgG half-life in β2m knockouts and heavy chain knockouts (1.4 days in the FcRn knockout vs. 1 days in the β2m knockouts compared to 9 days in the FcRn wild-type mice) (40).
Following intravenous administration of 125I-labeled 7E3, we found that control animals showed little distribution of the mAb to the brain, as the cumulative exposure of 7E3 in plasma was approximately 500-fold greater than the cumulative exposure of 7E3 in brain tissue. Relative to results observed in control animals, FcRn-deficient mice showed much lower 7E3 exposure in plasma, which is consistent with the absence of FcRn-mediated protection of IgG from catabolism. The ratio of brain and plasma 7E3 exposure in FcRn-deficient mice was nearly identical to the results found in wild-type mice (p = 0.3347 for the comparison of control vs. knockout AUC ratios). As such, this study strongly suggests that FcRn does not play a significant role in limiting or facilitating IgG distribution to the brain.
It is important to note, however, that the present investigation has not attempted to assess the significance of possible compensatory adaptations to FcRn deficiency. It is possible that FcRn-deficient animals up-regulate the expression of transport proteins that efflux IgG from the brain, such that the knockout mice fully compensate for the absence of FcRn-mediated IgG transport. Such functional adaptation, while clearly possible, is perhaps not very likely. FcRn inhibitors might allow for a more-direct evaluation of FcRn-mediated transport; however, specific inhibitors are not yet available.
CONCLUSIONS
In conclusion, this study provides direct evidence demonstrating that FcRn does not contribute significantly to the blood–brain barrier for IgG in mice, and the present data suggest that FcRn is not responsible for the low exposure of IgG in the brain relative to plasma.
Acknowledgments
This study was supported by grant AI60687 from the National Institutes of Health. Amit Garg was supported by a pre-doctoral fellowship from Eli Lilly and Company.
References
- 1.Borvak J, Richardson J, Medesan C, Antohe F, Radu C, Simionescu M, et al. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int Immunol. 1998;10(9):1289–98. doi: 10.1093/intimm/10.9.1289. [DOI] [PubMed] [Google Scholar]
- 2.Israel EJ, Taylor S, Wu Z, Mizoguchi E, Blumberg RS, Bhan A, et al. Expression of the neonatal Fc receptor, FcRn, on human intestinal epithelial cells. Immunology. 1997;92(1):69–74. doi: 10.1046/j.1365-2567.1997.00326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spiekermann GM, Finn PW, Ward ES, Dumont J, Dickinson BL, Blumberg RS, et al. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life: functional expression of FcRn in the mammalian lung. [erratum appears in J Exp Med. 2003 Jun 2;197(11):1601] J Exp Med. 2002;196(3):303–10. doi: 10.1084/jem.20020400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blumberg RS, Koss T, Story CM, Barisani D, Polischuk J, Lipin A, et al. A major histocompatibility complex class I-related Fc receptor for IgG on rat hepatocytes. J Clin Invest. 1995;95(5):2397–402. doi: 10.1172/JCI117934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu X, Meng G, Dickinson BL, Li X, Mizoguchi E, Miao L, et al. MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. J Immunol. 2001;166(5):3266–76. doi: 10.4049/jimmunol.166.5.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kristoffersen EK, Matre R. Co-localization of beta 2-microglobulin and IgG in human placental syncytiotrophoblasts. Eur J Immunol. 1996;26(2):505–7. doi: 10.1002/eji.1830260234. [DOI] [PubMed] [Google Scholar]
- 7.Leach JL, Sedmak DD, Osborne JM, Rahill B, Lairmore MD, Anderson CL. Isolation from human placenta of the IgG transporter, FcRn, and localization to the syncytiotrophoblast: implications for maternal–fetal antibody transport. J Immunol. 1996;157(8):3317–22. [PubMed] [Google Scholar]
- 8.Story CM, Mikulska JE, Simister NE. A major histocompatibility complex class I-like Fc receptor cloned from human placenta: possible role in transfer of immunoglobulin G from mother to fetus. J Exp Med. 1994;180(6):2377–81. doi: 10.1084/jem.180.6.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlachetzki F, Zhu C, Pardridge WM. Expression of the neonatal Fc receptor (FcRn) at the blood–brain barrier. J Neurochem. 2002;81(1):203–6. doi: 10.1046/j.1471-4159.2002.00840.x. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Pardridge WM. Mediated efflux of IgG molecules from brain to blood across the blood–brain barrier. J Neuroimmunol. 2001;114(1–2):168–72. doi: 10.1016/S0165-5728(01)00242-9. [DOI] [PubMed] [Google Scholar]
- 11.Boado RJ, Zhang Y, Zhang Y, Xia CF, Pardridge WM. Fusion antibody for Alzheimer's disease with bidirectional transport across the blood–brain barrier and abeta fibril disaggregation. Bioconjug Chem. 2007;18(2):447–55. doi: 10.1021/bc060349x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deane R, Sagare A, Hamm K, Parisi M, LaRue B, Guo H, et al. IgG-assisted age-dependent clearance of Alzheimer's amyloid beta peptide by the blood–brain barrier neonatal Fc receptor. J Neurosci. 2005;25(50):11495–503. doi: 10.1523/JNEUROSCI.3697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hansen RJ, Balthasar JP. Pharmacokinetics, pharmacodynamics, and platelet binding of an anti-glycoprotein IIb/IIIa monoclonal antibody (7E3) in the rat: a quantitative rat model of immune thrombocytopenic purpura. J Pharmacol Exp Ther. 2001;298(1):165–71. [PubMed] [Google Scholar]
- 14.Varner JA, Nakada MT, Jordan RE, Coller BS. Inhibition of angiogenesis and tumor growth by murine 7E3, the parent antibody of c7E3 Fab (abciximab; ReoPro) Angiogenesis. 1999;3(1):53–60. doi: 10.1023/A:1009019223744. [DOI] [PubMed] [Google Scholar]
- 15.Jensenius JC, Williams AF. The binding of anti-immunoglobulin antibodies to rat thymocytes and thoracic duct lymphocytes. Eur J Immunol. 1974;4(2):91–7. doi: 10.1002/eji.1830040207. [DOI] [PubMed] [Google Scholar]
- 16.Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn. 2007;34(5):687–709. doi: 10.1007/s10928-007-9065-1. [DOI] [PubMed] [Google Scholar]
- 17.International Committee for Standardization in Hematology (ICSH) Panel on Diagnostic Applications of Radioisotopes in Haematology Standard techniques for the measurement of red-cell and plasma volume. Br J Haematol. 1973;25(6):801–14. doi: 10.1111/j.1365-2141.1973.tb01792.x. [DOI] [PubMed] [Google Scholar]
- 18.International Committee for Standardization in Haematology (ICSH) Recommended methods for measurement of red-cell and plasma volume. J Nucl Med. 1980;21(8):793–800. [PubMed] [Google Scholar]
- 19.Gore CJ, Hopkins WG, Burge CM. Errors of measurement for blood volume parameters: a meta-analysis. J Appl Physiol. 2005;99(5):1745–58. doi: 10.1152/japplphysiol.00505.2005. [DOI] [PubMed] [Google Scholar]
- 20.Bernareggi A, Rowland M. Physiologic modeling of cyclosporin kinetics in rat and man. J Pharmacokinet Biopharm. 1991;19(1):21–50. doi: 10.1007/BF01062191. [DOI] [PubMed] [Google Scholar]
- 21.Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health. 1997;13(4):407–84. doi: 10.1177/074823379701300401. [DOI] [PubMed] [Google Scholar]
- 22.Bailer AJ. Testing for the equality of area under the curves when using destructive measurement techniques. J Pharmacokinet Biopharm. 1988;16(3):303–9. doi: 10.1007/BF01062139. [DOI] [PubMed] [Google Scholar]
- 23.Nedelman JR, Gibiansky E, Lau DT. Applying Bailer's method for AUC confidence intervals to sparse sampling. Pharm Res. 1995;12(1):124–8. doi: 10.1023/A:1016255124336. [DOI] [PubMed] [Google Scholar]
- 24.Hansen RJ, Balthasar JP. Intravenous immunoglobulin mediates an increase in anti-platelet antibody clearance via the FcRn receptor. [see comment] Thromb Haemost. 2002;88(6):898–9. [PubMed] [Google Scholar]
- 25.Brambell FW, Hemmings WA, Morris IG. A theoretical model of gamma-globulin catabolism. Nature. 1964;203:1352–4. doi: 10.1038/2031352a0. [DOI] [PubMed] [Google Scholar]
- 26.Rodewald R, Kraehenbuhl JP. Receptor-mediated transport of IgG. J Cell Biol. 1984;99(1 Pt 2):159s–64. doi: 10.1083/jcb.99.1.159s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simister NE, Mostov KE. An Fc receptor structurally related to MHC class I antigens. Nature. 1989;337(6203):184–7. doi: 10.1038/337184a0. [DOI] [PubMed] [Google Scholar]
- 28.Simister NE, Rees AR. Isolation and characterization of an Fc receptor from neonatal rat small intestine. Eur J Immunol. 1985;15(7):733–8. doi: 10.1002/eji.1830150718. [DOI] [PubMed] [Google Scholar]
- 29.Raghavan M, Bjorkman PJ. Fc receptors and their interactions with immunoglobulins. Annu Rev Cell Dev Biol. 1996;12:181–220. doi: 10.1146/annurev.cellbio.12.1.181. [DOI] [PubMed] [Google Scholar]
- 30.Dickinson BL, Badizadegan K, Wu Z, Ahouse JC, Zhu X, Simister NE, et al. Bidirectional FcRn-dependent IgG transport in a polarized human intestinal epithelial cell line. J Clin Invest. 1999;104(7):903–11. doi: 10.1172/JCI6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallo JM, Li S, Guo P, Reed K, Ma J. The effect of P-glycoprotein on paclitaxel brain and brain tumor distribution in mice. Cancer Res. 2003;63(16):5114–7. [PubMed] [Google Scholar]
- 32.Kemper EM, van Zandbergen AE, Cleypool C, Mos HA, Boogerd W, Beijnen JH, et al. Increased penetration of paclitaxel into the brain by inhibition of P-glycoprotein. Clin Cancer Res. 2003;9(7):2849–55. [PubMed] [Google Scholar]
- 33.Mayer U, Wagenaar E, Beijnen JH, Smit JW, Meijer DK, van Asperen J, et al. Substantial excretion of digoxin via the intestinal mucosa and prevention of long-term digoxin accumulation in the brain by the mdr 1a P-glycoprotein. Br J Pharmacol. 1996;119(5):1038–44. doi: 10.1111/j.1476-5381.1996.tb15775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood–brain barrier and to increased sensitivity to drugs. Cell. 1994;77(4):491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 35.Schinkel AH, Wagenaar E, Mol CA, van Deemter L. P-glycoprotein in the blood–brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996;97(11):2517–24. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Asperen J, Schinkel AH, Beijnen JH, Nooijen WJ, Borst P, van Tellingen O. Altered pharmacokinetics of vinblastine in Mdr1a P-glycoprotein-deficient mice. J Natl Cancer Inst. 1996;88(14):994–9. doi: 10.1093/jnci/88.14.994. [DOI] [PubMed] [Google Scholar]
- 37.Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES. Abnormally short serum half-lives of IgG in beta 2-microglobulin-deficient mice. Eur J Immunol. 1996;26(3):690–6. doi: 10.1002/eji.1830260327. [DOI] [PubMed] [Google Scholar]
- 38.Israel EJ, Wilsker DF, Hayes KC, Schoenfeld D, Simister NE. Increased clearance of IgG in mice that lack beta 2-microglobulin: possible protective role of FcRn. Immunology. 1996;89(4):573–8. doi: 10.1046/j.1365-2567.1996.d01-775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A. 1996;93(11):5512–6. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol. 2003;170(7):3528–33. doi: 10.4049/jimmunol.170.7.3528. [DOI] [PubMed] [Google Scholar]