

Abstract
Neurotrophins regulate development, maintenance, and function of vertebrate nervous systems. Neurotrophins activate two different classes of receptors, the Trk family of receptor tyrosine kinases and p75NTR, a member of the TNF receptor superfamily. Through these, neurotrophins activate many signaling pathways, including those mediated by ras and members of the cdc-42/ras/rho G protein families, and the MAP kinase, PI-3 kinase, and Jun kinase cascades. During development, limiting amounts of neurotrophins function as survival factors to ensure a match between the number of surviving neurons and the requirement for appropriate target innervation. They also regulate cell fate decisions, axon growth, dendrite pruning, the patterning of innervation and the expression of proteins crucial for normal neuronal function, such as neurotransmitters and ion channels. These proteins also regulate many aspects of neural function. In the mature nervous system, they control synaptic function and synaptic plasticity, while continuing to modulate neuronal survival.
Keywords: Trk receptor, nerve growth factor, apoptosis, plasticity, synapse, signaling, survival, differentiation
INTRODUCTION
Neurotrophins are important regulators of neural survival, development, function, and plasticity (for reviews, see Korsching 1993, Eide et al 1993, Segal & Greenberg 1996, Lewin & Barde 1996, Reichardt & Fariñas 1997, McAllister et al 1999, Sofroniew et al 2001). As the central concept of the neurotrophic factor hypothesis, targets of innervation were postulated to secrete limiting amounts of survival factors that function to ensure a balance between the size of a target organ and the number of innervating neurons (reviewed in Purves 1988). Nerve growth factor (NGF), the first such factor to be characterized, was discovered during a search for such survival factors (reviewed in Levi-Montalcini 1987). There are four neurotrophins characterized in mammals. NGF, brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4) are derived from a common ancestral gene, are similar in sequence and structure, and are therefore collectively named neurotrophins (e.g. Hallbook 1999). Although members of other families of proteins, most notably the glial cell–derived neurotrophic factor (GDNF) family and the neuropoietic cytokines, have been shown to also regulate survival, development, and function in the nervous system, this review focuses on the neurotrophins, examining mechanisms by which they signal and control development and function of the nervous system. A companion review by others describes the roles of these fascinating proteins in supporting the injured and aging nervous systems (Sofroniew et al 2001).
As the first neurotrophic factors to be discovered, the neurotrophins have had an unusually important influence on biology. The experiments leading to the discovery of NGF revealed the essential role of cellular interactions in development. Now almost all cells are believed to depend on their neighbors for survival (see Raff et al 1993). Almost a decade before endocytosis and transport were studied seriously in nonneural cells, NGF was shown to be internalized by receptor-dependent mechanisms and to be transported for vast distances along axons in small membrane vesicles by an energy and microtubule-dependent mechanism with eventual degradation of NGF in lysosomes. Now almost all cells are known to utilize similar mechanisms for trafficking of receptors and their ligands. Finally, neurotrophins have been shown to activate receptor tyrosine kinases. Within neural precursors and neurons, the pathways regulated by tyrosine kinases include proliferation and survival, axonal and dendritic growth and remodeling, assembly of the cytoskeleton, membrane trafficking and fusion, and synapse formation and function. Recent studies on the neurotrophins have shown that they regulated each of these functions and have increased our understanding of the molecular mechanisms underlying each. Thus, studies on these factors continue to provide insights of widespread interest to modern biologists.
Sources of Neurotrophins
NGF was purified as a factor able to support survival of sympathetic and sensory spinal neurons in culture (Levi-Montalcini 1987). Anti-NGF injections demonstrated that this factor is important in maintaining survival of sympathetic neurons in vivo as well as in vitro. Development of a two-site ELISA assay and of an NGF mRNA assay, using as probe the cloned NGF gene, made it possible to demonstrate that NGF is synthesized and secreted by sympathetic and sensory target organs (reviewed in Korsching 1993). From these sources, it is captured in nerve terminals by receptor-mediated endocytosis and is transported through axons to neuronal cell bodies where it acts to promote neuronal survival and differentiation. Within the target organs, synthesis of NGF and of other neurotrophins is associated with end organs, such as hair follicles, which become innervated by the axons of these neurons.
Subsequent work has demonstrated that there are other sources of neurotrophins. First, after peripheral nerve injury, macrophages infiltrate the nerve as part of an inflammatory response and release cytokines, which induce the synthesis of NGF in Schwann cells and fibroblasts within the injured nerve (reviewed by Korsching 1993). NGF is also synthesized in mast cells and is released following mast cell activation (reviewed in Levi-Montalcini et al 1996). NGF and other neurotrophic factors synthesized in damaged nerve are believed to be essential for survival and regeneration of injured neurons. Second, during development, neurotrophins are expressed in regions being invaded by sensory axons en route to their final targets, so they may provide trophic support to neurons that have not yet contacted their final targets (e.g. Fariñas et al 1996, 1998; Huang et al 1999a; Ringstedt et al 1999). Third, many neurons also synthesize neurotrophins. For example, several populations of sensory neurons have been shown to synthesize BDNF (e.g. Mannion et al 1999, Brady et al 1999). Although some evidence has been presented suggesting that BDNF may act in an autocrine or paracrine fashion to support dorsal root ganglion (DRG) sensory neurons (Acheson et al 1995, Robinson et al 1996), in other instances it may be transported anterogradely and act trans-synaptically on targets of the central afferents of these neurons within the brain (Brady et al 1999; see also Altar et al 1997, Fawcett et al 1998, von Bartheld et al 1996). Finally, when overexpressed in skin, sufficient target-derived NGF is released from the somata of trigeminal sensory neurons to support aberrant innervation by NGF-dependent sympathetic fibers (Davis et al 1998, Walsh et al 1999b). Thus, in some circumstances, a neurotrophin provided by one cell not only is effective at supporting neurons whose axons are in its vicinity, it also can provide support to more distant neurons via transcellular transport.
Neurotrophins and Their Receptors
Currently, six neurotrophins have been isolated: NGF, BDNF, NT-3, NT-4 (also known as NT-5), NT-6, and NT-7. There is substantial evidence that they all arose through successive duplications of the genome of an ancestral chordate (Hallbook 1999). The NT-6 and NT-7 genes have been identified only in fish and probably do not have mammalian or avian orthologues (Gotz et al 1994, Nilsson et al 1998). NT-4 has not been detected in avian species. Neurotrophins generally function as noncovalently associated homodimers, but at least some neurotrophin subunits are able to form heterodimers with other neurotrophin subunits. NGF, NT-6, and NT-7 appear to act on very similar and perhaps identical populations of neurons. BDNF and NT-4 have also very similar targets (e.g. Ip et al 1993). Thus the neurotrophins can be divided into three classes based upon target neuron populations, and all vertebrate species are likely to have at least one neurotrophin in each class. The structures of NGF, NT-3, and NT-4 and of NT-3/BDNF and NT-4/BDNF dimers have been solved and novel features of their structures—a tertiary fold and cystine knot—are present in several other growth factors, including platelet-derived growth factor and transforming growth factor β (McDonald et al 1991; Fandl et al 1994; Robinson et al 1995, 1999; Butte et al 1998; reviewed in McDonald & Chao 1995).
Initial efforts to identify NGF receptors resulted in discovery of a receptor now named p75NTR. For many years this was believed to be a low-affinity receptor specific for NGF. More recently, it has been shown to bind to all of the neurotrophins with a very similar affinity (Rodriguez-Tebar et al 1991). p75NTR is a distant member of the tumor necrosis factor receptor family (Chao 1994, Bothwell 1995). The cytoplasmic domain of this receptor contains a “death” domain structurally similar to those in other members of this receptor family (Liepinsch et al 1997). For many years after its discovery, it was not certain whether this receptor transmitted any signals or whether it functioned simply as a binding protein. Work during the past few years has shown, however, that this protein transmits signals important for determining which neurons survive during development. Signaling by this receptor is discussed at length below.
In a dramatic advance, the three members of the Trk (tropomyosin-related kinase) receptor tyrosine kinase family were shown to be a second class of neurotrophin receptors (reviewed in Bothwell 1995). The neurotrophins have been shown to directly bind and dimerize these receptors, which results in activation of the tyrosine kinases present in their cytoplasmic domains. NGF is specific for TrkA. BDNF and NT-4 are specific for TrkB. NT-3 activates TrkC and is also able to activate less efficiently each of the other Trk receptors. The most important site at which Trk receptors interact with neurotrophins has been localized to the most proximal immunoglobulin (Ig) domain of each receptor. The three-dimensional structures of each of these Ig domains has been solved (Ultsch et al 1999), and the structure of NGF bound to the TrkA membrane proximal Ig domain has also been determined (Wiesmann et al 1999). This exciting structural information has provided detailed information about interactions that regulate the strength and specificity of binding between neurotrophins and Trk receptors (e.g. Urfer et al 1998).
The unique actions of the neurotrophins made it seem likely that they would prove to have receptors and signal transduction pathways completely different from those of the mitogenic growth factors, such as platelet-derived growth factor or epidermal growth factor, whose receptors were known to be receptor tyrosine kinases. Thus, it was surprising when Trk receptors were identified as functional, survival-promoting receptors for neurotrophins. During the past few years, however, members of other neurotrophic factor families have also been shown to activate tyrosine kinases. These include GDNF and its relatives and ciliary neurotrophic factor (CNTF) and other neuropoietic cytokines (reviewed in Reichardt & Fariñas 1997). These tyrosine kinases activate many of the same intracellular signaling pathways regulated by the receptors for mitogens. Appreciation of this shared mechanism of action has been a major conceptual advance of the past decade.
Control of Neurotrophin Responsiveness by Trk Receptors
Tyrosine kinase–mediated signaling by endogenous Trk receptors appears to promote survival and/or differentiation in all neuronal populations examined to date. With a few exceptions, ectopic expression of a Trk receptor is sufficient to confer a neurotrophin-dependent survival and differentiation response (e.g. Allsopp et al 1994, Barrett & Bartlett 1994). Usually, endogenous expression of a Trk receptor confers responsiveness to the neurotrophins with which it binds, but this generalization is oversimplified for several reasons. First, differential splicing of the TrkA, TrkB, and TrkC mRNAs results in expression of proteins with differences in their extracellular domains that affect ligand interactions (Meakin et al 1992, Clary & Reichardt 1994, Shelton et al 1995, Garner et al 1996, Strohmaier et al 1996). The presence or absence of short amino acid sequences in the juxtamembrane domains of each receptor has been shown to affect the ability of some neurotrophins to activate these receptors. Although BDNF, NT-4, and NT-3 are capable of activating the TrkB isoform containing these amino acids, the TrkB isoform lacking them can only be activated by BDNF (Strohmaier et al 1996). These isoforms of TrkB have been shown to be expressed in nonoverlapping populations of avian sensory neurons, so splicing of this receptor almost certainly has important functional consequences (Boeshore et al 1999). Similarly, an isoform of TrkA containing a short juxtamembrane sequence is activated by both NGF and NT-3, whereas the isoform lacking these amino acids is much more specifically activated by NGF (Clary & Reichardt 1994). Although this short polypeptide sequence was not localized in the three-dimensional structure of the NGF-TrkA ligand binding domain complex, the organization of the interface between the two proteins is compatible with the possibility that these residues may directly participate in binding (Wiesmann et al 1999). The abilities of NT-3 to activate TrkA and of NT-3 and NT-4 to activate TrkB are also negatively regulated by high levels of the pan-neurotrophin receptor p75NTR (Bennedetti et al 1993, Lee et al 1994b, Clary & Reichardt 1994, Bibel et al 1999). Thus, factors that regulate differential splicing of extracellular exons in Trk receptor genes and signaling pathways that control expression of p75NTR affect the specificity of neuronal responsiveness to neurotrophins.
Important also has been the discovery of differential splicing of exons encoding portions of the Trk receptor cytoplasmic domains. Not all isoforms of TrkB and TrkC contain tyrosine kinase domains (reviewed in Reichardt & Fariñas 1997). Differential splicing generates isoforms of both TrkB and TrkC, which lack these domains. The functions of nonkinase-containing isoforms of TrkB and TrkC in nonneuronal cells may include presentation of neurotrophins to neurons. Within neurons, these same receptors are likely to inhibit productive dimerization and activation of full-length receptors, thereby attenuating responses to neurotrophins (e.g. Eide et al 1996). There is also evidence suggesting that ligand binding to truncated isoforms of TrkB and TrkC can modulate intracellular signaling pathways more directly (Baxter et al 1997, Hapner et al 1998). Differential splicing has also been shown to result in expression of an isoform of TrkC, which contains an amino acid insert within the tyrosine kinase domain. This insert does not eliminate the kinase activity of TrkC but does appear to modify its substrate specificity (e.g. Guiton et al 1995, Tsoulfas et al 1996, Meakin et al 1997).
Finally, in some central nervous system (CNS) projection neurons, Trk receptors appear to be largely sequestered in intracellular vesicles (Meyer-Franke et al 1998). Only in the presence of a second signal, such as cAMP or Ca2+, are the receptors inserted efficiently into the plasmalemma. In these neurons, expression of a kinase-containing isoform of a Trk receptor may not be sufficient to confer responsiveness to a neurotrophin if the neurons are not incorporated into a signaling network that results in production of these second messengers. Thus, neurotrophin responsiveness is controlled by many factors in addition to regulators of Trk receptor gene expression.
Control of Neurotrophin Responsiveness by the Pan-Neurotrophin Receptor p75NTR
Each neurotrophin also binds to the low-affinity neurotrophin receptor p75NTR, which is a member of the tumor necrosis factor receptor superfamily (see Frade & Barde 1998). In vitro studies on p75NTR have documented that it can potentiate activation of TrkA by subsaturating concentrations of NGF (e.g. Mahadeo et al 1994, Verdi et al 1994). What is surprising is that it does not appear to potentiate activation of the other Trk receptors by their ligands in vitro, even though these also bind to p75NTR. A role for p75NTR in potentiating actions of neurotrophins in vivo, however, provides one possible explanation of the deficits in multiple classes of sensory neurons observed in the p75NTR mutant (Stucky & Koltzenburg 1997, Bergmann et al 1997, Kinkelin et al 1999). As discussed above, studies in cell culture also indicate that p75NTR reduces responsiveness of Trk receptors to noncognate ligands (Benedetti et al 1993, Clary & Reichardt 1994, Lee et al 1994b). Recently, NT-3 has also been shown to maintain survival of TrkA-expressing sympathetic neurons in vivo more effectively in the absence than in the presence of p75NTR (Brennan et al 1999). The presence of p75NTR has also been shown to promote retrograde transport of several neurotrophins (e.g. Curtis et al 1995, Ryden et al 1995, Harrison et al 2000). Most intriguing, both in vitro and in vivo evidence now indicates that ligand engagement of p75NTR can directly induce neuronal death via apoptosis (reviewed in Frade & Barde 1998; see also Friedman, 2000). Analysis of the p75NTR mutant phenotype has demonstrated that regulation of apoptosis by ligand engagement of p75NTR is important during peripheral nervous system as well as CNS development in vivo (e.g. Bamji et al 1998, Casademunt et al 1999). Finally, absence of p75NTR signaling perturbs axon growth in vitro and both axon growth and target innervation in vivo (e.g. Lee et al 1994a, Yamashita et al 1999b, Bentley & Lee 2000, Walsh et al 1999a,b).
REGULATION OF SIGNALING BY NEUROTROPHINS
Trk Receptor-Mediated Signaling Mechanisms
Ligand engagement of Trk receptors has been shown to result in phosphorylation of cytoplasmic tyrosine residues on the cytoplasmic domains of these receptors (Figure 1). Trk receptors contain 10 evolutionarily conserved tyrosines in their cytoplasmic domains, of which three–Y670, Y674, and Y675 (human TrkA sequence nomenclature)–are present in the autoregulatory loop of the kinase domain that controls tyrosine kinase activity (e.g. Stephens et al 1994, Inagaki et al 1995). Phosphorylation of these residues further activates the receptor. Phosphorylation of the other tyrosine residues promotes signaling by creating docking sites for adapter proteins containing phosphotyrosine-binding (PTB) or src-homology-2 (SH-2) motifs (reviewed in Pawson & Nash 2000). These adapter proteins couple Trk receptors to intracellular signaling cascades, which include the Ras/ERK (extracellular signal–regulated kinase) protein kinase pathway, the phosphatidylinositol-3-kinase (PI-3 kinase)/Akt kinase pathway, and phospholipase C (PLC)-γ1 (see Reichardt & Fariñas 1997, Kaplan & Miller 2000). Two tyrosines not in the kinase activation domain (Y490 and Y785) are major sites of endogenous phosphorylation, and most research has focused on interactions mediated by these sites with Shc and PLC-γ1, respectively (Stephens et al 1994). Five of the remaining seven conserved tyrosines also contribute to NGF-induced neurite outgrowth, however, so interactions mediated by Y490 and Y785 can mediate only a subset of Trk receptor interactions important in neurotrophin-activated signaling (Inagaki et al 1995). Recent work has resulted in identification of additional adapter proteins that interact with Trk receptors at different sites and has demonstrated that transfer of Trk receptors to various membrane compartments controls the efficiency with which these receptors can associate with and activate adapter proteins and intracellular signaling pathways (e.g. Qian et al 1998; Saragovi et al 1998; York et al 2000; C Wu, C-F Lai, WC Mobley, unpublished observations).
Figure 1.
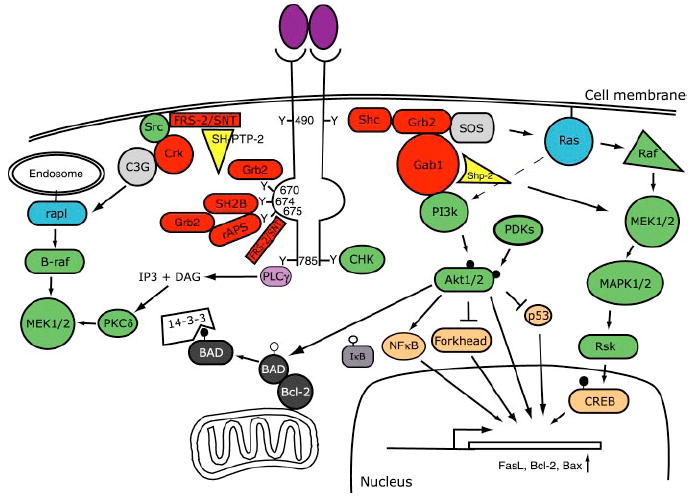
Schematic diagram of Trk receptor-mediated signal transduction pathways. Binding of neurotrophins to Trk receptors leads to the recruitment of proteins that interact with specific phosphotyrosine residues in the cytoplasmic domains of Trk receptors. These interactions lead to the activation of signaling pathways, such as the Ras, phosphatidylinositol-3-kinase (PI3k), and phospholipase C (PLC)-γ pathways, and ultimately result in activation of gene expression, neuronal survival, and neurite outgrowth (see text for detailed discussions and abbreviations). The nomenclature for tyrosine residues in the cytoplasmic domains of Trk receptors are based on the human sequence for TrkA. In this diagram, adaptor proteins are red, kinase green, small G proteins blue, and transcription factors brown.
PLC-γ1 Signaling
Phosphorylation of Y785 on TrkA has been shown to recruit PLC-γ1 directly, which is activated by phosphorylation and then acts to hydrolyse phosphatidyl inositides to generate inositol tris-phosphate and diacylglycerol (DAG) (Vetter et al 1991). Inositol tris-phosphate induces release of Ca2+ stores, increasing levels of cytoplasmic Ca2+. This results in activation of various enzymes regulated by cytoplasmic Ca2+, including Ca2+-calmodulin–regulated protein kinases and phosphatases and Ca2+-regulated isoforms of protein kinase C. Formation of DAG stimulates the activity of DAG-regulated protein kinase C isoforms. In PC12 cells, protein kinase C (PKC)δ, a DAG-regulated PKC, is activated by NGF and is required for neurite outgrowth and for activation of the ERK cascade (Corbit et al 1999). Inhibition of PKCδ has been shown to inhibit activation of MEK [mitogen-activated protein kinase kinase (MAPKK)/ERK kinase)] but not of c-raf, so PKCδ appears to act between Raf and MEK in the ERK kinase cascade.
RAS-ERK Signaling
Activation of Ras is essential for normal differentiation of PC12 cells and neurons. In many cells, Ras activation also promotes survival of neurons, either by activation of PI-3 kinase or through activation of the ERK family of MAP kinases. Transient vs prolonged activation of the MAP kinase pathway has been closely associated, respectively, with a proliferation-inducing vs a differentiation-promoting response to neurotrophin application (e.g. Grewal et al 1999).
The pathways leading to activation of Ras are surprisingly complex. In the first pathway to be characterized, phosphorylation on Y490 was shown to result in recruitment and phosphorylation of the adapter protein Shc, with binding mediated by the Shc PTB domain (Stephens et al 1994; reviewed in Kaplan & Miller 2000). Shc is then phosphorylated by Trk, resulting in recruitment of a complex of the adapter protein Grb-2 and the Ras exchange factor SOS. Activation of Ras by SOS has many downstream consequences, including stimulation of PI-3 kinase, activation of the c-raf/ERK pathway, and stimulation of the p38 MAP kinase/MAP kinase-activated protein kinase 2 pathway (e.g. Xing et al 1996). Downstream targets of the ERK kinases include the RSK kinases (ribosomal S6 kinase). Both RSK and MAP kinase-activated protein kinase 2 phosphorylate CREB (cAMP-regulated enhancer binding protein) and other transcription factors (Xing et al 1998). These transcription factors in turn control expression of many genes known to be regulated by NGF and other neurotrophins. Among these, CREB regulates genes whose products are essential for prolonged neurotrophin-dependent survival of neurons (Bonni et al 1999, Riccio et al 1999).
Neurotrophin signaling through Shc/Grb-2/SOS mediates transient, but not prolonged, activation of ERK signaling pathways (e.g Grewal et al 1999). Prolonged ERK activation has been shown to depend on a distinct signaling pathway involving the adapter protein Crk, the exchange factor C3G, the small G protein rap1, and the serine-threonine kinase B-raf (York et al 1998). Neurotrophins activate this signaling pathway by utilization of a distinct adapter named FRS-2 (fibroblast growth factor receptor substrate-2) or SNT(suc-associated neurotrophic factor-induced tyrosine-phosphorylated target), which competes with Shc for phosphorylated Y490 on TrkA (Meakin et al 1999). FRS-2 is phosphorylated by Trk activation and has been shown to have binding sites for several additional proteins, including the adapter proteins Grb-2 and Crk, the cytoplasmic tyrosine kinase Src, the cyclin-dependent kinase substrate p13suc1, and the protein phosphatase SH-PTP-2 (e.g. Meakin et al 1999). Crk associates with phosphorylated FRS-2 and then binds and activates the exchange factor C3G (e.g. Nosaka et al 1999). Activation by C3G of the small G protein Rap1 results in stimulation of B-raf, which activates the ERK kinase cascade. As predicted by this model, overexpression of FRS-2 or Crk results in differentiation of pheochromocytoma (PC)-12 cells (Tanaka et al 1993, Matsuda et al 1994, Hempstead et al 1994, Meakin et al 1999). In addition to providing a crucial link to a pathway that appears to be essential for prolonged MAP kinase activation, FRS-2 provides a mechanism not dependent on Shc for activation of the Grb-2/SOS/Ras pathway. This adapter protein also provides a link to the Src family tyrosine kinases, which have been implicated in receptor endocytosis and other cellular responses (e.g. Wilde et al 1999, Beattie et al 2000). Finally, binding to FRS-2 of the protein phosphatase SH-PTP-2 also facilitates activation of the ERK pathway, probably by inactivation of an inhibitor, such as Ras-GAP or MAPK phosphatase (Wright et al 1997).
PI-3 Kinase Signaling
Activation of phosphatidylinositol-3-kinase (PI-3 kinase) is essential for survival of many populations of neurons. In collaboration with the phosphatidylinositide-dependent kinases, phosphatidyl inositides generated by PI-3 kinase activate the protein kinase Akt/protein kinase B. Akt then phosphorylates and controls the biological functions of several proteins important in modulating cell survival (reviewed in Datta et al 1999, Yuan & Yankner 2000). Among the substrates of Akt are BAD, a Bcl-2 family member that promotes apoptosis by binding to Bcl-xL, which in the absence of binding would inhibit the proapoptotic activity of Bax. Phosphorylation of BAD results in its association with 14-3-3 proteins and prevents it from promoting apoptosis (Datta et al 1997). BAD is also a substrate for MAP kinases, which similarly inactivate its apoptosis-promoting function (Bonni et al 1999). Another demonstrated target of Akt is IκB (reviewed in Datta et al 1999). Phosphorylation of IκB results in its degradation and activation of NFκB, which is normally sequestered by IκB in the cytoplasm. Transcription activated by nuclear NFκB has been shown to promote neuronal survival (e.g. Middleton et al 2000). A third Akt substrate of potential relevance for neuronal survival is the forkhead transcription factor FKHRL1, which controls expression of apoptosis-promoting gene products, such as FasL (Brunet et al 1999). Another Akt substrate is human but not mouse caspase-9 (Brunet et al 1999). Glycogen synthase kinase 3-β (GSK3β) is also stimulated by trophic factor withdrawal and negatively regulated by Akt phosphorylation (Hetman et al 1999). In cultured cortical neurons, elevated GSK3β promotes apoptosis (Hetman et al 1999). Many additional proteins in the cell death cascade, including Bcl-2, Apaf-1, caspase inhibitors, and caspases, have a consensus site for Akt phosphorylation but have not been shown to be phosphorylated by this kinase (Datta et al 1999). Analyses of mouse mutants have documented the importance of many, but not all, of these proteins (reviewed in Yuan & Yankner 2000). Mutants lacking caspase-9 or Bax have reductions in neuronal apoptosis, whereas a mutant lacking Bcl-X-L has an increase in neuronal apoptosis during development (e.g. Deckwerth et al 1996, Shindler et al 1998). Absence of BAD, however, does not detectably alter neuronal apoptosis during CNS development, which suggests that Akt-mediated phosphorylation of this protein is not an essential link in the PI-3 kinase–dependent survival cascade in vivo (Shindler et al 1998). It is important to note that not all substrates of Akt are involved in cell survival. S6 kinase, for example, is important for promoting translation of a subset of mRNAs, including certain cyclins essential for cell cycle progression.
PI-3 kinase is activated by Ras. In many but not all neurons, Ras-dependent activation of PI-3 kinase is the major pathway by which neurotrophins convey survival-promoting signals (e.g. Vaillant et al 1999). PI-3 kinase and signaling pathways dependent on PI-3 kinase function can also be activated through Shc and Grb-2 by a Ras-independent mechanism. Recruitment by phosphorylated Grb-2 of the adaptor protein Gab-1 results in subsequent binding to this complex of PI-3 kinase, which is then activated (Holgado-Madruga et al 1997, reviewed by Kaplan & Miller 2000). In some cells, but not in PC-12 cells, insulin receptor substrate (IRS)-1 has been shown to be phosphorylated in response to neurotrophins and in turn to recruit and activate PI-3 kinase (Yamada et al 1997).
In addition to providing an adapter that facilitates activation of PI-3 kinase, Gab-1 has also been shown to function as an adapter that nucleates formation of a complex that includes the protein tyrosine phosphatase Shp-2 (Shi et al 2000). Shp-2 has been shown to enhance activation of the RAS-RAF-MEK-ERK pathway by a mechanism that is not clear, but that appears to involve dephosphorylation of a 90-kDa protein that is also associated with the Gab-1 complex.
Control of the Actin Cytoskeleton
The neurotrophins induce rapid ruffling and cytoskeletal rearrangements similar to those induced by other growth factors (e.g. Connolly et al 1979). These have been shown by many laboratories to involve small G proteins of the Cdc-42/Rac/Rho family, which regulate the polymerization and turnover of F-actin (reviewed in Kjoller & Hall 1999, Bishop & Hall 2000). Several exchange factors for this family of G proteins are known to be expressed in neurons and to be regulated by tyrosine phosphorylation and/or phosphatidyl inositides generated by PI-3 kinase activity (e.g. Liu & Burridge 2000). Many of these are undoubtedly regulated by Trk receptor signaling. SOS also has a latent activity as an exchange factor for rac in addition to its activity as an exchange factor for ras (Nimnual et al 1998). Activated ras has been shown to activate the exchange factor activity of SOS for rac through a mechanism dependent on PI-3 kinase. Thus SOS provides a mechanism for the coordination of ras and rac activities.
Control of Trk Signaling by Membrane Trafficking
Recent work has added complexity to the scheme presented above by providing evidence that the ability of Trk receptors to activate specific signaling pathways is regulated by endocytosis and membrane sorting. It has long been appreciated that communication of survival signals from nerve terminals to neuronal cell bodies requires retrograde transport (e.g. Thoenen & Barde 1980). Several groups have demonstrated during the past few years that NGF and activated Trk receptors are transported together in endocytotic vesicles (e.g. Grimes et al 1997, Riccio et al 1997, Tsui-Pierchala & Ginty 1999; CL Howe, E Beattie, JS Valletta, WC Mobley, unpublished observations). More recently, evidence has accumulated indicating that membrane sorting determines which pathways are activated by Trk receptors. In one set of experiments, cells were exposed to a complex of NGF and a monoclonal antibody (mAb) that does not interfere with receptor binding but induces unusually rapid internalization of the mAb-NGF-TrkA complex (Saragovi et al 1998). This NGF-mAb complex was shown to promote transient MAP kinase activation, Shc phosphorylation, and PC12 cell survival. In contrast, FRS-2 was not phosphorylated and the cells did not differentiate normally. The results suggest that recruitment of FRS-2 by ligand receptor complexes occurs on the cell surface with comparatively slow kinetics. Perhaps FRS-2, which is myristoylated, is segregated into a compartment that is not immediately accessible to the TrkA receptor.
In another set of experiments, a thermosensitive dynamin that functions as a dominant negative protein at high temperature was used to reversibly inhibit ligand-receptor internalization (Zhang et al 2000). Inhibition of internalization did not inhibit survival of PC12 cells but did strongly inhibit their differentiation. This observation suggests that ligand-receptor complexes must be internalized to activate efficiently pathways essential for differentiation. As previous work described above has strongly suggested that FRS-2 signaling through Crk is essential for prolonged MAP kinase activation and normal differentiation, the data suggest that activation of this pathway requires internalization of the NGF-TrkA signaling complex.
Consistent with the involvement of PI-3 kinase products in endocytosis (Wendland et al 1998), inhibitors of PI-3 kinase have been shown to reduce retrograde transport and to affect the activation of NGF-dependent intracellular signaling pathways (Kuruvilla et al 2000, York et al 2000). Activation of Ras has been shown to occur in the absence of TrkA internalization and absence of PI-3 kinase activity (York et al 2000). In contrast, activation of Rap-1 and B-raf and sustained ERK activation require internalization and PI-3 kinase activity (York et al 2000). To activate B-raf, TrkA must be transported to a brefeldin-A–sensitive population of endosomes (C Wu, C-F Lai, WC Mobley, unpublished observations). Examination of the distributions of Ras and Rap-1 provide a possible explanation. Although there is prominent expression of Ras on the cell surface, expression of Rap-1 appears to be restricted to small intracellular vesicles. Thus, the data suggest that for TrkA to activate Rap-1, which in turn activates B-raf and the ERK kinase cascade, it must be internalized into membrane vesicles that fuse with vesicles containing Rap-1 (York et al 2000, C Wu, C-F Lai, WC Mobley, unpublished observations). Thus, sustained activation of the ERK pathway, which is essential for normal differentiation, is regulated by both the kinetics and specificity of membrane transport and sorting. Because there are so many mechanisms for regulating membrane transport and sorting, these recent papers suggest many interesting directions for future research.
Control of Trk Signaling by Other Adapters
Results described above predict that both survival and differentiation pathways will depend on interactions of Shc or Frs-2 with the phosphorylated Y490 site. Despite this, mice homozygous for a targeted Y to F mutation of this site in TrkB are viable and have a much milder phenotype than is observed in mice lacking the TrkB kinase domain (Minichiello et al 1998). Clearly, other sites in TrkB must be capable of activating intracellular signaling pathways important for neuronal survival and differentiation. Recent results of particular interest have suggested that the phosphorylated tyrosines in the activation loop of the Trk tyrosine kinase domain have dual functions. In addition to controlling activity of the kinase, they appear to function as docking sites for adapter proteins. Grb-2 has been shown to interact directly with a phosphorylated tyrosine residue in 2-hybrid assays and by coimmunoprecipitation (MacDonald et al 2000). Grb-2 also interacts with other sites, including the PLC-γ1 site Y785, but it is not certain these interactions are direct. Two additional adapters, rAPS and SH2-B, are similar proteins that contain a PH domain, an SH2 domain, and tyrosines phosphorylated in response to Trk activation. Both have been shown to interact with phosphorylated tyrosines in the activation loops of all three Trk receptors (Qian et al 1998). Both of these adapter proteins may also interact with other sites in the Trk receptor cytoplasmic domains. These two proteins form homodimers and also associate with each other. Both also bind to Grb-2, providing a potential link to the PI-3 kinase and Ras signaling cascades. Antibody perturbation and transfections using dominant negative constructs implicate rAPS in NGF-dependent survival, MAP kinase activation, and neurite outgrowth in neonatal sympathetic neurons (Qian et al 1998). Taken together, these results indicate that initial models of Trk receptor signaling pathways were far simpler than Trk receptor signaling is in reality.
Our current understanding of Trk receptor signaling is incomplete. First, not all functionally important interactions with Trk receptors may depend on phosphotyrosine-dependent associations. Recent work suggests that the c-abl tyrosine kinase interacts with the juxtamembrane domain of TrkA, whether or not the tyrosines in this region are phosphorylated (Yano et al 2000). A deletion in this region has been shown to block differentiation of PC12 cells without preventing mitotic responses or phosphorylation of SHC or FRS-2 (Meakin & MacDonald 1998). As c-abl is involved in many aspects of neuronal differentiation (e.g. see Hu & Reichardt 1999), it will be interesting to determine whether it has a role in Trk-mediated signaling that is perturbed by this juxtamembrane deletion.
To provide a few more examples of proteins whose roles in signaling pathways are poorly understood, in cultured cortical neurons, the insulin receptor substrates (IRS)-1 and -2 are phosphorylated in response to BDNF, which promotes sustained association with and activation of PI-3 kinase (Yamada et al 1997, 1999). This is not seen after ligand engagement of Trk receptors in PC12 cells, the most popular cellular model for neurotrophin signaling studies, which suggests that a critical adapter protein is missing from these cells. As another example, CHK, a cytoplasmic protein kinase that is a homologue of CSK (control of src kinase), has been shown to interact with TrkA in PC12 cells and to enhance ERK pathway-dependent responses, including neurite outgrowth (Yamashita et al 1999a). The pathway by which CHK affects ERK activation is not understood. Finally, a transmembrane protein with three extracellular immunoglobulin and four cytoplasmic tyrosine motifs has been shown to provide a docking site for recruitment of the protein phosphatase Shp-2 and to enhance BDNF-dependent activation of the PI-3 kinase pathway by mechanisms not prevented by mutation of the four tyrosine residues (Araki et al 2000a,b). Again, mechanisms are not understood.
In summary, although there has been rapid progress in understanding many pathways controlled by Trk receptor signaling, there are still many loose ends. This discussion has proceeded as if all signaling molecules were present in all cells, but this is certainly not so. Differences in their concentrations within different neuronal populations undoubtedly contribute to the diversity of responses seen in different neuronal populations. From the discussion above, it would be obvious to assume that TrkA, TrkB, and TrkC each activate very similar signaling pathways because of the very high similarities between them in their cytoplasmic domains. Although probably true, examples are described later in this review where it is clear that signaling through different Trk receptors has quite different actions on the same cell, as assessed by nonredundant effects on survival, differentiation, or axon guidance (e.g. Carroll et al 1998, Ming et al 1999). In some tumor cells, neurotrophin-activated Trk receptor signaling has even been shown to induce apoptosis (e.g. Kim et al 1999). Clearly, many of the most interesting details of signaling by these receptors remain to be discovered.
p75NTR Receptor-Mediated Signaling Mechanisms: NFκB Activation
As mentioned previously, p75NTR binds with approximately equal affinity to each of the neurotrophins. Ligand engagement of p75NTR has been shown to promote survival of some cells and apoptosis of others (e.g. Barrett & Bartlett 1994). p75NTR-mediated signaling also affects axonal outgrowth both in vivo and in vitro (e.g. Bentley & Lee 2000, Yamashita et al 1999b; Walsh et al 1999a,b).
Several signaling pathways are activated by p75NTR and in some cases the pathways are known in detail (see Figure 2). An important pathway promoting cell survival of many cell populations involves activation of NFκB. For example, cytokines promote neuronal survival by activation of the NFκB signaling pathway (Middleton et al 2000). In both embryonic sensory and sympathetic neurons, neurotrophins have been shown to promote p75NTR-dependent activation of NFκB and NFκB–dependent neuronal survival (Maggirwar et al 1998, Hamanoue et al 1999). All neurotrophins have been shown to promote association of p75NTR with the adapter protein TRAF-6 (Khursigara et al 1999). In other systems, TRAF-6 has been shown to activate the protein kinase NIK (NFκB-interacting kinase), which phosphorylates IKK (inhibitor of IκB kinase), which in turn phosphorylates IκB, resulting in release and nuclear translocation of NFκB (reviewed in Arch et al 1998). It is interesting that although all neurotrophins bind to p75NTR, in rat Schwann cells, only NGF is able to induce NFκB nuclear translocation (Carter et al 1996).
Figure 2.
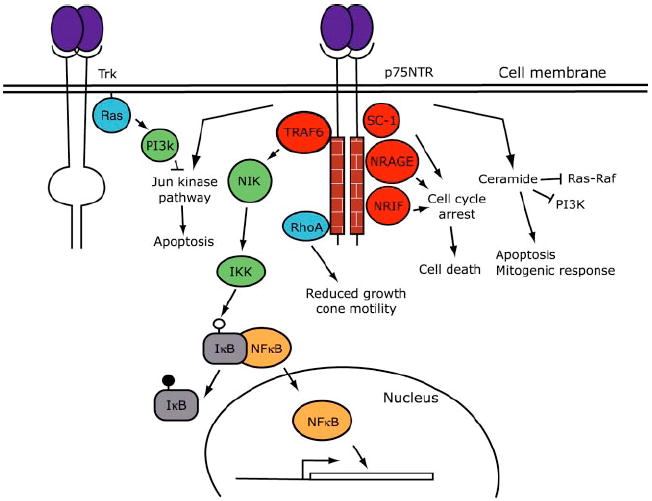
Schematic diagram of p75NTR-mediated signal transduction pathways. P75NTR interacts with proteins, including TRAF6, RhoA, NRAGE (neurotrophin receptor-interacting MAGE homologue), SC-1, and NRIF, and regulates gene expression, the cell cycle, apoptosis, mitogenic responses, and growth cone motility. Binding of neurotrophins to p75NTR has also been shown to activate the Jun kinase pathway, which can be inhibited by activation of the Ras-phosphatidylinositol-3-kinase (PI3K) pathway by Trk receptors. Similar to Figure 1, adaptor proteins are red, kinase green, small G proteins blue, and transcription factors brown. (See text for abbreviations.)
p75NTR Receptor-Mediated Signaling Mechanisms: Jun Kinase Activation
The Jun kinase signaling cascade is activated following NGF withdrawal and by binding of neurotrophins to p75NTR (Xia et al 1995, Eilers et al 1998, Aloyz et al 1998). Apoptosis mediated by p75NTR requires activation of p53 through the Jun kinase–mediated signaling pathway (Aloyz et al 1998). P53 controls cell survival in many cells besides neurons (e.g. Agarwal et al 1998). Among its targets, the activation of the Jun kinase cascade has been shown to induce expression of Fas ligand in neuronal cells, which promotes apoptosis by binding to the Fas receptor (Le-Niculescu et al 1999). p53 has many gene targets, including the proapoptic gene Bax. In both PC12 cells and sympathetic neurons, activation of the Jun kinase cascade and apoptosis following trophic withdrawal involve Cdc-42 because apoptosis is strongly inhibited by a dominant negative Cdc-42 (Bazenet et al 1998). The MAP kinase kinase kinase named apoptosis signal-regulating kinase-1 (ASK1) is in the pathway controlled by Cdc-42 because over-expression of a kinase-inactive mutant of ASK-1 strongly inhibits cell death promoted by either NGF withdrawal or expression of a constitutively active Cdc-42 (Kanamoto et al 2000). The kinase providing a link between ASK-1 and Jun kinase has not been identified but may be the Jun kinase kinase named MKK7 in sympathetic neurons (Kanamoto et al 2000). Embryos lacking both JNK1 and JNK2 show aberrant, region-specific perturbations of neuronal cell apoptosis in early brain development (Kuan et al 1999), whereas the neurons of animals lacking JNK3 are resistant to excitoxicity-induced apoptosis (Yang et al 1997). Thus, the Jun kinase cascade is important in regulating apoptosis of neurons in vivo.
p75NTR Receptor-Mediated Stimulation of Sphingolipid Turnover
Ligand engagement of p75NTR has also been shown to activate acidic sphingomyelinase, which results in generation of ceramide (Dobrowsky et al 1995). Ceramide has been shown to promote apoptosis and mitogenic responses in different cell types through control of many signaling pathways, including the ERK and Jun kinase cascades and NFκB. For example, ceramide binds to Raf and may induce formation of inactive Ras-Raf complexes, effectively inhibiting the ERK signaling cascade (Muller et al 1998). Many groups have shown that ceramide also inhibits signaling mediated through PI-3 kinase (e.g. Zhou et al 1998). Recent experiments suggest that ceramide inhibits the activity of PI-3 kinase in cells by modifying the association of receptor tyrosine kinases and PI-3 kinase with caveolin-1 in lipid rafts (Zundel et al 2000). In fibroblasts, the sensitivity of growth factor–stimulated PI-3 kinase activity to ceramide inhibition was increased and decreased by over-expression and reduced expression of caveolin-1, respectively. Ceramide may also inhibit directly PI-3 kinase activity (Zhou et al 1998). Thus, ceramide inhibits at least two of the survival and differentiation-promoting pathways activated by Trk receptor signaling.
Adapter Proteins That Bind to p75NTR
In addition to TRAF-6, several additional proteins that interact with p75NTR have been identified, each of which is a candidate to mediate Jun kinase activation or sphingolipid turnover. NRIF (neurotrophin receptor interacting factor) is a widely expressed Zn-finger–containing protein that interacts with both the juxtamembrane and death domains of p75NTR (Casademunt et al 1999). Over-expression of NRIF has been shown to kill cells in culture, and in mice lacking NRIF, there are reductions in developmentally regulated cell death among neuronal populations that are very similar to the reductions observed in mice lacking p75NTR. At this time, it is not known whether NRIF’s activity or association with p75NTR is regulated by neurotrophins. It is also unclear which downstream signaling pathways are activated by this interesting protein. Another protein named NRAGE [neurotrophin receptor–interacting MAGE (melanoma-associated antigen) homologue] has recently been shown to associate with p75NTR and to be recruited to the plasma membrane when NGF is bound to p75NTR (Salehi et al 2000). NRAGE prevents the association of p75NTR with TrkA, and overexpression of NRAGE promotes NGF-stimulated, p75NTR-dependent cell cycle arrest and death of MAH (v-myc-infected, adrenal-derived, HNK-1-positive) cells. SC-1 (Schwann cell-1) is a distinct Zn-finger–containing protein, which has been shown to associate with p75NTR and to redistribute from the cytoplasm to the nucleus after treatment of p75-expressing cos cells with NGF (Chittka & Chao 1999). Nuclear expression of SC-1 correlates with cell cycle arrest, which suggests that nuclear localization of this protein may be involved causally in growth arrest. Thus, both NRAGE and SC-1 appear to be interesting proteins involved in the signaling events promoted by p75NTR. Finally, when overexpressed in 293 cells, several (TNF receptor-associated factor) proteins in addition to TRAF-2 can associate with either monomeric or dimeric p75NTR, and some of these promote apoptosis of these cells (Ye et al 1999). Although the interactions of these adapter proteins with p75NTR are interesting, much work remains to be done to characterize their expression patterns and signaling mechanisms in neurons.
Control of the Cytoskeleton by p75NTR
In addition to regulating neuronal cell survival, ligand engagement of p75NTR has been reported to directly enhance neurite outgrowth by ciliary neurons in culture (e.g. Yamashita et al 1999b). In contrast, it inhibits neurite outgrowth by sympathetic neurons in culture (Kohn et al 1999). Sensory and motor neurons extend axons more slowly toward their peripheral targets in mouse embryos lacking p75NTR (Yamashita et al 1999b, Bentley & Lee 2000). In adult animals, perturbations of target innervation patterns are also seen in these mice with some, but not all, targets lacking normal innervation (e.g. Lee et al 1994a, Peterson et al 1999, Kohn et al 1999). Recent work has demonstrated an interaction between p75NTR and RhoA (Yamashita et al 1999b). p75NTR was observed to activate RhoA. Neurotrophin binding to p75NTR eliminated activation of RhoA by p75NTR. Pharmacological inactivation of RhoA and ligand engagement of p75NTR have similar stimulatory effects on neurite outgrowth by ciliary ganglion neurons, a neuronal population that does not express Trk receptors. Results from this interesting paper suggest that unliganded p75NTR tonically activates RhoA, which in turn is known to reduce growth cone motility. This observation does not provide an immediately obvious explanation for the reduced axon outgrowth by sensory and motor neurons observed in embryonic p75NTR–/– animals. Perhaps the presence of ligand-engaged p75 effectively sequesters RhoA in its inactive form. Alternatively, the presence of p75NTR has been shown to promote retrograde transport of NGF, BDNF, and NT-4 (Curtis et al 1995, Harrison et al 2000). Reductions in retrograde transport may result in reduced axon growth and neuronal survival. As a third possibility, Schwann cell migration has been shown to depend on p75NTR-mediated signaling and is clearly deficient in this mutant (Anton et al 1994, Bentley & Lee 2000). Perhaps deficits in Schwann cell migration indirectly reduce the rate of axonal outgrowth.
Reciprocal Regulation of Signaling by Trk Receptors and p75NTR
Activation of Trk receptors has profound effects on p75NTR-dependent signaling. Neurotrophins are much more effective at inducing apoptosis through p75NTR in the absence than in the presence of Trk receptor activation (e.g. Davey & Davies 1998, Yoon et al 1998). In the initial experiments demonstrating that NGF induced sphingomyelin hydrolysis and ceramide production, activation of a Trk receptor was shown to completely suppress this response (Dobrowsky et al 1995). Trk receptor activation also suppresses activation of the Jun kinase cascade (Yoon et al 1998). Activation of Ras in sympathetic neurons has been shown to suppress the Jun kinase cascade (Mazzoni et al 1999). In these neurons, activation by Ras of PI-3 kinase is essential for efficient suppression of this cascade. In recent studies utilizing nonneural cells, c-raf has been shown to bind, phosphorylate, and inactivate ASK-1 (Chen & Fu 2000). If this pathway functions efficiently in neurons, it provides a mechanism by which activation of Trk receptors may suppress p75NTR-mediated signaling through the Jun kinase cascade. It is notable that although Trk receptor kinase-mediated signaling suppresses proapoptotic responses mediated by p75NTR, Trk signaling does not inhibit induction by p75NTR of the NFκB cascade (Yoon et al 1998). Thus, in the presence of Trk signaling, activation of the NFκB cascade makes a synergistic contribution to survival (Maggirwar et al 1998, Hamanoue et al 1999).
Although kinase activity of Trk receptors suppresses signaling pathways mediated by p75NTR, Trk signaling is not invariably completely efficient at suppressing p75NTR-mediated apoptosis. NGF is able to increase apoptosis of cultured motoneurons from wild-type but not from p75NTR–/– embryos (Wiese et al 1999). In PC12 cells, BDNF binding to p75NTR has been reported to reduce NGF-dependent autophosphorylation of TrkA, possibly by promoting phosphorylation of a serine residue in the TrkA cytoplasmic domain (MacPhee & Barker 1997).
The overall picture that emerges from these studies is that the proapoptotic signals of p75NTR are largely suppressed by activation of Ras and PI-3 kinase by neurotrophins. Thus, p75NTR appears to refine the ligand-specificity of Trk receptors and may promote elimination of neurons not exposed to an appropriate neurotrophic factor environment. Consistent with this possibility, reductions in apoptosis have been observed in the retina and spinal cord in p75NTR–/– embryos (Frade & Barde 1999). It is less obvious why there are fewer sensory neurons in p75NTR mutant animals (Stucky & Koltzenburg 1997). The reductions in retrograde transport of many neurotrophins observed in the p75NTR mutant may increase apoptosis. In addition, the reduced rate of sensory axon outgrowth observed in these animals may create situations where axons are not exposed to adequate levels of neurotrophins. As neurotrophin expression is regulated during development by local tissue interactions not dependent on innervation (Patapoutian et al 1999), delays in innervation could have catastrophic consequences.
REGULATION OF NEURONAL SURVIVAL BY NEUROTROPHINS
The important experiments described above of Hamburger, Levi-Montalcini, and later workers demonstrated that NGF has essential roles in maintaining the viability of nociceptive sensory and sympathetic neurons in vivo (see Levi-Montalcini 1987, Purves 1988). These results suggested that all neurons may depend on trophic support derived from their targets for continued survival, not only during development but also in the adult nervous system. The observations also raised the possibility that neural precursors and developing neurons whose axons have not contacted their ultimate targets may also require trophic support.
Major extensions of this work have been made possible by development of gene targeting technology. With this technology, mice with deletions in genes encoding each of the neurotrophins and their receptors have been generated. Mice with deletions in almost all of the genes encoding GDNF, GDNF family members, and their receptors are also available and the few exceptions will almost certainly become available shortly. A summary of neuronal losses in the various knockouts of the neurotrophic factors and their receptors is presented in Table 1 and Table 2. In addition, Figure 3 provides a schematic overview of these losses in the peripheral nervous system. In many instances, a particular ganglion has not been examined in an individual mutant, frequently because the known expression of the receptor or responsiveness of the neurons in cell culture made a phenotype appear very unlikely. In other instances, the initial report focused on the most obvious phenotype, and examination of other possible phenotypes was deferred, often indefinitely. To summarize briefly, cranial ganglia neurons that transmit different modalities of sensory information tend to be segregated into different ganglia, so neurons in specific ganglia are often dramatically affected by loss of an individual signaling pathway. Within DRG sensory ganglia, modalities are mixed and mutants typically have severe effects only on functionally distinct subpopulations of cells.
TABLE 1.
Neuronal losses in neurotrophin and Trk-deficient micea
| Determinant | TrkA | NGF | TrkB | BDNFb | NT-4/5c | TrkC | NT-3 | TrkB/TrkCd | NT-3/BDNF | NT4/BDNFe | NT-3/BDNF/NT-4/5f | p75NTRg |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sensory ganglia | ||||||||||||
| Trigeminal | 70% | 75% | 60% | 30% | NS | 21% | 60% | ND | 74% | 9% | 88% | ND |
| N-P | ND | ND | 90% | 45% | 40% | 14% | 30% | ND | 62% | 90% | 96% | ND |
| Vestibular | NS | ND | 60% | 85% | NS | 15% | 20% | 100% | 100% | 89% | 100% | ND |
| Cochlear | NS | NS | 15% | 7% | ND | 50% | 85% | 65% | 100% | ND | ND | ND |
| Dorsal root | 70–90% | 70% | 30% | 35% | NS | 20% | 60% | 41% | 83% | NS | 92% | Small |
| Geniculate | ND | ND | ND | ND | ND | 11% | 35% | ND | ND | ND | 100% | ND |
| TMNh | ND | ND | 38% | 41% | 8% | 45% | 57% | ND | 88% | 46% | 95% | ND |
| Comments | —i | —i | —j | —k | —l | —m | —n | |||||
| Sympathetic ganglia | ||||||||||||
| Superior cervical | >95% | >95% | ND | ND | NS | NS | 50% | ND | ND | 47% | NS | |
| Motor | ||||||||||||
| Facial | ND | ND | ND | NS | ND | NS | 22% | ND | ||||
| Spinal cord | ND | ND | NS | NS | NS | ND | ND | ND | ND | NS | 20% | ND |
| CNS | —o | ND | —p | ND | ND | —q | —r | —s | ||||
| Viability | P | P | VP | PM | G | M | VP | VP | VP | VP | G | |
Note: Neuronal losses are expressed as the percentage of neurons lost in the mutants compared with the wild-type controls. This table is updated from a similar table in Reichardt & Fariñas (1997), in which original references for older papers is provided. N-P, Nodose-petrosal; TMN, Trigeminal mesencephalic nucleus neurons; ND, not done; NS, not significant; P, poor; VP, very poor; PM, poor to moderate; M, moderate; G, good.
Small CGRP (nociceptive) and BS1 (thermoceptive) positive neurons missing.
Myelinated and nonmyelinated axon lost. Ia afferents present. Complete loss of nodose-petrosal innervation of carotid body.
D-hair afferents completely lost.
Proprioceptive neurons missing.
Proprioceptive and cutaneous mechano-receptors missing. Partial losses of nociceptors. Partial losses of D-hair and SA fibers.
Partial deficits in all neurons.
Cholinergic basal forebrain neurons present. Reduced hippocampal innervation.
Deficits in NPY, calbindin, and parvalbumin expression. Cerebellar foliation defect.
No clear deficits.
Increased apoptosis in hippocampal and cerebellar granule neurons.
Increase in the number of forebrain cholinergic neurons.
TABLE 2.
Neuronal losses in other neurotrophic factor and receptor-deficient micea
| Determinant | GDNFg | Neurturinb, g | GFRα1c, g | GFRα2d | GFRα3e | c-retf, g | CNTFRα | LIFR |
|---|---|---|---|---|---|---|---|---|
| Sensory ganglia | NS | 70% reduction of GFRα2(+)neurons | NS | NS | ||||
| Trigeminal | NS | NS | ||||||
| N-P | 40% | NS | 15% | NS | ||||
| Vestibular | NS | NS | ||||||
| Cochlear | ND | |||||||
| Dorsal root | 23% | 45% reduction of pfGFRα2(+)neurons | NS | NS | NS | NS | ||
| Comments | GFRα1 neurons lost in trigeminal ganglion | 68% and 45% loss of GFRα2- expressing neurons in trigeminal and DRG | Only 10% of trigeminal neurons express GFRα2 | |||||
| Sympathetic ganglia | ||||||||
| SC | 35% | NS | NS | ND | ML | ML | NS | |
| Parasympathetic ganglia | ||||||||
| Ciliary | 40% | 48% | ND | NS | 48% | |||
| Submandibular | 36% | 45% | 33% | 81% | NS | 30% | ||
| Otic | 86% | NS (reductions in neuronal size) | NS | 99% | ||||
| Enteric nervous system | ||||||||
| Stomach | ML | ML | NS | |||||
| Intestine/colon | 100% | NS | 100% | NS | ||||
| Reduced VIPC & SPC fibers; reduced neuronal size | Reduced fiber density | |||||||
| Motor | ||||||||
| Facial | NS | NS | 40% | 35% | ||||
| Trigeminal | 19% | 22% | 35% | |||||
| Spinal cord | 22% | NS | 24% | 40% | ||||
| CNS | No deficit in TH+ neurons | No deficit in TH+ neurons | ||||||
| Viability | VP | G | VP | PM | G | G | VP | VP |
Note: Similar to Table 1, neuronal losses are expressed as the percentage of neurons lost in the mutants compared with the wild-type controls. See Reichardt & Fariñas (1997) for references of older papers. NS, Not significant; ND, not done; N-P, nodose-petrosal; SC, superior cervical; DRG, dorsal root ganglia; ML, most lost; VP, very poor; G, good, PM, poor to moderate.
Figure 3.
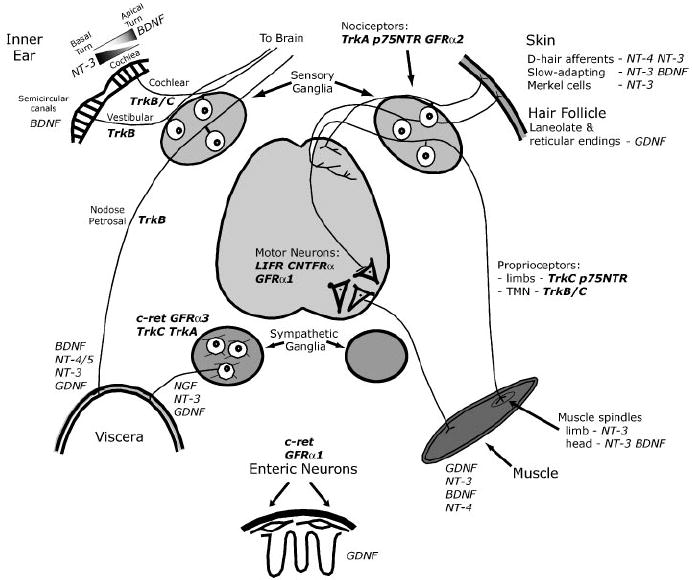
Summary of survival functions of neurotrophins in the peripheral nervous system. This diagram illustrates various components in the peripheral nervous system, including sensory ganglia, sympathetic ganglia, and enteric neurons. Only ligands or receptors with definitive loss of function phenotype are indicated in this figure. Ligands are indicated in italics and receptors are indicated in bold italics. (See text for abbreviations.)
Sensory Ganglia Survival
Trigeminal and Dorsal Root Ganglia
In DRG and trigeminal ganglia, neurons conveying different modalities of sensory information are present in the same ganglion, and substantial progress has been made in demonstrating differential neurotrophic factor dependencies of neurons with different sensory modalities. Thus, almost all nociceptive neurons express TrkA at some time during their development, and essentially all of these neurons are lost in the TrkA and NGF mutants (Crowley et al 1994, Smeyne et al 1994). A second major population of DRG neurons expresses TrkC from the time of initial neurogenesis. Most of these neurons differentiate into proprioceptive neurons conveying information from end organs, such as muscle spindles and tendon organs. At spinal cord levels, these neurons are completely lost in NT-3 and TrkC mutants together with end organs, such as muscle spindles, whose morphogenesis requires the presence of sensory axons. NT-3 expression is observed in both muscle spindles and the ventral spinal cord, both targets of proprioceptive Ia afferents, consistent with NT-3 functioning as a target-derived trophic factor. These neurons are lost almost immediately after neurogenesis, however, which suggests that they depend on NT-3 provided initially by intermediate targets (e.g. Fariñas et al 1996).
The expression patterns of Trk receptors in sensory neurons of mouse DRG and trigeminal ganglia have been characterized and correlate with the neuronal deficits in these ganglia. It has become clear that most neurons in both ganglia in mice, with few exceptions, express one Trk receptor during neurogenesis (Fariñas et al 1998, Huang et al 1999a). Although expression of Trk receptors remains unchanged in most sensory neurons, a small fraction of neurons show dynamic changes in switching of neurotrophin receptors and neurotrophin dependence (see, e.g. Enokido et al 1999).
Several transcription factors have been shown to regulate expression of Trk receptors in sensory ganglia. For instance, targeted deletion of the POU domain transcription factor Pou4f1 (Brn-3a/Brn-3.0) prevents initiation of TrkC expression in the trigeminal ganglion and results in downregulation of TrkA and TrkB in this ganglion at later times, resulting in apoptosis of neurotrophin-dependent neurons (McEvilly et al 1996, Huang et al 1999b). Absence of Pou4f1 also prevents normal expression of TrkC in the spiral ganglion (W Liu, EJ Huang, B Fritzsch, LF Reichardt, M Xiang, unpublished observations). The basic helix-loop-helix (bHLH) factor NeuroD also controls regulation of Trk receptor expression. Its absence results in severely reduced expression of TrkB and of TrkC in the embryonic vestibulocochlear ganglion (Kim et al 2001). Expression of TrkB and TrkC appears to be relatively normal in the trigeminal ganglion in this mutant, so the effects of this mutation on Trk receptor expression are surprisingly specific. Consistent with this, a recent analysis of cis-elements in the TrkA enhancer has identified sites required for global expression and sites that are specifically required for expression within sympathetic, DRG, or trigeminal neurons (Ma et al 2000). Thus, the transcriptional machinery that specifies TrkA expression is not the same in each of these neuronal populations. The specificity in the phenotypes of the mutants described above implies that transcriptional control of TrkB and TrkC must be equally complex. In summary, these results show that accurate control of the transcription of neurotrophin receptor genes is essential for ensuring normal neuronal survival and differentiation. In order to extend this work, it will be necessary to characterize the transcriptional machinery in each population of neurotrophin-responsive neurons.
Cutaneous Sensory Receptors and Innervation
Within both DRG and trigeminal ganglia, a well-defined subpopulation of the nociceptive neurons initiates expression of c-ret plus one or more of the GFR (GDNF family receptor)-α adapter subunits, eventually losing expression of TrkA (Molliver et al 1997, Huang et al 1999b). These neurons appear to be affected in mutants lacking constituents of these signaling pathways. In some instances, mutations have been shown to result in almost complete deficits in innervation of peripheral targets. For example, there are specific deficits in transverse lanceolate endings and reticular endings within the hair follicle in GDNF mutant heterozygotes, whereas other endings are not detectably affected (Fundin et al 1999).
Analyses of neurotrophin mutants also provided important information regarding the roles of neurotrophins in the development of cutaneous receptors. For example, in addition to the deficit in proprioceptors, NT-3 deficiency has been shown to result in deficits in specific cutaneous [D-hair (Down hair receptor) and SA (slow-adapting)] innervation and the development of Merkel cells (Airaksinen et al 1996), which are manifested only postnatally. The time course of development of these deficits indicates that NT-3 functions as a target-derived trophic factor for these neurons. NT-4 also functions as an essential survival factor for D-hair afferents (Stucky et al 1998). The dependence of these neurons on NT-4 appears to follow their dependence on NT-3. Unlike NT-4, BDNF is required for the survival of mechanical functions of SA fibers but plays no role in the survival of D-hair receptors (Carroll et al 1998). Together, these data indicate that BDNF and NT-4 have distinct, nonoverlapping roles in cutaneous innervation and that the expression of these neurotrophins may show spatial or temporal differences during the development of cutaneous receptors.
The presence of NT-4 has also been shown recently to be required for normal survival of TrkB-expressing DRG sensory neurons at the time of DRG formation (Liebl et al 2000). Since D-hair afferents are present during the first few postnatal weeks in the NT-4 mutant (Stucky et al 1998), they cannot be derived from this embryonic population of TrkB-expressing neurons.
Trigeminal Mesencephalic Nucleus
The trigeminal mesencephalic neurons are a group of neural crest–derived sensory neurons that reside in the brainstem at the pontomedullary junction. These neurons morphologically resemble sensory neurons in the peripheral ganglia and convey proprioceptive information from the head region. However, unlike proprioceptive neurons at the trunk level, which are completely dependent on NT-3, trigeminal mesencephalic neurons are only partially lost in the absence of NT-3 or TrkC, and partial neuronal deficits are also generated by the loss of BDNF or TrkB (Fan et al 2000, Matsuo et al 2000). It is interesting that the majority of these neurons are lost in double mutants lacking NT-3 and BDNF, and all these neurons are lost in triple mutants lacking NT-3, BDNF, and NT-4 (Fan et al 2000). Expression of a BDNFlacZ reporter has been detected in a subset of muscle spindles in one target of these neurons, the masseter muscle. It seems likely that each of the neurotrophins will prove to be expressed in subsets of these spindles, and that this explains why each neurotrophin supports the survival of a subset of these neurons.
Vestibular and Cochlear (Spiral) Ganglia
During development, these two ganglia first emerge as one single ganglion, which subsequently separates into two distinct ganglia that innervate the semicircular canals and cochlea. Because of their well-documented axon projection patterns, the vestibular and cochlear ganglia are an ideal system in which to investigate the effect of neurotrophins. The initial analyses of neurotrophin mutants showed that almost all neurons in the vestibular ganglion depend on BDNF for survival, whereas the vast majority of neurons in the cochlear (spiral) ganglion require NT-3 (Ernfors et al 1994 a,b; Jones et al 1994; Fariñas et al 1994). In a double NT-3/BDNF mutant, essentially all neurons in both ganglia are lost (Ernfors et al 1995).
The apparent dependence of separate populations of cochlear neurons on these two neurotrophins has stimulated investigations to understand the differences in the neurons or cochlea that explain the phenotypes. The first publications suggested that cell type–specific expression of NT-3 in inner hair cells and BDNF in outer hair cells controls the survival of the type I and type II neurons, which are responsible, respectively, for innervating each of these two hair cell populations (Ernfors et al 1995). Absence of TrkC was reported to result in preferential loss of innervation of inner hair cells, whereas deficiency in TrkB appeared to cause loss of innervation to outer hair cells (Schimmang et al 1995). Later investigations challenged these data, however, because neuronal losses and innervation deficits in these two sets of mutants were shown not to be distributed uniformly throughout the cochlea but to be distributed in gradients along the cochlear turns (Fritzsch et al 1997, 1998). Neurons in the basal turn of the cochlea were completely missing whereas those in the apical turn were much more mildly affected in the NT-3 and TrkC mutants. In the BDNF and TrkB mutants, the only obvious deficits were observed among neurons in the apical turn (Bianchi et al 1996, Fritzsch et al 1997, 1998).
Recent observations demonstrate that all neurons in the cochlear (spiral) ganglion express both TrkB and TrkC, indicating that they can be supported by either neurotrophin (I Fariñas, KR Jones, L Tessarollo, AJ Vigers, E Huang, M Kirstein, DC De Caprona, V Coppola, C Backus, LF Reichardt, B Fritzsch, unpublished observations). The phenotype of the NT-3 mutant can be explained by a spatial apical-to-basal gradient of BDNF expression, which in the absence of NT-3 causes a complete absence of trophic support for these neurons in the basal turn during a brief, but crucial, period of development. Using a β-galactosidase (LacZ) reporter integrated into either the NT-3 (NT-3lacZ) or BDNF (BDNFlacZ) locus to monitor gene expression, rapid changes in the expression patterns of both neurotrophins were seen as development proceeded. Approximately one day before the loss of neurons in the NT-3 mutant, however, expression of BDNF was barely detectable in the cochlea, with only weak expression in the apical turn and no detectable expression in the developing middle and basal turns. This suggested that in the NT-3 mutant, neurons were lost in the basal turn because BDNF was not present there to compensate for its absence, whereas neurons were partially spared in the apical turn because of the presence of low levels of BDNF. This model predicts that expression of BDNF under control of the NT-3 gene promoter and regulatory elements will rescue neuronal losses in the NT-3 mutant. This mouse has been generated and homozygotes are completely deficient in NT-3 (V Coppola, J Kucera, ME Palko, J Martinez-De Velasco, WE Lyons, B Fritzsch, L Tessarollo, unpublished observations). As predicted, neurons innervate normally all regions of the cochlea, including the basal turn, at E13.5 and P0, and there is almost complete rescue of basal turn spiral neurons (I Fariñas, KR Jones, L Tessarollo, AJ Vigers, E Huang, M Kirstein, DC De Caprona, V Coppola, C Backus, LF Reichardt, B Fritzsch, unpublished observations; V Coppola, J Kucera, ME Palko, J Martinez-De Velasco, WE Lyons, B Fritzsch, L Tessarollo, unpublished observations). Thus, there is convincing evidence that a spatial-temporal gradient of neurotrophin expression controls survival of these cells.
Nodose-Petrosal Ganglion
The nodose-petrosal ganglion contains neurons that are responsible for visceral sensory innervation. In this ganglion, all neurons express TrkB (Huang et al 1999a) and are lost in the BDNF–NT-4 double mutant or in the TrkB mutant (Conover et al 1995). Approximately half of the nodose-petrosal neurons are lost in the absence of either BDNF or NT-4 alone. The dopaminergic neurons responsible for innervation of the carotid body and other sensors of blood pH and pressure are completely dependent on BDNF, which is synthesized by these target organs during the initial period of innervation (Erickson et al 1996, Brady et al 1999). Thus, mice lacking BDNF show lack of innervation to chemo- and baroreceptors, resulting in deficits in control of breathing. In this ganglion, a neuron appears to depend on either BDNF or NT-4 alone, depending on which neurotrophin is expressed in the target that it innervates.
Sympathetic Ganglia Survival
Other populations of peripheral neurons also show strong dependencies on particular signaling pathways. As expected from the work of Levi-Montalcini, sympathetic neurons are almost completely lost in the absence of NGF to TrkA signaling (Crowley et al 1994, Smeyne et al 1994). Consistent with the expression patterns of TrkA in the sympathetic neurons, extensive cell death occurs perinatally in the sympathetic ganglion of TrkA mutants. In fact, a significant deficit is already present at E17.5 and develops progressively after birth (Fagan et al 1996). Unlike the prominent effects of NGF/TrkA on sympathetic neurons, the view on how NT-3 affects the development of sympathetic neurons has undergone a major revision in recent years. A number of experiments demonstrate that NT-3 is able to support the survival of early sympathetic neuroblasts in vitro (Birren et al 1993, Verdi & Anderson 1994, Verdi et al 1996). Consistent with these results, TrkC mRNA has been detected in early sympathetic ganglia. It is interesting that the level of TrkC mRNA decreases in sympathetic ganglion during late embryogenesis as the expression of TrkA increases. These studies suggested that NT-3 signaling through TrkC followed by NGF signaling through TrkA provides sequential support for cells in sympathetic ganglia during early and late stages of development, respectively. Therefore, it was not surprising when an initial analysis of the NT-3 mutant reported an increase of cell death among proliferating sympathetic precursor cells during initial formation of this ganglion (ElShamy et al 1996). However, no deficit was detected in a TrkC mutant (Fagan et al 1996), and later workers examining the NT-3 mutant have not detected an early phenotype in either precursors or neurons (Wyatt et al 1997, Francis et al 1999). Instead, the deficits in the NT-3 and NGF mutants appeared after E15.5 and developed during very similar stages of development (Wyatt et al 1997, Francis et al 1999). In agreement with these findings, NT-3lacZ is expressed in the target tissues of sympathetic innervation but is not present in or around the ganglion before E15.5 (Francis et al 1999). Thus, these data indicate that NT-3 and NGF are both required for the survival of sympathetic neurons, not neuroblasts. Because no deficits are seen in the TrkC mutant, the survival-promoting effects of NT-3 must be transmitted through TrkA. The ability of TrkA to mediate NT-3 signaling during development is promoted by the absence of p75NTR (Brennan et al 1999).
Although recent results do not support the concept that NT-3 and NGF act sequentially during development of sympathetic ganglia, members of the GDNF family of ligands do play essential early roles in development of one sympathetic ganglion, the superior cervical ganglion (SCG). The requirement for GDNF family members clearly precedes the dependence of SCG neurons on NT-3, NGF, and TrkA. Initially, mice lacking c-ret, the tyrosine kinase activated by GDNF, were shown to lack all neurons in the SCG without any obvious phenotype in the sympathetic chain at the trunk level (Durbec et al 1996). The absence of c-ret was shown to result in loss of a common set of neural crest–derived precursors for both the SCG and the enteric nervous system. In the absence of c-ret, these cells failed to survive and migrate to the future site of the SCG from the vicinity of the postotic hindbrain. More recently, mice lacking the GFRα3 binding subunit have been shown to have a somewhat similar, but significantly less severe, deficit attributable in part to an effect on early precursors of the SCG (Nishino et al 1999). GFRα3 mediates activation of c-ret by Artemin, a protein closely related to GDNF. In mice lacking GFRα3, the initial ventral migration to the aorta of SCG precursors is not obviously perturbed, but a later rostral migration to the future site of the SCG does not occur. Although a majority of the precursors exit the cell cycle and differentiate into immature neurons, these neurons do not mature, are not successful in contacting their normal targets, and undergo progressive, largely postnatal apoptosis. Artemin is expressed in the vicinity of the SCG precursors at E12.5, so a deficit in Artemin-mediated signaling is very likely to account for the early deficit in migration and may explain the later phenotype also. Because the deficit in the GFRα3 mutant is clearly less severe than that in the c-ret mutant, signaling through other adapter subunits must contribute to early development of SCG precursors. No deficit within the SCG has been reported in mice lacking the GFRα1 or GFRα2 subunits (Cacalano et al 1998, Rossi et al 1999). The phenotype of mice lacking the GFRα4 subunit has not been reported. It will be interesting to see whether double or triple mutants in GFRα subunit genes develop a phenotype in the SCG that is as severe as that observed in the c-ret mutant. Mice lacking GDNF have approximately 35% fewer SCG neurons at birth than do control animals (Moore et al 1996). Although the embryonic development of this phenotype has not been studied, it seems likely that a deficit in GDNF-mediated signaling contributes to the very early phenotype seen in the c-ret mutant.
Parasympathetic Ganglia and Enteric Neuron Survival
Enteric neurons in the intestine are almost completely lost in the absence of GDNF, GFRα1, or c-ret, and mice lacking neurturin or GFRα2 have neurons of reduced size with less fiber density (Table 2) (Sanchez et al 1996, Pichel et al 1996, Moore et al 1996, Durbec et al 1996, Cacalano et al 1998, Enomoto et al 1998). Absence of c-ret appears to affect not only early survival and migration of precursors, but also later differentiation of the precursors and the neurons arising from them (Pachnis et al 1998, Taraviras et al 1999, Natarajan et al 1999). C-ret is also required for normal development of the human enteric nervous system. Patients with Hirschsprung’s disease (or congenital megacolon) have been shown to have mutations in c-ret, which results in impaired migration of enteric neurons into the colon (Romeo et al 1994, Edery et al 1994).
The trophic factor dependencies of parasympathetic neurons are less completely defined. The GFRα2 subunit and c-ret are expressed in parasympathetic neurons (e.g. Nishino et al 1999), and major deficits in several parasympathetic ganglia have been detected in mice lacking Neurturin or GFRα2 (Rossi et al 1999, Heuckeroth et al 1999). No deficits have been reported in mice lacking GFRα1, GFRα3, or GDNF. However, parasympathetic ganglia may have been examined closely only in the GFRα3 mutants (Nishino et al 1999). These neurons are not affected by mutations in any of the neurotrophin or Trk receptor genes.
Motor Neuron Survival
With the exception of NGF, each of the neurotrophins is able to promote survival of purified motor neurons in vitro (reviewed in Reichardt & Fariñas 1997). GDNF, CNTF, and other CNTF-related cytokines are also potent survival factors for these neurons in vitro. Despite this, the vast majority of motor neurons are spared in mice lacking any single one of these factors in vivo. For example, no single neurotrophin or Trk mutant has a significant reduction in total number of motor neurons. Even a triple mutant lacking BDNF, NT-3, and NT-4 has only a 20% deficit in facial and spinal motor neurons (Liu & Jaenisch 2000). Deficits in the GDNF and GFRα1 mutants are small, approximately 20% in the spinal motor neuron population. The most severe deficits were seen in mice lacking either the CNTF receptor-α or the leukemia inhibitory factor receptor-β subunit, where deficits of approximately 40% were reported in the facial motor nucleus (see Reichardt & Fariñas 1997). Until recently, these results were believed to reflect functional redundancy, i.e. the concept that any motor neuron has access to multiple trophic factors in vivo. However, more recent data suggest strongly that different motor neuron pools are dependent on different trophic factors and that the deficits observed in mutants are small because there is so much diversity in the trophic factor dependence of the different pools. Analysis of the NT-3 mutant argued several years ago that continued survival of γ-motor neurons requires the presence of this neurotrophin (Kucera et al 1995). These efferents innervate muscle spindles, which express NT-3. These spindles are also innervated by NT-3–dependent sensory neurons, so motor neurons and sensory neurons that innervate the same end organ appear to require the same neurotrophin. In contrast, survival of α-motor neurons that innervate skeletal myotubes is not detectably reduced in the NT-3 mutant (Kucera et al 1995). During the past several years, different pools of motor neurons have been shown to express different combinations of transcription factors (e.g. Tanabe & Jessell 1996) and receptors for neurotrophic substances (e.g. Oppenheim et al 2000, Garces et al 2000). Recently, analyses of the GDNF and GFRα1 mutants have shown that specific pools and subpopulations of motor neurons are severely affected in each mutant, whereas other pools and subpopulations are not detectably affected (e.g. Oppenheim et al 2000, Garces et al 2000). When these studies are extended to other mutants, it now seems likely that mice lacking individual trophic factors or their receptors will prove to have severe deficits in individual populations of motor neurons.
CNS Neuron Survival
Perhaps most striking of all is the paucity of survival deficits observed in populations of CNS neurons, many of which are responsive to these same factors in cell culture. For example, CNS neurons responsive to NGF in vitro include basal forebrain and striatal cholinergic neurons. Although differentiation of these neurons is affected (Smeyne et al 1994), survival of these populations is not detectably affected perinatally in the NGF or TrkA knockout animals. Postnatal atrophy of NGF-dependent populations of cholinergic forebrain neurons has been observed in adult NGF mutant heterozygotes, however; thus, these neurons appear to retain some dependence on this neurotrophin (Chen et al 1997).
BDNF and NT-4–responsive neurons include cerebellar granule cells, mesencephalic dopaminergic neurons, and retinal ganglion cells. Although a modest increase in postnatal apoptosis of hippocampal and cerebellar granule cells is observed in TrkB and TrkB/TrkC mutants (Minichiello & Klein 1996, Alcantara et al 1997), it is not comparable to the dramatic losses observed in these same mutant animals in sensory neurons. To examine the roles of TrkB in the maintenance and function of adult nervous system, mice with cell type–specific deletions of TrkB have been generated using the Cre/loxP recombination system (Minichiello et al 1999, Xu et al 2000b). Selective deletion of TrkB in the pyramidal neurons of the neocortex leads to altered dendritic arborization and compression in cortical layers II/III and V (Xu et al 2000b). At later times, loss of TrkB also results in progressive elimination of SCIP (suppressed, cAMP-inducible POU)-expressing neurons in the somatosensory and visual cortices, whereas the Otx-1–expressing neurons are not lost (Xu et al 2000b). Taken together, the data described above indicate that neurotrophin-mediated signaling through TrkB is important for maintaining survival of neuronal populations in the CNS. Because weeks, not hours, are required to observe significant losses of neurons, it is uncertain whether interruption of the same signaling pathways important for survival of peripheral neurons accounts for these phenotypes.
Multiple Neurotrophic Factor Dependence
In summarizing dependencies of sensory neurons on neurotrophins, it is worth noting that, in general, the neurotrophin and Trk receptor mutant phenotypes are consistent with in vitro observations of ligand and receptor specificities. Analyses of the mutant phenotypes have provided valuable information for reconstructing at least four models that demonstrate unique receptor-ligand interactions in individual sensory ganglia. In the first model, a single neurotrophin appears to interact with a sole receptor. As a result, absence of either the ligand or receptor results in a similar phenotype. As one example, both the NGF and TrkA mutants appear to lack the same sensory neuron populations in the DRG and cranial ganglia (Table 1). In the second model, deficits in neuron numbers are larger in receptor-deficient than in neurotrophin-deficient mice. In the nodose-petrosal ganglion, the great majority of neurons express TrkB, and targeted deletion of TrkB leads to an almost complete absence of these neurons. However, BDNF and NT-4 appear to be expressed in different target fields, so each neurotrophin supports a separate subpopulation of neurons within this ganglion. In the third model, a single ligand interacts with multiple receptors. Several reports have documented the promiscuous role in vitro of NT-3 in activating TrkA and TrkB, in addition to activating TrkC (e.g. Ip et al 1993, Clary & Reichardt 1994, Davies et al 1995). In the DRG and trigeminal ganglia in an NT-3 mutant, many TrkA- and TrkB-expressing neurons undergo apoptotic cell death shortly after they are born (Fariñas et al 1998, Huang et al 1999a). These neurons are not killed in the TrkC mutant, so NT-3 must be directly activating the other Trk receptors. Thus, there must be high enough local concentrations of NT-3 in vivo to support neurons expressing TrkA or TrkB. Indeed, expression of NT-3 has been detected in the immediate vicinity of these ganglia during the period of neurogenesis (Fariñas et al 1998, Huang et al 1999a). By examining the expression of both NT-3lacZ and BDNFlacZ, we have identified distinct regions in the embryonic branchial arches where only the NT-3 gene is expressed between E11.5 and E12.5 (EJ Huang, unpublished data). Presumably, TrkB-expressing neurons that project to these regions will require NT-3 for their survival because BDNF is not available. This model predicts that replacement of NT-3 by BDNF should rescue some TrkB-expressing neurons. Consistent with this prediction, the phenotype in the DRG of mice in which BDNF is inserted into the NT-3 gene has recently been reported (V Coppola, J Kucera, ME Palko, J Martinez-De Velasco, WE Lyons, B Fritzsch, L Tessarollo, unpublished observations). In the complete absence of NT-3, the expression of BDNF in the same pattern as NT-3 can indeed delay and partially rescue neuronal deficits in the DRG. Finally, in the fourth model, survival of neurons that express more than one Trk receptor will be controlled by the patterns of neurotrophin expression. As described above, in the cochlear ganglion, neuronal survival in the NT-3 mutant is determined by an apical-to-basal gradient of BDNF. In the trigeminal mesencephalic ganglion, survival depends on whether NT-3 or BDNF is present in a particular muscle spindle (Fan et al 2000).
The data in Table 1 and Table 2, although incomplete, indicate that there is excellent correspondence between the specificities of ligand interactions with receptors as defined in vitro and the phenotypes of mutants lacking these proteins in vivo. This is evident from the analyses of mice lacking neurotrophins or Trk receptors. There are also striking similarities in the phenotypes of the GDNF and GFR-α1 and Neurturin and GFR-α2 mutants, which also agrees with the majority of biochemical studies characterizing interactions of these ligands with these two receptor subunits.
Switching of Neurotrophic Factor Dependence
The data in Table 1 also make it clear that many neurons require more than one neurotrophic factor-receptor signaling pathway to survive. In the vast majority of instances, this appears to reflect requirements at different developmental stages, made necessary by changes in ligand accessibility or in receptor expression. Examination in more detail of the development of the phenotypes of these mutants has suggested that ligand activation of tyrosine kinases is important for survival of neural precursors and immature neurons, as well as of mature neurons in contact with their final targets. In some instances, the same factor is essential at many stages of development. In others, different factors become important as development proceeds. For example, the SCG shows sequential dependency on c-ret–mediated followed by TrkA-mediated signaling during embryogenesis. Thus, the SCG is completely absent in the c-ret, NGF, and TrkA mutants, and less-severe deficiencies are seen in mice lacking GDNF, GFRα3, or NT-3. As discussed above, the requirement for c-ret clearly precedes that for the neurotrophins. A similar transient dependence on NT-3 of TrkA- and TrkB-expressing trigeminal neurons also appears to reflect the presence of NT-3, but not other neurotrophins, in the mesenchyme invaded by the axons of these neurons immediately after neurogenesis (Huang et al 1999a). Expression of NT-3 in the mesenchyme has been shown to be induced by epithelial-mesenchymal interactions, which are likely to be mediated by Wnt proteins secreted by the epithelium (Patapoutian et al 1999, O’Connor & Tessier-Lavigne 1999).
Although much attention has been focused on switches in neurotrophin responsiveness during early development of the trigeminal ganglion, more recent work suggests that there is comparatively little switching in expression of Trk receptors (Huang et al 1999a; but see also Enokido et al 1999). Instead, similar to the DRG (Ma et al 1999), neurons expressing different Trk receptors are born in waves that peak at different times. Much later in development, however, defined subsets of TrkA-expressing DRG and trigeminal ganglion neurons become responsive to GDNF family members, acquire expression of c-ret and one or more GFR adapter subunits, and lose responsiveness to NGF (e.g. Molliver et al 1997, Huang et al 1999b, Fundin et al 1999). Specific expression patterns of GDNF family ligands in targets and GFR-α adapter subunits in both targets and neurons indicate that these changes reflect the sorting out of subsets of sensory neurons specialized for innervation of specific targets and transmission of specific modalities of sensory information (Snider & McMahon 1998, Fundin et al 1999). There are many other examples, too numerous to review here, in which changes in ligand or receptor expression have been shown to explain the requirement for more than one neurotrophic factor by a single population of neurons.
Precursor Cell Survival and Proliferation
A large number of studies characterizing effects of neurotrophins on CNS neuroepithelial precursors, neural crest cells, or precursors of the enteric nervous system have demonstrated effects on both proliferation and differentiation of these cells in vitro and, in some instances, in vivo. Some of these have been described above, most notably the effect of c-ret–mediated signaling on SCG and enteric precursors. Only a few examples are described here, but they are representative of the interesting actions of these proteins on immature cells of the nervous system (for more extended descriptions, see Reichardt & Fariñas 1997).
In cell culture, many populations of CNS precursors are regulated by neurotrophins. Nestin-positive cells from the rat striatum can be induced to proliferate with NGF (Cattaneo & McKay 1990). The proliferation and survival of oligodendrocyte precursors (O2A progenitors) have been shown to be promoted by NT-3 in vitro and in vivo (Barres et al 1994). In other instances, neurotrophin application has been shown to induce differentiation of precursors. For example, NT-3, but not other neurotrophins, promotes differentiation of rodent cortical precursors (Ghosh & Greenberg 1995). The differentiation of hippocampal neuron precursors is promoted by BDNF, NT-3, and NT-4 (Vicario-Abejóon et al 1995). Although these observations suggest that neurotrophins regulate the size of precursor pools and neurogenesis in vivo, analyses of the various neurotrophin and Trk receptor knockouts has provided no evidence that development of these cell populations within the CNS is perturbed during embryogenesis.
NT-3 has been shown to promote proliferation of cultured neural crest cells (e.g. Sieber-Blum 1991, 1999). In vivo, however, initial formation of sensory and sympathetic ganglia appears to occur normally in mouse embryos (Fariñas et al 1996, Wilkinson et al 1996). Only after initiation of neurogenesis is elevated apoptosis observed in the NT-3 mutant, and apoptosis appears to be restricted to cells that have left the cell cycle and express neuronal markers (Wilkinson et al 1996; Fariñas et al 1996, 1998; Huang et al 1999a). This is consistent with evidence in murine sensory ganglia that expression of Trk receptors follows and does not precede withdrawal from the cell cycle (Fariñas et al 1998, Huang et al 1999a). Although direct effects of neurotrophin deficiency appear to be restricted to neurons, evidence for an indirect effect on precursors has been obtained in the DRG sensory ganglia of both NT-3 and NGF mutants (Fariñas et al 1996; I Fariñas, unpublished data). In these ganglia, the loss of neurons through apoptosis stimulates the differentiation of precursors into neurons without affecting their survival or rate of proliferation. As a result, the precursor pool is depleted during late stages of neurogenesis, and many fewer neurons than normal are born. Increased differentiation of precursors in these mutants could potentially be caused by a reduction in lateral inhibition mediated by the Delta-to-Notch signaling pathway.
The failure to detect Trk receptor expression in murine sensory ganglia precursors does not mean that these receptors are not expressed in any proliferating neural crest cell derivatives in mouse embryos. The mesenchymal cells derived from cardiac neural crest have been shown to be important in cardiac development in avian embryos, and a similar population has recently been identified in murine embryos (Jiang et al 2000). NT-3 and TrkC mutant embryos have severe cardiovascular abnormalities, including atrial and ventricular septal defects and pulmonary stenosis, which seemed likely to be caused by deficits in a neural crest–derived population of cells (Donovan et al 1996, Tessarollo et al 1997). More recently, however, expression of both NT-3 and TrkC have been detected in embryonic cardiac myocytes, and TrkC receptor-dependent signaling has been shown to promote proliferation of these myocytes during early development (Lin et al 2000). This raises the possibility that abnormalities in valve and outflow tracts observed in NT-3 and TrkC mutants are not caused by a signaling deficit in neural crest cell progeny. Effects could potentially be indirect. It is interesting that BDNF deficiency also results in abnormalities in the heart (Donovan et al 2000). BDNF activation of TrkB receptors expressed in the cardiac vasculature is essential for maintaining the survival of cardiac endothelial cells and integrity of the cardiac vascular bed in early postnatal animals.
ESSENTIAL ROLES IN DIFFERENTIATION AND FUNCTION
Sensory and Sympathetic Neuron Development
In a few instances, neurotrophins have been shown to regulate the pathways of differentiation selected by neural precursors and to regulate the differentiation process, helping to determine the levels of expression of proteins essential for the normal physiological functions of differentiated neurons, such as neurotransmitters, ion channels, and receptors. For example, in vitro and in vivo, NGF promotes the differentiation of sympathoadrenal precursors into sympathetic neurons as opposed to adrenal chromaffin cells (Levi-Montalcini 1987, Anderson 1993). In contrast, glucocorticoids have been shown to suppress these responses to NGF, inhibiting differentiation into neurons and promoting differentiation into mature adrenal chromaffin cells. As glucocorticoids are present in high concentrations in the adrenal gland, they seem likely to regulate differentiation of sympathoadrenal precursors similarly in vivo. In vitro and in vivo, the actions of NGF and glucocorticoids on these precursors are largely irreversible and dramatic. The sympathetic neurons formed are permanently dependent on NGF for survival, whereas the chromaffin cells are not. The two differentiated cell types differ in their predominant transmitter (norepinephrine vs epinephrine). Morphologically they are distinct, and this clearly reflects differences in many molecular constituents, which are regulated either directly or indirectly by NGF and the glucocorticoids. In PC12 cells, a very brief exposure to NGF has been shown to result in long-term induction of a sodium channel gene (Toledo-Aral et al 1995). This may serve as a model system for investigating how neurotrophins can cause irreversible fate changes within neurons.
As described above, precursors of murine sensory neurons do not appear to express Trk receptors in vivo or be affected directly by deficiencies in neurotrophins (Fariñas et al 1998, 2000; Huang et al 1999a). In mouse DRG, at least, initial generation of TrkB- and TrkC-expressing versus TrkA-expressing neurons appears to occur in two waves, dependent on sequential expression of neurogenins 2 and 1 (Ma et al 1999). The neurotrophins function to maintain the viability of these neurons. They do not appear to guide their initial determination. There is some evidence indicating that the situation may be more complicated in the chicken embryo. There, evidence has been obtained that suggests a subpopulation of DRG precursors expresses TrkC, and available evidence suggests there is more dynamic regulation of Trk receptor expression after neurogenesis than is observed in mouse sensory ganglia (e.g. Rifkin et al 2000). It seems unlikely, however, that the role of neurotrophins is very different there than in murine sensory ganglia.
Neurotrophins are important in regulating aspects of later sensory neuron development that, in some instances, control important aspects of neuronal phenotype. For example, NGF-responsive sensory neurons primarily convey nociceptive information and extend either unmyelinated C-fibers or thinly myelinated Aδ fibers. They express small peptide transmitters, such as CGRP and substance P, specific receptors such as the capsaicin receptor, and distinct Na+ channels isoforms (e.g. Amaya et al 2000, reviewed by Mendell et al 1999). Expression levels of most of these proteins are regulated by NGF (e.g. Fjell et al 1999; reviewed in Lewin & Barde 1996, Mendell 1999; Mendell et al 1999). Although the absence of NGF during embryogenesis results in loss of almost all nociceptive neurons, at later ages withdrawal of NGF no longer kills these neurons. Instead, perturbation of NGF levels results in phenotypic changes. When NGF is sequestered during the early postnatal period, the properties of Aδ fibers are dramatically changed (Ritter et al 1991). Normally, many of these fibers are responsive to high threshold mechanical stimulation and are classified as high-threshold mechanoreceptors (HTMRs). Postnatal deficiency in NGF results in almost complete loss of HTMRs with a proportional increase in D-hair fibers, which respond to light touch. As this change is not associated with elevated apoptosis, the results suggest that the neurons of origin for the HTMR fibers are not lost but instead undergo a change in phenotype, becoming D-hair fibers that function as touch, but not pain, receptors. The central projections of D-hair and HTMR fibers have different termination zones in the substantia gelatinosa, and it is interesting that the phenotypic conversion induced by withdrawal of NGF does not result in inappropriate innervation by D-hair fibers of zones normally innervated by HTMR fibers (Lewin & Mendell 1996). Central projections appear to be regulated to maintain appropriate modality connectivity.
Overexpression of NGF in skin using the keratin-14 promoter results in increased survival of both the C and Aδ classes of nociceptive neurons and, in addition, affects the functional properties of these neurons (Stucky et al 1999, Mendell et al 1999, Stucky & Lewin 1999). The percentage of Aδ fibers responsive to nociceptive stimuli increases from 65% to 97%, which may reflect selective survival of these neurons. Even more notably, the percentage of C-fibers responsive to heat increases from 42% to 96%, an effect too large to be accounted for by their selective survival. In addition, the chronic presence of NGF in skin also affects the functional properties of heat-sensitive C-fibers, increasing their thermal responsiveness and lowering their mechanical responsiveness. It seems likely that regulation of VR-1, the capsaicin receptor, may be involved in these phenotypic changes.
NGF is not the only neurotrophic factor to regulate the phenotype of nociceptive neurons. Although all nociceptors are believed initially to express TrkA, a proportion of these begin subsequently to express c-ret together with one or more of the GFR adapter subunits (e.g. Snider & McMahon 1998). In addition to expressing c-ret, these neurons are distinguished by expression of a binding site for the plant lectin isolectin B4. As development proceeds, their survival becomes dependent on GDNF family members (Molliver et al 1997). Targeted disruption of the GFR-α2 gene does not cause loss of cells expressing the isolectin B4 ligand but does result in a three-fold reduction in the percentage of isolectin B4 ligand-expressing neurons sensitive to heat (CL Stucky, J Rossi, MS Airaksinen, GR Lewin, unpublished observations). The results indicate that signaling mediated by a GDNF family member, most likely neurturin, is necessary for these neurons to manifest a nociceptive phenotype.
Continued presence of D-hair afferents has been shown to depend on NT-3 in early postnatal development and on NT-4 at later times (Airaksinen et al 1996, Stucky et al 1998). It is not certain whether these phenotypes are caused by neuronal loss or changes in neuronal phenotype.
Control of Target Innervation
Each of the neurotrophins has been shown to promote neurite outgrowth by responsive neurons in vitro. Elegant experiments have demonstrated that local NGF regulates the advance of sympathetic neuron growth cones (Campenot 1977). The presence of NGF within a compartment was shown to be essential for axons to grow into that compartment, even when neurons received adequate trophic support. When neurons were seeded between two chambers, one with NGF and one with no neurotrophin, axons invaded only the chamber containing NGF. If at a later time NGF was withdrawn from a chamber, the axons stopped growing and slowly retracted. In addition to promoting growth, gradients of neurotrophins are able to steer growth cones in vitro (Gundersen & Barrett 1979). It is intriguing that in these assays, whether a neurotrophin acts as a chemoattractant or a chemorepellent depends on cyclic nucleotide levels within neurons (Song et al 1997, Song & Poo 1999). The chemoattractive activities of NGF and BDNF, acting through TrkA and TrkB, respectively, are converted to chemorepellent activities by inhibitors of the cAMP signaling cascade. Effects of a PI-3 kinase inhibitor and of NGF signaling through a TrkA mutant lacking a putative PI-3 kinase docking site suggest that activation of PI-3 kinase is required for the chemoattractive response (Ming et al 1999). It is intriguing that although the different Trk receptors are believed to function through similar signal transduction pathways, the chemoattractive activity of NT-3, acting through TrkC, is not affected by agents that affect cAMP-mediated signaling. Instead, inhibitors of cGMP signaling convert this chemotrophic response from attractive to repulsive (Song & Poo 1999). These observations argue persuasively that there are fundamental differences in the signaling mediated by different Trk receptors.
Since the discovery of NGF, it has been appreciated that systematically applied neurotrophins affect innervation patterns in vivo (e.g. Levi-Montalcini 1987). NGF was shown to increase innervation of tissues that receive sympathetic or sensory innervation normally and to induce aberrant innervation of tissues that normally are not innervated. In adults, neurotrophins are generally concentrated in targets of sensory and sympathetic targets (e.g. see Reichardt & Fariñas 1997). Analyses of transgenic animals either lacking or expressing ectopically neurotrophins have provided many examples where disruptions of normal expression patterns of a neurotrophin results in perturbations of innervation, including aberrant routing of axons and interference with innervation of specific targets. For example, elevation of NGF in pancreatic islets using the insulin promoter induces dense sympathetic innervation of cells within the pancreatic islets, which normally are not innervated (Edwards et al 1989). Elevation of NGF in the epidermis using the keratin-14 promoter induces similarly dense sympathetic innervation of the epidermis (Guidry et al 1998). In this case also, the pattern of innervation is perturbed. Sympathetic innervation of the footpad vasculature and sweat glands is strongly inhibited. Instead, the sympathetic fibers are found in a plexus together with sensory fibers in the dermis. Sympathetic innervation is also distributed aberrantly in the vicinity of the mystacial pads (Davis et al 1997). Overexpression of NGF under control of the keratin-14 promoter also increases greatly the density of sensory innervation, selectively promoting innervation by NGF-dependent nociceptors (Stucky et al 1999). In contrast to sympathetic fibers, aberrant targeting of these endings was not detected. As a final example, in mice that overexpress BDNF under control of the nestin promoter, sensory fibers dependent on this neurotrophin appear to stall at sites of ectopic BDNF expression at the base of the tongue and fail to reach the gustatory papillae (Ringstedt et al 1999). Fibers that do not traverse these sites of ectopic BDNF expression are able to reach and innervate their targets normally. Taken together, the results of these studies indicate that elevated expression of a neurotrophin in a region usually results in an increased density of innervation by axons from neurons that normally innervate that region. Overexpression in regions that are normally not innervated often, but not always, results in aberrant innervation by neurons responsive to that neurotrophin. This suggests that guidance and targeting clues are either masked or overridden by the presence of high levels of a neurotrophin. Consistent with this concept, recent studies in tissue culture have shown that uniform exposure of TrkA-expressing neurons to NGF results in a desensitization of chemotactic responses to gradients of netrin, BDNF, or myelin-associated glycoprotein, which suggests that these factors share common cytosolic signaling pathways (Ming et al 1999).
In some instances, deficits or aberrancies in innervation observed in transgenic animals may reflect competitive phenomena. In analyses of innervation by sensory neuron fibers of mystacial pads, elevated innervation by TrkA-dependent sympathetic fibers was observed in mutants lacking BDNF or TrkB (Rice et al 1998). Excessive innervation by TrkA-dependent sensory endings was seen in mice lacking BDNF, NT-4, or TrkB. When innervation of different classes of endings that detect mechanosensation was examined in these mutants, each ligand and Trk receptor was shown to support innervation of at least one type of mechanoreceptor (Fundin et al 1997). Innervation of some endings is dependent on more than one neurotrophin or Trk receptor. For example, NT-3 is important for formation of all types of endings, but it may signal through different Trk receptors as development proceeds. In addition, the results suggested that BDNF signaling through TrkB may suppress Merkel innervation whereas NT-3 signaling through TrkC suppresses Ruffini innervation. There is not a single compelling explanation for these observations. Some sprouting may be attributable to loss of competition for a neurotrophin. Absence of TrkC-dependent endings, for example, may result in less NT-3 being transported out of the region. The elevated level of NT-3 remaining in the region may mediate sprouting of endings otherwise supported by NGF alone. Not all observations appear compatible with this model, however. Instead, the results suggest that neurotrophin-mediated signaling regulates, both positively and negatively, responses to other factors involved in sensory fiber targeting and differentiation.
Sensory Neuron Function
Neurotrophins have multiple interesting effects on the functional properties of sensory neurons extending beyond regulation of their survival. Essentially all modalities of sensory information are modulated in different ways by changes in the levels of these factors.
During early postnatal rodent development, both NT-3 and BDNF have been shown to regulate the development of the synapses formed between Ia afferents and motor neurons (Seebach et al 1999). Chronic NT-3 results in larger monosynaptic excitatory postsynaptic potentials (EPSPs) and reduced polysynaptic components, whereas BDNF actually reduces the size of the monosynaptic EPSPs and increases the contribution of polysynaptic signaling. Infusion with TrkB-Ig also results in the appearance of larger monosynaptic EPSPs, which suggests that endogenous BDNF is an important modulator of development of these synapses. Infusion with TrkC-Ig had little effect. These data argue that levels of endogenous BDNF within the spinal cord control the comparative efficiencies of monosynaptic and polysynaptic signaling between Ia afferents and motor neurons. The role of endogenous NT-3 is less certain.
During the first postnatal week, but not subsequently, direct application of NT-3 has been shown to acutely potentiate the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptor-mediated monosynaptic EPSP at the synapses formed by Ia afferents on motor neurons (Arvanov et al 2000). Potentiation is long lasting and is prevented by an inhibitor of Trk receptor kinase activity. Initiation, but not maintenance, of potentiation requires N-methyl-d-aspartate (NMDA) receptors and postsynaptic Ca2+. The dependence on NMDA receptor function and Ca2+ are similar to the requirements for generation of long-term potentiaion (LTP) in the hippocampus (see Malenka & Nicoll 1999). It is intriguing that EPSPs with similar properties evoked by stimulation of a different innervation pathway are not potentiated by NT-3, so the effects of NT-3 are synapse specific. NT-3 is effective at potentiating the strength of the Ia afferent-motor neuron synapse only during the first week, very likely because NMDA receptors are downregulated at later times.
Effects of peripheral nerve transection on monosynaptic Ia EPSPs, however, suggest that NT-3 from peripheral sources is also important in regulating the efficiency of synaptic function in adult animals. Peripheral transection results in long-term declines in conduction velocity and monosynaptic EPSP amplitude (Mendell et al 1999). These declines can be prevented by infusion of NT-3 in the vicinity of the cut nerve ending, which suggests that interruption of NT-3 transport is the cause of the synaptic deficiencies observed after transection.
Although overexpression of NGF induces the changes described in the previous section that are likely to involve gene expression, acute NGF also has striking effects on nociceptors. Application of NGF to the undersurface of a patch of skin acutely sensitizes nociceptive C and Aδ fibers to heat within 10 min (e.g. Shu & Mendell 1999a). Sensitization is not seen in the skin of animals depleted of mast cells, so a major pathway mediating this response is believed to involve activation of mast cells by NGF, resulting in secretion from these cells of serotonin, histamine, and other agents, including NGF (e.g. see Levi-Montalcini et al 1996). Acute application of NGF also sensitized the subsequent response of sensory neurons to capsaicin (Shu & Mendell 1999b). As sensitization is seen in dissociated sensory neuron cultures, NGF must be acting directly on the sensory neurons.
One of the proteins known to be upregulated by NGF in sensory neurons is the neurotrophin BDNF (Michael et al 1997). There is evidence that BDNF is transported to both peripheral and central terminals of nociceptive sensory neurons. In the periphery, BDNF and NT-4 have been shown to acutely sensitize nociceptive fibers by a pathway that requires the presence of mast cells (Shu et al 1999, Rueff & Mendell 1996). Sensory neuron–derived BDNF also appears to act centrally (Mannion et al 1999, Woolf & Costigan 1999). Perfusion of the spinal cord with TrkB-IgG has been shown to prevent the progressive hypersensitivity elicited by low-intensity tactile stimulation of inflamed tissues.
Cortical Circuitry and Function
Several neurotrophins are expressed in the neocortex and hippocampus during development, and their expression continues in adult animals, which suggests that they have functions extending beyond initial development. NGF, for example, is widely expressed in both the developing and adult neocortex (e.g. Large et al 1986). Projections from the cholinergic basal forebrain extend throughout the neocortex and hippocampus (e.g. Mesulam et al 1983). The fibers of these projections express TrkA (e.g. Sobreviela et al 1994), and expression in these neurons of proteins associated with cholinergic function, such as choline-o-acetyl transferase, is increased by infusion of NGF (e.g. Hefti et al 1989). NGF infusion has been shown to attenuate the behavioral deficits associated with cholinergic atrophy (e.g. Fischer et al 1987). Maintenance of normal function of these neurons in adult animals is sensitive to small perturbations of NGF levels. For example, animals heterozygous for a mutation in the NGF gene express approximately half the normal level of NGF mRNA and protein and have significant deficits in memory acquisition and retention, which can be corrected by prolonged infusion of NGF (Chen et al 1997). Mice lacking TrkA have also been shown to have deficits in cholinergic projections from the basal forebrain (Smeyne et al 1994).
Both BDNF and TrkB are widely expressed in the developing and adult hippocampus and neocortex (e.g. Cellerino et al 1996). BDNF mRNA is present in excitatory pyramidal neurons, but not in GABAergic inhibitory interneurons. TrkB is expressed by both classes of neurons, although its expression is higher in inhibitory interneurons. In addition, expression of BDNF is regulated by both sensory input and electrical activity. For example, induction of seizures in the hippocampus strongly induces BDNF expression (e.g. Kornblum et al 1997). In the visual and somatosensory cortices, expression has been shown to be regulated by sensory inputs, with deprivation reducing expression of this neurotrophin (e.g. Castren et al 1992, Rocamora et al 1996, Singh et al 1997). Several promoters control expression of BDNF mRNA, and one of these is regulated by Ca2+ acting through Ca2+-calmodulin–dependent protein kinase IV to phosphorylate and activate the transcription factor CREB (Tao et al 1998, Shieh et al 1998). BDNF is sorted into a regulated secretory pathway in hippocampal neurons (e.g. Farhadi et al 2000), so increases in neuronal activity should both activate transcription of the BDNF gene and increase secretion of the BDNF protein.
Expression of TrkB has also been shown to be modestly increased by activity (e.g. Castren et al 1992). Equally important, surface expression of TrkB is also regulated by activity (Meyer-Franke et al 1998). In the absence of activity, this protein appears to be largely sequestered into cytoplasmic vesicles. This result suggests that neurons become more responsive to BDNF as a result of activity. At the subcellular level, regulation of TrkB distribution may provide a mechanism by which active and inactive synapses differ in their responsiveness to BDNF, thereby regulating actin dynamics, glutamate receptor activity, and other functions important for adjusting synaptic function. In the absence of a direct demonstration of activity-regulated TrkB trafficking in vivo, however, this should be considered only an intriguing possibility.
In the neocortex, BDNF signaling through TrkB has been implicated in both development and maintainance of cortical circuitry. BDNF expression in excitatory neurons is promoted by activity, whereas increased release of BDNF can be expected to enhance the effectiveness of inhibitory interneurons. This has raised the possibility that BDNF-to-TrkB signaling modulates an autoregulatory circuit between excitatory pyramidal cells and inhibitory interneurons. In mixed cultures of postnatal rat cortical neurons, activity blockage has been shown to reduce reversibly GABA expression in interneurons and to reduce GABA-mediated inhibition on pyramidal cells (Rutherford et al 1997). In these cultures, the rates of firing are stabilized by scaling of the amplitude of AMPA receptor-mediated synaptic inputs (Rutherford et al 1998). These effects appear to be modulated by endogenous BDNF, as effects of activity blockade can be prevented by exogenous BDNF and effects of activity blockade are mimicked by a BDNF scavenger (Desai et al 1999). It is attractive to imagine that this autoregulatory circuit functions in vivo.
Formation of Ocular Dominance Columns
The density of innervation of layer IV by afferents from the thalamus is increased by exogenous BDNF and reduced by a scavenger of endogenous BDNF, TrkB-IgG (Cabelli et al 1997). Both agents appear to interfere with sorting of these afferents into ocular dominance columns, raising the possibility that competition for limiting amounts of BDNF by these afferents is involved in some manner in the sorting mechanism. In addition, infusion of NT-4 into the visual cortex during the critical period has been shown to prevent many of the consequences of monocular deprivation (Gillespie et al 2000). In the presence of NT-4, neurons remain responsive to stimuli from the deprived eye. Even after responses to the deprived eye are lost, infusion of NT-4 is able to restore them. These observations suggest that TrkB activation during the critical period promotes connectivity independent of correlated activity.
The function of inhibitory interneurons is essential for formation of ocular dominance columns (e.g. Hensch et al 1998), and maturation of these neurons is regulated by BDNF in vitro and in vivo. In initial analysis of the BDNF mutant, several deficits in interneuron maturation were detected, including expression of Ca2+-binding proteins and peptide neurotransmitters (Jones et al 1994). Recently, BDNF has been overexpressed in excitatory pyramidal neurons by use of the Ca2+-calmodulin–dependent protein kinase II promoter (Huang et al 1999c). In these animals, maturation of interneurons is accelerated, as assessed by expression and synaptic localization of glutamate decarboxylase, expression of parvalbumin, and the strength of inhibitory postsynaptic potentials. In addition, the critical period of ocular dominance plasticity begins and terminates precociously, and the acuity of vision increases on an accelerated time course (Huang et al 1999c, Hanover et al 1999). As visual stimulation also increased expression of BDNF within pyramidal neurons, the results suggest that early sensory stimulation acts to promote maturation of interneurons through BDNF-to-TrkB signaling. Interneurons in turn promote the refinements in synaptic connectivity needed for maturation of the cortex. The results suggest that refinement of cortical circuitry is driven by intracortical mechanisms, which then drive the sorting of thalamic afferents. Recent work supports this model (e.g. Trachtenberg et al 2000).
Another mechanism by which neurotrophins may control development and changes in cortical circuitry is through control of dendritic and axonal arbors. Neurotrophins affect neuronal morphologies at many levels in the visual pathway. Local application of BDNF or of a BDNF scavenger, for example, has been shown to decrease and increase, respectively, the complexity of Xenopus retinal ganglion cell dendritic arbors (Lom & Cohen-Cory 1999). Local application to the optic tectum of these same agents has different effects on axonal branching patterns, with BDNF increasing the complexity of retinal ganglion cell–derived axonal arbors and a BDNF scavenger having the opposite effect. NT-4 has been shown to prevent the atrophy of lateral geniculate neurons seen after monocular visual deprivation (Riddle et al 1995). Applications of BDNF, NT-3, or NT-4 to slices of neonatal neocortex have been shown to regulate the dendritic morphologies of pyramidal cells over comparatively short time spans (e.g. Horch et al 1999; reviewed in McAllister et al 1999). The effects are distinct, cell specific, and layer specific. For example, BDNF was observed to promote dendritic arborization of neurons in layers IV and V, but BDNF actually inhibited arborization by neurons in layer VI (McAllister et al 1997; see also Castellani & Boltz 1999). In layer IV, NT-3 was shown to oppose the stimulation of dendritic arborization promoted by BDNF. In layer VI, BDNF inhibited the stimulation of arborization induced by NT-3. Apical and basal dendrites of the same neurons responded differently to the same neurotrophin (e.g. McAllister et al 1995). Many of the observed effects were prevented by blocking electrical activity (e.g. McAllister et al 1996). Specific deletion of the TrkB gene in pyramidal neurons also results in striking changes in these cells during postnatal development, with significant retraction of dendrites observed at 6 weeks and loss of many neurons seen at 10 weeks of age (Xu et al 2000b). Although the changes in dendritic morphology and subsequent loss of neurons were cell autonomous, requiring deletion of the TrkB gene within the affected neurons, changes in gene expression were also seen that were not cell autonomous, i.e. they were observed in neurons that continued to express TrkB. It seems likely that circuit perturbation as a result of alterations of dendritic morphologies accounts for these changes.
Taken together, the results described above indicate that neurotrophins are important in regulating establishment and function of cortical circuits. Within the visual system, they regulate development of retinal ganglion cell axonal and dendritic arbors, thalamic afferents, cortical pyramidal cells, and cortical interneurons, with profound effects on cortical function. Although mechanisms by which neurotrophins influence axonal and dendritic morphologies in vivo have not be examined, they almost certainly involve regulation of the Cdc-42/Rac/Rho family of small GTPases. Aberrant expression of constitutively active and dominant negative mutants of these proteins have been shown to have dramatic effects on dendritic branch patterns and spine density (e.g. Li et al 2000, Nakayama et al 2000).
Synaptic Strength and Plasticity
Mechanisms underlying establishment of LTP between afferents from CA3 pyramidal cells and postsynaptic CA1 pyramidal neurons in the hippocampus have been of intense interest, as these mechanisms are believed to provide a paradigm for regulation of synaptic strength and plasticity (reviewed in Malenka & Nicoll 1999). BDNF is expressed in CA3 and CA1 pyramidal neurons within the hippocampus, and TrkB is expressed by almost all hippocampal neurons, including dentate granule cells, CA3 and CA1 pyramidal cells, and inhibitory interneurons. It is interesting that LTP is greatly reduced in BDNF mutants, both in homozygotes and in heterozygotes (e.g. Korte et al 1995, Patterson et al 1996). Long-lasting, protein synthesis-dependent LTP is not seen in these mutants (Korte et al 1998). There are also deficits in long-lasting LTP and in memory consolidation in the hippocampus in mutant mice lacking NT-4 (Xie et al 2000). CA1 LTP is also reduced in TrkB mutant heterozygotes and in a mouse mutant that expresses reduced levels of TrkB (Minichiello et al 1999, Xu et al 2000a). Signaling through p75NTR does not appear to be important, because there is very little expression of this receptor within the hippocampus and because functional antibodies to p75NTR do not affect LTP (Xu et al 2000a). Loss of TrkB from excitatory pyramidal neurons in the hippocampus and forebrain interferes with memory acquisition and consolidation in many learning paradigms (Minichiello et al 1999).
In these mutants, the observed reductions in synaptic plasticity probably reflect functional, not developmental, deficits. First, the hippocampi of these animals appear to be morphologically normal. Second, a very similar inhibition of LTP can be seen following acute application of the BDNF/NT-4 scavenger TrkB-IgG to hippocampal slices (e.g. Chen et al 1999). Finally, the deficits observed in BDNF mutant heterozygotes and homozygotes can be rescued by exposure of hippocampal slices to BDNF (Korte et al 1995, Patterson et al 1996). The enhanced efficiency of synaptic transmission observed after induction of LTP is largely mediated by an NMDA receptor-dependent increase in AMPA receptor function (reviewed in Malenka & Nicoll 1999). Most evidence suggests, however, that BDNF-to-TrkB signaling is not directly involved in the biochemical changes underlying LTP within the postsynaptic cells, but instead modulates the competence of presynaptic nerve terminals to generate the repetitive exocytotic events needed to modify the responses of these postsynaptic neurons. In one set of experiments, LTP was generated normally in a TrkB hypomorph by a low-frequency–paired depolarization protocol that specifically assesses properties of postsynaptic cells and puts minimal demands on presynaptic terminal function (Xu et al 2000a). In addition, AMPA and NMDA receptor functions appeared to be normal in the postsynaptic neurons. In contrast, the ability of presynaptic nerve terminals to respond to repetitive pulses of stimulation was clearly impaired. Consistent with a presynaptic deficit, BDNF has been shown to enhance synaptic vesicle release in response to tetanic stimulation, possibly by promoting docking of synaptic vesicles to the presynaptic membrane (e.g. Gottschalk et al 1998, Pozzo-Miller et al 1999). Also consistent with a presynaptic deficit, LTP is further reduced in a TrkB mutant heterozygote by elimination of the remaining functional TrkB gene from excitatory pyramidal cells in both the CA3 and CA1 regions, eliminating TrkB from both presynaptic and postsynaptic neurons (Minichiello et al 1999). LTP is not further reduced, however, by deletion of the TrkB gene solely within the postsynaptic CA1 pyramidal neurons in the mouse mutant that expressed reduced TrkB levels (Xu et al 2000a). All these data argue that BDNF-to-TrkB signaling is crucial in presynaptic nerve terminals in CA1 that are derived from neurons in CA3. In addition, BDNF has been shown to decrease inhibitory postsynaptic currents on CA1 pyramidal cells (Tanaka et al 1997, Frerking et al 1998), so it is possible that part of the LTP deficit reflects an increase in inhibitory signaling by GABAergic interneurons in the absence of normal TrkB function within these cells. BDNF has been shown to affect NMDA function in hippocampal neurons in culture by increasing the open probability of their channels (Levine et al 1998). The experiments described above suggest that TrkB did not modify the function of these channels during tetanic stimulation.
At many developing and mature synapses, application of a neurotrophin acutely stimulates neurotransmitter release. This has been most intensely studied at the CA1 synapse in the hippocampus and in developing Xenopus neuromuscular synapses (e.g. Kang & Schuman 1996, Wang & Poo 1997). In the developing neuromuscular cultures, the neurotrophins have both pre- and postsynaptic effects, increasing spontaneous and evoked release of synaptic vesicles and changing the kinetics of opening of the acetylcholine receptor (e.g. Wang & Poo 1997; Schinder et al 2000). Low levels of neurotrophin act synergistically with synaptic terminal depolarization (e.g. Boulanger & Poo 1999b). To be effective, the cell must be depolarized during the period of neurotrophin exposure. A cAMP agonist also synergizes with BDNF to potentiate spontaneous and action potential–evoked neurotransmitter release (Boulanger & Poo 1999a). The synergistic effect of depolarization appears to be caused by an increase in the level of cAMP. What is surprising is that a very similar presynaptic potentiation induced by NT-3 application is not affected by agents that inhibit cAMP-mediated signaling.
In hippocampal cultures, BDNF also potentiates release from presynaptic nerve terminals, but potentiation depends on inositol tris-phosphate gated Ca2+ stores in presynaptic nerve terminals (Li et al 1998a,b). LTP of synaptic transmission at the CA1 synapse in hippocampal slices has been seen by some, but not all, workers (e.g. Kang & Schuman 1995, Figurov et al 1996). The difference appears to be caused by differences in slice culture and conditions of application of BDNF. BDNF must be applied rapidly to slices to observe potentiation (Kang et al 1996). In conditions where it is observed, LTP also depends on inositol tris-phosphate gated Ca2+ stores, but in addition it requires local protein synthesis (Kang & Schuman 1996, 2000). In studies using slices of postnatal rat visual cortex, potentiation of synaptic transmission from layer IV cells to cells in layers II/III was seen only with very high concentrations of BDNF (Akaneya et al 1997). At lower concentrations, BDNF enhanced the magnitude of LTP without potentiating basal synaptic transmission. In both the hippocampus and postnatal visual cortex, BDNF enhances LTP in conditions where it does not potentiate synaptic transmission. The acute changes in BDNF concentration needed to potentiate synaptic transmission probably occur only rarely in vivo. BDNF also potentiates transmitter release from brain synaptosomes (Jovanovic et al 2000). In this case, it has been elegantly demonstrated that a MAP kinase phosphorylation of synapsin I mediates this response. The response is not seen in synaptosomes isolated from a synapsin I mutant and is prevented by inhibitors of the MAP kinase cascade. Phosphorylation of synapsins by MAP kinase has been shown to regulate their interactions with the actin cytoskeleton (Jovanovic et al 1996), so the MAP kinase cascade may potentiate synaptic transmission by releasing synaptic vesicles from the cytoskeleton, facilitating their entry into a exocytosis-competent pool. It will be interesting to determine whether BDNF-to-TrkB signaling regulates generation of LTP in mice lacking synapsin I.
CONCLUSION
The discovery of NGF and the neurotrophins was made possible by the observation that many populations of neurons depend on cell-cell interactions, specifically neuron-target interactions, for survival during embryonic development. The concept that limiting amounts of survival factors ensured a match between the number of neurons and the requirement for their functions was attractive. This model also provided a potential means for eliminating mistakes through death of neurons with aberrantly projecting axons. Thus, this was visualized as a mechanism for constructing the complex nervous systems of vertebrates. Subsequent studies have shown that these proteins are involved in many more aspects of neural development and function. Cell fate decisions, axon growth, dendrite pruning, synaptic function, and plasticity are all regulated by the neurotrophins. Studies of this small group of proteins have provided illuminating insights into most areas of contemporary neuroscience research and will almost certainly continue to do so in the future.
Acknowledgments
We wish to thank our many colleagues who shared papers and preprints with us. Without their help, this review could not have been written. We apologize to many of these same colleagues for our inability to discuss all work in this field because of space and time constraints. We thank Ardem Patapoutian and Michael Stryker for comments on the manuscript. Work from our laboratories has been supported by the National Institutes of Health and by the Howard Hughes Medical Institute.
Footnotes
The US Government has the right to retain a nonexclusive, royalty-free license in and to any copyright covering this paper.
NOTE ADDED IN PROOF In a recent study of the roles of GDNF and neurturin in the development of parasympathetic ganglia, GDNF has been shown to be required for precursor development, while neurturin regulates neuronal survival in the sphenopalatine and otic ganglia (Enomoto H et al 2000). A change in expression of GFRα receptors provides the most likely explanation for this switch.
LITERATURE CITED
- Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, et al. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–53. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- Agarwal ML, Taylor WR, Chernov MV, Chernova OB, Stark GR. The p53 network. J Biol Chem. 1998;273:1–4. doi: 10.1074/jbc.273.1.1. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Koltzenburg M, Lewin GR, Masu Y, Helbig C, et al. Specific subtypes of cutaneous mechanoreceptors require neurotrophin-3 following peripheral target innervation. Neuron. 1996;16:287–95. doi: 10.1016/s0896-6273(00)80047-1. [DOI] [PubMed] [Google Scholar]
- Akaneya Y, Tsumoto T, Kinoshinta S, Hatanaka H. Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. J Neurosci. 1997;17:6707–16. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara S, Frisen J, del Rio JA, Soriano E, Barbacic M, Silos-Santiago I. TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J Neurosci. 1997;17:3623–33. doi: 10.1523/JNEUROSCI.17-10-03623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allsopp TE, Robinson M, Wyatt S, Davies AM. TrkA mediates an NGF survival response in NGF-independent sensory neurons but not in parasympathetic neurons. Gene Ther. 1994;1(Suppl 1):S59. [PubMed] [Google Scholar]
- Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, et al. P53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol. 1998;143:1691–703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, et al. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–60. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Amaya F, Decosterd I, Samad TA, Plumpton C, Tate S, et al. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;16:331–42. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- Anderson DJ. Cell fate determination in the peripheral nervous system: the sympathoadrenal progenitor. J Neurobiol. 1993;24:185–98. doi: 10.1002/neu.480240206. [DOI] [PubMed] [Google Scholar]
- Anton ES, Weskamp G, Reichardt LF, Matthew WD. Nerve growth factor and its low-affinity receptor promote Schwann cell migration. Proc Natl Acad Sci USA. 1994;91:2795–99. doi: 10.1073/pnas.91.7.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Yamada M, Ohnishi H, Sano SI, Hatanaka H. BIT/SHPD-1 enhances brain-derived neurotrophic factor-promoted neuronal survival in cultured cerebral cortical neurons. J Neurochem. 2000a;75:1502–10. doi: 10.1046/j.1471-4159.2000.0751502.x. [DOI] [PubMed] [Google Scholar]
- Araki T, Yamada M, Ohnishi H, Sano SI, Uetsuki T, Hatanaka H. Shp-2 specifically regulates several tyrosine-phosphorylated proteins in brain-derived neurotrophic factor signaling in cultured cerebral cortical neurons. J Neurochem. 2000b;74:659–68. doi: 10.1046/j.1471-4159.2000.740659.x. [DOI] [PubMed] [Google Scholar]
- Arch RH, Gedrich RW, Thompson CB. Tumor necrosis factor receptor-associated factors (TRAFs)-a family of adapter proteins that regulates life and death. Genes Dev. 1998;12:2821–30. doi: 10.1101/gad.12.18.2821. [DOI] [PubMed] [Google Scholar]
- Arvanov VL, Seebach BS, Mendell LM. NT-3 evokes an LTP-like facilitation of AMPA/kainate receptor-mediated synaptic transmission in the neonatal rat spinal cord. J Neurophysiol. 2000;84:752–58. doi: 10.1152/jn.2000.84.2.752. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, et al. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–23. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Raff MC, Gaese F, Bartke I, Dechant G, Barde YA. A crucial role for neurotrophin-3 in oligodendrocyte development. Nature. 1994;367:371–75. doi: 10.1038/367371a0. [DOI] [PubMed] [Google Scholar]
- Barrett GL, Bartlett PF. The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. Proc Natl Acad Sci USA. 1994;91:6501–5. doi: 10.1073/pnas.91.14.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter GT, Radeke MJ, Kuo RC, Makrides V, Hinkle B, et al. Signal transduction mediated by the truncated TrkB receptor isoforms, TrkB.T1 and TrkB.T2. J Neurosci. 1997;17:2683–90. doi: 10.1523/JNEUROSCI.17-08-02683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazenet CE, Mota MA, Rubin LL. The small GTP-binding protein Cdc42 is required for nerve growth factor withdrawal-induced neuronal death. Proc Natl Acad Sci USA. 1998;95:3984–89. doi: 10.1073/pnas.95.7.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Howe CL, Wilde A, Brodsky FM, Mobley WC. NGF signals through TrkA to increase clathrin at the plasma membrane and enhance clathrin-mediated membrane trafficking. J Neurosci. 2000;20:7325–33. doi: 10.1523/JNEUROSCI.20-19-07325.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti M, Levi A, Chao MV. Differential expression of nerve growth factor receptors leads to altered binding affinity and neurotrophin responsiveness. Proc Natl Acad Sci USA. 1993;90:7859–63. doi: 10.1073/pnas.90.16.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley CA, Lee K-F. p75 is important for axon growth and Schwann cell migration during development. J Neurosci. 2000;20:7706–15. doi: 10.1523/JNEUROSCI.20-20-07706.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann I, Priestley JV, McMahon SB, Bröcker EB, Toyka KV, Koltzenburg M. Analysis of cutaneous sensory neurons in transgenic mice lacking the low affinity neurotrophin receptor p75. Eur J Neurosci. 1997;9:18–28. doi: 10.1111/j.1460-9568.1997.tb01349.x. [DOI] [PubMed] [Google Scholar]
- Bianchi LM, Conover JC, Fritzsch B, DeChiara T, Lindsay RM, Yancopoulos GD. Degeneration of vestibular neurons in late embryogenesis of both heterozygous and homozygous BDNF null mutant mice. Development. 1996;122:1965–73. doi: 10.1242/dev.122.6.1965. [DOI] [PubMed] [Google Scholar]
- Bibel M, Hoppe E, Barde YA. Biochemical and functional interactions between the neurotrophin receptors Trk and p75NTR. EMBO J. 1999;18:616–22. doi: 10.1093/emboj/18.3.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birren SJ, Lo L, Anderson DJ. Sympathetic neuroblasts undergo a developmental switch in trophic dependence. Development. 1993;119:597–610. doi: 10.1242/dev.119.3.597. [DOI] [PubMed] [Google Scholar]
- Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348(Part 2):241–55. [PMC free article] [PubMed] [Google Scholar]
- Boeshore KL, Luckey CN, Zigmond RE, Large TH. TrkB isoforms with distinct neurotrophin specificities are expressed in predominantly nonoverlapping populations of avian dorsal root ganglion neurons. J Neurosci. 1999;19:4739–47. doi: 10.1523/JNEUROSCI.19-12-04739.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–62. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223–53. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- Boulanger L, Poo MM. Gating of BDNF-induced synaptic potentiation by cAMP. Science. 1999a;284:1982–84. doi: 10.1126/science.284.5422.1982. [DOI] [PubMed] [Google Scholar]
- Boulanger L, Poo MM. Presynaptic depolarization facilitates neurotrophin-induced synaptic potentiation. Nat Neurosci. 1999b;2:346–51. doi: 10.1038/7258. [DOI] [PubMed] [Google Scholar]
- Brady R, Zaidi SI, Mayer C, Katz DM. BDNF is a target-derived survival factor for arterial baroreceptor and chemoafferent primary sensory neurons. J Neurosci. 1999;19:2131–42. doi: 10.1523/JNEUROSCI.19-06-02131.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan C, Rivas-Plata K, Landis SC. The p75 neurotrophin receptor influences NT-3 responsiveness of sympathetic neurons in vivo. Nat Neurosci. 1999;2:699–705. doi: 10.1038/11158. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Butte MJ, Hwang PK, Mobley WC, Fletterick RJ. Crystal structure of neurotrophin-3 homodimer shows distinct regions are used to bind its receptors. Biochemistry. 1998;37:16846–52. doi: 10.1021/bi981254o. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of TrkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- Cacalano G, Fariñas I, Wang LC, Hagler K, Forgie A, et al. GFRalpha1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 1998;21:53–62. doi: 10.1016/s0896-6273(00)80514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenot RB. Local control of neurite development by nerve growth factor. Proc Natl Acad Sci USA. 1977;74:4516–19. doi: 10.1073/pnas.74.10.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll P, Lewin GR, Koltzenberg M, Toyka KV, Thoenen H. A role for BDNF in mechanosensation. Nat Neurosci. 1998;1:42–46. doi: 10.1038/242. [DOI] [PubMed] [Google Scholar]
- Carter BD, Kalschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R, et al. Selective activation of NF-kB by nerve growth factor through neurotrophin receptor p75. Science. 1996;272:542–45. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- Casademunt E, Carter BD, Benzel I, Frade JM, Dechant G, Barde YA. The zinc finger protein NRIF interacts with the neurotrophin receptor p75NTR and participates in programmed cell death. EMBO J. 1999;18:6050–61. doi: 10.1093/emboj/18.21.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani V, Bolz J. Opposing roles for neurotrophin-3 in targeting and collateral formation of distinct sets of developing cortical neurons. Development. 1999;126:3335–45. doi: 10.1242/dev.126.15.3335. [DOI] [PubMed] [Google Scholar]
- Castren E, Zafra F, Thoenen H, Lindholm D. Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc Natl Acad Sci USA. 1992;89:9444–48. doi: 10.1073/pnas.89.20.9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo E, McKay R. Proliferation and differentiation of neuronal stem cells regulated by nerve growth factor. Nature. 1990;347:762–65. doi: 10.1038/347762a0. [DOI] [PubMed] [Google Scholar]
- Cellerino A, Maffei L, Domenici L. The distribution of brain-derived neurotrophic factor and its receptor TrkB in parvalbumin-containing neurons of the rat visual cortex. Eur J Neurosci. 1996;8:1190–97. doi: 10.1111/j.1460-9568.1996.tb01287.x. [DOI] [PubMed] [Google Scholar]
- Chao MV. The p75 neurotrophin receptor. J Neurobiol. 1994;25:1373–85. doi: 10.1002/neu.480251106. [DOI] [PubMed] [Google Scholar]
- Chen J, Fu H. Regulation of apoptosis signal-regulating kinase (ASK)1-induced cell death by Raf-1. Molec Biol Cell. 2000;11(S):1807. Abstr. [Google Scholar]
- Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci. 1999;19:7983–90. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KS, Nishimura MC, Armanini MP, Crowley C, Spencer SD, Phillips HS. Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurons and memory deficits. J Neurosci. 1997;17:7288–96. doi: 10.1523/JNEUROSCI.17-19-07288.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittka A, Chao MV. Identification of a zinc finger protein whose subcellular distribution is regulated by serum and nerve growth factor. Proc Natl Acad Sci USA. 1999;96:10705–10. doi: 10.1073/pnas.96.19.10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary DO, Reichardt LF. An alternatively spliced form of the nerve growth factor receptor TrkA confers an enhanced response to neurotrophin 3. Proc Natl Acad Sci USA. 1994;91:11133–37. doi: 10.1073/pnas.91.23.11133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly JL, Greene LA, Viscarello RR, Riley WD. Rapid, sequential changes in surface morphology of PC12 pheochromocytoma cells in response to nerve growth factor. J Cell Biol. 1979;82:820–27. doi: 10.1083/jcb.82.3.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover JC, Erickson JT, Katz DM, Bianchi LM, Poueymirou WT, et al. Neuronal deficits, not involving motor neurons, in mice lacking BDNF and/or NT4. Nature. 1995;375:235–38. doi: 10.1038/375235a0. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Foster DA, Rosner MR. Protein kinase Cδ mediates neurogenic but not mitogenic activation of mitogen-activated protein kinase in neuronal cells. Mol Cell Biol. 1999;19:4209–18. doi: 10.1128/mcb.19.6.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, et al. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–11. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- Curtis R, Adryan KM, Stark JL, Park JS, Compton DL, et al. Differential role of the low affinity neurotrophin receptor (p75) in retrograde axonal transport of the neurotrophins. Neuron. 1995;14:1201–11. doi: 10.1016/0896-6273(95)90267-8. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–27. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:1–20. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Davey F, Davies AM. TrkB signalling inhibits p75-mediated apoptosis induced by nerve growth factor in embryonic proprioceptive neurons. Curr Biol. 1998;8:915–18. doi: 10.1016/s0960-9822(07)00371-5. [DOI] [PubMed] [Google Scholar]
- Davies AM, Minichiello L, Klein R. Developmental changes in NT3 signalling via TrkA and TrkB in embryonic neurons. EMBO J. 1995;14:4482–89. doi: 10.1002/j.1460-2075.1995.tb00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BM, Fundin BT, Albers KM, Goodness TP, Cronk KM, Rice FL. Overexpression of nerve growth factor in skin causes preferential increases among innervation to specific sensory targets. J Comp Neurol. 1997;387:489–506. doi: 10.1002/(sici)1096-9861(19971103)387:4<489::aid-cne2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Davis BM, Goodness TP, Soria A, Albers KM. Over-expression of NGF in skin causes formation of novel sympathetic projections to TrkA-positive sensory neurons. NeuroReport. 1998;9:1103–7. doi: 10.1097/00001756-199804200-00027. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–11. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. BDNF regulates the intrinsic excitability of cortical neurons. Learn Mem. 1999;6:284–91. [PMC free article] [PubMed] [Google Scholar]
- Dobrowsky RT, Jenkins GM, Hannun YA. Neurotrophins induce sphingomyelin hydrolysis. Modulation by co-expression of p75NTR with Trk receptors. J Biol Chem. 1995;270:22135–42. doi: 10.1074/jbc.270.38.22135. [DOI] [PubMed] [Google Scholar]
- Donovan MJ, Hahn R, Tessarollo L, Hempstead BL. Identification of an essential nonneuronal function of neurotrophin 3 in mammalian cardiac development. Nat Genet. 1996;14:210–13. doi: 10.1038/ng1096-210. [DOI] [PubMed] [Google Scholar]
- Donovan MJ, Lin MI, Wiege P, Ringstedt T, Kraemer R, et al. Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development. 2000;127:4531–40. doi: 10.1242/dev.127.21.4531. [DOI] [PubMed] [Google Scholar]
- Durbec PL, Larsson-Blomberg LB, Schuchardt A, Costantini F, Pachnis V. Common origin and developmental dependence on c-ret of subsets of enteric and sympathetic neuroblasts. Development. 1996;122:349–58. doi: 10.1242/dev.122.1.349. [DOI] [PubMed] [Google Scholar]
- Edery P, Lyonnet S, Mulligan LM, Pelet A, Dow E, et al. Mutations of the RET protooncogene in Hirschsprung’s disease. Nature. 1994;367:378–80. doi: 10.1038/367378a0. [DOI] [PubMed] [Google Scholar]
- Edwards RH, Rutter WJ, Hanahan D. Directed expression of NGF to pancreatic beta cells in transgenic mice to selective hyperinnervation of the islets. Cell. 1989;58:161–70. doi: 10.1016/0092-8674(89)90412-1. [DOI] [PubMed] [Google Scholar]
- Eide FF, Lowenstein DH, Reichardt LF. Neurotrophins and their receptors: Current concepts and implications for neurologic disease. Exp Neurol. 1993;121:200–14. doi: 10.1006/exnr.1993.1087. [DOI] [PubMed] [Google Scholar]
- Eide FF, Vining ER, Eide BL, Zang K, Wang XY, Reichardt LF. Naturally occurring truncated TrkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci. 1996;16:3123–29. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers A, Whitfield J, Babji C, Rubin LL, Ham J. Role of the jun kinase pathway in the regulation of c-jun expression and apoptosis in sympathetic neurons. J Neurosci. 1998;18:1713–24. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElShamy WM, Linnarsson S, Lee K-F, Jaenisch R, Ernfors P. Prenatal and postnatal requirements of NT-3 for sympathetic neuroblast survival and innervation of specific targets. Development. 1996;122:491–500. doi: 10.1242/dev.122.2.491. [DOI] [PubMed] [Google Scholar]
- Enokido Y, Wyatt S, Davies AM. Developmental changes in the response of trigeminal neurons to neurotrophins: influence of birth date and the ganglion environment. Development. 1999;126:4365–73. doi: 10.1242/dev.126.19.4365. [DOI] [PubMed] [Google Scholar]
- Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, et al. GFR alpha1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron. 1998;21:317–24. doi: 10.1016/s0896-6273(00)80541-3. [DOI] [PubMed] [Google Scholar]
- Enomoto H, Heuckeroth RO, Golden JP, Johnson EM, Jr, Milbrandt J. Development of cranial parasympathetic ganglia requires sequential actions of GDNF and neurturin. Development. 2000;127:4877–89. doi: 10.1242/dev.127.22.4877. [DOI] [PubMed] [Google Scholar]
- Erickson JT, Conover JC, Borday V, Champagnat J, Barbacid M, et al. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental deficit in control of breathing. J Neurosci. 1996;16:5361–71. doi: 10.1523/JNEUROSCI.16-17-05361.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994a;368:147–50. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Kucera J, Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994b;77:503–12. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Van De Water T, Loring J, Jaenisch R. Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron. 1995;14:1153–64. doi: 10.1016/0896-6273(95)90263-5. Erratum. 1995. Neuron 15(3):739. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Zhang H, Landis SC, Smeyne RJ, Silos-Santiago I, Barbacid M. TrkA, but not TrkC, receptors are essential for survival of sympathetic neurons in vivo. J Neurosci. 1996;16:6208–18. doi: 10.1523/JNEUROSCI.16-19-06208.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Copray S, Huang EJ, Jones K, Yan Q, et al. Formation of a full complement of cranial proprioceptors requires multiple neurotrophins. Dev Dyn. 2000;218(2):359–70. doi: 10.1002/(SICI)1097-0177(200006)218:2<359::AID-DVDY9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Fandl JP, Tobkes NJ, McDonald NQ, Hendrickson WA, Ryan TE, et al. Characterization and crystallization of recombinant human neurotrophin-4. J Biol Chem. 1994;269:755–59. [PubMed] [Google Scholar]
- Farhadi HF, Mowla SJ, Petrecca K, Morris SJ, Seidah NG, Murphy RA. Neurotrophin-3 sorts to the constitutive secretory pathway of hippocampal neurons and is diverted to the regulated secretory pathway by coexpression with brain-derived neurotrophic factor. J Neurosci. 2000;20:4059–68. doi: 10.1523/JNEUROSCI.20-11-04059.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fariñas I, Jones KR, Backus C, Wang XY, Reichardt LF. Severe sensory and sympathetic deficits in mice lacking neurotrophin-3. Nature. 1994;369:658–61. doi: 10.1038/369658a0. [DOI] [PubMed] [Google Scholar]
- Fariñas I, Wilkinson GA, Backus C, Reichardt LF, Patapoutian A. Characterization of neurotrophin and Trk receptor functions in developing sensory ganglia: direct NT-3 activation of TrkB neurons in vivo. Neuron. 1998;21:325–34. doi: 10.1016/s0896-6273(00)80542-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fariñas I, Yoshida CK, Backus C, Reichardt LF. Lack of neurotrophin-3 results in death of spinal sensory neurons and premature differentiation of their precursors. Neuron. 1996;17:1065–78. doi: 10.1016/s0896-6273(00)80240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JP, Bamji SX, Causing CG, Aloyz R, Ase AR, et al. Functional evidence that BDNF is an anterograde neuronal trophic factor in the CNS. J Neurosci. 1998;18:2808–21. doi: 10.1523/JNEUROSCI.18-08-02808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Want T, Lu B. Regulation of synaptic resposnes to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;318:706–9. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Fischer W, Wictorin K, Bjorklund A, Williams LR, Varon S, Gage FH. Amelioration of cholinergic neurons atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature. 1987;329:65–68. doi: 10.1038/329065a0. [DOI] [PubMed] [Google Scholar]
- Fjell J, Cummins TR, Davis BM, Albers KM, Fried K, et al. Sodium channel expression in NGF-overexpressing transgenic mice. J Neurosci Res. 1999;57:39–47. doi: 10.1002/(SICI)1097-4547(19990701)57:1<39::AID-JNR5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Frade JM, Barde YA. Nerve growth factor: two receptors, multiple functions. BioEssays. 1998;20:137–45. doi: 10.1002/(SICI)1521-1878(199802)20:2<137::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Frade JM, Barde YA. Genetic evidence for cell death mediated by nerve-growth factor and the neurotrophin receptor p75 in the developing mouse retina and spinal cord. Development. 1999;126:683–90. doi: 10.1242/dev.126.4.683. [DOI] [PubMed] [Google Scholar]
- Francis N, Fariñas I, Rivas-Plata K, Backus C, Reichardt LF, Landis S. NT-3, like NGF, is required for survival of sympathetic neurons, but not their precursors. J Dev Biol. 1999;210:411–27. doi: 10.1006/dbio.1999.9269. [DOI] [PubMed] [Google Scholar]
- Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol. 1998;80:3383–86. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- Friedman W. Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci. 2000;20:6340–46. doi: 10.1523/JNEUROSCI.20-17-06340.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Barbacid M, Silos-Santiago I. The combined effects of TrkB and TrkC mutations on the innervation of the inner ear. Int J Dev Neurosci. 1998;16:493–505. doi: 10.1016/s0736-5748(98)00043-4. [DOI] [PubMed] [Google Scholar]
- Fritzsch B, Fariñas I, Reichardt LF. Lack of neurotrophin 3 causes losses of both classes of spiral ganglion neurons in the cochlea in a region-specific fashion. J Neurosci. 1997;17:6213–25. doi: 10.1523/JNEUROSCI.17-16-06213.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fundin BT, Mikaels A, Westphal H, Ernfors P. A rapid and dynamic regulation of GDNF-family ligands and receptors correlate with the developmental dependency of cutaneous sensory innervation. Development. 1999;126:2597–610. doi: 10.1242/dev.126.12.2597. [DOI] [PubMed] [Google Scholar]
- Fundin BT, Silos-Santiago I, Ernfors P, Fagan AM, Aldskogius H, et al. Differential dependency of cutaneous mechanoreceptors on neurotrophins, Trk receptors, and P75 LNGFR. Dev Biol. 1997;190:94–116. doi: 10.1006/dbio.1997.8658. [DOI] [PubMed] [Google Scholar]
- Garces A, Haase G, Airaksiner MS, Livet J, Filippi P, de Lypeyriere O. GFRα1 is required for differentiation of distinct sub-populations of motoneuron. J Neurosci. 2000;20:4992–5000. doi: 10.1523/JNEUROSCI.20-13-04992.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner AS, Menegay HJ, Boeshore KL, Xie XY, Voci JM, et al. Expression of TrkB receptor isoforms in the developing avian visual system. J Neurosci. 1996;16:1740–52. doi: 10.1523/JNEUROSCI.16-05-01740.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME. Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron. 1995;15:89–103. doi: 10.1016/0896-6273(95)90067-5. [DOI] [PubMed] [Google Scholar]
- Gillespie DC, Crair MC, Stryker MP. Neurotrophin-4/5 alters responses and blocks the effect of monocular deprivation in cat visual cortex during the critical period. J Neurosci. 2000;20:9174–86. doi: 10.1523/JNEUROSCI.20-24-09174.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–39. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz R, Koster R, Winkler C, Raulf F, Lottspeich F, et al. Neurotrophin-6 is a new member of the nerve growth factor family. Nature. 1994;372:266–69. doi: 10.1038/372266a0. [DOI] [PubMed] [Google Scholar]
- Grewal SS, York R, Stork PJS. Extracellular signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–53. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- Grimes ML, Beattie E, Mobley WC. A signaling organelle containing the nerve growth factor-activated receptor tyrosine kinase, TrkA. Proc Natl Acad Sci USA. 1997;94:9909–14. doi: 10.1073/pnas.94.18.9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidry G, Landis SC, Davis BM, Albers KM. Overexpression of nerve growth factor in epidermis disrupts the distribution and properties of sympathetic innervation in footpads. J Comp Neurol. 1998;393:231–43. doi: 10.1002/(sici)1096-9861(19980406)393:2<231::aid-cne7>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Guiton M, Gunn-Moore FJ, Glass DJ, Geis DR, Yancopoulos GD, Tavare JM. Naturally occurring tyrosine kinase inserts block high affinity binding of phospholipase C gamma and Shc to TrkC and neurotrophin-3 signaling. J Biol Chem. 1995;270:20384–90. doi: 10.1074/jbc.270.35.20384. [DOI] [PubMed] [Google Scholar]
- Gundersen RW, Barrett JN. Neuronal hemotaxis: chick dorsal root ganglion axons turn toward high concentrations of nerve growth factor. Science. 1979;206:1079–80. doi: 10.1126/science.493992. [DOI] [PubMed] [Google Scholar]
- Hallbook F. Evolution of the vertebrate neurotrophin and Trk receptor gene families. Curr Opin Neurobiol. 1999;9:616–21. doi: 10.1016/S0959-4388(99)00011-2. [DOI] [PubMed] [Google Scholar]
- Hamanoue M, Middleton G, Wyatt S, Jaffray E, Hay RT, Davies AM. p75-mediated NF-kB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol Cell Neurosci. 1999;14:28–40. doi: 10.1006/mcne.1999.0770. [DOI] [PubMed] [Google Scholar]
- Hanover JL, Huang ZJ, Tonegawa S, Stryker MP. Brain-derived neurotrophic factor overexpression induces precocious critical period in mouse visual cortex. J Neurosci. 1999;19:U12–16. doi: 10.1523/JNEUROSCI.19-22-j0003.1999. Online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hapner SJ, Boeshore KL, Large TH, Lefcort F. Neural differentiation promoted by truncated TrkC receptors in collaboration with p75(NTR) Dev Biol. 1998;201:90–100. doi: 10.1006/dbio.1998.8970. [DOI] [PubMed] [Google Scholar]
- Harrison SMW, Jones ME, Uecker S, Albers KM, Kudrycki KE, Davis BM. Levels of nerve growth factor and neurotrophin-3 are affected differentially by the presence of p75 in sympathetic neurons in vivo. J Comp Neurol. 2000;424:99–110. doi: 10.1002/1096-9861(20000814)424:1<99::aid-cne8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Hefti F, Hartikka J, Knussel B. Function of neruotrophic factors in the adult and aging brain and their possible use in the treatment of neurodegenerative diseases. Neurobiol Aging. 1989;10:515–33. doi: 10.1016/0197-4580(89)90118-8. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Birge RB, Fajardo JE, Glass-man R, Mahadeo D, et al. Expression of the v-crk oncogene product in PC12 cells results in rapid differentiation by both nerve growth factor- and epidermal growth factor-dependent pathways. Mol Cell Biol. 1994;14:1964–71. doi: 10.1128/mcb.14.3.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch T, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF. Local GABA circuit control of expreience-dependent plasticity in developing visual cortex. Science. 1998;282:1504–8. doi: 10.1126/science.282.5393.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z. Role of glycogen synthase kinase-3β in neuronal apoptosis induced by tophic withdrawal. J Neurosci. 1999;20:2567–74. doi: 10.1523/JNEUROSCI.20-07-02567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuckeroth RO, Enomoto H, Grider JR, Golden JP, Hanke JA, et al. Gene targeting reveals a critical role for neurturin in the development and maintenance of enteric, sensory, and parasympathetic neurons. Neuron. 1999;22:253–63. doi: 10.1016/s0896-6273(00)81087-9. [DOI] [PubMed] [Google Scholar]
- Holgado-Madruga M, Moscatello DK, Emlet DR, Dieterich R, Wong AJ. Grb2-associated binder-1 mediates phosphatidylinositol-3-OH kinase activation and the promotion of cell survival by nerve growth factor. Proc Natl Acad Sci USA. 1997;94:12419–24. doi: 10.1073/pnas.94.23.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horch HW, Kruttgen A, Portbury SD, Katz LC. Destabilization of cortical dendrites and spines by BDNF. Neuron. 1999;23:353–64. doi: 10.1016/s0896-6273(00)80785-0. [DOI] [PubMed] [Google Scholar]
- Hu S, Reichardt LF. From membrane to cytoskeleton: enabling a connection. Neuron. 1999;22:419–22. doi: 10.1016/s0896-6273(00)80696-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Wilkinson GA, Fariñas I, Backus C, Zang K, et al. Expression of Trk receptors in the developing mouse trigeminal ganglion: In vivo evidence for NT-3 activation of TrkA and TrkB in addition to TrkC. Development. 1999a;126:2191–2203. doi: 10.1242/dev.126.10.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Zang K, Schmidt A, Saulys A, Xiang M, Reichardt LF. POU domain factor Brn-3a controls the differentiation and survival of trigeminal neurons by regulating Trk receptor expression. Development. 1999b;126:2869–82. doi: 10.1242/dev.126.13.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, et al. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999c;98:739–55. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Thoenen H, Lindholm D. TrkA tyrosine residues involved in NGF-induced neurite outgrowth of PC12 cells. Eur J Neurosci. 1995;7:1125–33. doi: 10.1111/j.1460-9568.1995.tb01102.x. [DOI] [PubMed] [Google Scholar]
- Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, et al. Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron. 1993;10:137–49. doi: 10.1016/0896-6273(93)90306-c. [DOI] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–16. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Jones KR, Fariñas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–99. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, et al. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci USA. 1996;93:3679–83. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci. 2000;3:323–29. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- Kanamoto T, Mota MA, Takeda K, Rubin LL, Miyazopo K, et al. Role of apoptosis signal-regulating kinase in regulation of the c-Jun N-terminal kinase pathway and apoptosis in sympathetic neurons. Mol Cell Biol. 2000;20:196–204. doi: 10.1128/mcb.20.1.196-204.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Jia LZ, Suh KY, Tang L, Schuman EM. Determinants of BDNF-induced hippocampal synaptic plasticity: Role of the TrkB receptor and the kinetics of neurotrophin delivery. Learn Mem. 1996;3:188–96. doi: 10.1101/lm.3.2-3.188. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–62. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–6. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Intracellular Ca(2+) signaling is required for neurotrophin-induced potentiation in the adult rat hippocampus. Neurosci Lett. 2000;282:141–44. doi: 10.1016/s0304-3940(00)00893-4. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–91. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Khursigara G, Orlinick JR, Chao MV. Association of the p75 neurotrophin receptor with TRAF6. J Biol Chem. 1999;274:2597–600. doi: 10.1074/jbc.274.5.2597. [DOI] [PubMed] [Google Scholar]
- Kim W-Y, Fritzsch B, Serls A, Bakel LA, Huang EJ, et al. NeuroD-null mice are deaf due to a severe loss of the inner ear sensory neurons during development. Development. 2001;128:417–26. doi: 10.1242/dev.128.3.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JYH, Sutton ME, Lu DT, Cho TA, Goumnerova LC, et al. Activation of neurotrophin-3 receptor TrkC induces apoptosis in medulloblastomas. Cancer Res. 1999;59:711–19. [PubMed] [Google Scholar]
- Kinkelin I, Stucky CL, Koltzenburg M. Postnatal loss of Merkel cells, but not of slowly adapting mechanoreceptors in mice lacking the neurotrophin receptor p75. Eur J Neurosci. 1999;11:3963–69. doi: 10.1046/j.1460-9568.1999.00822.x. [DOI] [PubMed] [Google Scholar]
- Kjoller L, Hall A. Signaling to Rho GT-Pases. Exp Cell Res. 1999;253:166–79. doi: 10.1006/excr.1999.4674. [DOI] [PubMed] [Google Scholar]
- Kohn J, Aloyz RS, Toma JG, Haak-Frendsho M, Miller FD. Functionally antagonistic interactions between the TrkA and p75 neurotrophin receptors regulate sympathetic neuron growth and target innervation. J Neurosci. 1999;19:5393–408. doi: 10.1523/JNEUROSCI.19-13-05393.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblum HI, Sankar R, Shin DH, Wasterlain CG, Gall CM. Induction of brain derived neurotrophic factor mRNA by seizures in neonatal and juvenile rat brain. Brain Res Mol Brain Res. 1997;44:219–28. doi: 10.1016/s0169-328x(96)00224-0. [DOI] [PubMed] [Google Scholar]
- Korsching S. The neurotrophic factor concept: a reexamination. J Neurosci. 1993;13:2739–48. doi: 10.1523/JNEUROSCI.13-07-02739.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–60. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Kang H, Bonhoeffer T, Schuman E. A role for BDNF in the late-phase of hippocampal long-term potentiation. Neropharmacology. 1998;37:553–59. doi: 10.1016/s0028-3908(98)00035-5. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Roy DRS, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–76. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Kucera J, Ernfors P, Walro J, Jaenisch R. Reduction in the number of spinal motor neurons in neurotrophin-3-deficient mice. Neuroscience. 1995;69:321–30. doi: 10.1016/0306-4522(95)00221-4. [DOI] [PubMed] [Google Scholar]
- Kuruvilla R, Ye H, Ginty DD. Spatially and functionally distinct roles of the P13-K effector pathway during NGF signaling in sympathetic neurons. Neuron. 2000;27:499–512. doi: 10.1016/s0896-6273(00)00061-1. [DOI] [PubMed] [Google Scholar]
- Large TH, Bodary SC, Clegg DO, Weskamp G, Otten U, Reichardt LF. Nerve growth factor gene expression in the developing rat brain. Science. 1986;234:352–55. doi: 10.1126/science.3764415. [DOI] [PubMed] [Google Scholar]
- Lee KF, Bachman K, Landis S, Jaenisch R. Dependence on p75 for innervation of some sympathetic targets. Science. 1994a;263:1447–49. doi: 10.1126/science.8128229. [DOI] [PubMed] [Google Scholar]
- Lee KF, Davies AM, Jaenisch R. p75-deficient embryonic dorsal root sensory and neonatal sympathetic neurons display a decreased sensitivity to NGF. Development. 1994b;120:1027–33. doi: 10.1242/dev.120.4.1027. [DOI] [PubMed] [Google Scholar]
- Le-Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green D, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19:751–63. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–62. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R, Skaper SD, Dal Toso R, Petrelli L, Leon A. Nerve growth factor: from neurotrophin to neurokine. Trends Neurosci. 1996;19:514–20. doi: 10.1016/S0166-2236(96)10058-8. [DOI] [PubMed] [Google Scholar]
- Levine ES, Crozier R, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci USA. 1998;95:10235–9. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Mendell LM. Maintenance of modality-specific connections in the spinal cord after neonatal nerve growth factor deprivation. Eur J Neurosci. 1996;8:1677–84. doi: 10.1111/j.1460-9568.1996.tb01311.x. [DOI] [PubMed] [Google Scholar]
- Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM. Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci USA. 1998a;95:10884–89. doi: 10.1073/pnas.95.18.10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998b;18:10231–40. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Van Aelst L, Cline HT. Rho GT-Pases regulate distinct aspects of dendritic arbor growth in Xenopus central neurons in vivo. Nat Neurosci. 2000;3:217–25. doi: 10.1038/72920. [DOI] [PubMed] [Google Scholar]
- Liebl DJ, Klesse LJ, Tessarollo L, Wohlman T, Parada LF. Loss of brain-derived neurotrophic factor-dependent neural crest-derived sensory neurons in neurotrophin-4 mutant mice. Proc Natl Acad Sci USA. 2000;97(5):2297–302. doi: 10.1073/pnas.040562597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liepinsh E, Ilag LL, Otting G, Ibanez CF. NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J. 1997;16:4999–5005. doi: 10.1093/emboj/16.16.4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MI, Das I, Schwartz GM, Tsoulfas P, Mikawa T, Hempstead BL. Trk C receptor signaling regulates cardiac myocyte proliferation during early heart development in vivo. Dev Biol. 2000;226:180–91. doi: 10.1006/dbio.2000.9850. [DOI] [PubMed] [Google Scholar]
- Liu BP, Burridge K. Vav2 activates rac1, cdc42, and RhoA downstream from growth factor receptors but not beta1 integrins. Mol Cell Biol. 2000;20:7160–69. doi: 10.1128/mcb.20.19.7160-7169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jaenisch R. Severe peripheral sensory neuron loss and modest motor neuron reduction in mice with combined deficiency of brain-derived neurotrophic factor, neurotrophin 3 and neurotrophin 4/5. Dev Dyn. 2000;218:94–101. doi: 10.1002/(SICI)1097-0177(200005)218:1<94::AID-DVDY8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Lom B, Cohen-Cory S. Brain-derived neurotrophic factor differentially regulates retinal ganglion cell dendritic and axonal arborization in vivo. J Neurosci. 1999;19:9928–38. doi: 10.1523/JNEUROSCI.19-22-09928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Fode C, Guillemot F, Anderson DJ. Neurogenin-1 and neurogenin-2 control two distinct waves of neurogenesis in developing dorsal root ganglia. Genes Dev. 1999;13:1717–28. doi: 10.1101/gad.13.13.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Merenmies J, Parada LF. Molecular characterization of the TrkA/NGF receptor minimal enhancer reveals regulation by multiple cis elements to drive embryonic neuron expression. Development. 2000;127:3777–88. doi: 10.1242/dev.127.17.3777. [DOI] [PubMed] [Google Scholar]
- MacDonald JI, Gryz EA, Kubu CJ, Verdi JM, Meakin SO. Direct binding of the signaling adapter protein Grb2 to the activation loop tyrosines on the nerve growth factor receptor tyrosine kinase, TrkA. J Biol Chem. 2000;275:18225–33. doi: 10.1074/jbc.M001862200. [DOI] [PubMed] [Google Scholar]
- MacPhee IJ, Barker PA. Brain-derived neurotrophic factor binding to the p75 neurotrophin receptor reduces TrkA signaling whereas increasing serine phosphorylation in the TrkA intracellular domain. J Biol Chem. 1997;272:23547–51. doi: 10.1074/jbc.272.38.23547. [DOI] [PubMed] [Google Scholar]
- Maggirwar SDB, Sarmiere PD, Dewhurst S, Freeman RS. Nerve growth factor-dependent activation of NF-kb contributes to survival of sympathetic neurons. J Neurosci. 1998;18:10356–65. doi: 10.1523/JNEUROSCI.18-24-10356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadeo D, Kaplan L, Chao MV, Hempstead BL. High affinity nerve growth factor binding displays a faster rate of association than p140Trk binding. Implications for multi-subunit polypeptide receptors. J Biol Chem. 1994;269:6884–91. [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation–a decade of progress? Science. 1999;285:1870–74. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, et al. Neurotrophins: peripherally and centrrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci USA. 1999;96:9385–90. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M, Hashimoto Y, Muroya K, Hasegawa H, Kurata T, et al. CRK protein binds to two guanine nucleotide-releasing proteins for the Ras family and modulates nerve growth factor-induced activation of Ras in PC12 cells. Mol Cell Biol. 1994;14:5495–500. doi: 10.1128/mcb.14.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo S, Ichikawa H, Silos-Santiago I, Arends JJ, Henderson TA, et al. Proprioceptive afferents survive in the masseter muscle of TrkC knockout mice. Neuroscience. 2000;95:209–16. doi: 10.1016/s0306-4522(99)00424-8. [DOI] [PubMed] [Google Scholar]
- Mazzoni IE, Said FA, Aloyz R, Miller FD, Kaplan D. Ras regulates sympathetic neuron survival by suppressing the p53-mediated cell death pathway. J Neurosci. 1999;19:9716–27. doi: 10.1523/JNEUROSCI.19-22-09716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–64. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–78. doi: 10.1016/s0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- McDonald NQ, Chao MV. Structural determinants of neurotrophin action. J Biol Chem. 1995;270:19669–72. doi: 10.1074/jbc.270.34.19669. [DOI] [PubMed] [Google Scholar]
- McDonald NQ, Lapatto R, Murray-Rust J, Gunning J, Wlodawer A, Blundell TL. New protein fold revealed by a 2.3-A resolution crystal structure of nerve growth factor. Nature. 1991;354:411–14. doi: 10.1038/354411a0. [DOI] [PubMed] [Google Scholar]
- McEvilly RJ, Erkman L, Luo L, Sawchenko PE, Ryan AF, Rosenfeld MG. Requirement for Brn-3.0 in differentiation and survival of sensory and motor neurons. Nature. 1996;384:574–77. doi: 10.1038/384574a0. [DOI] [PubMed] [Google Scholar]
- Meakin SO, MacDonald JI. A novel juxtamembrane deletion in rat TrkA blocks differentiative but not mitogenic cell signaling in response to nerve growth factor. J Neurochem. 1998;71:1875–88. doi: 10.1046/j.1471-4159.1998.71051875.x. [DOI] [PubMed] [Google Scholar]
- Meakin SO, Gryz EA, MacDonald JI. A kinase insert isoform of rat TrkA supports nerve growth factor-dependent cell survival but not neurite outgrowth. J Neurochem. 1997;69:954–67. doi: 10.1046/j.1471-4159.1997.69030954.x. [DOI] [PubMed] [Google Scholar]
- Meakin SO, MacDonald JIS, Gryz EA, Kubu CJ, Verdi JM. The signaling adapter FRS-2 competes with Shc for binding to the nerve growth factor receptor TrkA. J Biol Chem. 1999;274:9861–70. doi: 10.1074/jbc.274.14.9861. [DOI] [PubMed] [Google Scholar]
- Meakin SO, Suter U, Drinkwater CC, Welcher AA, Shooter EM. The rat Trk protooncogene product exhibits properties characteristic of the slow nerve growth factor receptor. Proc Natl Acad Sci USA. 1992;89:2374–78. doi: 10.1073/pnas.89.6.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell LM. Neurotrophin action on sensory neurons in adults: an extension of the neurotrophic hypothesis. Pain. 1999;(Suppl 6):S127–32. doi: 10.1016/S0304-3959(99)00146-3. [DOI] [PubMed] [Google Scholar]
- Mendell LM, Albers KM, Davis BM. Neurotrophins, nociceptors, and pain. Microsc Res Tech. 1999;45:252–61. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<252::AID-JEMT9>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Wainer BH, Levey AI. Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch10Ch6) Neuroscience. 1983;10:1185–201. doi: 10.1016/0306-4522(83)90108-2. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, et al. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–93. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, et al. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci. 1997;17:8476–90. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton G, Hamanoue M, Enokido Y, Wyatt S, Pennica D, et al. Cytokine-induced nuclear factor kappa B activation promotes the survival of developing neurons. J Cell Biol. 2000;148:325–32. doi: 10.1083/jcb.148.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming G, Song H, Berninger B, Inagaki N, Tessier-Lavigne M, Poo MM. Phospholipase C-gamma and phosphoinositide 3-kinase mediate cytoplasmic signaling in nerve growth cone guidance. Neuron. 1999;23:139–48. doi: 10.1016/s0896-6273(00)80760-6. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Casagranda F, Tatche RS, Stucky CL, Postigo A, et al. Point mutation in TrkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron. 1998;21:335–45. doi: 10.1016/s0896-6273(00)80543-7. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Klein R. TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes Dev. 1996;10:2849–58. doi: 10.1101/gad.10.22.2849. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Huhn R, Unsicker K, et al. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–14. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Piehl F, Vazquez E, Schimmang T, Hokfelt T, et al. Differential effects of combined Trk receptor mutations on dorsal root ganglion and inner ear sensory neurons. Development. 1995;121:4067–75. doi: 10.1242/dev.121.12.4067. [DOI] [PubMed] [Google Scholar]
- Molliver DC, Wright DE, Leitner M, Parsadanian AS, Doster K, et al. IB4-binding DRG neurons switch from NGF to GDNF dependence in early postnatal life. Neuron. 1997;19:849–61. doi: 10.1016/s0896-6273(00)80966-6. [DOI] [PubMed] [Google Scholar]
- Moore MW, Klein RD, Fariñas I, Sauer H, Armanini M, et al. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- Muller G, Storkz P, Bourtelle S, Doppler H, Pfizenmaier K, et al. Regulation of Raf-1 kinase by TNF via its second messenger ceramide and crosstalk with mitogenic signalling. EMBO J. 1998;17:732–42. doi: 10.1093/emboj/17.3.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Smal GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–38. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan D, Grigoriou M, Marcos-Gutierrez CV, Atkins C, Pachnis V. Multipotential progenitors of the mammalian enteric nervous system capable of colonising aganglionic bowel in organ culture. Development. 1999;126:157–68. doi: 10.1242/dev.126.1.157. [DOI] [PubMed] [Google Scholar]
- Nilsson AS, Fainzilber M, Falck P, Ibanez CF. Neurotrophin-7: a novel member of the neurotrophin family from the zebrafish. FEBS Lett. 1998;424:285–90. doi: 10.1016/s0014-5793(98)00192-6. [DOI] [PubMed] [Google Scholar]
- Nimnual AS, Yatsula BA, Bar-Sagi D. Coupling of Ras and Rac guanosine triphosphatases through the Ras exchanger Sos. Science. 1998;279:560–63. doi: 10.1126/science.279.5350.560. [DOI] [PubMed] [Google Scholar]
- Nishino J, Mochida K, Ohfuji Y, Shimazaki T, Meno C, et al. GFR-alpha-3, a component of the artemin receptor, is required for migration and survival of the superior cervical ganglion. Neuron. 1999;23:725–36. doi: 10.1016/s0896-6273(01)80031-3. [DOI] [PubMed] [Google Scholar]
- Nosaka Y, Arai A, Miyasaka N, Miura O. CrkL mediates ras-dependent activation of the Raf/ERK pathway through the guanine nucleotide exchange factor C3G in hematopoietic cells stimulated with erythropoietin or interleukin-3. J Biol Chem. 1999;274:30154–62. doi: 10.1074/jbc.274.42.30154. [DOI] [PubMed] [Google Scholar]
- O’Connor R, Tessier-Lavigne M. Identification of maxillary factor, a maxillary process-derived chemoattractant for developing trigeminal sensory axons. Neuron. 1999;24:165–78. doi: 10.1016/s0896-6273(00)80830-2. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Houenou LJ, Parsadanian AS, Prevette D, Snider WD, Shen L. Glial-cell line-derived neurotrophic factor and developing mammalian motoneurons: regulation of programmed cell death among motoneuron subtypes. J Neurosci. 2000;20:5001–11. doi: 10.1523/JNEUROSCI.20-13-05001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachnis V, Durbec P, Taraviras S, Grigoriou M, Natarajan D. III. Role of the RET signal transduction pathway in development of the mammalian enteric nervous system. Am J Physiol Gastrointest Liver Physiol. 1998;275:G183–86. doi: 10.1152/ajpgi.1998.275.2.G183. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Backus C, Kispert A, Reichardt LF. Regulation of neurotrophin-3 expression by epithelial-mesenchymal interactions: The role of wnt factors. Science. 1999;283:1180–83. doi: 10.1126/science.283.5405.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TAS, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–45. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000;14:1027–47. [PubMed] [Google Scholar]
- Peterson DA, Dickinson-Anson HA, Leppert JT, Lee KF, Gage FH. Ceptral neuronal loss and behavioural impairment in mice lacking neurotrophin receptor p75. J Comp Neurol. 1999;404:1–20. doi: 10.1002/(sici)1096-9861(19990201)404:1<1::aid-cne1>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, et al. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Gottschalk W, Zhang L, Mc-Dermott K, Du J, et al. Impairments in high frequency transmission, synaptic vesicle docking and synaptic protein distribution in the hippocampus of BDNF knockout mice. J Neurosci. 1999;19:4972–83. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves D. Body and Brain A. Trophic Theory of Neural Connections. Cambridge, MA: Harvard Univ Press; 1988. [DOI] [PubMed] [Google Scholar]
- Qian X, Riccio A, Zhang Y, Ginty DD. Identification and characterization of novel substrates of Trk receptors in developing neurons. Neuron. 1998;21:1017–29. doi: 10.1016/s0896-6273(00)80620-0. [DOI] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- Reichardt LF, Fariñas I. Neurotrophic factors and their receptors. Roles in neuronal development and function. In: Cowan WM, Jessell TM, Zipursky SL, editors. Molecular and Cellular Approaches to Neural Development. New York: Oxford Univ. Press; 1997. pp. 220–63. [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–61. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- Riccio A, Pierchala BA, Ciarallo CL, Ginty DD. An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science. 1997;277:1097–1100. doi: 10.1126/science.277.5329.1097. [DOI] [PubMed] [Google Scholar]
- Rice FL, Albers KM, Davis BM, Silos-Santiago I, Wilkinson GA, et al. Differential dependency of unmyelinated and A delta epidermal and upper dermal innervation on neurotrophins, Trk receptors, and p75LNGFR. Dev Biol. 1998;198:57–81. [PubMed] [Google Scholar]
- Riddle DR, Lo DC, Katz LC. NT-4-mediated rescue of lateral geniculate neurons from effects of monocular deprivation. Nature. 1995;378:189–91. doi: 10.1038/378189a0. [DOI] [PubMed] [Google Scholar]
- Rifkin JT, Todd VJ, Anderson LW, Lefcort F. Dynamic expression of neurotrophin receptors during sensory neuron genesis and differentiation. Dev Biol. 2000;227:465–80. doi: 10.1006/dbio.2000.9841. [DOI] [PubMed] [Google Scholar]
- Ringstedt T, Ibáñez CF, Nosrat CA. Role of brain-derived neurotrophic factor in target invasion in the gustatory system. J Neurosci. 1999;19:3507–18. doi: 10.1523/JNEUROSCI.19-09-03507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter AM, Lewin GR, Kremer NE, Mendell LM. Requirement for nerve growth factor in the development of myelinated nociceptors in vivo. Nature. 1991;350:500–2. doi: 10.1038/350500a0. [DOI] [PubMed] [Google Scholar]
- Robinson M, Buj-Bello A, Davies AM. Paracrine interactions of BDNF involving NGF-dependent embryonic sensory neurons. Mol Cell Neurosci. 1996;7:143–51. doi: 10.1006/mcne.1996.0011. [DOI] [PubMed] [Google Scholar]
- Robinson RC, Radziejewski C, Spraggon G, Greenwald J, Kostura MR, et al. The structures of the neurotrophin 4 homodimer and the brain-derived neurotrophic factor/neurotrophin 4 heterodimer reveal a common Trk-binding site. Protein Sci. 1999;8:2589–97. doi: 10.1110/ps.8.12.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RC, Radziejewski C, Stuart DI, Jones EY. Structure of the brain-derived neurotrophic factor/neurotrophin 3 heterodimer. Biochemistry. 1995;34:4139–46. doi: 10.1021/bi00013a001. [DOI] [PubMed] [Google Scholar]
- Rocamora N, Welker E, Pascual M, Soriano E. Upregulation of BDNF mRNA expression in the barrel cortex of adult mice after sensory stimulation. J Neurosci. 1996;16:4411–19. doi: 10.1523/JNEUROSCI.16-14-04411.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Tebar A, Dechant G, Barde YA. Neurotrophins: structural relatedness and receptor interactions. Philos Trans R Soc London Ser B. 1991;331:255–8. doi: 10.1098/rstb.1991.0013. [DOI] [PubMed] [Google Scholar]
- Romeo G, Ronchetto P, Luo Y, Barone V, Seri M, et al. Point mutations affecting the tyrosine kinase domain of the RET protooncogene in Hirschsprung’s disease. Nature. 1994;367:377–8. doi: 10.1038/367377a0. [DOI] [PubMed] [Google Scholar]
- Rossi J, Luukko K, Poteryaev D, Laurikainen A, Sun YF, et al. Retarded growth and deficits in the enteric and parasympathetic nervous system in mice lacking GFR-alpha-2, a functional neurturin receptor. Neuron. 1999;22:243–52. doi: 10.1016/s0896-6273(00)81086-7. [DOI] [PubMed] [Google Scholar]
- Rueff A, Mendell LM. Nerve growth factor NT-5 induce increased thermal sensitivity of cutaneous nociceptors in vitro. J Neurophysiol. 1996;76:3593–96. doi: 10.1152/jn.1996.76.5.3593. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG. Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci. 1997;17:4527–35. doi: 10.1523/JNEUROSCI.17-12-04527.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–30. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Ryden M, Murray-Rust J, Glass D, Ilag LL, Trupp M, et al. Functional analysis of mutant neurotrophins deficient in low-affinity binding reveals a role for p75LNGFR in NT-4 signalling. EMBO J. 1995;14:1979–90. doi: 10.1002/j.1460-2075.1995.tb07190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi AH, Roux PP, Kubu CJ, Zeindleer C, Bhakar A, et al. NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron. 2000;27:279–88. doi: 10.1016/s0896-6273(00)00036-2. [DOI] [PubMed] [Google Scholar]
- Sanchez MP, Silos-Santiago I, Frisen J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382:70–73. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- Saragovi HU, Zheng W, Maliartchouk S, DiGugliemo GM, Mawal YR, et al. A TrkA-selective, fast internalizing nerve growth factor-antibody complex induces trophic but not neuritogenic signals. J Biol Chem. 1998;273:34933–40. doi: 10.1074/jbc.273.52.34933. [DOI] [PubMed] [Google Scholar]
- Schimmang T, Minichiello L, Vazquez E, San Jose I, Giraldez F, et al. Developing inner ear sensory neurons require TrkB and TrkC receptors for innervation of their peripheral targets. Development. 1995;121:3381–91. doi: 10.1242/dev.121.10.3381. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Berninger B, Poo MM. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron. 2000;25:151–63. doi: 10.1016/s0896-6273(00)80879-x. [DOI] [PubMed] [Google Scholar]
- Seebach BS, Arvanov V, Mendell LM. Effects of BDNF and NT-3 on development of Ia/motoneuron functional connectivity in neonatal rats. J Neurophysiol. 1999;81:2398–2405. doi: 10.1152/jn.1999.81.5.2398. [DOI] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Res Neurosci. 1996;19:463–89. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, et al. Human Trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci. 1995;15:477–91. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi ZQ, Yu DH, Park M, Marshall M, Feng GS. Molecular mechanism for the Shp-2 tyrosine phosphatase function in promoting growth factor stimulation of Erk activity. Mol Cell Biol. 2000;20:1526–36. doi: 10.1128/mcb.20.5.1526-1536.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–40. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Shindler KS, Yunker AM, Cahn R, Zha J, Korsmeyer SJ, Roth KA. Trophic support promotes survival of bcl-x-deficient telencephalic cells in vitro. Cell Death Differ. 1998;5:901–10. doi: 10.1038/sj.cdd.4400421. [DOI] [PubMed] [Google Scholar]
- Shu XQ, Llinas A, Mendell LM. Effects of TrkB and TrkC neurotrophin receptor agonists on thermal nociception: a behavioral and electrophysiological study. Pain. 1999;80:463–70. doi: 10.1016/S0304-3959(99)00042-1. [DOI] [PubMed] [Google Scholar]
- Shu XQ, Mendell LM. Neurotrophins and hyperalgesia. Proc Natl Acad Sci USA. 1999a;96:7693–96. doi: 10.1073/pnas.96.14.7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu XQ, Mendell LM. Nerve growth factor acutely sensitizes the response of adult rat sensory neurons to capsaicin. Neurosci Lett. 1999b;274:159–62. doi: 10.1016/s0304-3940(99)00701-6. [DOI] [PubMed] [Google Scholar]
- Sieber-Blum M. Role of the neurotrophic factors BDNF and NGF in the commitment of pluripotent neural crest cells. Neuron. 1991;6:949–55. doi: 10.1016/0896-6273(91)90235-r. [DOI] [PubMed] [Google Scholar]
- Sieber-Blum M. The neural crest colony assay: assessing molecular influences in development in culture. In: Haynes LW, editor. The Neuron in Tissue Culture. New York: Wiley; 1999. pp. 5–22. [Google Scholar]
- Singh TD, Mizuno K, Kohno T, Nakamura S. BDNF and TrkB mRNA expression in neurons of the neonatal mouse barrel field cortex: normal development and plasticity after cauterizing facial vibrissae. Neurochem Res. 1997;22:791–97. doi: 10.1023/a:1022075508176. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, et al. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246–409. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- Snider WD, McMahon SB. Tackling pain at the source: new ideas about nociceptors. Neuron. 1998;20:629–32. doi: 10.1016/s0896-6273(00)81003-x. [DOI] [PubMed] [Google Scholar]
- Sobreviela T, Clary DO, Reichardt LF, Brandabur MM, Kordower JH, Mufson EJ. TrkA-immunoreactive profiles in the central nervous system: colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J Comp Neurol. 1994;350:587–611. doi: 10.1002/cne.903500407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection and neural repair. Annu Rev Neurosci. 2001;24:1217–81. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- Song HJ, Ming GL, Poo MM. cAMP-induced switching in turning direction of nerve growth cones. Nature. 1997;388:275–79. doi: 10.1038/40864. [DOI] [PubMed] [Google Scholar]
- Song HJ, Poo MM. Signal transduction underlying growth cone guidance by diffusible factors. Curr Opin Neurobiol. 1999;9:355–63. doi: 10.1016/s0959-4388(99)80052-x. [DOI] [PubMed] [Google Scholar]
- Stephens RM, Loeb DM, Copeland TD, Pawson T, Greene LA, Kaplan DR. Trk receptors use redundant signal transduction pathways involving SHC and PLC-gamma 1 to mediate NGF responses. Neuron. 1994;12:691–705. doi: 10.1016/0896-6273(94)90223-2. [DOI] [PubMed] [Google Scholar]
- Strohmaier C, Carter B, Urfer R, Barde YA, Dechant G. A splice variant of the neurotrophin receptor TrkB with increased specificity for brain-derived neurotrophic factor. EMBO J. 1996;15:3332–37. [PMC free article] [PubMed] [Google Scholar]
- Stucky CL, Koltzenburg M. The low-affinity neurotrophin receptor p75 regulates the function but not the selective survival of specific subpopulations of sensory neurons. J Neurosci. 1997;17:4398–4405. doi: 10.1523/JNEUROSCI.17-11-04398.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucky CL, Koltzenburg M, DeChiara T, Lindsay RM, Yancopoulos GD. Neurotrophin 4 is required for the survival of a subclass of hair follicle receptors. J Neurosci. 1998;18:7040–46. doi: 10.1523/JNEUROSCI.18-17-07040.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucky CL, Koltzenburg M, Schneider M, Engle MG, Albers KM, Davis BM. Over-expression of nerve growth factor in skin selectively affects the survival and functional properties of nociceptors. J Neurosci. 1999;19:8509–16. doi: 10.1523/JNEUROSCI.19-19-08509.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucky CL, Lewin GR. Isolectin B4-positive and -negative nociceptors are functionally distinct. J Neurosci. 1999;19:6497–6505. doi: 10.1523/JNEUROSCI.19-15-06497.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y, Jessell TM. Diversity and pattern in the developing spinal cord. Science. 1996;274:1115–23. doi: 10.1126/science.274.5290.1115. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Hattori S, Kurata T, Nagashima K, Fukui Y, et al. Both the SH2 and SH3 domains of human CRK protein are required for neuronal differentiation of PC12 cells. Mol Cell Biol. 1993;13:4409–15. doi: 10.1128/mcb.13.7.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Saito H, Matsuki N. Inhibition of GABAa synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–66. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–26. doi: 10.1016/s0896-6273(00)81010-7. Erratum. 1998. Neuron 20(6):1297. [DOI] [PubMed] [Google Scholar]
- Taraviras S, Marcos-Gutierrez CV, Durbec P, Jani H, Grigoriou M, et al. Signalling by the RET receptor tyrosine kinase and its role in the development of the mammalian enteric nervous system. Development. 1999;126:2785–97. doi: 10.1242/dev.126.12.2785. [DOI] [PubMed] [Google Scholar]
- Tessarollo L, Tsoulfas P, Donovan MJ, Palko ME, Blair-Flynn J, et al. Targeted deletion of all isoforms of the TrkC gene suggests the use of alternate receptors by its ligand neurotrophin-3 in neuronal development and implicates TrkC in normal cardiogenesis. Proc Natl Acad Sci USA. 1997;94:14776–81. doi: 10.1073/pnas.94.26.14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoenen H, Barde YA. Physiology of nerve growth factor. Physiol Rev. 1980;60:1284–335. doi: 10.1152/physrev.1980.60.4.1284. [DOI] [PubMed] [Google Scholar]
- Toledo-Aral JJ, Brehm P, Halegoua S, Mandel G. A single pulse of nerve growth factor triggers long-term neuronal excitability through sodium channel gene induction. Neuron. 1995;14:607–11. doi: 10.1016/0896-6273(95)90317-8. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Trepel C, Stryker MP. Rapid extragranular plasticity in the absence of thalamocortical plasticity in the developing primary visual cortex. Science. 2000;287:2029–32. doi: 10.1126/science.287.5460.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoulfas P, Stephens RM, Kaplan DR, Parada LF. TrkC isoforms with inserts in the kinase domain show impaired signaling responses. J Biol Chem. 1996;271:5691–97. doi: 10.1074/jbc.271.10.5691. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Ginty DD. Characterization of an NGF-P-TrkA retrograde-signaling complex and age-dependent regulation of TrkA phosphorylation in sympathetic neurons. J Neurosci. 1999;19:8207–18. doi: 10.1523/JNEUROSCI.19-19-08207.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ultsch MH, Wiesmann C, Simmons LC, Henrich J, Yang M, et al. Crystal structures of the neurotrophin-binding domain of TrkA, TrkB and TrkC. J Mol Biol. 1999;290:149–59. doi: 10.1006/jmbi.1999.2816. [DOI] [PubMed] [Google Scholar]
- Urfer R, Tsoulfas P, O’Connell L, Hongo JA, Zhao W, Presta LG. High resolution mapping of the binding site of TrkA for nerve growth factor and TrkC for neurotrophin-3 on the second immunoglobulin-like domain of the Trk receptors. J Biol Chem. 1998;273:5829–40. doi: 10.1074/jbc.273.10.5829. [DOI] [PubMed] [Google Scholar]
- Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD. Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-Akt pathway to synergistically regulate neuronal survival. J Cell Biol. 1999;146:955–66. doi: 10.1083/jcb.146.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdi JM, Anderson DJ. Neurotrophins regulate sequential changes in neurotrophin receptor expression by sympathetic neuroblasts. Neuron. 1994;13:1359–72. doi: 10.1016/0896-6273(94)90421-9. [DOI] [PubMed] [Google Scholar]
- Verdi JM, Birren SJ, Ibanez CF, Persson H, Kaplan DR, et al. p75LNGFR regulates Trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron. 1994;12:733–45. doi: 10.1016/0896-6273(94)90327-1. [DOI] [PubMed] [Google Scholar]
- Verdi JM, Groves AK, Fariñas I, Jones K, Marchionni MA, et al. A reciprocal cell-cell interaction mediated by NT-3 and neuregulins controls the early survival and development of sympathetic neuroblasts. Neuron. 1996;16:515–27. doi: 10.1016/s0896-6273(00)80071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter ML, Martin-Zanca D, Parada LF, Bishop JM, Kaplan DR. Nerve growth factor rapidly stimulates tyrosine phosphorylation of phospholipase C-gamma 1 by a kinase activity associated with the product of the Trk protooncogene. Proc Natl Acad Sci USA. 1991;88:5650–54. doi: 10.1073/pnas.88.13.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario-Abejón C, Johe KK, Hazel TG, Collazo D, McKay RDG. Functions of basic fibroblast growth factor and neurotrophins in the differentiation of hippocampal neurons. Neuron. 1995;15:105–14. doi: 10.1016/0896-6273(95)90068-3. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS, Byers MR, Williams R, Bothwell M. Anterograde transport of neurotrophins and axodendritic transfer in the developing visual system. Nature. 1996;379:830–33. doi: 10.1038/379830a0. [DOI] [PubMed] [Google Scholar]
- Walsh GS, Krol KM, Crutcher KA, Kawaja MD. Enhanced neurotrophin-induced axon growth in myelinated portions of the CNS in mice lacking the p75 neurotrophin receptor. J Neurosci. 1999a;19:4155–68. doi: 10.1523/JNEUROSCI.19-10-04155.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh GS, Krol KM, Kawaja MD. Absence of the p75 neurotrophin receptor alters the pattern of sympathosensory sprouting in the trigeminal ganglia of mice overexpressing nerve growth factor. J Neurosci. 1999b;19:258–73. doi: 10.1523/JNEUROSCI.19-01-00258.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XH, Poo MM. Potentiation of developing synapses by postsynaptic release of neurotrophin-4. Neuron. 1997;19:825–35. doi: 10.1016/s0896-6273(00)80964-2. [DOI] [PubMed] [Google Scholar]
- Wendland B, Emr SD, Riezman H. Protein traffic in the yeast endocytic and vacuolar protein sorting pathways. Curr Opin Cell Biol. 1998;10:513–22. doi: 10.1016/s0955-0674(98)80067-7. [DOI] [PubMed] [Google Scholar]
- Wiese S, Metzger F, Holtmann B, Sendtner M. The role of p75NTR in modulating neurotrophin survival effects in developing motoneurons. Eur J Neurosci. 1999;11:1668–76. doi: 10.1046/j.1460-9568.1999.00585.x. [DOI] [PubMed] [Google Scholar]
- Wiesmann C, Ultsch MH, Bass SH, de Vos AM. Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature. 1999;401:184–88. doi: 10.1038/43705. [DOI] [PubMed] [Google Scholar]
- Wilde A, Beattie EC, Lem L, Riethof DA, Liu SH, et al. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell. 1999;96:677–87. doi: 10.1016/s0092-8674(00)80578-4. [DOI] [PubMed] [Google Scholar]
- Wilkinson GA, Fariñas I, Backus C, Yoshida CK, Reichardt LF. Neurotrophin-3 is a survival factor in vivo for early mouse trigeminal neurons. J Neurosci. 1996;16:7661–69. doi: 10.1523/JNEUROSCI.16-23-07661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Costigan M. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci USA. 1999;96:7723–30. doi: 10.1073/pnas.96.14.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JH, Drueckes P, Bartoe J, Zhao Z, Shen SH, Krebs EG. A role for the SHP-2 tyrosine phosphatase in nerve growth-induced PC12 cell differentiation. Mol Biol Cell. 1997;8:1575–85. doi: 10.1091/mbc.8.8.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt S, Pinon LGP, Ernfors P, Davies AM. Sympathetic neuron survival and TrkA expression in NT3-deficient mouse embryos. EMBO J. 1997;16:3115–23. doi: 10.1093/emboj/16.11.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–31. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Xie CW, Sayah D, Chen QS, Wei WZ, Smith D, Liu X. Deficient long-term memory and long-lasting long-term potentiation in mice with a targeted deletion of neurotrophin-4 gene. Proc Natl Acad Sci USA. 2000;97:8116–21. doi: 10.1073/pnas.140204597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation of RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–63. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol. 1998;18:1946–55. doi: 10.1128/mcb.18.4.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson R, Schnell E, et al. The role of BDNF receptors in the mature hippocampus: Modulation of long term potentiation through a presynaptic mechanism. J Neurosci. 2000a;20:6888–97. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, et al. Cortical degeneration in the absence of neurotrophin signaling: Dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000b;26:233–45. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- Yamada M, Enokido Y, Ikeuchi T, Hatanaka H. Epidermal growth factor prevents oxygen-triggered apoptosis and induces sustained signalling in cultured rat cerebral cortical neurons. Eur J Neurosci. 1995;7:2130–38. doi: 10.1111/j.1460-9568.1995.tb00635.x. [DOI] [PubMed] [Google Scholar]
- Yamada M, Ohnishi H, Sano SI, Araki T, Nakatami A, et al. Brain-derived neurotrophic factor stimulates interactions of Shp2 with phosphatidylinositol 3-kinase and Grb2 in cultured cerebral cortical neurons. J Neurochem. 1999;73:41–49. doi: 10.1046/j.1471-4159.1999.0730041.x. [DOI] [PubMed] [Google Scholar]
- Yamada M, Ohnishi H, Sano SI, Nakatami A, Ikeuchi T, Hatanaka H. Insulin receptor substrate (IRS)-2 and IRS-2 are tyrosine-phosphorylated and associated with phosphatidylinositol 3-kinase in response to brain-derived neurotrophic factor in cultured cerebral cortical neurons. J Biol Chem. 1997;48:30334–39. doi: 10.1074/jbc.272.48.30334. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Avraham S, Jiang S, Dikic I, Avraham H. The Csk homologous kinase associates with TrkA receptors and is involved in neurite outgrowth of PC12 cells. J Biol Chem. 1999a;274:15059–65. doi: 10.1074/jbc.274.21.15059. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tucker KL, Barde YA. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron. 1999b;24:585–93. doi: 10.1016/s0896-6273(00)81114-9. [DOI] [PubMed] [Google Scholar]
- Yang DD, Kuan C, Whitmarsh HJ, Rincon M, Zheng TS, et al. Absence of excitotoxicity-reduced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–70. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Yano H, Cong F, Birge RB, Goff SP, Chao MW. Association of the Abl tyrosine kinase with the Trk nerve growth factor receptor. J Neurosci Res. 2000;59:356–64. doi: 10.1002/(sici)1097-4547(20000201)59:3<356::aid-jnr9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Ye X, Mehlen P, Rabizadeh S, VanArsdale T, Zhang H, et al. TRAF family proteins interact with the common neurotrophin receptor and modulate apoptosis induction. J Biol Chem. 1999;274:30202–8. doi: 10.1074/jbc.274.42.30202. [DOI] [PubMed] [Google Scholar]
- Yoon SO, Carter BD, Cassaccia-Bonnefil P, Chao MV. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci. 1998;18:3273–81. doi: 10.1523/JNEUROSCI.18-09-03273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York RD, Mollivar DC, Grewal SS, Steinberg PE, McCleskey EW, Stork PJS. Role of phosphoinositide 3-kinase and endocytosis in NGF induced extracellular signal-regulated kinase activation via Ras and Rap1. Mol Cell Biol. 2000;20:18069–83. doi: 10.1128/mcb.20.21.8069-8083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York RD, Yao H, Dillon T, Ellig CL, Eckert SP, et al. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature. 1998;393:622–26. doi: 10.1038/33451. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–9. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Moheban DB, Conway BR, Bhattacharyya A, Segal RA. Cell surface Trk receptors mediate NGF-induced survival while internalized receptors regulate NGF-induced differentiation. J Neurosci. 2000;20:5671–78. doi: 10.1523/JNEUROSCI.20-15-05671.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Summers S, Birnbaum MJ, Pittman RN. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J Biol Chem. 1998;273:16568–75. doi: 10.1074/jbc.273.26.16568. [DOI] [PubMed] [Google Scholar]
- Zundel W, Swiersz LM, Giaccia A. Caveolin 1-mediated regulation of receptor tyrosine kinase-associated phosphatidylinosi-tol 3-kinase activity by ceramide. Mol Cell Biol. 2000;20:1507–14. doi: 10.1128/mcb.20.5.1507-1514.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]