

Abstract
Background
We previously established that neutrophils play a critical role in the development of experimental abdominal aortic aneurysm (AAA). The signal that initiates the influx of neutrophils to the aortic wall, however, remains unknown. In this study we tested the hypothesis that complement participates in the development of AAA by providing the necessary chemotactic signal that recruits neutrophils to the aortic wall.
Methods and Results
Using an elastase-induced model of AAA we showed that pre-treatment of C57BL/6 mice with cobra venom factor (CVF) that depleted serum of complement activity protected mice from AAA development. Whereas control mice exhibited a mean aortic diameter (AD) of 156 ± 2% on day 14 after elastase perfusion, CVF-treated mice exhibited a mean AD of 90 ± 4% (P < 0.001). Examination of mice deficient in factor B (fB) further indicated that the alternative pathway of complement played a major role in this process (mean AD of 105 ± 4%, P < 0.001 compared with controls). Activation of the alternative pathway led to the generation of the anaphylatoxins C3a and C5a that recruited neutrophils to the aortic wall. Moreover, antagonism of both C3a and C5a activities was required to block AAA, suggesting that each can independently promote the aneurysmal phenotype. In addition, we demonstrated that complement alternative pathway involvement was not restricted to this experimental model but was also evident in human AAAs.
Conclusion
The identification of the complement system involvement in the pathophysiology of AAA provides a new target for therapeutic intervention in this common disease.
Keywords: aneurysm, immune system, inflammation, leukocytes, complement system
Human AAA is a dynamic remodeling process involving chronic aortic wall inflammation and connective tissue remodeling.1 Studies suggest that neutrophils and neutrophil-associated proteases are important in the pathogenesis of human AAA2, 3 and neutrophil depletion protects against AAA development in rodents.4, 5 We recently reported that dipeptidyl peptidase I (DPPI) plays a critical role in the development of elastase-induced experimental AAA.5 DPPI is a cysteine protease that plays a key role in regulating neutrophil recruitment at sites of inflammation.5, 6 Using an elastase-induced experimental AAA model,7 we established that DPPI-/- mice were resistant to experimental AAA, due in part to diminished recruitment of neutrophils to the aortic wall and impaired local production of CXC chemokines.5 It is not clear, however, what signal initiates the influx of neutrophils to the aortic wall.
The complement system plays a central role in innate immunity and is an effector arm of humoral immunity. The role of complement in host defense, including clearance of immune complexes, opsonization, and lytic activity, is well recognized.8, 9 It is now understood that complement activity extends beyond host defense as complement participates in autoimmunity, debris removal, and response to tissue injury.10 Complement activation generates the anaphylatoxins C3a and C5a, which are potent leukocyte chemoattractants.11 In this study, we tested the hypothesis that complement participates in the development of elastase-induced AAA by providing the necessary chemotactic signal that recruits neutrophils to the aortic wall.
Methods
Animals
WT (C57BL/6 and C57BL/10Sn) and C5-/- (B10.D2-Hc0 H2d H2-T18c/oSnJ) mice were obtained from The Jackson Laboratory. C3-/- and fB-/- mice were backcrossed to C57BL/6 for 11 and 9 generations respectively.12, 13 C57BL/6 C4-/- mice were obtained from Dr. Michael Carroll (Harvard Medical School).14 Mice were kept in a pathogen free condition at Washington University Specialized Research Facility, and all the experiments were performed according to protocols approved by the Division of Comparative Medicine.
Elastase-induced model of AAA
AAA was induced in 8 to 12-week old mice as previously described.5 Briefly, mice were anesthetized with 55-60 mg/kg intraperitoneal sodium pentobarbital. A laparotomy was performed under sterile conditions. With the assistance of an operating stereomicroscope, the abdominal aorta was isolated and the pre-perfused AD was measured with a calibrated ocular grid. Temporary 7-0 silk ligatures were placed around the proximal and distal aorta. The proximal ligature was closed to interrupt proximal flow. An aortotomy was created at the inferior ligature using the tip of a 30-gauge needle, and a heat-tapered segment of PE10 polyethylene tubing was introduced and secured in position with 7-0 silk. The aortic lumen was perfused for 5 min at 100 mmHg with a solution containing 0.145 U/ml type 1 porcine pancreatic elastase (Sigma). After removal of the catheter, the aortotomy was repaired without constriction of the lumen to restore the flow. At different time points, a second laparotomy was performed and the perfused segment of the abdominal aorta was re-exposed and measured in situ prior to euthanasia and tissue procurement.
Complement depletion
To achieve maximum reduction of serum complement mice were injected intravenously with 9 U of CVF (Quidel) per mouse in two boluses 4 h apart 1 day before surgery, or on day 1, 3, or 6 post-surgery.
Western Blot
Serum samples (1:100 dilution) were fractionated by SDS-PAGE under reducing condition and blotted with goat anti-mouse C3 (1:10,000 dilution, Valeant Pharmaceuticals International) for 2 h at RT followed by incubation with HRP-conjugated rabbit anti-goat IgG (Southern Biotechnology Associates) for 1 h at 37°C. The membrane was stripped and re-probed with HRP-conjugated anti-mouse IgG (Jackson ImmunoResearch Laboratories) to control for protein loading. The bands were visualized using a SuperSignal Western Blotting Kit (Pierce).
Analysis of neutrophils
Aortas were harvested and digested with collagenase IV (291 U/ml, Worthington Biochemical Corporation) and dispase (0.625 U/ml, BD Biosciences) at 37°C for 1 h. The cells were passed through a 70 μm cell strainer to remove debris, counted, stained with FITC-conjugated anti-CD45 and PE-conjugated anti-Gr1 antibodies (BD Biosciences) and analyzed by flow cytometry.
Serum reconstitution
Pooled sera were obtained from WT and fB-/- mice. Mice were injected intravenously with 200 μl WT or fB-/- pooled sera immediately and 24 h after surgery.
Treatment with C5 receptor antagonist
The C5a receptor antagonist (C5aRA) consists of the cyclic hexapeptide AcF[OpdChaWR], designed from the COOH terminus of C5a15 and synthesized as previously described.16 Mice were injected intravenously with three doses of C5aRA (1mg/kg in 0.2 ml PBS/mouse/injection) immediately prior to surgery, and on days 1 and 3 after surgery. Control mice received the same doses of a control peptide, C5RC (AcF[OpdChaAdr]).
C3a and C5a ELISA
ELISA plates were coated overnight with anti-mouse C3a (4 μg/ml) or anti-mouse C5a (5 μg/ml) monoclonal antibody (BD Pharmingen). After blocking with 10% FCS, the plates were washed, and incubated with sera (100 μl of serum diluted 1:20) for 2 h at RT followed by biotinylated anti-mouse C3a (250 ng/ml) or C5a (500 ng/ml) monoclonal antibody (BD Pharmingen). Following incubation with strepavidin-peroxidase (400 ng/ml, Sigma) 100 μl of 1-Step™ Turbo TMB-ELISA (Pierce) was added to each well and color development was read at 450 nm using a SpectraMax Plus reader (Molecular Devices). Mouse recombinant C3a and C5a (BD Pharmingen) were used to establish the standard curve.
Immunohistochemistry
Mouse abdominal aorta was dissected, snap-frozen in OCT compound, and sectioned at 5 μm. Elastin was stained with Verhoeff-van Gieson (VVG) using an Accustain Elastic Stain Kit (Sigma). Elastin degradation was graded on a scale of 1-4: 1 = less than 25% degradation, 2 = 25-50% degradation, 3 = 50-75% degradation, 4 = greater than 75% degradation. Smooth muscle cell (SMC) content was evaluated using an alkaline phosphatase-conjugated antibody to α-smooth muscle (1:200 dilution, Sigma). Color was visualized using an alkaline phosphatase substrate kit (Vector Laboratories). SMC was graded on a scale of 1-4: 1 = less than 25% loss, 2 = 25-50% loss, 3 = 50-75% loss, 4 = greater than 75% loss. Macrophages and neutrophils were visualized with biotinylated anti-Mac-3 (1:200 dilution, Cedarlane Laboratories) and anti-Gr-1 monoclonal antibody (1:100 dilution, BD Biosciences) respectively. Following incubation in 8% BSA and strepavidin-HRP, color development was revealed using the Histomark Orange Peroxidase Substrate Kit (Kirkegaard & Perry Laboratories). Mast cells were visualized with polychrome methylene blue and differentiated in glycerin-ether solution. All sections were counterstained with 1% methyl green.
Immunofluorescence
Human abdominal aorta specimens were obtained at time of elective surgery through a protocol approved by the Washington University School of Medicine Institutional Review Board. Cross-sections of aortic tissues (5 μm) were fixed in methanol, blocked in 3% dry milk in PBS, and incubated with a goat polyclonal antibody to human C3 (1:1600 dilution, CompTech Complement Technology), human C4 (1:800 dilution, CompTech), human fB (1:800 dilution, CompTech), human properdin (1:800 dilution, CompTech), or monoclonal antibody to C5-9 neoantigen (1:100 dilution, Quidel) for 1 h at RT, washed, then incubated with a biotinylated anti-goat antibody (Vector Laboratories) for 1 h at RT, followed by incubation with streptavidin-PE (BD Biosciences) or rhodamine-conjugated goat anti-mouse antibody (Jackson ImmunoResearch). Normal goat serum at the same dilutions as above was used for control and showed no specific fluorescence on normal or AAA tissues.
Statistics
Comparisons between groups were made by one-way ANOVA followed by Bonferroni's post-test to compare all groups of data. Data are presented as the mean ± SEM. P values less than 0.05 were considered significant.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Elastase-induced AAA is complement dependent
Although no single animal model reproduces all aspects of human AAA, the elastase-induced experimental AAA model recapitulates many features of this disease.17 Transient perfusion of the abdominal aorta with a porcine elastase solution reproducibly leads to AAA in 100% of C57BL/6 WT animals.7 Mild aortic dilatation was observed immediately following the elastase perfusion. This AD remained relatively stable until day 3, after which there was rapid secondary increase in AD to a maximum difference in AD of 0.82 ± 0.01 mm on day 14 (Figure 1A). In this model, AAA is defined on day 14 as an overall increase in the AD of at least 100% over the pre-perfused parameters.7 Consistent with previous studies,5, 7 WT animals exhibited a mean increase in AD of 156 ± 2% on day 14 after elastase perfusion (Figure 1B).
Figure 1.
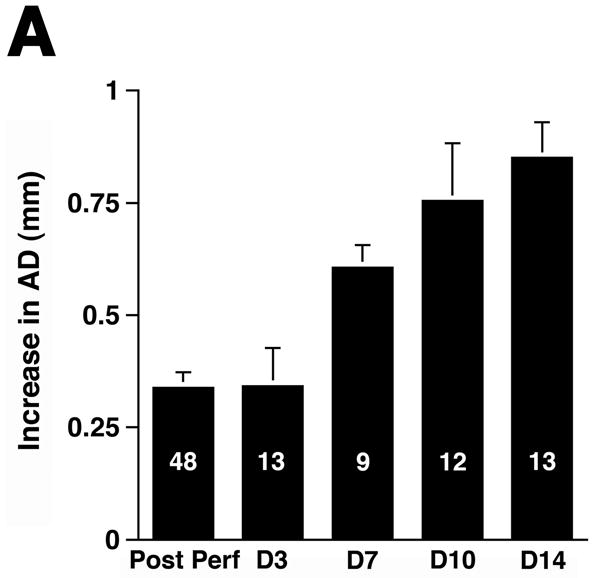
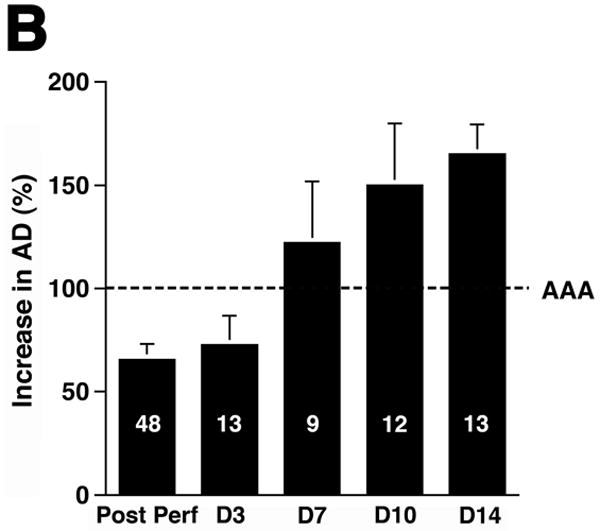
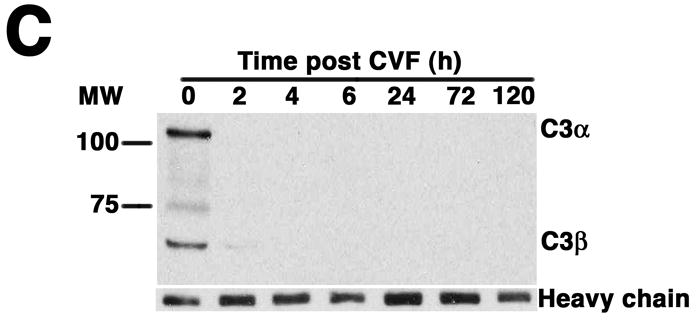
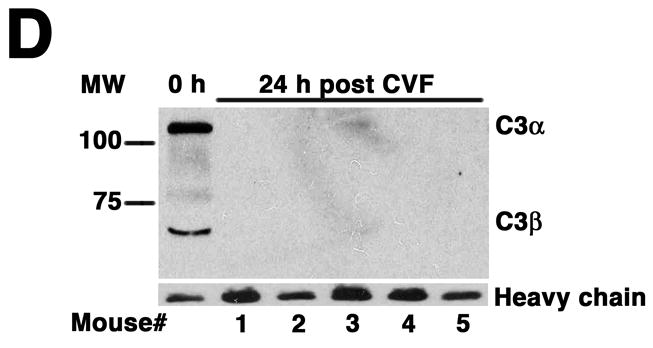
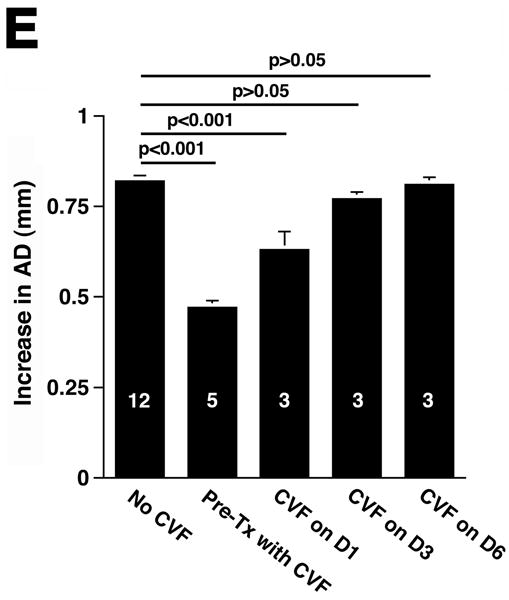
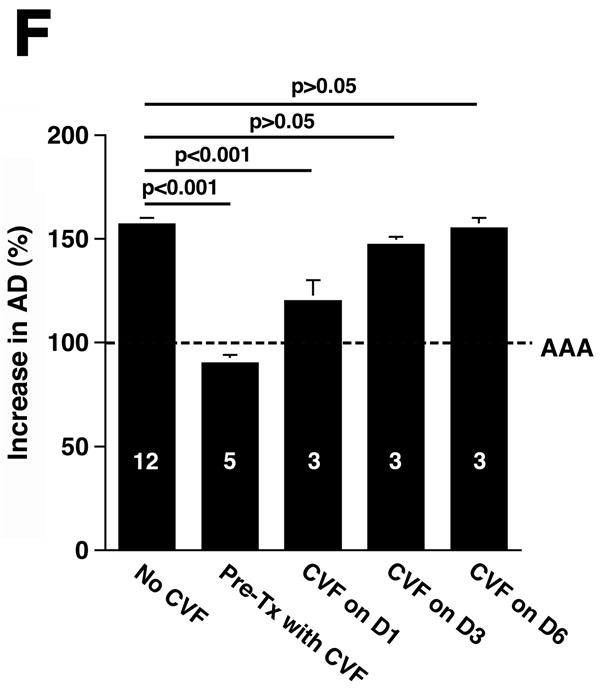
Complement depletion protects against AAA development. Increase in AD on day 14 was expressed in mm (A) or percentage (B) over the baseline AD measured prior to elastase perfusion. AAA was defined by an increase in AD of 100% or greater than that measured before elastase perfusion. Western blots demonstrate the decrease in C3 (α and β chains) over time (C) and in the mice pre-treated with CVF (D). Heavy chain serves as control for protein content. Mice were pre-treated with CVF 24 h prior to elastase perfusion or at the time indicated. The increase in AD on day 14 was expressed in mm (E) or percentage (F). The number of animals per time point or treatment type is indicated for each group.
CVF, a functional analogue of C3b, forms a stable convertase that within hours depletes serum of its key complement activation component C3,18 an effect that lasts up to 5 days post treatment (Figure 1C). Pre-treatment with CVF 24 h prior to elastase perfusion consistently depleted serum of complement activity (Figure 1D) and prevented AAA development in all animals (increase in AD of 0.47 ± 0.02 mm or 90 ± 4%, P < 0.001, Figure 1E-F). Mice that received CVF 24 h after elastase perfusion developed smaller aneurysms (increase in AD of 0.63 ± 0.05 mm or 120 ± 10%, P < 0.001). However, complement depletion on day 3 (or later) after elastase perfusion had no effect on the extent of aortic dilatation (Figure 1D-E). These results confirm that complement activation plays a pivotal role in elastase-induced AAA development.
Histological analysis of day 14 aortas from animals treated with CVF prior to elastase perfusion showed well-preserved elastic fibers (Figure 2A) and significantly less SMC depletion (Figure 2B) compared with aortas of untreated animals. Immunohistochemistry of day 14 CVF-treated aortas showed significantly reduced numbers of macrophages (9.8 ± 1.4 cells/cross-section of CVF-treated aortas versus 84.9 ± 16.7 cells/cross-section of untreated aortas, P < 0.001, Figure 2C) and mast cells (6.7 ± 1.2 cells/cross-section of CVF-treated aortas versus 12.6 ± 1.9 cells/cross-section of untreated aortas, P < 0.001, Figure 2D). Previous studies suggested that mast cell activation modulates AAA development in mice.19 Consistent with these results, we found that a majority of mast cells from aortas of untreated mice had undergone degranulation while only half of the mast cells in CVF-treated aortas showed signs of degranulation (P < 0.001, Figure 2D). Taken together, these results indicate a significant reduction in aortic wall inflammation following CVF treatment, which helps preserve the elastin and SMC content.
Figure 2.
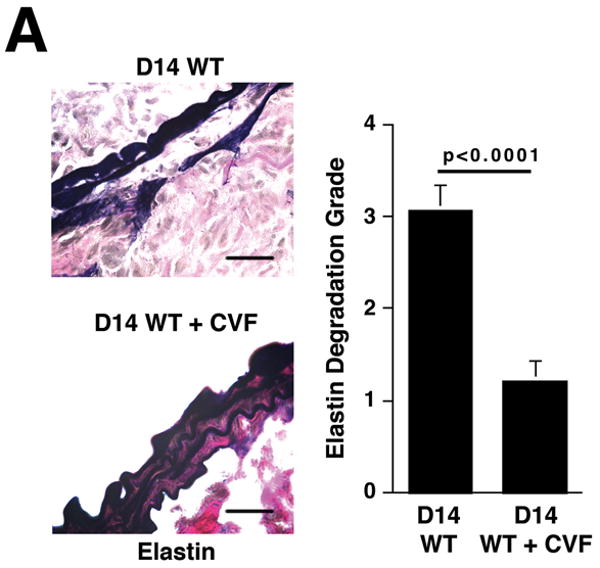
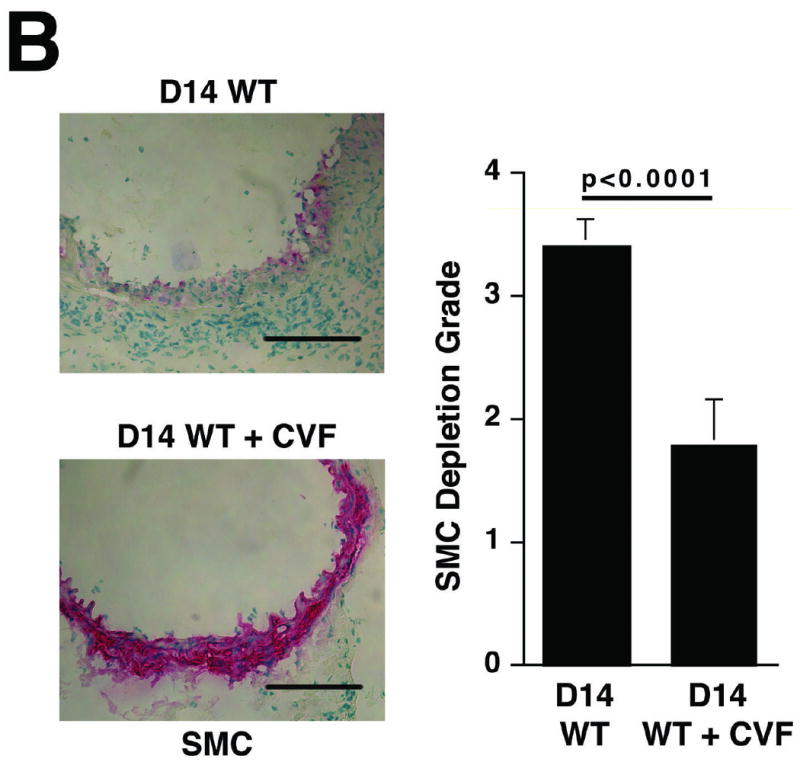
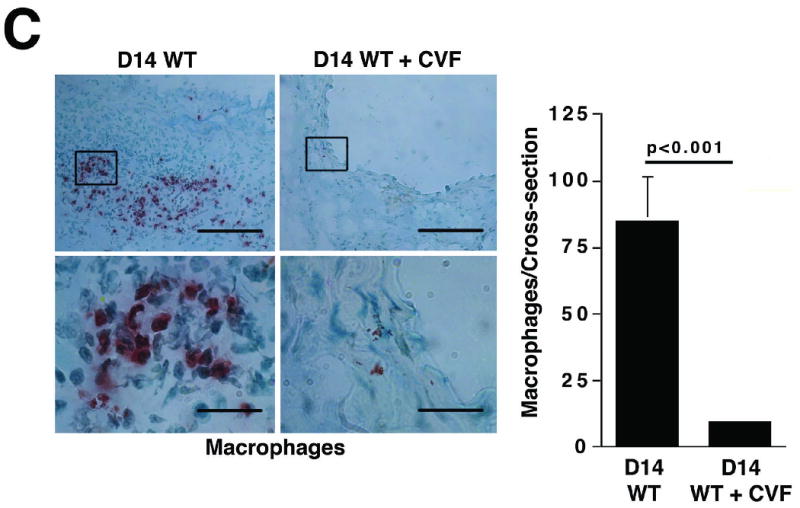
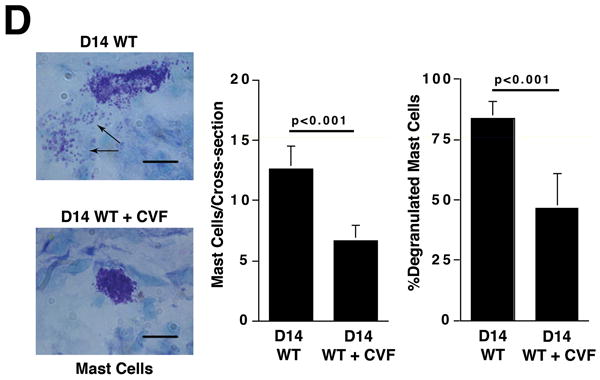
Complement depletion protects against elastin degradation and smooth muscle depletion by limiting the inflammatory response. Elastin (A) was detected with VVG staining and SMC (B) were detected with an antibody to α -actin. Scale bar, 0.02 mm. Serial sections from untreated and CVF-treated (24 h prior to elastase perfusion) aortas were stained for Mac-3 and with methylene blue to show the presence of macrophages (C) and mast cells (D), respectively. Insets from upper panels in (C) are shown at higher magnification in lower panels. Scale bar, 0.1 mm (upper panels), 0.02 mm (lower panels). Degranulated mast cells show abundant extracellular granules (arrows in D). Percent of degranulated mast cells was calculated by the formula: (degranulated cells)/(compact cells + degranulated cells) × 100. Scale bar, 0.01 mm. Histological grading in A, B, C, and D was performed on 6 serial cross-sections per aorta, 3 aortas per genotype/treatment type, by two independent blinded observers.
The alternative pathway is critical for AAA development
We next determined which complement pathways are responsible for the elastase-induced AAA phenotype. There are three initiator pathways of complement activation: the classical, the alternative, and the lectin pathways.8, 9 The importance of the standard classical and lectin pathways was assessed in mice lacking C4.13 C4-/- mice developed AAA normally (increase in AD of 163 ± 5%), indicating that the standard classical and lectin pathways are likely dispensable in this model (Figure 3A). To assess the importance of the alternative pathway, we induced AAA in mice lacking fB.14 We found that the fB-/- mice were largely resistant to the development of AAA (increase in AD of 105 ± 4%, Figure 3A). In addition, histological analysis revealed minimal degree of elastin degradation in day 14 fB-/- aortas, implying that the development of AAA requires activation of the alternative pathway.
Figure 3.
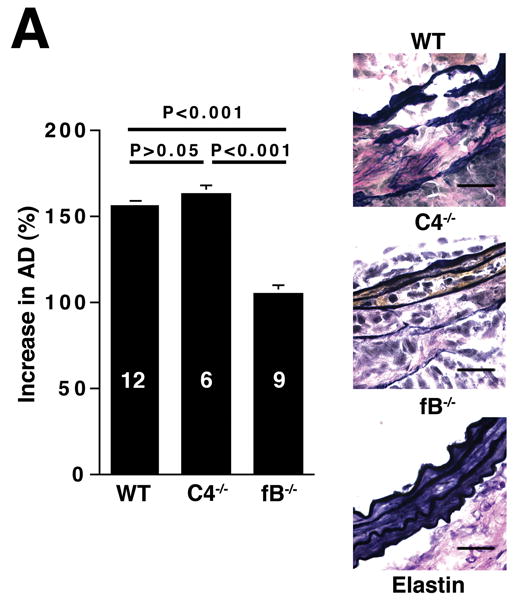
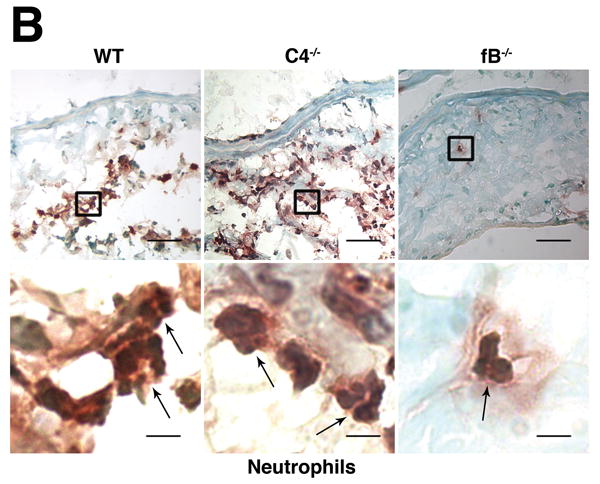
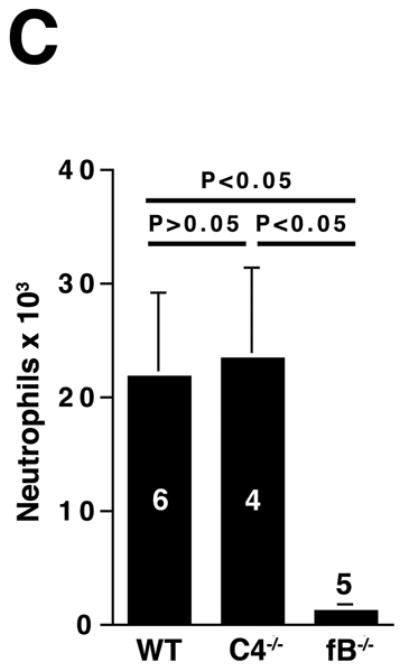
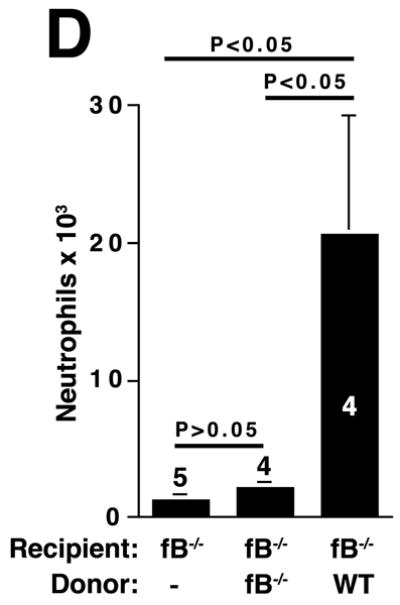
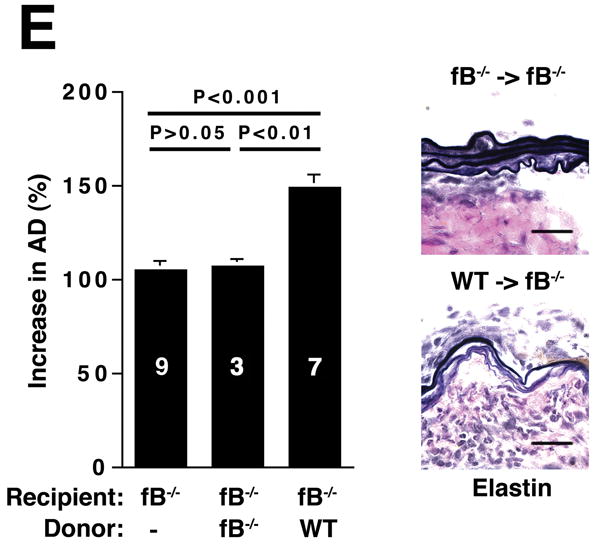
The alternative pathway is essential for AAA development. A) AD increase in WT, C4-/-, and fB-/- mice on day 14. VVG staining showed intact elastic fibers in fB-/- mice. Scale bar, 0.02 mm. B) Neutrophils in aortic wall were stained with Gr-1 (brown) and identified by their segmented nuclei (arrows). Insets from upper panels are shown at higher magnification in lower panels. Scale bar, 0.025 mm (upper panels), 0.01 mm (lower panels). C) The absolute number of neutrophils was calculated by multiplying the total number of cells per aorta by the percentage of CD45+/Gr-1+ cells. D) 24 h following the last WT or fB-/- serum injection, aortas were harvested and the number of neutrophils per aorta was calculated (D). Another group of mice were sacrificed on day 14 and the AD was assessed (E). Reconstitution with WT sera led to AAA development and fragmentation of elastic fibers in fB-/- mice. Scale bar, 0.02 mm. The number of animals per genotype is indicated for each group.
Alternative pathway activation leads to neutrophil recruitment in elastase-induced AAA
We next sought to determine the mechanism by which the alternative pathway contributes to AAA development. Previous studies showed that neutrophils and mast cells modulate the development of experimental AAA.4, 5, 19 However, mast cells did not accumulate in significant numbers in the aortic wall until day 7 while neutrophil presence peaked on day 3 post elastase perfusion.19, 5 Given the fact that CVF treatment 3 days after perfusion did not offer protection against AAA development (Figure 1E), we reasoned that the protective effect against AAA development seen in CVF-treated and fB-/- mice was more likely due to an impaired influx of neutrophils to the aortic wall. To test this hypothesis, we analyzed disease induction in fB-/- mice. Immunostaining for Gr-1 (a marker for neutrophils) in WT and C4-/- aortas obtained on day 3 post elastase perfusion revealed abundant Gr-1+ neutrophils while these cells were rarely seen in fB-/- aortas (Figure 3B). For a more quantitative measurement, we digested the aortas and enumerated the neutrophils recruited to the aortic wall post elastase perfusion by flow cytometry. We confirmed that resistance to AAA in fB-/- mice was accompanied by greater than 90% decrease in neutrophil recruitment in the aortic wall (21,820 ± 7,392 neutrophils per WT aorta versus 23,410 ± 8,065 neutrophils per C4-/- aorta versus 1,223 ± 336 neutrophils per fB-/- aorta, Figure 3C).
Based on these above findings, we hypothesized that alternative pathway complement activation led to the production of mediators that sustained the recruitment of neutrophils to the aortic wall. Therefore, restoring fB should lead to alternative pathway complement activation and neutrophil recruitment. To this end, animals were treated with an exogenous source of fB after elastase perfustion. Reconstitution of fB-/- mice with WT serum as a source of fB restored the influx of neutrophils to the aorta (Figure 3D) and rendered the fB-/- mice susceptible to AAA development while fB-/- mice reconstituted with fB-/- serum did not develop AAA (Figure 3E). Taken together these results suggest that activation of the alternative pathway directs the recruitment of neutrophils to the aortic wall. These neutrophils in turn propagate the inflammatory response, which leads to the aneurysmal dilatation associated with AAA.
Both C3a and C5a contribute to AAA development
The three pathways of complement activation converge at the cleavage of C3, generating C3a and C3b. C3a is an anaphylatoxin with bacterocidal, cell activating, and leukocyte chemoattractant properties11 while C3b promotes the assembly of the membrane attack complex and initiates the alternative pathway amplification loop.20 Given the central role of C3, we examined AAA development in C3-/- mice (Figure 4A). Unexpectedly, AAA was not abolished (increase in AD of 133 ± 9% in C3-/- animals versus increase in AD of 105 ± 4% in fB-/- animals, P < 0.05). These results together with those derived from CVF-treated and fB-/- mice suggest the possibility of a C3-independent pathway of C5a generation. Huber-Lang et al. have shown that thrombin is overexpressed in C3-/- mice and can substitute for C3 convertase in the generation of C5a in injured lung tissue.21 Thus to determine whether C5a was generated in C3-/- mice the animals were perfused with elastase, their sera were collected and tested for the presence of C5a. Consistent with this hypothesis, C5a was detected in C3-/- mice (Figure 4B) and the mean C5a levels in C3-/- mice were comparable to the levels detected in WT animals (348 ± 189 ng/ml in WT mice versus 210 ± 92 ng/ml in C3-/- mice). In contrast, C5a levels were low in fB-/- mice 24 h post elastase perfusion (18 ± 7 ng/ml), suggesting that porcine pancreatic elastase (used during perfusion) did not cleave C5. Moreover, these results indicate that there was no C3-independent C5a generation in fB-/- mice. Next we determined whether C5a activity was required for aneurysm formation, by examining the susceptibility of C5-/- mice to AAA development. We found that C5-/- mice developed AAA to a similar extent as WT mice (increase in AD of 136 ±13% in C5-/- mice versus increase in AD of 150 ± 4% in WT mice, P > 0.05, Figure 4A), suggesting that in the absence of C5a, C3a may be sufficient to recruit neutrophils and confer susceptibility to the aneurysmal phenotype. In fact, we confirmed that C3a was produced at normal levels in C5-/- mice (Figure 4C) and the production of either C3a or C5a was sufficient to recruit neutrophils to the aortic wall (Figure 4D).
Figure 4.
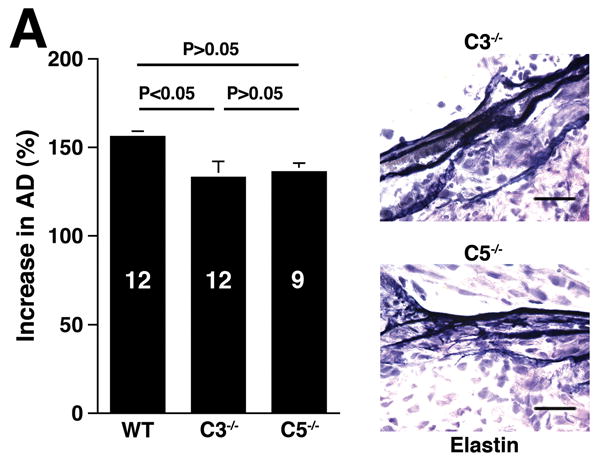
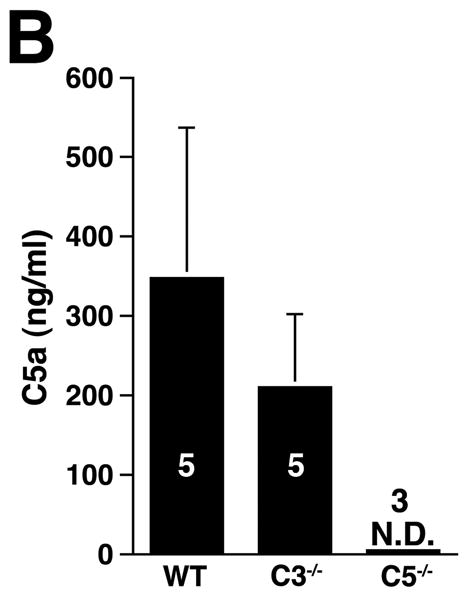
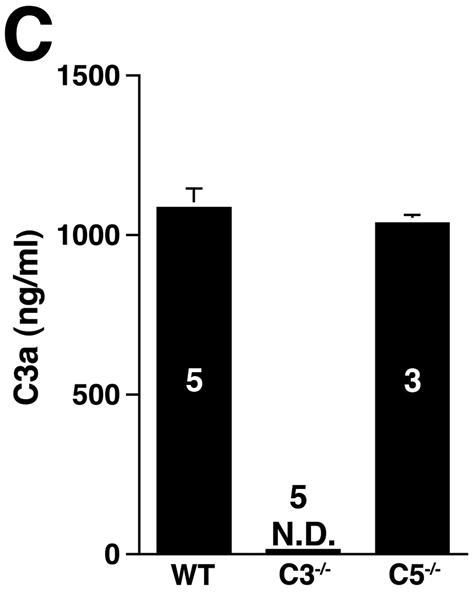
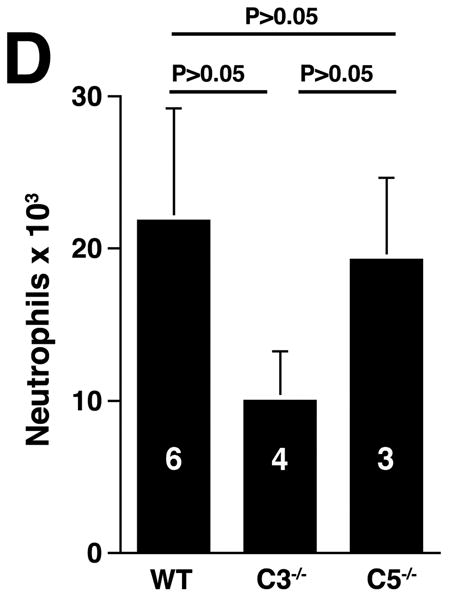
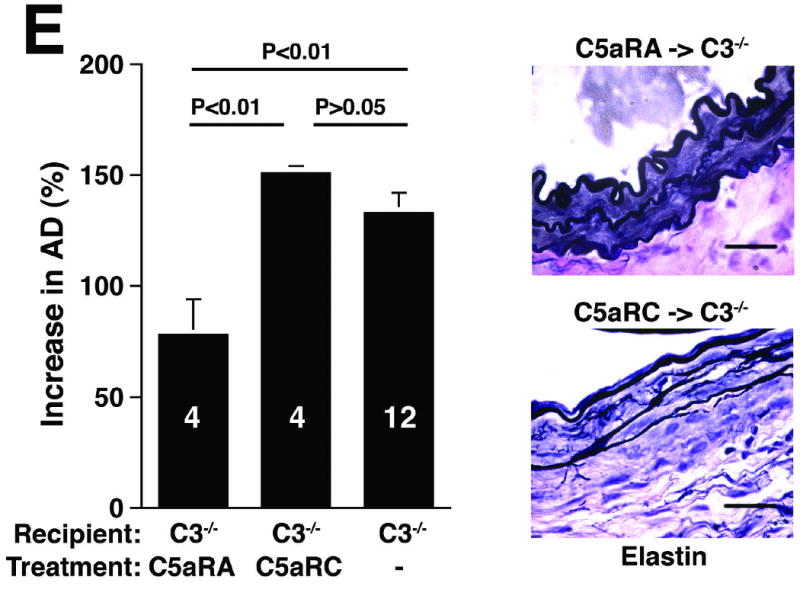
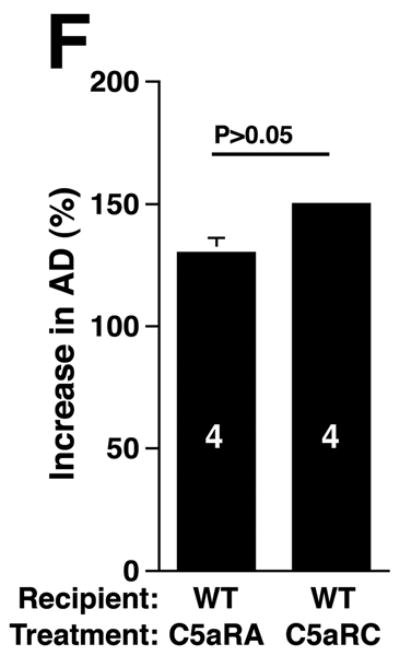
C3 and C5 activation products independently promote AAA development. AD increase and elastin staining C3-/- and C5-/- mice (A). C5a (B) and C3a (C) levels in sera 24 h after elastase perfustion. C5a levels in C5-/- sera and C3a levels in C3-/- sera were below the limit of detection (N.D., not detected). C3a and C5a levels prior to elastase perfusion were low (C5a = 10 ± 5 ng/ml, C3a = 17 ± 10 ng/ml). D) Neutrophil numbers in WT, C3-/-, and C5-/- aortas on day 3 were equivalent. E) C3-/- mice were injected with C5aRA or C5aRC, a control peptide, and AD increase and elastin degradation were assessed on day 14. F) WT mice received either C5aRA or C5aRC and the AD was assessed on day 14. Scale bar, 0.02 mm. The number of animals per genotype or treatment type is indicated for each group.
Blocking both C3a and C5a activity protects against AAA development
If either C3a or C5a were sufficient to sustain AAA, then blocking the activity of both would be expected to suppress AAA. To block both C3a and C5a activities, we administered a C5aRA to C3-/- mice one day prior to and on days 1 and 3 after elastase perfusion. C5aRA treatment led to complete protection against AAA development in C3-/- mice while treatment with C5aRC did not (increase in AD of 78 ± 8% in C5aRA-treated animals versus increase in AD of 151 ± 3% in C5aRC-treated animals, P < 0.01, Figure 4E). However, blockade of C5a:C5aR interaction with C5aRA did not suppress AAA in WT mice (Figure 4F). These results and the data obtained from C5-deficient mice (Figure 4A) confirm that C3 and C5 activation products independently promote aneurysm formation.
The alternative pathway of complement is activated in human AAA
Previous studies demonstrated the presence of complement-fixing immunoglobulin subclasses and C3 deposition in the aortic wall of AAAs, prompting the investigators to suggest that activation of the classical pathway may contribute to the pathogenesis of human AAA.22, 23 While our results do not preclude classical pathway activity, our findings suggest alternative pathway involvement in human AAA as well. Immunostaining of AAA aortic wall tissues for C3, C4, fB, properdin (a component of the alternative pathway convertase)24, and C5-9 neoantigen (a complement lytic pathway marker) confirmed the presence of these complement proteins throughout all layers of the aortic wall (Figure 5). C3, C4, and fB, showed similar pattern of fluorescence mainly along the medial and adventitial layers while properdin and C5-9 neoantigen were also seen prominently along the luminal side. Thus these samples obtained from patients undergoing elective surgical repair point to the activation of both classical and alternative pathways in human AAAs.
Figure 5.
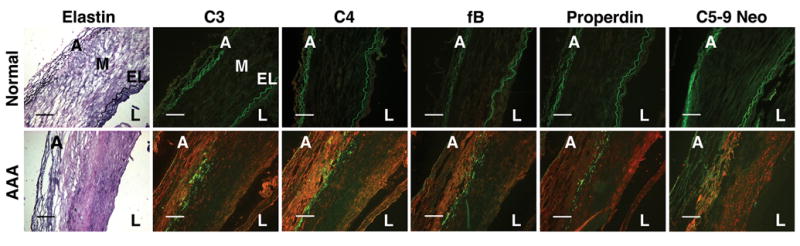
The alternative pathway is activated in human AAA. Immunohistochemistry of human aortic tissues revealed abundant C3, C4, fB, properdin, and C5-9 neoantigen (red) throughout all layers of aortic tissues in AAA but not in normal aortas. A, adventitia; M, media; EL, elastic lamella; L, lumen. Elastin in the aortic wall was detected by VVG stain. The extensive remodeling in AAA obscures the distinct layers of the aortic wall. The elastic fibers autofluoresce (green). Scale bar, 0.2 mm. Photomicrographs shown are representative of 3 human AAA specimens.
Discussion
In this study we identified complement as a critical mediator of neutrophil recruitment and subsequent development of elastase-induced AAA. We made several new findings: 1) AAA development in the mouse model was dependent on the activation of the complement cascade; 2) the alternative pathway played a major role in the process while the standard classical and lectin pathways together were not required; and 3) either C3a or C5a could independently promote the aneurysmal phenotype (Figure 6, Supplementary Table 1). In addition, we showed that complement alternative pathway involvement was not restricted to our mouse model but is also seen in established human AAAs.
Figure 6.
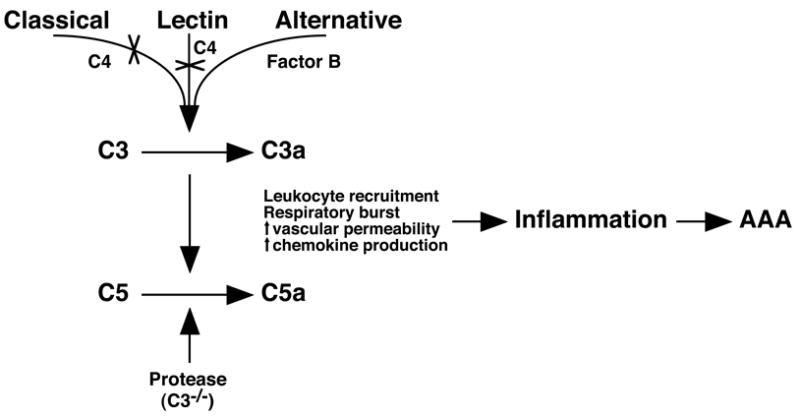
Alternative pathway of complement activation generates anaphylatoxins that propagate inflammation. Activation of the alternative pathway in the elastase-induced AAA model leads to the generation of anaphylatoxins C3a and C5a that recruit leukocytes and perpetuate inflammation, leading to AAA. The classical and lectin pathways are dispensable (X) in this model. In C3-/- mice, another protease can substitute for C3-convertase activity to cleave C5 to generate C5a.
In view of the results with CVF and given the central role of C3 and C3 activation products in inducing the downstream activation of C5, we expected that a deficiency in C3 would be sufficient to halt the development of AAA. The results we obtained with C3-/- mice were not as anticipated and prompted us to consider alternative means of C5a production. That C5 might be cleaved in C3-/- mice has been recently described. In a study by Huber-Lang et al, the authors showed that, while thrombin had limited function in the presence of C3, it became the major C5 convertase in C3-/- mice.21 In the present study, we demonstrated that a compensatory pathway also exists to generate C5a in the absence of C3. Moreover, interruption of the C5a:C5aR interaction by a receptor antagonist suppressed AAA development in C3-/- mice, confirming that C5a was indeed active in the absence of C3. However, contrary to other complement- and neutrophil-dependent animal models, where C5a is pivotal in disease induction21, 25-27, we found that elastase-induced AAA was not suppressed in C5-/- mice, suggesting that in the absence of C5a, C3-mediated activity and activation products were sufficient to sustain the inflammatory process that led to aneurysmal dilatation.
C3a and C5a have been shown to have overlapping functions, including their ability to chemoattract and activate many cell types, to trigger oxidative bursts, and to increase endothelial permeability.11 However, the prominent role of C5a in other disease models was attributed to its unique ability to attract neutrophils while C3a was unable to do so.28 Perhaps the role of C3 in AAA development can be explained by recent studies showing that C3a was required for the production of neutrophil-specific CXC chemokines while C5a did not have the same effect.29 Therefore, C3a may provide the necessary signal to recruit neutrophils indirectly, through the elaboration of CXC chemokines. Taken together, these results suggest that the functions of C3a and C5a are important and antagonism of both factors is required to halt progression of AAA.
How does one explain the critical involvement of complement in this model of AAA? The likely scenario is that chemical injury elicited by elastase perfusion provides a protected site for complement activation. In the presence of complement activation, C3 and C5 convertases are generated, leading to the release anaphylatoxins that recruit inflammatory cells, specifically neutrophils. Neutrophils may amplify complement activation through the release of properdin that stabilizes the alternative pathway convertase.30 Properdin may also bind to target cell surfaces directly, thus providing a platform for the assembly of the alternative pathway convertase.24 The strong staining for properdin observed on the luminal surface of the AAA specimens suggests that properdin may indeed serve as a focal point for the initiation of the alternative pathway complement activation. The direct role of properdin in this model of AAA is currently under investigation. The alternative pathway has also been shown to proceed directly via the lectin pathway, bypassing the standard C4 requirement.31, 32 The significance of this pathway in vivo and to this AAA model awaits further investigation. Regardless of the initiation mechanism, the alternative pathway plays a critical role in recruiting the neutrophils that are needed to sustain the inflammation in elastase-induced AAA.
Lastly, there is scant literature regarding the role of complement in human AAAs. So far only two reports described deposition of C3 and antibodies in human AAA tissues.22, 23 Presently, evidence for direct complement participation in AAA development is still lacking. The elastase-induced model of experimental AAA allowed us to definitively establish that the alternative pathway of complement directly controls aneurysmal development in mice. However, C5 cleavage in the mouse has been shown to proceed mainly via the alternative pathway 33. Thus the importance of the alternative pathway in human AAA may be less and remains to be determined. Nonetheless, we present evidence that the classical (or lectin) and alternative pathways of complement are activated in human AAA tissues, which strengthens the hypothesis that these pathways indeed play a role in human AAA. Whether complement activation in human AAA contributes to the initiation and ongoing destruction of aortic wall tissue or merely reflects a fixed injury requires further studies.
In summary, the wall of a large blood vessel like the aorta represents an example of a tissue site where chronic inflammation is certainly undesirable. How innate immune responses are generated, maintained, and modulated at this particular tissue site in humans are yet to be discerned. This report is an attempt to analyze the complement system in the development of AAA. The results suggest that inhibition of the chronic inflammatory response in AAA through complement-targeting strategies merits further exploration.
Supplementary Material
Acknowledgments
Funding Sources: This work was supported by grants from the NIH (AI068730 to J.D.L., AI041592 to J.P.A., AI051436 to D.E.H, AI049261 to C.T.N.P., and HL056701 and P50HL083762 to R.W.T.). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Clinical Summary: Abdominal aortic aneurysm (AAA) is a disease characterized by chronic inflammation and remodeling of aortic wall tissue. Studies using “end-stage” human AAA tissues procured at surgery have identified a number of candidate molecules; however, these may or may not contribute to the initiation and progression of AAA. To better understand the mechanisms that promote AAA, we turn to an elastase-induced mouse model that recapitulates many features of human AAA. In this model, neutrophils are identified as critical mediators of AAA development. Neutrophil depletion or impaired neutrophil recruitment protect against AAA development. However, the signal that initiates the influx of neutrophils to the aortic wall remains undefined. We hypothesize that complement participates in the development of elastase-induced AAA, possibly by providing the chemotactic signal that recruits neutrophils to the aortic wall. In the present experiments we show that complement depletion abrogates AAA development. We also demonstrate that the alternative pathway of the complement system plays a major role in this process by generating the potent anaphylatoxins C3a and C5a that recruit neutrophils to the aortic wall. Ruptured AAA is the cause of death in 1-3% of men over the age of 65. While elective surgical repair is usually definitive, this operation is reserved for large aneurysms. At present, there are no therapies that alter the progressive growth of small aneuryms. The identification of the involvement of the complement system in the pathophysiology of AAA provides a new target for therapeutic intervention in this common disease.
Conflict of Interest Disclosures: J.D.L is a consultant to Acusphere Inc. on biomaterial induced complement activation and has several issued and pending patents on complement inhibitors. R.W.T. has received speaker fees for speaking at scientific meetings and serves on the National Advisory Board, University of Michigan Cardiovascular Center. R.W.T.'s spouse holds stock in Abbot Laboratories.
Contributor Information
Monica B. Pagano, Department of Surgery, Section of Vascular Surgery, Washington University School of Medicine, Saint Louis, MO 63110.
Hui-fang Zhou, Department of Medicine, Division of Rheumatology, Washington University School of Medicine, Saint Louis, MO 63110.
Terri L. Ennis, Department of Surgery, Section of Vascular Surgery, Washington University School of Medicine, Saint Louis, MO 63110.
Xiaobo Wu, Department of Medicine, Division of Rheumatology, Washington University School of Medicine, Saint Louis, MO 63110.
John D. Lambris, Department of Pathology and Laboratory Medicine, University of Pennsylvania School of Medicine, Philadelphia, PA 19104.
John P. Atkinson, Department of Medicine, Division of Rheumatology, Washington University School of Medicine, Saint Louis, MO 63110.
Robert W. Thompson, Department of Surgery, Section of Vascular Surgery, Washington University School of Medicine, Saint Louis, MO 63110.
Dennis E. Hourcade, Department of Medicine, Division of Rheumatology, Washington University School of Medicine, Saint Louis, MO 63110.
Christine T. N. Pham, Department of Medicine, Division of Rheumatology, Washington University School of Medicine, Saint Louis, MO 63110.
References
- 1.Thompson RW, Geraghty PJ, Lee JK. Abdominal aortic aneurysms: basic mechanisms and clinical implications. Curr Probl Surg. 2002;39:110–230. doi: 10.1067/msg.2002.121421. [DOI] [PubMed] [Google Scholar]
- 2.Cohen JR, Parikh S, Grella L, Sarfati I, Corbie G, Danna D, Wise L. Role of the neutrophil in abdominal aortic aneurysm development. Cardiovasc Surg. 1993;1:373–376. [PubMed] [Google Scholar]
- 3.Rao SK, Reddy KV, Cohen JR. Role of serine proteases in aneurysm development. Ann N Y Acad Sci. 1996;800:131–137. doi: 10.1111/j.1749-6632.1996.tb33304.x. [DOI] [PubMed] [Google Scholar]
- 4.Eliason JL, Hannawa KK, Ailawadi G, Sinha I, Ford JW, Deogracias MP, Roelofs KJ, Woodrum DT, Ennis TL, Henke PK, Stanley JC, Thompson RW, Upchurch GR., Jr Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation. 2005;112:232–240. doi: 10.1161/CIRCULATIONAHA.104.517391. [DOI] [PubMed] [Google Scholar]
- 5.Pagano MB, Bartoli MA, Ennis TL, Mao D, Simmons PM, Thompson RW, Pham CT. Critical role of dipeptidyl peptidase I in neutrophil recruitment during the development of experimental abdominal aortic aneurysms. Proc Natl Acad Sci U S A. 2007;104:2855–2860. doi: 10.1073/pnas.0606091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson RW, Curci JA, Ennis TL, Mao D, Pagano MB, Pham CT. Pathophysiology of abdominal aortic aneurysms: insights from the elastase-induced model in mice with different genetic backgrounds. Ann N Y Acad Sci. 2006;1085:59–73. doi: 10.1196/annals.1383.029. [DOI] [PubMed] [Google Scholar]
- 8.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 9.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 10.Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. 2007;171:715–727. doi: 10.2353/ajpath.2007.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haas PJ, van Strijp J. Anaphylatoxins: their role in bacterial infection and inflammation. Immunol Res. 2007;37:161–175. doi: 10.1007/BF02697367. [DOI] [PubMed] [Google Scholar]
- 12.Circolo A, Garnier G, Fukuda W, Wang X, Hidvegi T, Szalai AJ, Briles DE, Volanakis JE, Wetsel RA, Colten HR. Genetic disruption of the murine complement C3 promoter region generates deficient mice with extrahepatic expression of C3 mRNA. Immunopharmacology. 1999;42:135–149. doi: 10.1016/s0162-3109(99)00021-1. [DOI] [PubMed] [Google Scholar]
- 13.Fischer MB, Ma M, Goerg S, Zhou X, Xia J, Finco O, Han S, Kelsoe G, Howard RG, Rothstein TL, Kremmer E, Rosen FS, Carroll MC. Regulation of the B cell response to T-dependent antigens by classical pathway complement. J Immunol. 1996;157:549–556. [PubMed] [Google Scholar]
- 14.Matsumoto M, Fukuda W, Circolo A, Goellner J, Strauss-Schoenberger J, Wang X, Fujita S, Hidvegi T, Chaplin DD, Colten HR. Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc Natl Acad Sci U S A. 1997;94:8720–8725. doi: 10.1073/pnas.94.16.8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scola AM, Higginbottom A, Partridge LJ, Reid RC, Woodruff T, Taylor SM, Fairlie DP, Monk PN. The role of the N-terminal domain of the complement fragment receptor C5L2 in ligand binding. J Biol Chem. 2007;282:3664–3671. doi: 10.1074/jbc.M609178200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, Lambris JD. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003;198:913–923. doi: 10.1084/jem.20030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anidjar S, Salzmann JL, Gentric D, Lagneau P, Camilleri JP, Michel JB. Elastase-induced experimental aneurysms in rats. Circulation. 1990;82:973–981. doi: 10.1161/01.cir.82.3.973. [DOI] [PubMed] [Google Scholar]
- 18.Alper CA, Balavitch D. Cobra venom factor: evidence for its being altered cobra C3 (the third component of complement) Science. 1976;191:1275–1276. doi: 10.1126/science.56780. [DOI] [PubMed] [Google Scholar]
- 19.Sun J, Sukhova GK, Yang M, Wolters PJ, MacFarlane LA, Libby P, Sun C, Zhang Y, Liu J, Ennis TL, Knispel R, Xiong W, Thompson RW, Baxter BT, Shi GP. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J Clin Invest. 2007;117:3359–3368. doi: 10.1172/JCI31311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leslie RG, Nielsen CH. The classical and alternative pathways of complement activation play distinct roles in spontaneous C3 fragment deposition and membrane attack complex (MAC) formation on human B lymphocytes. Immunology. 2004;111:86–90. doi: 10.1111/j.1365-2567.2003.01780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 22.Capella JF, Paik DC, Yin NX, Gervasoni JE, Tilson MD. Complement activation and subclassification of tissue immunoglobulin G in the abdominal aortic aneurysm. J Surg Res. 1996;65:31–33. doi: 10.1006/jsre.1996.0339. [DOI] [PubMed] [Google Scholar]
- 23.Gregory AK, Yin NX, Capella J, Xia S, Newman KM, Tilson MD. Features of autoimmunity in the abdominal aortic aneurysm. Arch Surg. 1996;131:85–88. doi: 10.1001/archsurg.1996.01430130087017. [DOI] [PubMed] [Google Scholar]
- 24.Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. 2007;179:2600–2608. doi: 10.4049/jimmunol.179.4.2600. [DOI] [PubMed] [Google Scholar]
- 25.Wada K, Montalto MC, Stahl GL. Inhibition of complement C5 reduces local and remote organ injury after intestinal ischemia/reperfusion in the rat. Gastroenterology. 2001;120:126–133. doi: 10.1053/gast.2001.20873. [DOI] [PubMed] [Google Scholar]
- 26.Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, Takahashi K, Holers VM, Walport M, Gerard C, Ezekowitz A, Carroll MC, Brenner M, Weissleder R, Verbeek JS, Duchatelle V, Degott C, Benoist C, Mathis D. Arthritis critically dependent on innate immune system players. Immunity. 2002;16:157–168. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- 27.Huugen D, van Esch A, Xiao H, Peutz-Kootstra CJ, Buurman WA, Tervaert JW, Jennette JC, Heeringa P. Inhibition of complement factor C5 protects against anti-myeloperoxidase antibody-mediated glomerulonephritis in mice. Kidney Int. 2007;71:646–654. doi: 10.1038/sj.ki.5002103. [DOI] [PubMed] [Google Scholar]
- 28.Daffern PJ, Pfeifer PH, Ember JA, Hugli TE. C3a is a chemotaxin for human eosinophils but not for neutrophils. I. C3a stimulation of neutrophils is secondary to eosinophil activation. J Exp Med. 1995;181:2119–2127. doi: 10.1084/jem.181.6.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thurman JM, Lenderink AM, Royer PA, Coleman KE, Zhou J, Lambris JD, Nemenoff RA, Quigg RJ, Holers VM. C3a is required for the production of CXC chemokines by tubular epithelial cells after renal ishemia/reperfusion. J Immunol. 2007;178:1819–1828. doi: 10.4049/jimmunol.178.3.1819. [DOI] [PubMed] [Google Scholar]
- 30.Wirthmueller U, Dewald B, Thelen M, Schafer MK, Stover C, Whaley K, North J, Eggleton P, Reid KB, Schwaeble WJ. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J Immunol. 1997;158:4444–4451. [PubMed] [Google Scholar]
- 31.Schweinle JE, Ezekowitz RA, Tenner AJ, Kuhlman M, Joiner KA. Human mannose-binding protein activates the alternative complement pathway and enhances serum bactericidal activity on a mannose-rich isolate of Salmonella. J Clin Invest. 1989;84:1821–1829. doi: 10.1172/JCI114367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Selander B, Martensson U, Weintraub A, Holmstrom E, Matsushita M, Thiel S, Jensenius JC, Truedsson L, Sjoholm AG. Mannan-binding lectin activates C3 and the alternative complement pathway without involvement of C2. J Clin Invest. 2006;116:1425–1434. doi: 10.1172/JCI25982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ebanks RO, Isenman DE. Mouse complement component C4 is devoid of classical pathway C5 convertase subunit activity. Mol Immunol. 1996;33:297–309. doi: 10.1016/0161-5890(95)00135-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.