

Abstract
Genome-wide association studies (GWAS) have gained considerable momentum over the last couple of years for the identification of novel complex disease genes. In the field of Alzheimer's disease (AD), there are currently eight published and two provisionally reported GWAS, highlighting over two dozen novel potential susceptibility loci beyond the well-established APOE association. On the basis of the data available at the time of this writing, the most compelling novel GWAS signal has been observed in GAB2 (GRB2-associated binding protein 2), followed by less consistently replicated signals in galanin-like peptide (GALP), piggyBac transposable element derived 1 (PGBD1), tyrosine kinase, non-receptor 1 (TNK1). Furthermore, consistent replication has been recently announced for CLU (clusterin, also known as apolipoprotein J). Finally, there are at least three replicated loci in hitherto uncharacterized genomic intervals on chromosomes 14q32.13, 14q31.2 and 6q24.1 likely implicating the existence of novel AD genes in these regions. In this review, we will discuss the characteristics and potential relevance to pathogenesis of the outcomes of all currently available GWAS in AD. A particular emphasis will be laid on findings with independent data in favor of the original association.
INTRODUCTION
Genetically, Alzheimer's disease (AD), the most common form of dementia in the elderly, is a characteristic ‘complex’ disease. It can be divided into two major forms, (i) cases with strong familial clustering, often showing Mendelian disease transmission and typically exhibiting an early (<65 years) or very early (<50 years) age of onset, and (ii) cases of later-onset age (typically well beyond 65 years), showing no obvious familial aggregation. The Mendelian forms of AD are caused by rare and usually highly penetrant mutations in three genes (APP, PSEN1 and PSEN2), all of which alter production of the amyloid-β peptide (Aβ), the principal component of β-amyloid in senile plaques (1). Although probably several additional disease-causing genes remain to be identified for this type of AD, early-onset familial AD accounts for only <5% of all AD cases (2,3). The vast majority of AD is of the second form which, genetically, is much less well characterized. The most common conception is that late-onset AD is likely to be governed by an array of low-penetrance common risk alleles across a number of different, currently only ill-defined loci. These genes likely affect a variety of pathways, many of which are believed to be involved in the production, aggregation and removal of Aβ. Although the total number of AD risk genes (and their precise identity) remains elusive, there is good evidence to suggest that, in combination, they have a substantial impact on disease predisposition and age of onset (reviewed in Ref. 4).
In the quest to uncover the late-onset AD genes, a vast body of data has been accrued over the past 30 years in well over 1200 studies assessing more than 500 different genes as potential risk factors, mostly using a candidate gene-based approach (5). However, with the exception of one genetic variant, the ε4-allele of the apolipoprotein E gene (APOE) (6,7) none of these candidates has been proven to consistently influence disease risk or onset age in more than a handful of samples. Instead, most reports of ‘novel AD genes’ have been followed by a large number of conflicting results, challenging prior claims that they may play an important role in contributing to disease risk. Because of the exceedingly large number of studies, it has become virtually impossible to systematically follow, evaluate or interpret these findings. To alleviate this problem, our group has created and continues to maintain a regularly updated online encyclopedia and meta-analysis resource for genetic association studies in AD (‘AlzGene’; URL: www.alzgene.org) (5). The AlzGene database currently lists a total of 32 loci that contain at least one genetic variant showing nominally significant association in allele-based, random-effects meta-analyses of all available published data.
GENOME-WIDE ASSOCIATION STUDIES IN AD
By nature of their design, candidate gene studies typically do not allow conclusions beyond the scope of the initial hypothesis, which usually consists of the genetic elucidation of potential pathogenetic pathways. Genome-wide association studies (GWAS) simultaneously test a very large number of genetic markers, normally several hundreds of thousands, in a largely hypothesis-free (or ‘unbiased’) fashion. The markers on a GWAS array consist of single nucleotide polymorphisms (SNPs) which are chosen based on their ability to cover common variation in the human genome (for reviews see Refs. 8,9). More recent arrays also include probes that allow a systematic assessment of copy-number variants, that is, deletions or multiplications of certain chromosomal segments of variable length. Other, less commonly used GWAS arrays only assay SNPs located in known or predicted coding regions (cSNPs). This leads to an enrichment of potentially functionally relevant variants at the expense of overall genome-wide coverage.
For many genetically complex diseases, the GWAS approach has yielded an unexpectedly large number of genome-wide significant findings that were confirmed by independent follow-up studies (9,10). In many instances, these findings promise to advance our understanding of the pathogenetic forces underlying the investigated phenotypes, enabling researchers and clinicians to not only improve diagnostic accuracy, but also to deliver a whole array of new drug targets hopefully leading to more efficient treatment and disease prevention options in the not too distant future. In AD, GWAS results have thus far proved to be less consistent, with the exception of the APOE locus, whose association with AD was identified in all but one study, and always found to be orders of magnitude more significant than any of the newly implicated loci to date. Regardless of the currently observed lack of consistency across studies, the jury is still out as to how many and which of the potential new AD loci will replicate in independent follow-up studies as most of the currently published GWAS have only appeared within the past year.
At the day of this writing (1 August 2009) a total of eight GWAS have been published in AD. Two additional GWAS (Amouyel et al. and Williams) have recently been reported at scientific conferences and are included in this review in anticipation of their forthcoming publication in a peer-reviewed journal. In the remainder of this review, we will discuss the characteristics and outcomes of these 10 AD GWAS, with a particular highlight on the main findings and their potential relevance for AD pathogenesis. An overview of these studies can be found in Table 1, an up-to-date version of this table can be found in the ‘GWAS’ section on AlzGene (URL: http://www.alzgene.org/largescale.asp). Most emphasis will be laid on findings with independent data available in favor of the original findings (Table 2).
Table 1.
Overview of all published GWAS in AD
| GWAS | Design | Population | No. SNPs | No. AD GWAS (Follow-up)a | No. CTRL GWAS (Follow-up)a | ‘Featured’ Genes |
|---|---|---|---|---|---|---|
| Grupe et al. (11) | Case–control | USA and UK | 17 343 | 380 (1428) | 396 (1666) | APOEb, ACAN, BCR, CTSS, EBF3, FAM63Ac, GALP, GWA_14q32.13, GWA_7p15.2, LMNA, LOC651924, MYH13, PCK1, PGBD1, TNK1, TRAK2, UBD |
| Coon et al. (18), Reiman et al. (19) | Case–control | USA, Netherlands | 502 627 | 446 (415) | 290 (260) | APOEb, GAB2 |
| Li et al. (22) | Case–control | Canada and UK | 469 438 | 753 (418) | 736 (249) | APOEb, GOLM1, GWA_15q21.2, GWA_9p24.3 |
| Poduslo et al. (23) | Family-based and case–control | USA | 489 218 | 9 (199) | 10 (225) | TRPC4AP |
| Abraham et al. (25)d | Case–control | UKe | 561 494 | 1082 (–) | 1239 (1400) | APOEb, LRAT |
| Bertram et al. (17) | Family-based | USA | 484 522 | 941 (1767) | 404 (838) | APOEb, ATXN1, CD33, GWA_14q31 |
| Beecham et al. (29) | Case–control | USA | 532 000 | 492 (238) | 496 (220) | APOEb, FAM113B |
| Carrasquillo et al. (30) | Case–control | USA | 313 504 | 844 (1547) | 1255 (1209) | APOEb, PCDH11X |
| Amouyel et al. (33)f | Case–control | Europee | ∼540 000 | 2035 (3978) | 5328 (3297) | APOEb, CLU (APOJ), CR1 |
| Williams (34)f | Case–control | USA and Europee | ∼610 000 | 3941 (2023) | 7848 (2340) | APOEb, CLU (APOJ), PICALM |
Modified after content on the AlzGene website (http://www.alzgene.org; current on 1st August 2009). Studies are listed in order of publication date (determined by PubMed ID number). ‘Featured Genes’ are those genes/loci that were declared as ‘associated’ in the original publication, note that criteria for declaring association may vary across studies.
aNumbers of ‘AD Cases’ and ‘Controls’ refers to sample sizes used in initial GWA screening, whereas ‘Total’ refers to initial sample plus any follow-up samples (where applicable); please consult AlzGene website for more details on these studies.
bIn many studies, surrogate marker were used for APOE.
cThis locus was originally named ‘THEM5’.
dThis study is based on a pooled genotyping approach which may lead to invalid allele frequency estimates and results (see text for details).
eSome sample overlap across studies these studies.
fUnpublished conference reports (see bibliography for details).
Table 2.
Replication attempts of published GWAS results (excluding APOE ε4-related effects)
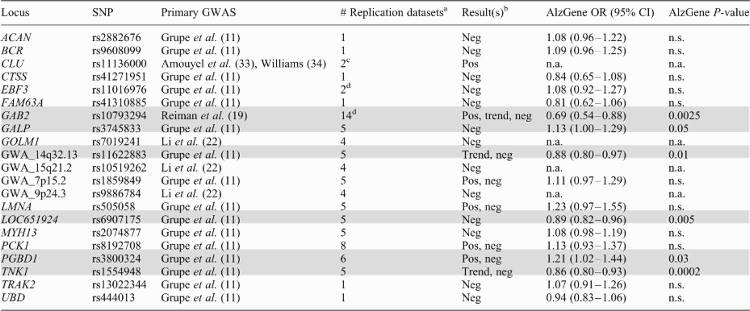 |
Modified after content on the AlzGene website (http://www.alzgene.org; current on 1st August 2009). All ‘featured’ genes from Table 1 not listed here currently have no independent replication data available. Shading indicates loci with at least one nominally significant polymorphism in Alzgene meta-analyses. ‘OR (95% CI)’ random effects allelic summary odds ratios and 95% confidence intervals; ‘n.a.’ not applicable because less than four independent datasets were available; ‘n.s.’ not significant (P-value >0.05).
aMarkers in replication datasets are not necessarily the same as in primary GWAS.
bOverall conclusion reached by authors of the original publications; ‘Neg’ no significant finding; ‘Pos’ significant or ‘Trend’ marginally significant findings; the latter two do not necessarily imply association with the same allele.
cSimultaneously reported in both primary GWAS; each can therefore be considered replication of the other study; note that neither study has yet been published in a peer-reviewed journal (see text for details and important caveats).
Grupe et al. (11)
This study, published online in February of 2007, represents the first GWAS in AD. However, owing to its focus on putative functional variants (cSNPs; see above), it is also the lowest-resolution GWAS in AD, and can therefore not be considered ‘unbiased’ in a strict sense. Overall, ∼17 000 cSNPs were genotyped first in a screening sample of nearly 800 combined AD cases and controls from the UK. Promising signals were then followed-up in four independent datasets from the UK and USA, totaling ∼3100 subjects. In addition to APOE ε4-linked markers, which were the only ones to exhibit genome-wide significance (smallest P-value <1 × 10−8), the analyses of this GWAS highlighted a total of 16 loci with P-values between 5 × 10−5 and 1 × 10−3 (Table 1). Four of these, located in GALP, TNK1, PCK1, and one in a hitherto uncharacterized locus on chromosome 14q32.13 (GWA_14q32.13), were particularly highlighted by the authors based on P-values <1 × 10−4 across all samples combined. However, none of the observed associations replicated in more than two of the five tested samples. Since its first publication, all of the 16 SNPs have been assessed in independent replication studies, with mixed results (Table 2). Five loci (GALP, GWA_14q32.13, LOC651924, PGBD1, TNK1) currently show significant association (P-value ≤0.05) in AlzGene, and are discussed in more detail below.
GALP encodes ‘galanin-like peptide’ (GALP), a member of the galanin family of neuropeptides. The associated SNP (rs3745833) codes for a non-synonymous substitution (Ile72Met) in exon 4 of the longest transcript. The common minor C-allele [minor allele frequency (MAF) in Caucasian populations ∼48%] increases the risk for AD by only ∼10%. GALP binds galanin receptors 1, 2 and 3 with the highest affinity for galanin receptor 3. Interestingly, galanin and its receptors have been shown to be over-expressed in limbic brain regions affected in AD. Galanin inhibits cholinergic neurotransmission and suppresses long-term potentiation in the hippocampus. Thus, galanin and its related peptides, e.g. GALP, when over-expressed could conceivably worsen AD symptoms (12). In support of this hypothesis, transgenic mice over-expressing galanin have been reported to display cognitive and neurochemical deficits characteristic of AD (13).
PGBD1 encodes ‘piggyBac transposable element derived 1’ (PGBD1), which belongs to the subfamily of piggyBac transposable element derived genes. The associated SNP (rs3800324) codes for a non-synonymous substitution (Gly244Glu) in exon 5. The relatively rare (MAF ≈ 6%) minor A (Glu) allele significantly increases risk for AD by ∼20% when all published data is combined. PGBD1 is specifically expressed in the brain, however, its exact function is not known. The gene is part of a complex locus encoding zinc finger protein 187 (ZNF187) and PGBD1. ZNF187 encodes a classic C2H2-zinc finger protein, which plays a role in transcriptional regulation (14). Whether PGBD1 genetically interacts with ZNF187 remains unclear.
TNK1 encodes ‘tyrosine kinase, non-receptor 1’ (TNK1), a non-receptor tyrosine kinase originally known as ‘thirty-eight-negative kinase 1’ (15). The AD-associated SNP (rs1554948) represents a synonymous base change at codon 24 in exon 2. The common minor allele (MAF ∼48%) confers a ∼15% reduction in AD risk on AlzGene. Although the effect size of this association is relatively modest, its significance (P-value = 2 × 10−4) on AlzGene is currently the strongest of any of the GWAS signals proposed in the Grupe et al. study. Furthermore, this association is one of the few showing ‘strong’ epidemiologic credibility when applying interim grading criteria of the Human Genome Epidemiology Network (16). As such, it currently is the highest ranking of all meta-analyzable GWAS SNPs in AD. Functionally, it could be interesting with regard to AD because when activated, TNK1 has been reported to enable tumor necrosis factor alpha (TNFα)-induced apoptosis (15). Thus, TNK1 may act as a novel molecular switch that can determine the properties of TNFα signaling and, potentially, neuronal cell death. Interestingly, the TNFα gene (TNF) is also one of AlzGene's current ‘Top Results’.
The remaining two loci from this GWAS showing significant meta-analysis results on AlzGene [LOC651924 (on chromosome 6q24.1) and GWA_14q32.13] have not yet been assigned to transcripts of known function, so their potential pathogenetic relevance in AD remains unclear. Note that the SNP underlying the latter signal on chromosome 14 (rs11622883) is located ∼10 Mb distal of another GWAS signal in this chromosomal region [rs11159647, identified by our group (17); Table 1]. However, owing to the low linkage disequilibrium (LD) between both markers (r2 = 0.05, based on CEU HapMap data), these two results very likely represent independent events.
Coon et al. (18) and Reiman et al. (19)
These studies from the Translational Genomics Research Institute (TGEN) refer to the same underlying dataset, and with ∼500 000 SNPs (from the Affymetrix 500K array) represent the first published high-resolution GWAS in AD. In the first wave of their analyses, Coon et al. reported that upon testing all genotyped SNPs in 1086 neuropathologically confirmed AD cases and controls, the only signal to reach genome-wide significance was elicited by a marker in strong LD with APOE ε4 (P-value = 5.3 × 10−34). A few months later, the same group reported a re-analysis of their GWAS data (19), for which they only considered ∼300 000 SNPs and divided their neuropathological sample into a ‘discovery’ (736 combined cases and controls), and ‘replication’ (321 subjects) cohort. This was supplemented by 364 AD cases and controls with ‘clinical’ (i.e. not neuropathologically confirmed) diagnoses. Upon stratification on APOE ε4 genotype, the authors identified genome-wide significant (smallest P-value = 9.7 × 10−11) association with five SNPs in GAB2. Whereas not statistically significant in all of the three analyzed cohorts, the direction of the observed associations were highly consistent across samples suggesting a ∼2–4-fold increase in risk for AD in carriers of the major alleles. In support of the genetic findings, the authors also reported the results of molecular analyses (see below) suggesting that the observed associations may be due to effects on the phosphorylation of tau protein, one of the two major histopathological hallmarks of AD.
Since the initial report, the potential association between GAB2 and AD risk was investigated in a number of independent case–control and family-based studies, the majority of which supported the notion that variants in GAB2 are associated with AD risk (Table 2). On AlzGene, all 10 of the meta-analyzed SNPs show evidence of significant association (P-values ranging from ∼0.03 to 0.0025). The strongest meta-analysis results in terms of effect size are currently observed with rs10793294, for which the minor allele indicates an almost 50% reduction in AD risk [OR = 0.69 (95% CI: 0.54–0.88); Table 2], in agreement with the originally proposed risk effect of the major allele. The case–control meta-analyses are confirmed by family-based analyses from our group which suggest a similar reduction of AD risk [OR = 0.76 (95% CI: 0.62–0.94)] in over 4000 subjects from nearly 1300 independent families (20). Although in most studies, the GAB2 effects appeared stronger in subjects carrying at least one ε4-APOE allele, the epidemiological (and molecular) basis of this potential interaction still needs to be further addressed. Overall, owing to the relative consistency across the up to 16 000 individuals in which this association has been tested to date, up to 90% of which originated from datasets independent of the GWAS samples, it appears relatively likely that GAB2 (or a locus in tight LD with it) represents a genuine AD susceptibility factor. As such, it would constitute the first proof-of-concept for the GWAS approach in AD.
GAB2 encodes ‘GRB2-associated binding protein 2’ (Gab2), a member of a evolutionarily highly conserved gene family characterized by their binding to GRB2 (growth factor receptor-bound protein 2). Of the 10 markers showing significant results on AlzGene, only one (rs1385600) is predicted to map within the coding region of GAB2, where it does not invoke a change in the amino acid sequence. GAB-family proteins function as scaffolding/adapter proteins involved in multiple signaling and transduction pathways. Gab2 is ubiquitously expressed, but is found at particularly high levels in the prefrontal cortex and the hypothalamus. The original GWAS article (19) suggested that changes in Gab2 expression could potentially affect glycogen synthase kinase 3 (Gsk3)-dependent phosphorylation of tau and the formation of neurofibrillary tangles. Moreover, growth factor receptor-bound protein (GRB2), which binds Gab2, also binds tau, APP, presenilin 1 and presenilin 2 (21). Interactions of these molecules with GRB2 have been proposed to regulate signal transduction (for example, via the extracellular signal-regulated kinase (ERK)1,2 pathway). Consequently, Gab2 could conceivably modulate APP processing and/or tau phosphorylation via its interaction with GRB2.
Li et al. (22)
The third AD GWAS studied the same 500 000 SNP panel as the TGEN study using about twice as many subjects, all of Caucasian ancestry originating from Canada (∼1500 combined cases and controls; Table 1). Suggestive signals were followed-up in 418 AD cases and 249 healthy controls from the UK. In addition to markers linked to APOE ε4 (P-value = 2.3 × 10−44), the authors highlighted four SNPs which showed consistent evidence of association in both investigated samples, none of which reached genome-wide significance (GWAS P-values ranging from ∼4 × 10−6 to 3 × 10−4). Three of these SNPs [rs10868366, rs7019241 (both in GOLM1), and rs9886784 (in an uncharacterized region on chromosome 9; GWA_9p24.3)] showed association with risk for AD, whereas one SNP [rs10519262 (in an uncharacterized region on chromosome 15; GWA_15q21.2)] showed the strongest association with onset age for AD. Thus far, the only independent replication attempt for these potential AD loci (20) failed to detect any association between disease risk or onset age for any of the implied variants (Table 2), despite good to excellent power (≥85%) to detect the originally suggested effect sizes.
GOLM1, which is also known as GOLPH2, encodes ‘golgi membrane protein 1’, type II Golgi transmembrane protein. Both SNPs implicated by Li et al. lie deep intronic without any known or obvious functional implication. Therefore, additional independent replication data should be awaited before hypotheses or molecular experiments on the functional relevance of this and the other two unknown loci in AD pathogenesis appear justified.
Poduslo et al. (23)
Owing to the small sample size analyzed, this GWAS probably represents the one with the highest uncertainty with respect to its outcome and relevance for AD. The authors studied a total of nine affected and ten unaffected individuals from two large, multiplex AD pedigrees on the Affymetrix 500K array. The only genome-wide significant association reported was with six SNPs in the TRPC4AP gene on chromosome 20q11 (smallest uncorrected P-value = 5.6 × 10−11). Possibly owing to the small number of individuals tested, no association was reported for markers within or in LD with APOE. Follow-up analyses in the remaining available members of the two pedigrees revealed association with a common (∼40%) 10-SNP haplotype spanning the entire coding region of the TRPC4AP locus. First, and foremost, for their initial subsequent analyses in 284 unrelated cases and controls suggested nominally significant association of the same haplotype with AD risk, however, no other group has yet reported an independent assessment of this association.
TRPC4AP encodes ‘transient receptor potential cation channel, subfamily C, member 4 associated protein’, which is also known as ‘tumor necrosis factor receptor-associated ubiquitous scaffolding and signaling protein’ (TRUSS). There is some evidence from in vitro assays that TRUSS may be involved in TNFα-related pathways (24). This could be of potential relevance to AD, as the regulation of inflammatory responses plays an important role in AD pathogenesis. Furthermore, several cytokines, including the gene encoding TNFα, currently show significant association with AD risk on AlzGene.
Abraham et al. (25)
This GWAS, assaying ∼560 000 HapMap-based SNPs (using Illumina technology) in a case–control dataset totaling ∼2300 subjects from the UK, differs from all other currently published AD GWAS by two characteristics. First, and most importantly, for their initial screening, this study utilized DNA pools, rather than individual DNA samples, for genotyping. Although this approach tremendously reduces the overall cost of the GWAS, this economical gain comes at the expense of scientific precision. The problem lies in the fact that genetic association studies, by design, compare genotype or allele frequencies in affected versus unaffected individuals. All statistical results and subsequent inferences will be flawed if the primary frequency estimations are incorrect, which may have appreciably affected the outcome of this study. For the most significant findings, comparisons of pooled GWAS versus individually generated allele frequency estimates averaged differences of ∼12%, for some SNPs exceeding 40%. Considering that the underlying allele frequency differences between cases versus controls only amount to 2–6% (depending on the MAF in the general population) for an allelic OR of ∼1.25 in a combined sample size equivalent to the one studied by Abraham et al., the observed differences between pooled and individual genotypes appear unacceptably large. Second, in the follow-up stage of their study, the authors only increased the number of controls by adding previously published ‘general disease’ controls from the Wellcome Trust Case-Control Consortium. Thus, this does not represent an independent assessment of the observed associations as the case-group remained unchanged compared to the GWAS screening. Regardless of these methodological limitations, the authors identified APOE ε4-related effects as their top finding (smallest P-value = 8.2 × 10−11), followed by nominally significant results for 109 other loci. Of these, the association between SNPs in the LRAT gene (encoding ‘lecithin retinol acyltransferase’) located on chromosome 4q32 was particularly highlighted. Owing to the potential methodological weaknesses of this study and the complete lack of independent follow-up data, it appears premature to speculate about the potential relevance of this association in AD pathogenesis. Note that the same group recently reported the outcome of a GWAS employing individual genotyping in a much larger, partially overlapping, dataset (Williams, see below), which supersedes the results of this study. In particular, LRAT was not one of the top results reported in the more recent GWAS.
Bertram et al. (17)
This GWAS was conducted by our group and represented the first to employ family-based methods for the initial screening and replication analyses. Overall, we studied DNA samples from 1345 subjects for the primary analyses using the Affymetrix 500K SNP panel, followed-up in 2605 individuals from three independent collections of AD families. Using a novel analytical approach developed by Lange and colleagues (26), the original GWAS dataset was first screened on the basis of the between-family information — which is statistically independent from the family-based association test-statistic — evaluating the evidence for association at a population level to estimate the conditional power for each marker. In a second step, the actual test statistic was computed for all markers using a compound phenotype constructed of affection status and onset age information. The significance of these tests was then assessed on the basis of individually adjusted alpha levels that maintain the overall type 1 error rate and that are weighted on the basis of the conditional power estimate for the corresponding marker from the first screening step. As for all other AD GWAS to date, the by far most significant signal was observed with a marker in strong LD with APOE ε4 (P-value 5.7 × 10−14). After correction for the number of tests performed, four non-APOE-related SNPs—none of which was previously described as potential modifier of AD risk or onset age—attained genome-wide significance at an overall alpha level of 5%. Three of these markers showed significant (GWA_14q31.2, CD33), or at least marginally significant (ATXN1; Table 1) association consistent with the GWAS findings in follow-up analyses on 2600 DNAs from family-based datasets. The top signal to emerge from these analyses (rs11159647 in a hitherto uncharacterized region on chromosome 14q; GWA_14q31.2) also showed consistent replication in one of the two publicly available GWAS datasets (TGEN, see above). Overall, the convergence of significant results in multiple independent family-based and at least one case–control sample provide compelling evidence implicating the presence of a putative AD locus on chromosome 14q31, and possibly additional loci on chromosomes 6p22 (in or near ATXN1) and 19q13 (in or near CD33).
The SNP at GWA_14q31 resides at position 83 844 962 bp on chromosome 14 in an intron of the Genscan-predicted gene, NT_026437.1360, which spans ∼723 kb. The coding region of this predicted gene in the region of rs11159647 reveals no significant homologies to other genes or coding regions. Interestingly, the 3′ end of this predicted gene contains exons with homology to the C2H2-type kruppel-like zinc-finger protein 268 (ZNF268) (27). However, the AD-associated SNP, rs11159647, is >350 kb from the ZNF268 homologous region, and SNPs in this area reveal no strong LD with rs11159647 on HapMap. There are three expressed sequence tags (ESTs) residing within 60 kb on either side of rs11159647, i.e. M85511, CA390254 and AI003603. All three ESTs are expressed in the brain and are encoded within the same region as the predicted gene. However, the predicted exon structure of these ESTs does not align with the predicted exons of NT_026437.1360. Thus, these ESTs may represent exons of separate gene(s) in this region, which are expressed in the brain.
With regard to the other two SNPs identified in our study, rs179943, on 6p22.3 resides within an intron of the ataxin 1 (ATXN1) gene, in which an elongated polyglutamine tract causes the progressive neurodegenerative disease, spinocerebellar ataxia (SCA1). SNP, rs3826656, on 19q33, resides less than 2 kb proximal of the transcription initiation site of CD33. This gene, also known as SIGLEC3, encodes a cell-surface receptor on cells of monocytic or myeloid lineage. It is also a member of the SIGLEC family of lectins that bind sialic acid and regulate the innate immune system via the activation of caspase-dependent and caspase-independent cell-death pathways.
Beecham et al. (28)
The seventh AD GWAS assayed ∼550 000 HapMap-based SNPs using Illumina technology on nearly 1000 combined AD cases and controls of Caucasian ancestry from the USA. Promising signals were followed-up in 458 independent subjects and by re-analysis of the publicly available TGEN data (see above). The second most significant finding after APOE (P-value <1 × 10−20), was elicited by a SNP in the FAM113B gene on chromosome 12q13 (P-value = 1.9 × 10−6), a region heavily studied in the AD genetics community based on genetic linkage data pointing slightly proximal to this general region on chromosome 12 (5). Although assessment in the much smaller follow-up sample suggested consistent and nominally significant (P-value = 0.05) association with the same marker, this SNP could not be tested in the TGEN dataset, and no other study has yet published independent association data at this locus. Of the remaining top ranking GWAS signals that were tested across the Beecham et al. dataset and the TGEN study, four showed consistent evidence of association across both studies [DISC1 on chromosome 1q42, several markers in ZNF224 on chromosome 19q13 (not linked to APOE), as well as two uncharacterized loci on chromosomes 4q28 and 6q14]. However, no results were presented for these latter signals in the authors' own replication sample, so that more data needs to be accrued before any further conclusions can be reached for these loci. Finally, the authors specifically re-analyzed both GWAS datasets for genes included in the AlzGene database, and found nominal evidence of association with a total of eight loci. However, no details were provided for these results, so that these loci, too, should be interpreted with caution until further assessments are published. After consideration of all analyses, the association of FAM113B with AD risk appears as the most meaningful outcome of this GWAS.
FAM113B encodes ‘family with sequence similarity 113, member B’, a hypothetical protein also known as ‘LOC91523’. The SNP found to be associated (rs11610206) maps ∼8 kb 3′ of this predicted protein into a chromosomal region that is evolutionarily not highly preserved. The authors also discussed the gene encoding vitamin D receptor as potentially underlying the association signal, however, this gene maps ∼600 kb proximal of the GWAS marker with no significant LD between the two loci. Meanwhile, little is known about the function of the putative FAM113B protein other than the fact that it contains a SGNH hydrolase-type esterase domain, which also occurs in some esterases and lipases (29).
Carrasquillo et al. (30)
Shortly after the Beecham et al. study, Younkin and colleagues reported the results of their GWAS on 2100 subjects from the Mayo clinic series using an Illumina array, which assays just over 300 000 HapMap-based markers. The only SNPs to attain genome-wide significance were located on chromosome 19 and showed strong LD with APOE ε4 (smallest P-value 4.8 × 10−46). The authors continued to test the 25 most strongly associated GWAS signals (10 in LD with APOE) in an independent series of nearly 2800 combined cases and controls. The only non-APOE-related marker to show Bonferroni-corrected significant P-values was rs5984894 which lies in PCDH11X located in the non-pseudoautosomal region on chromosome X. Accordingly, the association was strongest in females conferring ORs between 1.75 (homozygous) and 1.26 (heterozygous) in carriers versus non-carriers of the putative risk allele. For hemizygous males, a similar trend was observed (OR = 1.18), although this did not reach statistical significance (P-value 0.07). Genotyping of additional PCDH11X SNPs in the combined GWAS and replication series also yielded significant results, one (rs2573905) even slightly exceeding the degree of statistical significance observed for the GWAS marker in this region. This SNP was deemed as particularly interesting as it is located in a relatively well conserved 100 bp region, and also exhibits strong LD (r2 = 0.98) with the original GWAS marker. Since the publication of these results in February 2009, no other studies have assessed the potential association between PCDH11X and AD risk, so that it is currently impossible to evaluate the epidemiological relevance of this finding. Of all published and reported AD GWAS, however, it is the only to imply an X chromosome locus, which, if confirmed, could at least partially explain the well established increased disease prevalence in women versus men.
PCDH11X encodes ‘protocadherin 11 X-linked’ (PCHD11X), and belongs to a subfamily genes of the cadherin superfamily. With the possible exception of the 70% conservation between human and mouse of SNP rs2573905, none of the PCDH11X variants implied by Carrasquillo et al. have any proven or predicted functional consequences. Still, some lines of evidence could suggest a possible involvement in AD pathogenesis, namely that its Y-chromosome homologue, PCDH11Y, is a member of the cadherin family of cell surface receptors, which are involved in cell–cell adhesion and signaling, possibly in synaptic junctions (31). Since some protocadherins have been proposed as γ-secretase substrates (32), it would be interesting to test whether PCDH11X competes with APP for γ-secretase.
Amouyel et al. (33) and Williams (34)
The results of these independently performed GWAS were recently reported at the ‘2009 International Conference on Alzheimer's Disease’ in Vienna, Austria, and have not yet been published in any peer-reviewed journal. Notwithstanding the preliminary status of these GWAS at the day of this writing, they are included here for two reasons: (i) with ∼14 600 and ∼16 100 combined cases and controls, respectively, they each have studied at least 3-times more individuals than even the largest previous GWAS in AD; (ii) unlike any other two AD GWAS previously, they independently reported the same genome-wide significant non-APOE-related top result, namely association of AD risk with the same allele of one SNP (rs11136000) in the CLU gene on chromosome 8p21 (Tables 1 and 2). Methodologically, both studies employed a HapMap-based SNP genotyping approach using a combination of different Illumina arrays for their initial GWAS samples, resulting in similar uncorrected P-values of the CLU association (7.5 × 10−9 and 1.4 × 10−9, respectively). Interestingly, both studies estimated identical effect sizes (OR = 0.86) for the minor (MAF ∼30%) allele of rs11136000 when comparing carriers versus non-carriers and upon combining GWAS and follow-up datasets. Thus, in carriers of this allele the risk to develop AD would be reduced by ∼16%. According to calculations presented by the Williams group, however, this effect would explain only ∼2–3% of total AD risk in the general population, i.e. maximally one tenth of that estimated for APOE ε4 (5). Despite the consistency of the association, it should be noted that there was some overlap in datasets across both GWAS so that additional analyses will be necessary to determine the truly independent components of each study.
Due to its functional relatedness with APOE, CLU (also known as APOJ) actually represents one of first studied candidate genes in AD. However, the one published prior study on CLU only tested two SNPs in ∼920 AD cases and controls, but did not detect any consistent effects (35). This is not surprising, given that this study only had a theoretical power of ∼50% to detect an allelic OR of 0.86 at a P-value of 0.05 (in reality, power was likely much less due to the imperfect LD between the markers tested by Tycko et al. and rs11136000, and other study design-related issues). In addition to the association with CLU, each of the two GWAS also detected genome-wide association with one other locus (CR1 and PICALM, Table 1), which are not discussed here further owing of the lack of additional data.
CLU encodes ‘clusterin’ (also known as ‘apolipoprotein J’), which is a ∼75 kDa glycoprotein expressed in all tissues, including the CNS. The associated SNP lies deeply intronic with no known or implied functional consequences. According to data presented by the Williams group, it is in strong LD (r2 = 0.95) with a synonymous base-change in exon 5 of the CLU gene (rs7982; His315His), which—based on its location, and if confirmed as the underlying functional variant—might possibly be involved in alternative splicing or expressional regulation of the transcript. Of relevance to AD, clusterin has been proposed to bind soluble Aβ and transport it from plasma across the blood-brain barrier (36). Interestingly, apoE has been proposed to transport Aβ in the opposite direction, from the brain to the plasma (37). Thus, apoE and clusterin may both play major roles in regulating cerebral Aβ levels based on clearance, and more specifically, transport from and into the brain.
CONCLUSIONS
As for many genetically heterogeneous and complex diseases, the application of genome-wide association screening for the purpose of identifying novel susceptibility genes has gained considerable momentum in the field of AD over the last couple of years. To date, the results of eight published and two provisionally reported AD GWAS have been reported, highlighting over two dozen novel potential AD-associated loci. Whereas only few independent assessments of the reported GWAS signals have been performed yet, based on the data available at the time of this writing, the most compelling and genuine non-APOE-related published GWAS signals have been observed in GAB2, followed by less consistently replicated signals in GALP, PGBD1, TNK1. In addition, consistent replication has been announced at a public meeting for CLU. Finally, there are also at least three replicated loci in hitherto uncharacterized genomic regions on chromosomes 14q (2×) and 6q worthy of further investigation.
As additional GWAS are carried out on larger datasets and higher-resolution arrays, we can expect the list of novel AD gene candidates to keep growing over the coming years. For all of these putative associations, replication attempts and meta-analyses across multiple independent samples will be essential to determine the identity of bona fide AD susceptibility genes. Despite the rapid progress being made in these still early days of the GWAS era, it should be emphasized that for none of the novel AD candidate genes that have thus far emerged from genome-wide screening, do we have conclusive functional genetic evidence that would allow to unequivocally establish any of these loci as genuine AD risk genes. The emergence of such data will require considerable deep re-sequencing efforts in parallel with variant–activity relationship studies using suitable in vitro assays followed by validation in patient materials and/or relevant animal models. Only concurrent research programs involving more comprehensive GWAS of large datasets, replication assessments of GWAS hits in independent samples, and attempts to pinpoint pathogenic DNA variants/mutations in candidate loci will allow to establish ‘truly’ novel AD genes.
NOTE ADDED IN PROOF
During the typesetting of this manuscript the GWAS data earlier reported by Amouyel et al. (33) and Williams (34) were published. These papers have been added to the reference list as refs (38) and (39), respectively. Furthermore, a GWAS (40) using quantitative data from MR-imaging in AD patients and controls was published after the freeze date used for this article, highlighting a number of potential new candidate genes not related to APOE, including EFNA5 (chr. 5q21.3), CAND1 (chr. 12q14.3), MAGI2 (chr. 7q21.11), ARSB (chr. 5q14.1), and PRUNE2 (chr. 9q21.13). Please consult the AlzGene website (http://www.alzgene.org) for an up-to-date summary of these and future GWAS in AD.
FUNDING
This work was made possible by support from the Cure Alzheimer's Fund, NIMH, NIA, and the German Federal Ministry of Education and Research (BMBF). The AlzGene database is funded by the Cure Alzheimer's Fund.
ACKNOWLEDGEMENTS
We thank Dr. U.F. for his continuing inspiration and helpful comments on the manuscript.
Conflicts of Interest statement. L.B. reports no conflict of interest. R.E.T. serves as consultant to, and holds equity in Prana Biotechnology and Pathway Genomics. R.E.T. also serves as consultant to Eisai.
REFERENCES
- 1.Tanzi R.E., Bertram L. 20 years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Raux G., Guyant-Marechal L., Martin C., Bou J., Penet C., Brice A., Hannequin D., Frebourg T., Campion D. Molecular diagnosis of autosomal dominant early onset Alzheimer's disease: an update. J. Med. Genet. 2005;42:793–795. doi: 10.1136/jmg.2005.033456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janssen J.C., Beck J.A., Campbell T.A., Dickinson A., Fox N.C., Harvey R.J., Houlden H., Rossor M.N., Collinge J. Early onset familial Alzheimer's disease: Mutation frequency in 31 families. Neurology. 2003;60:235–239. doi: 10.1212/01.wnl.0000042088.22694.e3. [DOI] [PubMed] [Google Scholar]
- 4.Bertram L., Tanzi R.E. 30 years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat. Rev. Neurosci. 2008;9:768–778. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- 5.Bertram L., McQueen M.B., Mullin K., Blacker D., Tanzi R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 6.Strittmatter W.J., Saunders A.M., Schmechel D., Pericak-Vance M., Enghild J., Salvesen G.S., Roses A.D. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saunders A.M., Strittmatter W.J., Schmechel D., George-Hyslop P.H., Pericak-Vance M.A., Joo S.H., Rosi B.L., Gusella J.F., Crapper-MacLachlan D.R., Alberts M.J., et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 8.Barrett J.C., Cardon L.R. Evaluating coverage of genome-wide association studies. Nat. Genet. 2006;38:659–662. doi: 10.1038/ng1801. [DOI] [PubMed] [Google Scholar]
- 9.McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P., Hirschhorn J.N. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 10.Ioannidis J.P., Thomas G., Daly M.J. Validating, augmenting and refining genome-wide association signals. Nat. Rev. Genet. 2009;10:318–329. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grupe A., Abraham R., Li Y., Rowland C., Hollingworth P., Morgan A., Jehu L., Segurado R., Stone D., Schadt E., et al. Evidence for novel susceptibility genes for late-onset Alzheimer's disease from a genome-wide association study of putative functional variants. Hum. Mol. Genet. 2007;16:865–873. doi: 10.1093/hmg/ddm031. [DOI] [PubMed] [Google Scholar]
- 12.Counts S.E., He B., Che S., Ginsberg S.D., Mufson E.J. Galanin hyperinnervation upregulates choline acetyltransferase expression in cholinergic basal forebrain neurons in Alzheimer's disease. Neurodegener. Dis. 2008;5:228–231. doi: 10.1159/000113710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steiner R.A., Hohmann J.G., Holmes A., Wrenn C.C., Cadd G., Jureus A., Clifton D.K., Luo M., Gutshall M., Ma S.Y., et al. Galanin transgenic mice display cognitive and neurochemical deficits characteristic of Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2001;98:4184–4189. doi: 10.1073/pnas.061445598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Attar R.M., Gilman M.Z. Expression cloning of a novel zinc finger protein that binds to the c-fos serum response element. Mol. Cell. Biol. 1992;12:2432–2443. doi: 10.1128/mcb.12.5.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Azoitei N., Brey A., Busch T., Fulda S., Adler G., Seufferlein T. Thirty-eight-negative kinase 1 (TNK1) facilitates TNFalpha-induced apoptosis by blocking NF-kappaB activation. Oncogene. 2007;26:6536–6545. doi: 10.1038/sj.onc.1210476. [DOI] [PubMed] [Google Scholar]
- 16.Ioannidis J.P., Boffetta P., Little J., O'Brien T.R., Uitterlinden A.G., Vineis P., Balding D.J., Chokkalingam A., Dolan S.M., Flanders W.D., et al. Assessment of cumulative evidence on genetic associations: interim guidelines. Int. J. Epidemiol. 2008;37:120–132. doi: 10.1093/ije/dym159. [DOI] [PubMed] [Google Scholar]
- 17.Bertram L., Lange C., Mullin K., Parkinson M., Hsiao M., Hogan M.F., Schjeide B.M., Hooli B., Divito J., Ionita I., et al. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 2008;83:623–632. doi: 10.1016/j.ajhg.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coon K.D., Myers A.J., Craig D.W., Webster J.A., Pearson J.V., Lince D.H., Zismann V.L., Beach T.G., Leung D., Bryden L., et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer's disease. J. Clin. Psychiatry. 2007;68:613–618. doi: 10.4088/jcp.v68n0419. [DOI] [PubMed] [Google Scholar]
- 19.Reiman E.M., Webster J.A., Myers A.J., Hardy J., Dunckley T., Zismann V.L., Joshipura K.D., Pearson J.V., Hu-Lince D., Huentelman M.J., et al. GAB2 alleles modify Alzheimer's risk in APOE epsilon4 carriers. Neuron. 2007;54:713–720. doi: 10.1016/j.neuron.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schjeide B.M., Hooli B., Parkinson M., Hogan M.F., DiVito J., Mullin K., Blacker D., Tanzi R.E., Bertram L. GAB2 as an Alzheimer disease susceptibility gene: follow-up of genomewide association results. Arch. Neurol. 2009;66:250–254. doi: 10.1001/archneurol.2008.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nizzari M., Venezia V., Repetto E., Caorsi V., Magrassi R., Gagliani M.C., Carlo P., Florio T., Schettini G., Tacchetti C., et al. Amyloid precursor protein and Presenilin1 interact with the adaptor GRB2 and modulate ERK 1,2 signaling. J. Biol. Chem. 2007;282:13833–13844. doi: 10.1074/jbc.M610146200. [DOI] [PubMed] [Google Scholar]
- 22.Li H., Wetten S., Li L., St Jean P.L., Upmanyu R., Surh L., Hosford D., Barnes M.R., Briley J.D., Borrie M., et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch. Neurol. 2008;65:45–53. doi: 10.1001/archneurol.2007.3. [DOI] [PubMed] [Google Scholar]
- 23.Poduslo S.E., Huang R., Huang J., Smith S. Genome screen of late-onset Alzheimer's extended pedigrees identifies TRPC4AP by haplotype analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009;150B:50–55. doi: 10.1002/ajmg.b.30767. [DOI] [PubMed] [Google Scholar]
- 24.Soond S.M., Terry J.L., Riches D.W. TRUSS, a tumor necrosis factor receptor-1-interacting protein, activates c-Jun NH(2)-terminal kinase and transcription factor AP-1. FEBS Lett. 2006;580:4591–4596. doi: 10.1016/j.febslet.2006.06.098. [DOI] [PubMed] [Google Scholar]
- 25.Abraham R., Moskvina V., Sims R., Hollingworth P., Morgan A., Georgieva L., Dowzell K., Cichon S., Hillmer A.M., O'Donovan M.C., et al. A genome-wide association study for late-onset Alzheimer's disease using DNA pooling. BMC Med. Genomics. 2008;1:44. doi: 10.1186/1755-8794-1-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ionita-Laza I., McQueen M.B., Laird N.M., Lange C. Genomewide weighted hypothesis testing in family-based association studies, with an application to a 100K scan. Am. J. Hum. Genet. 2007;81:607–614. doi: 10.1086/519748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gou D.M., Sun Y., Gao L., Chow L.M., Huang J., Feng Y.D., Jiang D.H., Li W.X. Cloning and characterization of a novel Kruppel-like zinc finger gene, ZNF268, expressed in early human embryo. Biochim. Biophys. Acta. 2001;1518:306–310. doi: 10.1016/s0167-4781(01)00194-4. [DOI] [PubMed] [Google Scholar]
- 28.Beecham G.W., Martin E.R., Li Y.J., Slifer M.A., Gilbert J.R., Haines J.L., Pericak-Vance M.A. Genome-wide association study implicates a chromosome 12 risk locus for late-onset Alzheimer disease. Am. J. Hum. Genet. 2009;84:35–43. doi: 10.1016/j.ajhg.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molgaard A., Kauppinen S., Larsen S. Rhamnogalacturonan acetylesterase elucidates the structure and function of a new family of hydrolases. Structure. 2000;8:373–383. doi: 10.1016/s0969-2126(00)00118-0. [DOI] [PubMed] [Google Scholar]
- 30.Carrasquillo M.M., Zou F., Pankratz V.S., Wilcox S.L., Ma L., Walker L.P., Younkin S.G., Younkin C.S., Younkin L.H., Bisceglio G.D., et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer's disease. Nat. Genet. 2009;41:192–198. doi: 10.1038/ng.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanco P., Sargent C.A., Boucher C.A., Mitchell M., Affara N.A. Conservation of PCDHX in mammals; expression of human X/Y genes predominantly in brain. Mamm. Genome. 2000;11:906–914. doi: 10.1007/s003350010177. [DOI] [PubMed] [Google Scholar]
- 32.Haas I.G., Frank M., Veron N., Kemler R. Presenilin-dependent processing and nuclear function of gamma-protocadherins. J. Biol. Chem. 2005;280:9313–9319. doi: 10.1074/jbc.M412909200. [DOI] [PubMed] [Google Scholar]
- 33.Amouyel P., Lambert J.C. and the GWALZ Investigators. 2009 International Conference on Alzheimer's Disease. Vol. S5-03-05. Vienna, Austria: Alzheimer's Association; 2009. Genome wide association analysis of Alzheimer's disease. [Google Scholar]
- 34.Williams J. 2009 International Conference on Alzheimer's Disease. Vol. PL-05.Plenary 5. Vienna, Austria: Alzheimer's Association; 2009. Genome wide studies of Alzheimer's disease: the bigger the better. [Google Scholar]
- 35.Tycko B., Feng L., Nguyen L., Francis A., Hays A., Chung W.Y., Tang M.X., Stern Y., Sahota A., Hendrie H., et al. Polymorphisms in the human apolipoprotein-J/clusterin gene: ethnic variation and distribution in Alzheimer's disease. Hum. Genet. 1996;98:430–436. doi: 10.1007/s004390050234. [DOI] [PubMed] [Google Scholar]
- 36.Zlokovic B.V., Martel C.L., Matsubara E., McComb J.G., Zheng G., McCluskey R.T., Frangione B., Ghiso J. Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc. Natl. Acad. Sci. USA. 1996;93:4229–4234. doi: 10.1073/pnas.93.9.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanzi R.E., Moir R.D., Wagner S.L. Clearance of Alzheimer's Abeta peptide: the many roads to perdition. Neuron. 2004;43:605–608. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 38.Lambert J.C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M., Combarros O., Zelenika D., Bullido M.J., Tavernier B., et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009 doi: 10.1038/ng.439. Epub ahead of print September 6, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M.L., Pahwa J.S., Moskvina V., Dowzell K., Williams A., et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009 doi: 10.1038/ng.440. Epub ahead of print September 6, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Potkin S.G., Guffanti G., Lakatos A., Turner J.A., Kruggel F., Fallon J.H., Saykin A.J., Orro A., Lupoli S., Salvi E., et al. Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer's disease. PLoS One. 2009;7 doi: 10.1371/journal.pone.0006501. 4(8):e6501. [DOI] [PMC free article] [PubMed] [Google Scholar]