

Abstract
The human D5 monoclonal antibody binds to the highly conserved hydrophobic pocket on the N-terminal heptad repeat (NHR) trimer of HIV-1 gp41 and exhibits modest yet relatively broad neutralization activity. Both binding and neutralization depend on residues in the complementarity determining regions (CDRs) of the D5 IgG variable domains on heavy chain (VH) and light chain (VL). In an effort to increase neutralization activity to a wider range of HIV-1 strains, we have affinity matured the parental D5 scFv by randomizing selected residues in 5 of its 6 CDRs. The resulting scFv variants derived from four different CDR changes showed enhanced binding affinities to gp41 NHR mimetic (5-helix) which correlated to improved neutralization potencies by up to 8-fold. However, when converted to IgG1s, these D5 variants had up to a 12-fold reduction in neutralization potency over their corresponding scFvs despite their slightly enhanced in vitro binding affinities. Remarkably, D5 variant IgG1s bearing residue changes in CDRs that interact with epitope residues N-terminal to the hydrophobic pocket (such as VH CDR3 and VL CDR3) retained more neutralization potency than those containing residue changes in pocket-interacting CDRs (such as VH CDR2). These results provide compelling evidence for the existence of a steric block to an IgG that extends to the gp41 NHR hydrophobic pocket region, and can be a useful guide for developing therapeutic antibodies and vaccines circumventing this block.
Keywords: affinity maturation, monoclonal antibody, scFv, gp41, HIV-1, neutralization
Introduction
Eliciting highly potent and broadly neutralizing antibodies holds the key for the development of a successful prophylactic HIV-1 vaccine.1–3 Envelope (Env) glycoproteins present as spikes on the surface of virions have so far been the only target of choice for developing subunit vaccines, peptide-based vaccines, or even virus-like particle-based vaccines against HIV-1.1–3 Each Env spike on the virion surface is composed of a trimer of noncovalently linked gp120-gp41 heterodimers,4 and, as determined by cryoelectron microscopy tomography, there are approximately fourteen gp120-gp41 trimers present on each HIV-1 virion.5 However, the high degree of gp120 sequence variation and glycosylation, as well as conformational masking and occlusion of epitopes on gp41, allow HIV-1 to evade the humoral immune responses and pose great challenges for vaccine development.1,2 Therefore, despite decades of research, so far only a handful of monoclonal antibodies (mAbs) have proven to be able to effectively and broadly neutralize HIV-1 in vitro and to a lesser extent in vivo.2,6 The best characterized broadly neutralizing antibodies against HIV-1 include 2F5 and 4E10, which target the C-terminal portion of the C-terminal heptad repeat (CHR) region and the membrane-proximal external region (MPER) of gp41. Additional neutralizing antibodies include 2G12, which recognizes a specific conformation of oligomannose residues on native gp120, and b12, which overlaps the CD4-binding site on gp120.2,6
In general, the amino acid sequence of gp41 is far more conserved than that of gp120 which makes gp41 a target of great interest for developing specific monoclonal antibodies, peptide inhibitors and even small-molecule inhibitors.4,7 With the exception of the CHR and MPER regions, the bulk of gp41 is likely to be shielded by gp120, and therefore inaccessible to most inhibitors. However, upon the binding of gp120 to CD4 receptors, a series of conformational changes in gp41 brings it into a so-called ‘pre-hairpin’ intermediate state in which the N-terminal heptad repeat (NHR) helices of three gp41 subunits are exposed and associated to form a central three-stranded coiled-coil core, followed by antiparallel packing of the three CHR helices into grooves of the NHR trimer which forms a ‘trimer-of-hairpins’ (or ‘six-helix bundle’) structure. This six-helix bundle drives the viral and host cell membrane to fuse which enables viral entry.4,8 During a time frame of approximately 10–20 minutes,9,10 the exposed NHR coiled-coil core can be accessed by a synthetic CHR derived peptide T-20 (enfuvirtide, Fuzeon™) which has been successfully used in the clinic as an HIV-1 entry inhibitor, clearly validating the ‘pre-hairpin’ intermediate (and more specifically the NHR trimer in this case) as a target for antiviral intervention.
Antiserum from rabbits immunized with disulfide-linked trimeric NHR peptide N35CCG-N13 [a 48 residue peptide comprising N35CCG (gp41 NHR peptide N35 with Leu576, Gln577 and Ala578 substituted by Cys, Cys and Gly, respectively) immediately followed by N13] was shown to inhibit HIV-1 Env-mediated cell-cell fusion, and a proportion of IgG with strong affinity to this peptide was estimated to have neutralizing potency comparable to that of mAb 2G12.11 To directly validate the NHR trimer as a potential antibody and vaccine target, and to search for specific epitopes on the NHR trimer that are capable of eliciting neutralizing antibodies against HIV-1, we previously screened human B-cell-derived single chain variable fragment (scFv)-expressing phage display libraries sequentially against two NHR mimetics, 5-helix and IZN36.12,13 The rationale of this strategy was to identify scFvs that can bind to the common epitope components present on both mimetics, namely the hydrophobic groove in the inner coil of NHR trimer.14 One scFv that was identified using this approach was designated as ‘D5’. When converted to a whole IgG, D5 exhibited modest yet relatively broad neutralization against a panel of primary and laboratory-adapted HIV-1 isolates.14 NMR studies and infectivity assays using recombinant viruses containing single mutations mapped the D5-binding site to the highly-conserved hydrophobic pocket region on the NHR trimer.14 We further solved the 2.0 Å resolution X-ray structure of the complex between the Fab fragment of D5 and 5-helix, demonstrating that both binding and neutralization depend on residues in the D5 VH CDR2 loop that protrudes into the NHR hydrophobic pocket. Residues Trp571, Leu568, Lys574 and Gln575 mainly contribute to the interaction.15 Recently, using a similar screening approach, Nelson et al.16 identified a rabbit scFv, 8K8, and a human Fab, DN9, both of which appear to recognize an epitope on NHR that is overlapping, but distinct to that of D5. Other reported Fab or IgG that target to NHR include Fab 3674,17 and m46.18 The neutralization potencies of these anti-NHR mAbs are in the same range as that of D5, which are all 1 to 2 orders of magnitude lower than anti-gp41 mAb 2F5 or 4E10 and are simply too low to be clinically efficacious. Biochemical and functional characterization of a panel of D5 point mutants showed that neutralization potencies of D5 mutants correlated well with their binding affinities for NHR mimetic 5-helix.15 Therefore, identification of a D5-like antibody with a higher binding affinity for NHR and hence a higher neutralization potency is in theory possible. To this end, an effort was made to determine if affinity maturation of D5 could lead to an antibody with increased neutralization potency. An analysis of the resulting amino acid changes in D5 that either increase or decrease the neutralization potency will help to design further optimized immunogens as vaccine candidates.
Results
ScFv screen using ELISA binding assay.
Libraries were designed for the maturation of D5 by randomizing selected areas within 5 of the 6 CDRs focusing on VH CDR3 and VL CDR3. Only the VL CDR2 region was left unchanged. As antigen for the affinity maturation, we used a biotinylated version of a covalently stabilized trimeric form of IZN23, namely (Biotin-CCIZN23)3.19 All 7 of the libraries were panned separately against 10 nM (Biotin-CCIZN23)3 during Round 1 to prevent competition between the libraries. After Round 1, the two libraries for VH CDR3 and for VL CDR3 were combined and the remaining Rounds done with the five separate libraries. Three panning schemes were used to determine the optimal drop in antigen concentration to increase the chance for obtaining clones with the greatest affinity improvement while still retaining diversity. The concentrations of (Biotin-CCIZN23)3 used in the different rounds of panning are shown in Table 2.
Table 2.
Lead optimization selection outline
| Library panning scheme | Round | Antigen concentration | Phage input | Percent positive (%) |
| 7 separately (H1, H2, H3A, H3B, L1, L3A, L3B) | 1 | 10 nM | 1 × 1011 | 80–90 |
| 5 separately (H1, H2, H3A&B, L1, L3A&B) | 2A | 1 nM | 1 × 1010 from R1 | 95–100 |
| 5 separately (H1, H2, H3A&B, L1, L3A&B) | 2B | 0.1 nM | 1 × 1010 from R1 | 86–98 |
| 5 separately (H1, H2, H3A&B, L1, L3A&B) | 3A | 0.1 nM | 1 × 109 from R2A | 98–100 |
| 5 separately (H1, H2, H3A&B, L1, L3A&B) | 3B | 0.01 nM | 1 × 109 from R2B | 95–100 |
| 5 separately (H1, H2, H3A&B, L1, L3A&B) | 3C | 0.01 nM | 1 × 109 from R2B | 84–95 |
| 5 separately (H1, H2, H3A&B, L1, L3A&B) | 4 | 0.01 nM | 1 × 109 from R3A | 98–100 |
The primary screen of the maturation output plates was done by phage ELISA to the peptide (Biotin-CCIZN23)3. A total of 44 clones from each library were picked at each stage of panning (about 1,300 total), and screened in the ELISA. The percent positive phage found in each library, even after only one round of panning, was high. After Round 1, each of the libraries showed 80–90% positive samples (Table 2, last column). After Round 2A panning, which involved a 10-fold drop in antigen concentration, the number of positives were >95%. Further rounds of panning using the output from Round 2A continued to show the same high number of positives. The number of positive phage resulting from Round 2B, which involved a 100-fold drop in antigen concentration, increased to 86–98%, which was somewhat lower than seen in Round 2A. Of the positive output phages identified in the primary ELISA screen (about 1200), sequences of over 800 clones were determined. These included positive clones from all the libraries so that changes would be identified in all CDRs. Almost all of the sequences were unique, with only 12 sequences found more than once. These 12 sequences were found only as duplicates or triplicates (Table 3, highlighted in yellow), suggesting that a wide diversity of sequence can bind to the (Biotin-CCIZN23)3 peptide and that minimal selection occurred. The majority of repeat sequences were found in the VH CDR2 (6) and VH CDR3 (4) libraries.
Table 3.

Clones from first screen tested in single-cycle infectivity assay
A total of 22 scFv clones were selected for functional characterization in the single-cycle infectivity assay to determine if the maturation changes improved the scFv ability to inhibit HIV-1 entry into cells. In addition to the 12 enriched clones, ten more clones were selected from clusters of mutants which share high sequence homologies. These 22 clones contain at least one variation in one of the 5 of the CDRs (Table 3).
ScFvs for each of the 22 clones were prepared manually and purified by IMAC. Samples were tested in the single-cycle HIV-1 infectivity assay for their ability to block HIV-1 entry into cells. Only 6 of the 22 clones showed activity in this assay (Table 3). Most of the active clones (5 out of 6) were identified from the enriched sequences, and occurred in the VH CDR1 and VH CDR2 domains.
ScFv screen using single-cycle HIV-1 infectivity assay.
A second screen of the maturation output plates was designed to select clones by function (i.e., by neutralization potency) instead of peptide binding. In this screen, scFvs were directly tested in the single-cycle infectivity assay. Small-scale (3 ml) automated expression and purification using the His-tag on the scFvs20 enabled the preparation of scFvs from all ten of the output plates generated from rounds 3A, 3B, 3C and 4 of the panning scheme (Table 2). After purification on nickel resin, the samples were buffer exchanged into the cell medium to allow testing of neat samples. This enabled all the samples to be tested over four dilutions.
Two control scFv preparations were included in all the plates to establish a known range of inhibition. D5 scFv was used to define the parental inhibitory activity and X5 scFv was used to determine a level of inhibition that is more potent than D5. The X5 Fab was identified from libraries screened against the HIV-1 trimolecular gp120-CD4-CCR5 complex.21 X5 was found to potently neutralize several strains of HIV-1 as an Fab and an scFv, but lost activity when converted to an IgG.22 In the single-cycle infectivity assay, the X5 scFv was previously shown to be more potent than the D5 scFv.14
From the first screen of 880 clones, multiple clones showed activity similar to D5 and X5 in the assay. Since the concentration of these scFv samples was not determined, the clones were scored as either positive or negative. Positive clones (176) were picked for a second round of screening, ensuring that there were candidates from each of the HC and LC libraries and from all rounds of panning. All 176 clones were sequenced and used to prepare scFv samples in duplicate by the small-scale automated protocol. Each set of scFv samples was run in the single-cycle infectivity assay and the reproducibility of the results compared. Figure 2 shows a comparison of 11 scFv samples in two separate assays runs (2A and 2B). Although the exact titers of each clone varied, the trends of inhibition were reproducible between the assays. This showed that both the production of the scFv samples and the infectivity assay results were reproducible, validating this screening approach to identify HIV-1 entry inhibitors.
Figure 2.

Reproducibility of single-cycle HIV-1 infectivity assay in functional screening. Two sets of scFv were made and tested in separate infectivity assays 2A and 2B. Results of 11 clones are shown from each of the two runs. S1 to S4 indicate the dilution of each sample. The columns indicate the amount of HIV-1 that enters the P4/R5 cells. Clones D5 and D7 show inhibition at all 4 dilutions. Clone D9 shows the least amount of viral inhibition.
Twenty-six clones that showed dose-dependent inhibition in 3 of the 4 dilutions were chosen for further analysis. Another five clones that showed no improvement or were less potent than the parental D5 scFv were also chosen. Table 4 shows the list of 31 clones that were made by a large-scale scFv automated protocol. The majority of the improved clones (19) came from the VH CDR2 library. Three improved clones were also found in each of the VH CDR1 and VH CDR3 libraries. Only one improved clone was found in VL CDR3 and none from VL CDR1.
Table 4.

scFv screening results from duplicate runs in the single-cycle HIV-1 infectivity assay
ScFv preparations of the above selected clones were then generated by the large-scale automated purification protocol, and concentration of the samples determined. These samples were tested twice in the single-cycle infectivity assay, and IC50 values calculated. The values are given in Table 4. All 26 of the clones that showed improved inhibition of infectivity from the initial screens were found to have IC50 values lower than the parental D5, and 15 of them also showed IC50 values lower than X5. Clone 2–6 (VH-CDR1), which did not appear to be more potent than D5 in the initial screen, was found to have an IC50 lower than the parental D5.
Structural analysis on in vitro binding between D5 scFv variants and 5-helix.
The mutations in D5 identified following in vitro affinity maturation led to both enhanced and diminished neutralization activity. D5 variants lacking neutralization activity following affinity maturation are shown in Table 5. A subset of these mutations affected VH CDR2 loop residues that enter the gp41 hydrophobic pocket (Fig. 3A and B). For example, key hydrophobic interactions with gp41 are likely disrupted by mutation of the CDR2 residue Ile53 to Lys or Arg. Second, mutations in VH CDR3 loop that perturbed neutralization largely affect Leu100, which normally makes several Van der Waals contacts with gp41 Trp571 (Fig. 3C). The mutation of Leu100 to Arg was observed in nearly half of the non-neutralizing clones (Table 5). This mutation would likely cause a steric clash between the upper portion of the D5 pocket surrounding the critical gp41 and the critical residue Trp571. Finally, mutations in VL CDR3 loop that ablated neutralization all lack the side chain for D5 residue Tyr94, which forms critical hydrogen bonds with a solvent molecule in the D5/gp41 interface (ref. 15 and Fig. 3C). In fact, we previously observed that mutation of LC Tyr94Ala significantly reduced D5 neutralization potency.15
Table 5.

D5 scFv variants with undetectable functional activity
Figure 3.

Structural rationale for D5 scFv affinity maturation clones. (A) D5 CDR loops (colored and labeled) contacting the gp41 N-peptide trimeric inner core (shades of yellow and labeled). (B) D5 HC CDR2 loop entering gp41 hydrophobic pocket. Core D5 residues are labeled in blue and gp41 residues are labeled in black. (C) D5-derived ‘Trp 571’ pocket. Several residues from five D5 CDR loops surround the gp41 N-peptide. Loops and specific contact residues are colored and labeled as follows: H1 (green), H2 (purple), H3 (cyan), L1 (orange), L3 (peach). Solvent molecules are labeled in the interface and hydrogen bonds are shown as dotted lines. (D) Model of D5 mutant with alanine at position HC 33 substituted for glutamine. In this model, the water molecules w1 and w2 are replaced by the glutamine side chain such that new hydrogen bonds with gp41 Gln575, D5 HC Asp95 and D5 LC Tyr94 may exist. (E) The region of D5 HC CDR1 behind HC CDR2 contains conserved aromatic side chains (Phe29 and Trp36). In D5, the 34 position is an isoleucine, however several affinity matured clones contain a tyrosine at this residue.
Mutations that enhanced neutralization are also well rationalized given structural information of the complex. The two major sites of improvement are correlated with the two amino acid positions in D5 that are normally found in contact with large protein antigens,23 but they are not within Van der Waals contact distance to gp41.15 These two positions, Ala33 and Gly50, are mutated in nearly every affinity matured clone from VH CDR1 and CDR2 respectively (Table 4). The displacement of solvent molecules in the interface by extending these amino acids, and in some cases the putative formation of hydrogen bonds across the interface, likely result in increased neutralization potency of the D5 variants (Fig. 3D shows model of putative Ala33Gln mutation). One additional mutation that may be beneficial is Ile34Tyr in VH CDR1, which occurs in three of four purified CDR1 scFv clones. This mutation allows for an aromatic residue to enter a highly aromatic pocket sitting behind VH CDR2 (Fig. 3E). This pocket commonly contains a set of three aromatic residues at the 29 (Phe), 34 (Ile), and 36 (Trp) positions of the VH CDR1 loop. The Ile34Tyr mutation likely enhances the stability of the VH CDR2/gp41 hydrophobic pocket interaction.
Characterization of IgG1s converted from selected D5 scFv variants.
A set of 4 clones, representing the best scFv inhibitors in each of the CDR groups, was converted to IgG1 by cloning the scFv variable regions into human IgG1 HC vectors and kappa LC vectors. The four clones (1–25, 2–16, 2–50 and 2–75) are highlighted in yellow in Table 4. Both IgG1 and scFv preparations of the 4 clones were tested head-to-head in the infectivity assay to determine if the improvements seen with the scFv would translate into the IgG1 format. The results in Table 6 show that when converted to IgG1s from their corresponding scFvs, there was a loss of neutralization potency by 3 to 12-fold with all the variants despite the fact that the IgG1s had a 1.5 to 3-fold increase over their scFvs in in vitro binding affinity to 5-helix. Among the four matured D5 variants, 2–16 IgG1 (VH CDR1) and 2–50 IgG1 (VH CDR2) lost neutralization potency by 11 to 12-fold, followed by 1–25 IgG1 (VH CDR3, loss by 6-fold) and 2–75 IgG1 (VL CDR3, loss by 3-fold). Importantly, there was a correlation between in vitro binding affinity and neutralization potency for scFvs (R2 = 0.50), but not for corresponding IgGs (R2 = 0.07) (Fig. 4). Overall, these data suggest that when converted from a small scFv molecule to a larger IgG molecule, there is a significant loss of neutralizing potency in the context of virus-cell interaction which is not correlated with changes of binding affinity to the target protein in vitro.
Table 6.
Comparison between selected D5 scFv variants and their matched IgG1s
| D5 Variant | scFv data | IgG data | IgG IC50 | IgG Kd | ||||||
| IC50 (nM) | -fold | Kd (pM) | -fold | IC50 (nM) | -fold | Kd (pM) | -fold | scFv IC50 | scFv Kd | |
| Parental D5 | 121 ± 23 | 1.0 | 150 | 1.0 | 424 ± 150 | 1.0 | 50 | 1.0 | 3.5 | 0.33 |
| 1–25 (VH CDR3) | 61 ± 8 | 2.0 | 180 | 0.8 | 362 ± 68 | 1.2 | 110 | 0.5 | 5.9 | 0.61 |
| 2–16 (VH CDR1) | 19 ± 1 | 6.4 | 90 | 1.7 | 209 ± 62 | 2.0 | 60 | 0.8 | 11 | 0.67 |
| 2–50 (VH CDR2) | 16 ± 3 | 7.6 | 80 | 1.9 | 195 ± 77 | 2.2 | 40 | 1.3 | 12.2 | 0.5 |
| 2–75 (VL CDR3) | 48 ± 13 | 2.5 | 120 | 1.3 | 147 ± 31 | 2.9 | 70 | 0.7 | 3.1 | 0.58 |
Figure 4.

Correlations between neutralization potency (IC50) and in vitro binding affinity (Kd) for scFvs vs. IgGs of selected D5 variants. IC50 for the single-cycle HIV-1 infectivity assay vs. in vitro binding affinity determined by surface plasmon resonance for (A) scFv variants and (B) corresponding IgG variants. Inhibition of infection and binding affinity correlate for scFvs (R2 = 0.50) but not for IgGs (R2 = 0.07). Note that X and Y-axis scales are different between panels A and B to show correlations.
Discussion
Phage display technology has become a powerful tool to select scFvs and Fabs with varied specificities and binding affinities from natural or synthetic libraries.24 Previously we identified scFvs that are inhibitors of HIV-1 entry by sequentially screening phage display libraries against two NHR mimetics, namely 5-helix and IZN36.12,13 The identified scFvs can bind to the common epitope components present on both mimetics, namely the hydrophobic groove in the inner coil of NHR trimer.14 In this study we used only one antigen for the affinity maturation, namely a covalently stabilized trimeric form of IZN23, (Biotin-CCIZN23)3.19 Inhibitory potency and thus N-region mimicry by IZN peptides13 is limited by the trimerization equilibrium in solution. Unlike disulfide stabilized trimers based on the N-region, (CCIZN)3 peptides19 are thermodynamically more stable, are more potent fusion inhibitors and represent more faithful mimetics of gp41 NHR region especially at the low concentration which is usually required for panning.19 By screening seven libraries with randomized amino acid changes within 5 of the 6 CDRs of parental D5 scFv, we have sought to identify scFvs with improved binding affinity for a gp41 NHR mimetic, hoping to identify corresponding D5 variant IgGs that can display enhanced neutralization potency. The results presented here confirm our prediction that changes of critical residues in certain CDRs of D5 could in fact enhance both its binding affinity and neutralization potency, and demonstrate that affinity maturation is an effective strategy in obtaining scFv variants with higher binding affinity and neutralization potency. Furthermore, we clearly demonstrate that there is a correlation between neutralization potencies of scFvs of D5 variants and their in vitro binding affinities to the target viral protein mimetic (5-helix), and that slight increases in in vitro binding affinity could lead to dramatic improvement in neutralization potency. For example, the best two scFvs (variant 2–16 and 2–50) identified from our current study had only modest (1.7 and 1.9-fold, respectively) increases in binding affinity over the parental D5 scFv, yet a remarkable improvement on HIV-1 neutralization potency (by 6.4 and7.6-fold, respectively). However, in this particular case where the binding affinity of parental D5 scFv is already rather high (with a Kd value of 150 pM), and only two variants (out of a total ∼1010 phage clones screened) with modestly enhanced binding affinity were identified, it seems there may be limited potential for further improvement on binding affinity for parental D5 scFv.
The four scFv variants finally selected represent the best hits in each of the following CDR groups: VH CDR1, VH CDR2, VH CDR3 and VL CDR3. When converted to IgG1s by cloning the scFv variable regions into human IgG1 HC vectors and kappa LC vectors, all the D5 variant IgG1s showed a 1.5 to 3-fold increase in binding affinity for 5-helix, most likely due to an increase in avidity as a result of the bivalent binding of the IgG molecule. However, the slight increases in IgGs' binding affinity did not translate into proportional enhancements in HIV-1 neutralization potency. Instead, when compared with their corresponding scFvs, the IgGs of all four D5 variants had a 3 to 12-fold reduction in neutralization potency. It is noteworthy that the reduction of neutralization potency is also seen with parental D5 when converted to IgG1, consistent with the recent result obtained by Eckert et al.25 Here we report that against HXB2, the IC50 of variant 2–75 IgG is 147 nM which is 2.9-fold more potent than parental D5 IgG (IC50 = 424 nM). As we reported elsewhere,26 a corresponding 2.5-fold improvement in variant 2–75 IgG neutralization potency was seen with the primary isolate 89.6 (2–75 IgG IC50 = 700 nM, parental D5 IgG IC50 = 1,750 nM). Therefore, we believe the improved neutralization potencies of affinity-matured D5 variants seen with HXB2 should also carry over to other primary isolates. Unfortunately, we were not able to test potency to any other primary isolate because 89.6 was the only one among 12 tested HIV-1 primary isolates (RW/92/026, RW/93/022, 89.6, SF-162, MW/93/959, UG/93/067, BR/93/020, BR/93/029 from Merck inventory and 6535, PAVO, Du156, Du172 provided by Dr. David Montefiori at Duke University), that can infect the P4/R5 cells (HeLa cells expressing endogenous CXCR4 and stably transfected to express CD4 and CCR5 which also contain an integrated β-galactosidase reporter gene under control of an HIV-1 LTR promoter) that we use for the infectivity assay and generate reproducible viral signals (SF-162 had some weak and variable viral signals).
In contrast to the situation where scFv neutralization potency correlates well with in vitro binding affinity, there is no such correlation seen with the corresponding IgGs. This led us to conclude that there are factor(s) that can limit the neutralization potency of D5-derived IgGs, and IgGs with even higher binding affinity against the same epitope(s) are unlikely to further improve HIV-1 neutralization.
The most straightforward explanation for a loss of neutralization potency with all the D5 variants when the scFvs are converted to IgG1s is that the larger IgG1 molecules may encounter steric access restrictions to the NHR epitope(s) in the context of native viral structural environment. Indeed, Hamburger et al.27 found that C37, a gp41 CHR peptide, when fused to cargo proteins of increasing sizes, gradually loses its inhibitory potency in viral infectivity assays yet maintains its ability to bind NHR mimetics in vitro. By connecting the cargo proteins to either the N-terminus or C-terminus of the C37 peptide, Eckert et al.25 concluded that the source of steric block mainly comes from the virus instead of the host cell side, and gp120 is the most likely blocker that occludes large molecules (such as IgG) from accessing the ‘pre-hairpin intermediate’ during its transient exposure. Thus, our current result further confirms that the steric block imposed by gp120 extends to the gp41 NHR hydrophobic pocket region.
Notably, among the four D5 variants, the degree of reduction of neutralization potency with its IgG form differs significantly, with VH CDR1 and VH CDR2 variants being the most affected (by ∼12-fold), followed by VH CDR3 variant (by 6-fold) and VL CDR3 variant (by 3-fold). Based on the crystal structure of D5/5-helix complex (ref. 15, Fig. 3A), VH CDR2 (abbreviated as H2 in the figure) of D5 inserts into the NHR hydrophobic pocket and interacts with the most C-terminal (bottom) portion of the epitope residues. The other CDRs interact with increasingly N-terminal portions of the epitope residues in the following order: VH CDR1 (H1), VH CDR3 (H3) and VL CDR3 (L3).15 Thus, the order of neutralization potency reduction for each D5 variant IgG1 appears to be coincident with the geometrical order of each CDR region on IgG1 relative to the NHR trimer. This pattern of neutralization potency reduction is fully consistent with the above-mentioned model in which the steric block extends to the hydrophobic pocket, and further indicates that the epitope residues N-terminal to the hydrophobic pocket are more accessible to D5-derived IgG1s compared to those that reside in the pocket. Could the scFv form of D5 variants also suffer to some extent from the same steric block mechanism? The data presented by Hamburger et al.27 indicate this might be the case. In their study, when C37 was linked with a GFP at its N-terminus (i.e., towards the virus side) to give rise to a fusion protein (GFP-C37) with a molecular size similar to a scFv molecule, an ∼40-fold loss of its antiviral potency was observed in a viral infectivity assay against HIV-1 strain HXB2,27 suggesting the putative steric block component can affect the access of a molecule as small as 25 KDa. Nevertheless, the correlation between in vitro binding affinity and neutralization potency of D5-derived scFvs suggests that they encounter little if any steric block.
Given our current result and the results presented by the Kay laboratory,25,27 it seems evident that the hydrophobic pocket region of gp41 NHR trimer, a target for antiviral D-peptide entry inhibitors, is protected by a steric block mechanism mainly derived from the virus side. This poses severe limitations and significant challenges for developing therapeutic antibodies and vaccines targeting this otherwise attractive epitope region. Further characterization of the steric block mechanism will certainly help the design of a better presented N-trimer mimetic to include the steric component with its native conformation, and the key reagents we generated in the current study will be valuable to validate such designs. If a more naturally-emulated N-trimer mimetic can be engineered biochemically, it may be used either as a selection antigen to screen the scFv phage display libraries or as a vaccine to elicit antibodies capable of overcoming the steric restriction. Alternatively, based on the previous finding that the steric block surrounding the NHR trimer is asymmetric with its cell side (i.e., the N-terminal side of NHR trimer) more accessible,27 and our current finding that the VH CDR3 and VL CDR3 of the D5 IgG1 (which interact with epitope residues N-terminal to the hydrophobic pocket) suffer much less steric block, a thorough searching for antibodies targeting the N-terminal side of the gp41 NHR hydrophobic pocket may bring breakthroughs. In the current study, to focus on only the hydrophobic pocket, we used the N-peptide mimetic (CCIZN23)3,19 as the selection antigen. NHR trimer mimetics with residues extended toward the N-terminal side, such as IZN36,13 and N51,28,29 could be used as a selection antigen to identify scFvs and IgGs targeting the NHR epitope(s) that are N-terminal to the hydrophobic pocket. If such-selected scFvs and IgGs turn out to have much improved neutralization potencies in infectivity assays, immunogens containing corresponding epitope(s) may be attempted and optimized to develop an effective HIV-1 vaccine.
Materials and Methods
Library construction.
A total of seven libraries were constructed in pCANTAB6,30 by randomizing selected residues within 5 of the 6 CDRs of D5. Randomization was achieved by PCR using degenerate primers. Figure 1 shows the design of the libraries. Two overlapping blocks of six consecutive codons were randomized in the CDR3 of the VH and the VL. The five codons of the VH CDR1 plus one adjacent codon, which codes for a Vernier residue at position 30 within the VH CDR1 loop were randomized for that library. Although the VH CDR2 and VL CDR1 span more than six codons respectively, only one library each was made. The functional size of the four HC libraries was 1 × 109. The functional size of the LC libraries ranged from 0.2 to 1.1 × 1010.
Figure 1.

Target residues for library construction. Amino acid sequence of the variable regions of D5 heavy chain and light chain are shown with the CDR regions underlined. The target residues that were randomized in the maturation libraries are shown in red bold type. Two libraries were designed for each of the CDR3 regions, with six overlapping codons in each. These libraries are indicated as VH CDR3A&B (DNPTLL, LLGSDY) and VL CDR3A&B (QQYSNY, SNYPLT).
Soluble selection process testing.
The selection process was developed by enriching the parent D5 scFv from a null background in a soluble selection format at low antigen concentrations, based on the method previously described.31 (CCIZN23)3, a covalently stabilized trimeric form of IZN23 in which the NHR peptide N23 is connected to helix-promoting scaffold sequences derived from trimerization domains of modified isoleucine zipper (IZ) proteins,19 with a biotin moiety added at the N-terminus, namely (Biotin-CCIZN23)3, was used as the antigen. A test selection was performed with a small, known quantity of parent phage (106 D5 scFv phage) spiked into a null pBSKSII population of phage that do not recognize the antigen (1011 phage). Enrichment of the D5 scFv was first detected by the increase of white colonies (D5 scFv expressing cells) over blue colonies (Table 1). Scfv displayed on phage and periplasmic preparations of scFv from white colonies were confirmed for binding to (Biotin-CCIZN23)3 by ELISA and by DNA sequencing.
Table 1.
Validation of the panning process
| Panning Round | # blue colonies | # white colonies | % white colonies | Fold enrichment |
| 1st | 60 | 185 | 75 | 1333333 (over 0 round) |
| 2nd | 5 | 159 | 97 | 1.3 (over 1st round) |
| 3rd | 0 | 208 | 100 | 8.3 (over 2nd round) |
Library selection.
To affinity and functionally mature anti-gp41 antibody D5, the seven lead optimization libraries were screened against (Biotin-CCIZN23)3 in the validated soluble selection format. In the first round of panning, the seven libraries were separately selected against 10 nM of the antigen. After Round 1, the outputs from H3A and H3B were combined and the same was done for L3A and L3B. Starting at Round 2, three different selection schemes were used to pan the five libraries in parallel. In the first scheme, the antigen concentration was dropped 10-fold for each round (Round 2A at 1 nM, Round 3A at 0.1 nM and Round 4 at 0.01 nM). In the second scheme, the antigen concentration was dropped 10-fold for Round 2A (1 nM) and 100-fold for Round 3B (0.01 nM). For the third scheme, the antigen concentration was dropped 100-fold for Round 2B (0.1 nM) and 10-fold for Round 3B (0.01 nM). Table 2 shows the selection process.
ELISA screen.
A total of 44 clones from each library were picked at each stage of panning (Rounds 2A, 2B, 3A, 3B, 3C and 4). Phage from these clones were prepared as previously described32 and tested in an ELISA against the (Biotin-CCIZN23)3 peptide.19 Streptavidin coated plates were bound with 50 ng/well of (Biotin-CCIZN23)3, the mutant (Biotin-CCIZN23-Q567R)3 or an irrelevant biotin-peptide. Phage preparations were added to the plates. The detection antibody was HRP-mouse anti-M13 pVIII. Positive samples were identified as a signal greater than three times the background signal seen with the irrelevant peptide.
Automated high-throughput expression and purification of scFv.
All scFv were generated from the vector pCANTAB6 and thus contain a hexahistidine-tag to allow rapid purification by immobilized metal affinity chromatography (IMAC).32 Small-scale (3 ml) scFv preparations were isolated by IMAC for the initial functional screen in the single-cycle HIV-1 infectivity assay.20,34 The ten stock 96-well plates generated from rounds 3A, 3B, 3C and 4 panning outputs were used to inoculate cultures for expression and purification of scFv. The automated growth, induction and purification of scFv were done as described.20 The final samples were buffer exchanged into phenol red-free Dulbecco's modified Eagles' medium (DMEM) to obtain the concentration of scFv needed in the single-cycle HIV-1 infectivity assay. Samples were stored short-term at −20°C before being tested.
A total of 176 positive clones (88 from rounds 3A and 3B, and 88 from rounds 3C and 4) from the initial screen were picked into compression plates and stored. These plates were used to inoculate cultures for expression and purification of scFv as described above. For each compression plate, duplicate automated preps of scFv were made to be tested in the infectivity assay. Samples were stored short-term at −20°C before testing.
Large-scale (500 ml) purified scFv preparations were made from 31 clones and three controls (D5 and X5 as positive controls, 8B4 as negative control) for final testing in the single-cycle HIV-1 infectivity assay. They were grown and purified as described.20 The concentrations of scFv in purified preparations were determined using the Protein Express 100 assay on a LabChip90 Automated Electrophoresis System (Caliper Life Sciences, Hopkinton, MA).20 Samples were stored frozen before testing.
Single-cycle HIV-1 infectivity assay.
HIV-1 infection of P4/R5 cells was measured as previously described.20,34 P4/R5 are HeLa cells expressing endogenous CXCR4 and stably transfected to express CD4, CCR5 and an integrated β-galactosidase reporter gene under the control of the HIV-1 LTR promoter. Briefly, P4/R5 cells maintained in phenol red-free DMEM, 10% fetal bovine serum, 1% penicillin/streptomycin were seeded in 96-well plates at 2.5 × 103 cells/well. Cells were infected the following day with the HXB2 strain of HIV-1 in the presence of increasing concentrations of scFvs or IgGs. Infected and uninfected cells were used to establish maximal and minimal infectivity signals, respectively. After 48 h of infection, cells were lysed and the β-galactosidase activity (which is indicative of the viral replication level) was measured. D5 scFvs and IgGs act specifically at a pre-entry step by binding to HIV-1 and inhibiting its entry into the host cell. Therefore in this study the single-cycle HIV-1 infectivity assay is used as a means to evaluate the effect of scFvs and IgGs in blocking HIV-1 entry specifically.
Sequence of scFv clones.
All scFv clones are in pCANTAB6 and sequence was obtained using the primers: 5′-TTA TTA TTC GCA ATT CCT TTA GTT GTT CCT-3′ and 5′-GTC GTC TTT CCA GAC GTT AGT-3′.
Cloning scFv variable regions into human IgG vectors.
The variable regions were PCR amplified from pCANTAB using heavy chain (HCG1) or light chain (LCK) specific primers.
HCG1-F: ACA GGT GTC CAC TCG CAG GTG CAG CTG GTG CAG TCT
HCG1-R: GCC CTT GGT GGA TGC AAC TCG AGA CGG TGA CCA GGG T
LCK-F: ACA GAT GCC AGA TGC GAC ATC CAG ATG ACC CAG TCT
LCK-R: TGC AGC CAC CGT ACG TTT GAT CTC CAG CTT GGT CCC
PCR reactions were done using Dry-Down Master mix (Clonetech) in a volume of 25 µl containing high fidelity PCR master mix, 10 ng template and 0.5 µM forward and reverse primers. PCR conditions were 1 cycle of 94°C, 60 sec; 22 cycles of 94°C, 90 sec; 67°C, 90 sec; 74°C, 90 sec followed by 74°C, 7 min and 4°C until ready. The PCR products were digested and purified with QIAquick plate kit (Qiagen). 100 ng of linearized heavy chain (HCG1) or light chain (LCK) vector and 10 ng of the PCR fragment underwent InFusion (Clontech) reaction, were transformed into XL2 Blue MRF' competent cells and plated overnight on LB agar plates containing 50 µg/ml Kanamycin. Clones were screened by restriction analysis and confirmed by sequencing. Plasmids were made from sequence confirmed positive clones and used for transfection.
Transfection, purification of antibody.
Light chain and heavy chain vectors were transfected in 293 Freestyle cells (Invitrogen) using 293fectin™ Transfection Reagent (Invitrogen). Transfected cells were incubated at 37°C/8% CO2 for 7 days. The medium was collected, spun down and filtered through 0.22 µm filtration system (Millipore). 1/10 volume of 10× PBS was added to the filtered medium and loaded onto a pre-equilibrated HI Trap Mab Select SuRe column (GE healthcare). The column was washed with PBS, eluted with IgG Elution Buffer (Pierce) and neutralized immediately with Ultrapure 1 M Tris-HCl pH 8.0 (Invitrogen). IgG preparations were dialyzed against 100 mM Arginine, 100 mM Histidine, 6% (W/V) sucrose buffer overnight at 4°C and concentrated with Amicon (Pierce). Protein concentration was determined by OD280 nm with the extinction coefficient of 1.34 mg/ml. Purified antibody was analyzed using SDS-PAGE.
Surface plasmon resonance.
All surface plasmon resonance (SPR) measurements and procedures were conducted on a Biacore 2000 instrument (GE Healthcare) at a controlled temperature of 25°C following the general guide provided.35 Data were acquired with the Biacore 2000 Control Software version 3.2.1 and analyzed by BIAevaluation 4.1 and custom code in the Matlab language (Mathworks).
Antibodies were diluted in HBS-P (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.005% P-20), which was also used for the running buffer. IgG1s of parental D5 and its matured variants were directly immobilized onto the CM5 BIAcore chips at 1 µg/ml in Na-acetate pH 5.0 to reach a target of approximately 800 SPR response units (RU), and then 1 M ethanolamine-HCl pH 8.5 was flowed over the surfaces to deactivate any residually reactive site. Using a flow rate of 100 µl/min, 5-helix (4-fold serial diluted in HBS-P buffer) was injected over the IgG1 surface or blank reference surface for 2.5 min with a 120 sec stabilization interval and then allowed to dissociate for 60 min. Remaining IgG1s were released by a 30 sec pulse of 20 mM HCl flowed at 60 µl/min followed by a 120 sec stabilization interval. ScFvs were measured by SPR in a similar manner to the IgG1s except that 5-helix was first immobilized to the CM5 chips at a RU of 1800.
Throughout the experiment, time-resolved SPR signals from the sample channels (5-helix vs. D5 IgG1 or scFv, or their respective variants) and blank channels were simultaneously acquired and signals from the blank surfaces were subtracted as negative controls. Before final analysis, similarly treated signals from buffer-only injections were also subtracted from the data curves to reduce artifacts, known as double referencing.35 Subtracted curves were fit to an 1:1 Langmuir binding model to obtain best-fit KD values. The association rate ka and dissociation rate kd are direct fit parameters in this model; the equilibrium affinity KD = kd/ka. Due to the fast experimental flow rate, data curves were relatively insensitive to the association rate ka, therefore, most of the reported variation between D5 variants in Table 6 derives from underlying variation in the dissociation rate kd.
Peptide production.
The peptide (Biotin-CCIZN23)3 used in this study was prepared by solid-phase synthesis, as previously described.14,19 In particular, we first assembled the CCIZN23 sequence which was further elongated at the N-terminus with an additional glycine residue and a Biotin. The peptide sequence is the following: Biotin-GCCGG IKKEI EAIKK EQEAI KKKIE AIEKE IEAQQ HLLQL TVWGI KQLQA RIL-NH2.
At the end of the synthesis and after purification, the peptide was assembled to a covalent trimer by spontaneous air oxidation of three peptide chains to form three interchain disulfide bridges. The resulting peptide (Biotin-CCIZN23)3 is a biotinylated analog of a covalently stabilized coiled-coil trimer as shown previously for peptides (CCIZN23)3 and (CCIZN17)3,14 which are accurate mimetics of gp41 NHR coiled-coil. A schematic model of the peptide antigen used is shown in the Figure 5.
Figure 5.
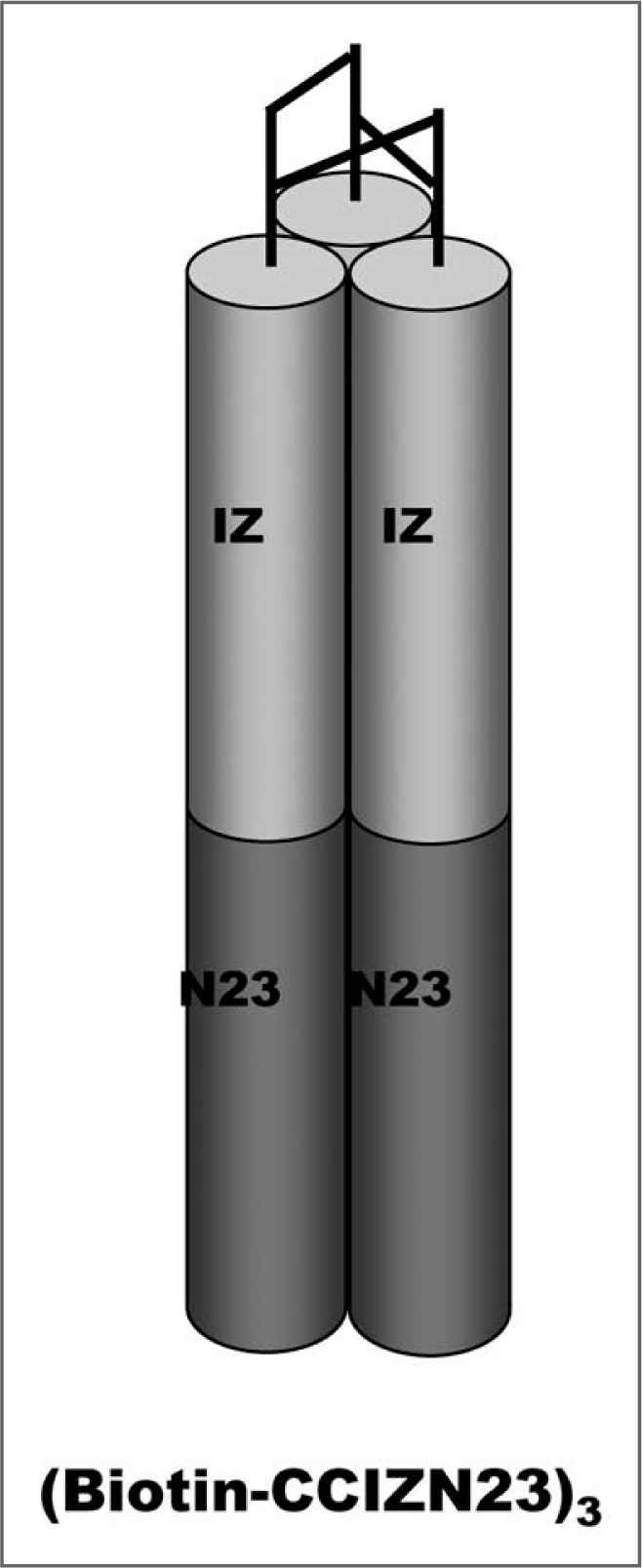
Schematic model of designed peptide antigen (Biotin-CCIZN23)3. The IZN helices forming a homotrimeric coiled-coil are represented by cylinders. The N peptide consists of an N-terminal designed trimeric coiled-coil (IZ, light gray) fused to a portion of the sequence from the NHR of gp41 (dark gray), namely N23 (residues 559–581 of HIV-HXB2). The (CCIZN23)3 helices are covalently stabilized at the N-termini by three interchain disulfides between pair of cysteines of each peptide chain. Only one of the possible combinations of the three disulfides is drawn. The Biotin-Gly residues (not shown in the model) are assembled at the N-terminus of the CC-IZN23 sequence.
Structural analysis.
The coordinates from the D5/gp41 three-dimensional X-ray structure published in Luftig et al.15 were used to model the mutations observed in the affinity maturation clones. Distances and molecular contact information were obtained using the CCP4 software package (Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994; 50:760–3). Modeling and image capture was performed using PyMol (DeLano WL. The PyMOL Molecular Graphics System (2002) DeLano Scientific, San Carlos, CA, USA. www.pymol.org).
Abbreviations
- CDR
complementarity determining region
- CHR
C-terminal heptad repeat
- DMEM
dulbecco's modified eagles' medium
- ELISA
enzyme-linked immunosorbent assay
- GFP
green fluorescent protein
- IgG
immunoglobulin G
- IMAC
immobilized metal affinity chromatography
- mAb
monoclonal antibody
- MPER
membrane-proximal external region
- NHR
N-terminal heptad repeat
- scFv
single chain variable fragment
- SDS-PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- SPR
surface plasmon resonance
- VH
variable region of immunoglobulin heavy chain
- VL
variable region of immunoglobulin light chain
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/9214
References
- 1.Barouch DH. Challenges in the development of an HIV-1 vaccine. Nature. 2008;455:613–619. doi: 10.1038/nature07352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burton DR, Desrosiers RC, Doms RW, Koff WC, Kwong PD, Moore JP, et al. HIV vaccine design and the neutralizing antibody problem. Nat Immunol. 2004;5:233–236. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- 3.Schiavone M, Quinto I, Scala G. Perspectives for a protective HIV-1 vaccine. Adv Pharmacol. 2008;56:423–452. doi: 10.1016/S1054-3589(07)56014-X. [DOI] [PubMed] [Google Scholar]
- 4.Root MJ, Steger HK. HIV-1 gp41 as a target for viral entry inhibition. Curr Pharm Des. 2004;10:1805–1825. doi: 10.2174/1381612043384448. [DOI] [PubMed] [Google Scholar]
- 5.Zhu P, Liu J, Bess J, Jr, Chertova E, Lifson JD, Grise H, et al. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441:847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
- 6.Choudhry V, Zhang MY, Dimitrova D, Prabakaran P, Dimitrov AS, Fouts TR, et al. Antibody-based inhibitors of HIV infection. Expert Opin Biol Ther. 2006;6:523–531. doi: 10.1517/14712598.6.5.523. [DOI] [PubMed] [Google Scholar]
- 7.Debnath AK. Progress in identifying peptides and small-molecule inhibitors targeted to gp41 of HIV-1. Expert Opin Investig Drugs. 2006;15:465–478. doi: 10.1517/13543784.15.5.465. [DOI] [PubMed] [Google Scholar]
- 8.Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 9.Munoz-Barroso I, Durell S, Sakaguchi K, Appella E, Blumenthal R. Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J Cell Biol. 1998;140:315–323. doi: 10.1083/jcb.140.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimitrov AS, Louis JM, Bewley CA, Clore GM, Blumenthal R. Conformational changes in HIV-1 gp41 in the course of HIV-1 envelope glycoprotein-mediated fusion and inactivation. Biochemistry. 2005;44:12471–12479. doi: 10.1021/bi051092d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Louis JM, Nesheiwat I, Chang L, Clore GM, Bewley CA. Covalent trimers of the internal N-terminal trimeric coiled-coil of gp41 and antibodies directed against them are potent inhibitors of HIV envelope-mediated cell fusion. J Biol Chem. 2003;278:20278–20285. doi: 10.1074/jbc.M301627200. [DOI] [PubMed] [Google Scholar]
- 12.Root MJ, Kay MS, Kim PS. Protein design of an HIV-1 entry inhibitor. Science. 2001;291:884–888. doi: 10.1126/science.1057453. [DOI] [PubMed] [Google Scholar]
- 13.Eckert DM, Kim PS. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc Natl Acad Sci USA. 2001;98:11187–11892. doi: 10.1073/pnas.201392898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller MD, Geleziunas R, Bianchi E, Lennard S, Hrin R, Zhang H, et al. A human monoclonal antibody neutralizes diverse HIV-1 isolates by binding a critical gp41 epitope. Proc Natl Acad Sci USA. 2005;102:14759–14764. doi: 10.1073/pnas.0506927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luftig MA, Mattu M, Di Giovine P, Geleziunas R, Hrin R, Barbato G, et al. Structural basis for HIV-1 neutralization by a gp41 fusion intermediate-directed antibody. Nat Struct Mol Biol. 2006;13:740–747. doi: 10.1038/nsmb1127. [DOI] [PubMed] [Google Scholar]
- 16.Nelson JD, Kinkead H, Brunel FM, Leaman D, Jensen R, Louis JM, et al. Antibody elicited against the gp41 N-heptad repeat (NHR) coiled-coil can neutralize HIV-1 with modest potency but non-neutralizing antibodies also bind to NHR mimetics. Virology. 2008;377:170–183. doi: 10.1016/j.virol.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gustchina E, Louis JM, Lam SN, Bewley CA, Clore GM. A monoclonal Fab derived from a human nonimmune phage library reveals a new epitope on gp41 and neutralizes diverse human immunodeficiency virus type 1 strains. J Virol. 2007;81:12946–12953. doi: 10.1128/JVI.01260-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choudhry V, Zhang MY, Sidorov IA, Louis JM, Harris I, Dimitrov AS, et al. Cross-reactive HIV-1 neutralizing monoclonal antibodies selected by screening of an immune human phage library against an envelope glycoprotein (gp140) isolated from a patient (R2) with broadly HIV-1 neutralizing antibodies. Virology. 2007;363:79–90. doi: 10.1016/j.virol.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bianchi E, Finotto M, Ingallinella P, Hrin R, Carella AV, Hou XS, et al. Covalent stabilization of coiled coils of the HIV gp41 N region yields extremely potent and broad inhibitors of viral infection. Proc Natl Acad Sci USA. 2005;102:12903–12908. doi: 10.1073/pnas.0502449102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su B, Hrin R, Harvey BR, Wang YJ, Ernst RE, Hampton RA, et al. Automated high-throughput purification of antibody fragments to facilitate evaluation in functional and kinetic based assays. J Immunol Methods. 2007;322:94–103. doi: 10.1016/j.jim.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Moulard M, Phogat SK, Shu Y, Labrijn AF, Xiao X, Binley JM, et al. Broadly cross-reactive HIV-1-neutralizing human monoclonal Fab selected for binding to gp120-CD4-CCR5 complexes. Proc Natl Acad Sci USA. 2002;99:6913–6918. doi: 10.1073/pnas.102562599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labrijn AF, Poignard P, Raja A, Zwick MB, Delgado K, Franti M, et al. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J Virol. 2003;77:10557–10565. doi: 10.1128/JVI.77.19.10557-10565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacCallum RM, Martin AC, Thornton JM. Antibody-antigen interactions: contact analysis and binding site topography. J Mol Biol. 1996;262:732–745. doi: 10.1006/jmbi.1996.0548. [DOI] [PubMed] [Google Scholar]
- 24.Pini A, Bracci L. Phage display of antibody fragments. Curr Protein Pept Sci. 2000;1:155–169. doi: 10.2174/1389203003381397. [DOI] [PubMed] [Google Scholar]
- 25.Eckert DM, Shi Y, Kim S, Welch BD, Kang E, Poff ES, et al. Characterization of the steric defense of the HIV-1 gp41 N-trimer region. Protein Sci. 2008;17:2091–2100. doi: 10.1110/ps.038273.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hrin R, Montgomery DL, Wang F, Condra J, An Z, Strohl WR, et al. In vitro synergy between peptides or neutralizing antibodies targeting the N- and C-terminal heptad repeats of HIV type 1 gp41. AIDS Res and Human Retroviruses. 2008;24:1537–1544. doi: 10.1089/aid.2008.0129. [DOI] [PubMed] [Google Scholar]
- 27.Hamburger AE, Kim S, Welch BD, Kay MS. Steric accessibility of the HIV-1 gp41 N-trimer region. J Biol Chem. 2005;280:12567–12572. doi: 10.1074/jbc.M412770200. [DOI] [PubMed] [Google Scholar]
- 28.Lu M, Blacklow SC, Kim PS. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 29.Kliger Y, Shai Y. Inhibition of HIV-1 entry before gp41 folds into its fusion-active conformation. J Mol Biol. 2000;295:163–168. doi: 10.1006/jmbi.1999.3368. [DOI] [PubMed] [Google Scholar]
- 30.McCafferty J, Fitzgerald KJ, Earnshaw J, Chiswell DJ, Link J, Smith R, et al. Selection and rapid purification of murine antibody fragments that bind a transition-state analog by phage display. Appl Biochem Biotechnol. 1994;47:157–171. doi: 10.1007/BF02787932. [DOI] [PubMed] [Google Scholar]
- 31.Hawkins RE, Russell SJ, Winter G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol. 1992;226:889–896. doi: 10.1016/0022-2836(92)90639-2. [DOI] [PubMed] [Google Scholar]
- 32.McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348:552–554. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 33.Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, et al. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 1996;14:309–314. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- 34.Joyce JG, Hurni WM, Bogusky MJ, Garsky VM, Liang X, Citron MP, et al. Enhancement of alpha-helicity in the HIV-1 inhibitory peptide DP178 leads to an increased affinity for human monoclonal antibody 2F5 but does not elicit neutralizing responses in vitro. Implications for vaccine design. J Biol Chem. 2002;277:45811–45820. doi: 10.1074/jbc.M205862200. [DOI] [PubMed] [Google Scholar]
- 35.Myszka DG. Improving biosensor analysis. J Mol Recognit. 1999;12:279–284. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<279::AID-JMR473>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]