

Abstract
Leukotriene (LT) B4 is generated in response to engagement of the Fcγ receptor (FcγR) and potently contributes to FcγR-mediated antimicrobial functions in pulmonary alveolar macrophages. In this study, we report that the LTB4 receptor leukotriene B4 receptor 1 (BLT1) redistributes from nonlipid raft (LR) to LR membrane microdomains upon immunoglobulin G–red blood cell, but not LTB4, challenge. Cholesterol depletion to disrupt LRs abolished LTB4-induced enhancement of phagocytosis, microbicidal activity, and signaling. The dependence on LR integrity for BLT1 signaling correlated with formation of a complex consisting of BLT1, its primary coupled G protein Gαi3, Src kinase, and FcγRI within LRs. This association was dependent on Src-mediated phosphorylation of BLT1. These data identify a novel form of regulation in which engagement of a macrophage immunoreceptor recruits a stimulatory G protein–coupled receptor into a LR microdomain with resultant enhanced antimicrobial signaling.
Introduction
Macrophages play a central role in antimicrobial immunity via their capacities for recognition, phagocytosis, and killing of opsonized and nonopsonized microbes. Nowhere are these functions more vigorously tested than in the lung, which comprises the largest internal interface with the outside environment and which is continuously exposed to microbes delivered via inhalation or oropharyngeal aspiration.1 Particles opsonized by immunoglobulin (Ig)G antibodies bind to receptors recognizing the Fc portion of IgG (Fcγ receptor [FcγR]).2 There are 3 general classes of FcγR (FcγRI, II, and III) that vary in their molecular structure, affinity for different IgG isotypes, signal transduction, and phagocyte effector functions.3 Not all FcγR classes are capable of eliciting phagocytosis.3 In the human pulmonary alveolar macrophage (AM), FcγRI is the most abundantly expressed class of phagocytic FcγRs, but low levels of FcγRIIa are also detected.4 On FcγR clustering, its immunoreceptor tyrosine-based activation motif (ITAM) is phosphorylated by receptor-associated protein tyrosine kinases (PTKs) of the Src family (Src, Lck, Lyn, Fgr, Hck, Fyn, and Yes). The phosphorylated ITAM then serves as a docking site for Src-homology 2 domain-containing adapter proteins and enzymes, including Syk PTK, phospholipase C, the Ras-activating enzyme Sos, and phosphoinositide 3-kinase.5 Upon their engagement by IgG-opsonized targets, both FcγRs as well as some of their associated signaling molecules redistribute to lipid rafts (LRs), highly dynamic cholesterol- and sphingolipid-enriched microdomains that can act as platforms on which signaling proteins are assembled and which serve to compartmentalize a variety of cellular processes.6 In addition to their roles in the membrane reorganization events of phagocytosis, these signaling molecules are also required for the induction of proinflammatory mediators involved in the amplification of phagocyte actions as well as the recruitment of leukocytes to the inflammatory milieu.7
Among such mediators, the 5-lipoxygenase–derived products of arachidonic acid, leukotriene (LT) B4, and the cysteinyl LTs (CysLTs), LTC4, LTD4, and LTE4, enhance a myriad of leukocyte functions.8 LTB4 and CysLTs each bind 2 different classes of G protein–coupled receptors (GPCRs), termed leukotriene B4 receptors 1 and 2 (BLT1 and BLT2)9,10 and CysLT receptors 1 and 2 (CysLT1 and CysLT2).11,12 GPCRs signal through the activation of small heterotrimeric G proteins that modulate the generation of second messengers, such as cyclic adenosine 5′-monophosphate and Ca2+.13 We have shown that exogenously added or endogenously generated LTB4 and LTD4 promote FcγR-dependent phagocytosis and microbial killing in AMs via BLT1 and CysLT1, respectively;14,15 however, there are key differences in the intracellular programs they use to enhance AM functions. First, LTB4/BLT1 is a more potent enhancer of AM phagocytosis and bacterial killing of IgG-opsonized targets than is LTD4/CysLT1.16 Second, BLT1 is functionally coupled primarily to Gαi, whereas CysLT1 is coupled only to Gαq.17 Third, LTB4 effects on FcγR-mediated phagocytosis are dependent on activation of Syk18 and protein kinase C (PKC)-α, whereas LTD4 effects are independent of Syk18 and PKC-α, but dependent on PKC-δ.19
Various FcγRs have also been shown to be differentially distributed to LRs. FcγRIIa and IIb translocate to LRs upon FcγR engagement in COS-1 and RBL cells as well as the macrophage cell lines U937 and RAW 264.7,20–23 whereas FcγRI is constitutively present in LRs in U937 and IIA1.6 cells.24 The recruitment of receptors and signaling molecules to the LR depends on a variety of signals, including acetylation and phosphorylation of proteins. The signals that govern the translocation of FcγRI to the LR are unknown.
In this study, we investigated the importance of LRs in the actions of LTB4 on FcγRI-mediated phagocytosis in AMs and the consequences of receptor localization and molecular interactions within LRs. Besides FcγRs, GPCRs, heterotrimeric G proteins, adenylate cyclases, small guanosine triphosphatases (GTPases), and kinases have all been observed to partition to LRs, either constitutively or after agonist challenge.25 We found that FcγRI engagement results in tyrosine phosphorylation of BLT1 by Src family kinases; this leads to a molecular complex within LRs comprising FcγRI, BLT1, Gαi3, and Src itself, which drives LTB4-enhanced signaling and phagocyte functions. To our knowledge, these findings are the first to demonstrate a functionally critical physical association between a GPCR and an immunoreceptor.
Methods
Animals
Wistar rats were purchased from Charles River Laboratories, and BLT1 knockout (KO; B6.129S4-Ltb4r1tm1Adl/J),26 and strain-matched wild-type C57BL/6 mice were purchased from Jackson ImmunoResearch Laboratories. All animals were treated according to National Institutes of Health guidelines for the use of experimental animals with the approval of the University of Michigan Committee for the Use and Care of Animals.
Reagents
A complete list and sources of reagents and antibodies can be found in supplemental materials (available on the Blood website; see the Supplemental Materials link at the top of the online article).
Isolation and culture of AMs
Resident AMs of more than 95% purity were obtained from rats via lung lavage and plated as described.27
Lipid raft fractionation
Membranes were isolated as described in supplemental materials. Separation of LR microdomains in OptiPrep gradient was performed according to the methods of MacDonald and Pike28 and Weerth et al,29 with modifications. The membrane pellets in 400 μL of HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid) were mixed with 550 μL of 60% OptiPrep to obtain a 45% solution. This mixture was overlaid with 950 μL of 35% and 475 μL of 5% OptiPrep/HEPES buffer without NaCl. Tubes were centrifuged for 18 hours at 100 000g. After centrifugation, a distinct band was apparent at the interface of the 35% end of the gradient and the 5% OptiPrep bottom layer. Ten fractions of equal volume were collected from the top of the tube. Based on pilot experiments, we routinely pooled fractions 3 to 6 as containing LR fractions and fractions 7 to 10 as containing non-LR fractions, and subjected each to immunoblot analysis as described.
Immunoprecipitation
For FcγRI and BLT1 immunoprecipitation, we performed detergent-free fractionation of the membranes, and both LR and non-LR fractions were precleared with protein A-Sepharose for 30 minutes and incubated overnight at 4°C with anti-FcγRI, anti-BLT1, or nonspecific IgG (1:80; CN:sc 2027; Santa Cruz Biotechnology). Protein A-Sepharose was added and incubated for 3 hours with rotation at 4°C, and immunoprecipitates were isolated and subjected to electrophoresis, as previously described.15
Immunoblotting
Immunoblot analysis was performed, as previously described,18 using primary antibodies described in supplemental materials. Densitometric analysis was as described;17 the density of the phosphorylated bands was divided by actin; and the degree of phosphorylation was taking into account the bands of untreated controls.
Confocal microscopy
A total of 2 × 105 AMs was plated in 4-well chamber slides (Nunc), incubated with or without 10:1 IgG–red blood cells (RBCs) for 10 minutes, and then washed with phosphate buffered saline. Patches of the LR marker GM1 ganglioside were identified by staining for 15 minutes with Alexa 488–cholera toxin B (CTxB; 1:1000) at 4°C. Rabbit anti-CTxB was used when AMs were stained for LT receptors, and mouse anti–CTxB was used when AMs were stained for FcγRI. For colocalization studies, slides were fixed with 3% paraformaldehyde, parasolubilized with 0.1% Triton X-100/1% bovine serum albumin, and blocked with 5% fetal bovine serum in phosphate-buffered saline before incubation with various combinations of primary antibodies against BLT1, CysLT1, or FcγRI (each used at 1:200). Rhodamine- or fluorescein isothiocyanate–conjugated goat anti–rabbit or anti–mouse secondary antibody (1:200) was used (Sigma-Aldrich; CN:F9887 and T5393). Simultaneous detection of GM1, BLT1, and FcγRI used Alexa 488-CTxB and rabbit anti-CTxB (Molecular Probes; 1:200; CN:V-34403), mouse Alexa 647 anti-BLT1 (AbD Serotec; CN:MCA2108A647T; 1:50), and mouse phycoerythrin anti-FcγRI (BD Biosciences; 1:50; CN:558455). Cells stained with isotype-matched control antibody were used to control for the background from each fluorophore. Slides were mounted in Prolong Gold mounting media (Molecular Probes) with DAPI (4,6 diamidino-2-phenylindole). Cells were imaged on a Zeiss LSM 510 confocal microscope with an inverted Axiovert 100 M microscope stand using a C-apochromat ×40/1.2 W corr. The 488-nm line from an Ar laser and the 543- and 633-nm lines from 2 He/Ne lasers were used for excitation. The images were analyzed using LS 2.5 image analysis software (Zeiss). Photographs were obtained only of AMs containing bound or internalized IgG-RBCs. Confocal images were taken with identical settings to allow comparison of staining. Single confocal sections of the cells were captured in multitrack. Each set of frames from a given treatment condition depicts a representative AM selected from 20 analyzed cells.
Cholesterol depletion
Cholesterol depletion was accomplished by treatment for 5 minutes with 5 mM MβCD and 2βΗPCD or the negative control MαCD, as described.30
Phagocytosis and bacterial killing assays
Phagocytosis of IgG-RBCs was assessed, as previously described.17 In one set of experiments, AMs were preincubated with anti-FcγRI, anti-FcγRII, anti-FcγRIII, or nonspecific rabbit IgG (1 μg/mL) for 30 minutes before 5-minute treatment plus or minus LTB4 (10 nM) and subsequent addition of IgG-RBCs. The ability of Klebsiella pneumoniae to survive intracellularly after phagocytosis was assessed, as previously described.15
Statistical analysis
Representative immunoblots from 3 to 5 independent experiments are depicted. Graphs represent the mean plus or minus SEM from at least 3 independent experiments. The means from different treatment groups were compared by analysis of variance, followed by Bonferroni analysis. Statistical significance was set at a P value of up to .05.
Results
BLT1 and FcγRI redistribute to LRs upon FcγR engagement
Using selective receptor antagonists, we previously demonstrated a critical role for BLT1 in the enhancement of FcγR-mediated phagocytosis.14–16 The importance of endogenous LTB4/BLT1-derived signals for optimal baseline IgG-RBC ingestion was verified in new studies using AMs from BLT1 KO mice, which ingested approximately 50% fewer IgG-RBCs at baseline than did wild-type AMs. Furthermore, AMs from BLT1 KO mice were entirely unable to potentiate phagocytosis when treated with LTB4, but thoroughly maintained their responsiveness to LTD4 (Figure 1A). The importance of FcγRI in phagocytosis by rat AMs has not been established previously. To determine which class of FcγR is involved in basal as well as LTB4-augmented IgG-RBC ingestion by rat AMs, we used blocking antibodies against FcγRI, II, and III,31 followed by the addition of LTB4 and then opsonized targets. Anti-FcγRI was the only blocking antibody that substantially inhibited basal IgG-RBC ingestion and completely inhibited LTB4 effects (Figure 1B). These findings provide the rationale for a focus on BLT1 and FcγRI in subsequent experiments.
Figure 1.

FcγRI mediates LTB4/BLT1 enhancement of IgG-RBC phagocytosis. (A) AMs from wild-type and BLT1 KO mice were pretreated with LTB4 (1 and 100 nM) or LTD4 (100 nM) for 5 minutes before challenge with 50:1 IgG-RBCs. Phagocytosis was determined, as described in “Phagocytosis and bacterial killing,” and expressed as the mean ± SEM from 3 independent experiments, each performed in quintuplicate. *P < .05 compared with control wild type, and #P < .05 compared with BLT1 KO. (B) Rat AMs were pretreated for 30 minutes with vehicle control or 1 μg/mL anti-FcγRI, anti-FcγRII, or anti-FcγRIII, followed by a 5-minute incubation with or without 10 nM LTB4. They were then incubated with IgG-RBCs (50:1), and phagocytosis was determined, as described in “Methods,” and expressed as the mean ± SEM from 3 independent experiments, each performed in quintuplicate. *P < .05 compared with control; #P < .001 vs LTB4-treated cells.
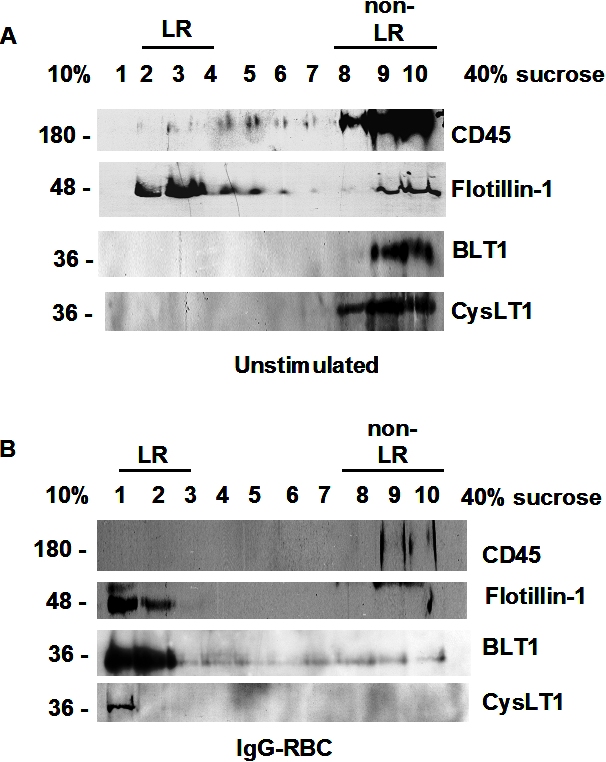
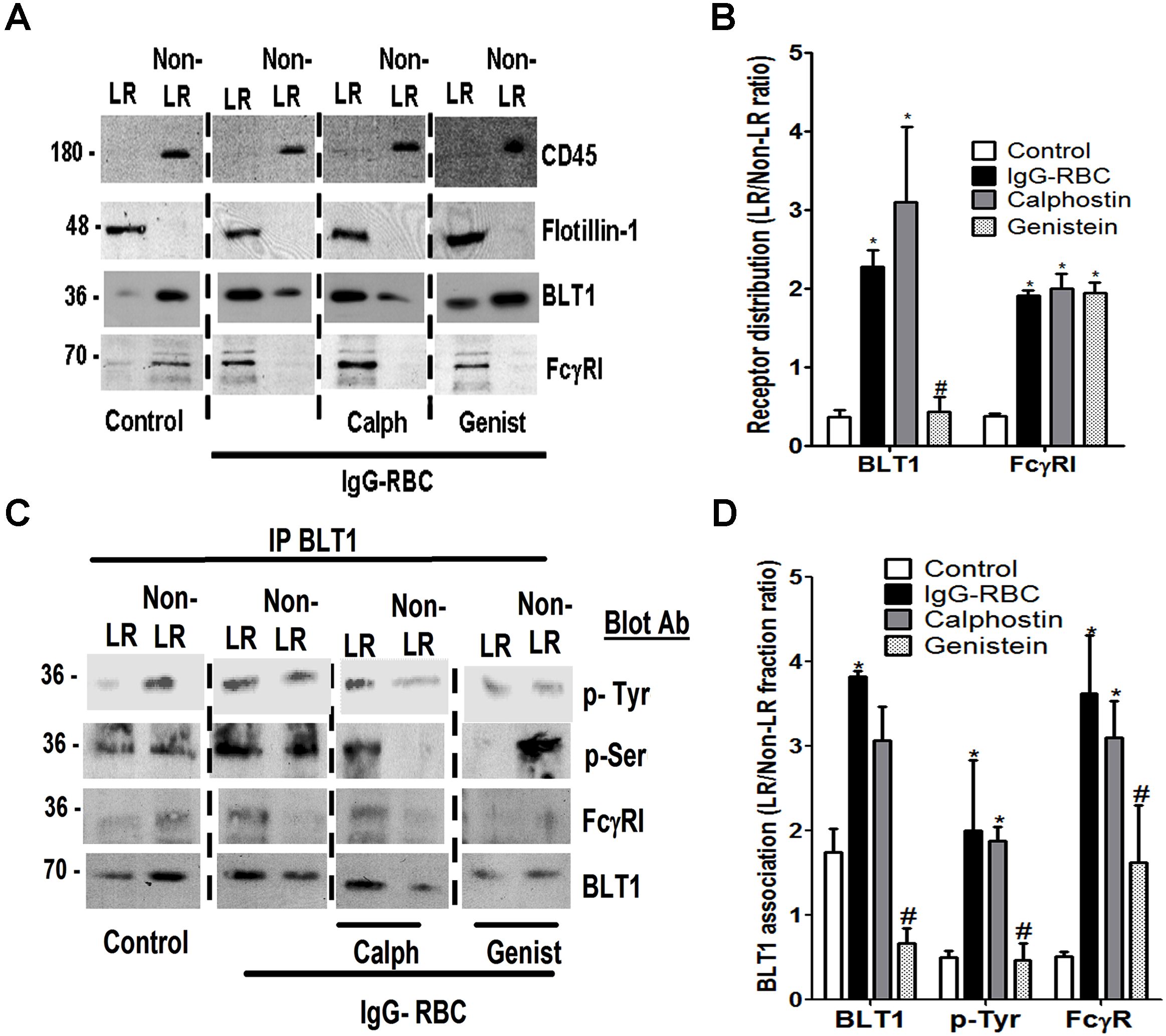
Because BLT1 signaling enhances FcγR-mediated processes in AMs, we asked whether both BLT1 and FcγRI are distributed to the LR upon FcγR engagement. To evaluate BLT1 and FcγRI receptor localization in membrane microdomains, we primarily used a detergent-free membrane fractionation protocol using Optiprep28,32 because detergent lysis can be subject to artifacts such as a propensity to preferentially solubilize outer leaflet membrane lipids.33 Using this technique, the LR marker flotillin-1 was enriched in fractions 3 to 6, whereas the non-LR marker CD45 was enriched in fractions 7 to 10 (Figure 2A). For subsequent experiments, the fractions enriched with flotillin-1 or CD45 were pooled and designated “LR fractions” and “non-LR fractions,” respectively. In resting cells, BLT1 was found predominantly in non-LR fractions, whereas FcγRI was observed in both LR and non-LR fractions. After a 10-minute incubation with IgG-RBCs at a target:cell ratio of 30:1, FcγRI as well as BLT1 were distributed predominantly in flotillin-1–positive LR fractions (Figure 2B). Discontinuous sucrose gradient centrifugation in 0.5% Triton X-10033 of membranes isolated from resting or IgG-RBC–stimulated AMs verified this redistribution of BLT1 to LRs upon FcγR engagement (supplemental Figure 1). We also investigated the distribution of FcγRIIa and b in AMs, but their expression levels were barely detectable in any fractions (data not shown); by contrast, they were detectable in membrane fractions of elicited mouse peritoneal macrophages and human monocytes, confirming the reactivity of the antibodies used (data not shown).
Figure 2.

FcγR engagement causes redistribution of BLT1 and FcγRI receptors to LRs. (A) Detergent-free fractionation of membranes into LR and non-LR fractions. A total of 100 000g membrane fractions from resting AMs was subjected to discontinuous Optiprep gradient centrifugation, as described in “Lipid raft fractionation.” Fractions were harvested and subjected to immunoblot analysis for the LR marker flotillin-1 and the non-LR marker CD45. (B) AMs were pretreated with or without 100 nM LTB4 for 5 minutes, followed by 10-minute incubation with or without 30:1 IgG-RBCs. Membrane fractions were prepared as in panel A, and the LR (fractions 3-6) and non-LR (fractions 7-10) fractions were pooled and subjected to immunoblot analysis for the proteins of interest shown on the right. The experiment depicted is representative of 3 to 5 independent experiments. (C) Relative microdomain distribution of BLT1, FcγRI, PKC-α, and PKC-δ was determined by densitometric analysis of immunoblots from 3 different experiments, as depicted in panel B, and expressed as the LR/non-LR ratio. *P < .05 vs control; #P < .001 vs IgG-RBC alone. (D) AMs were pretreated with 100 nM LTB4 alone for 5 minutes, membrane fractions were prepared as in panel A, and the LR (fractions 3-6) and non-LR (fractions 7-10) fractions were pooled and subjected to immunoblot analysis for the proteins of interest shown on the right. The experiment depicted is representative of 2 independent experiments. (E) AMs were incubated with (ii,iv) or without (i,iii) 10:1 IgG-RBCs for 10 minutes, and LR patches were stained with CTxB-Alexa 455/anti-CTxB. Anti-CTxB–patched cells were fixed and stained with anti-FcγRI (top) and anti-BLT1 (bottom) antibodies and visualized by confocal microcopy. Images are from one experiment representative of 3 independent experiments.
Pretreatment with exogenous LTB4 did not alter the IgG-triggered redistribution of BLT1 and FcγRI to LR fractions (Figure 2B). Importantly, 5-minute treatment with LTB4 in the absence of IgG targets was unable to promote redistribution of either BLT1 or FcγRI to LR fractions of AMs (Figure 2D), suggesting that signals required for BLT1 translocation to LRs are exclusively derived from FcγR. Next, the membrane localization of signaling molecules involved in BLT1 effects, including Gαi3 and Gαq,17 as well as PKC-α and PKC-δ,19 was examined. Both Gαi3 and Gαq were primarily localized to LR fractions in all conditions studied (Figure 2B). As expected, neither isoform of PKC was present in the membranes of resting cells; however, both isoforms accumulated in LR > non-LR membrane fractions upon FcγR engagement. Of note, LTB4 pretreatment increased the proportion of both PKC-α and PKC-δ within LRs compared with IgG-RBC challenge alone (Figure 2B-C).
To further exclude a potential role for BLT2 in LTB4 effects,9 we investigated the distribution of this receptor in AM membrane microdomains. BLT2 was identified only in non-LR domains in all conditions tested (Figure 2B). As a comparison, we also investigated the distribution of another class of LT receptor, namely CysLT1. In resting cells, CysLT1 was found in both LR and non-LR domains, and upon FcγR engagement, this receptor redistributed mainly to LRs (supplemental Figure 2A). Interestingly, pretreatment with the CysLT1 agonist LTD4, followed by IgG-RBCs, caused redistribution of signaling molecules involved in LTD4 enhancement of phagocytosis, such as Gαq, PKC-α, and PKC-δ, to the non-LR domain (supplemental Figure 2E). These data provide a striking contrast to those obtained for BLT1 and its associated signaling molecules.
Confocal microscopy was performed as an alternative approach to examining the localization of BLT1 and FcγRI, capitalizing on the fact that molecules of the constitutive LR ganglioside GM1 cluster when cross-linked and such clusters are large enough to be detected by microscopy.34 GM1 was cross-linked by CTxB, and anti-CTxB antibody detected clustering (patching) of GM1.34 Immunofluorescence detection indicated that a small proportion of FcγRI appeared to colocalize with GM1 in resting cells (Figure 2Ei), whereas BLT1 did not (Figure 2Eiii). The addition of IgG-RBCs for 10 minutes markedly increased the colocalization of FcγRI as well as of BLT1 with GM1 (Figure 2Eii-iv). In addition, CysLT1 colocalization with GM1 also increased upon FcγR engagement (supplemental Figures 2A and 4Bii). We observed no further increase in the colocalization of either BLT1 or FcγRI with GM1 when AMs were pretreated with LTB4 before the addition of IgG-coated targets (data not shown). A similar pattern of results was obtained when LRs were identified by immunofluorescence staining of flotillin-1 rather than by detection of GM1 (data not shown). These imaging experiments support the conclusions from immunoblot analysis that engagement of FcγR triggers redistribution of both the immunoreceptor as well as of BLT1 and CysLT1 to LRs.
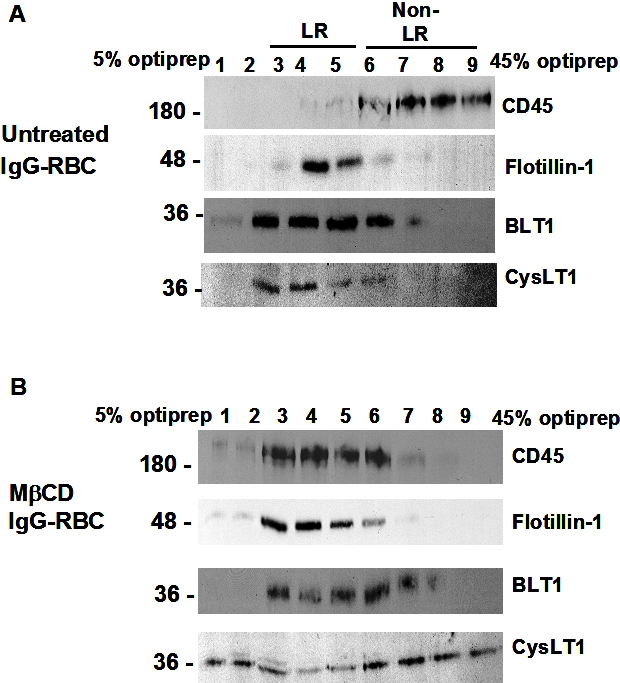
LRs mediate BLT1 effects on AM signaling and antimicrobial effector functions
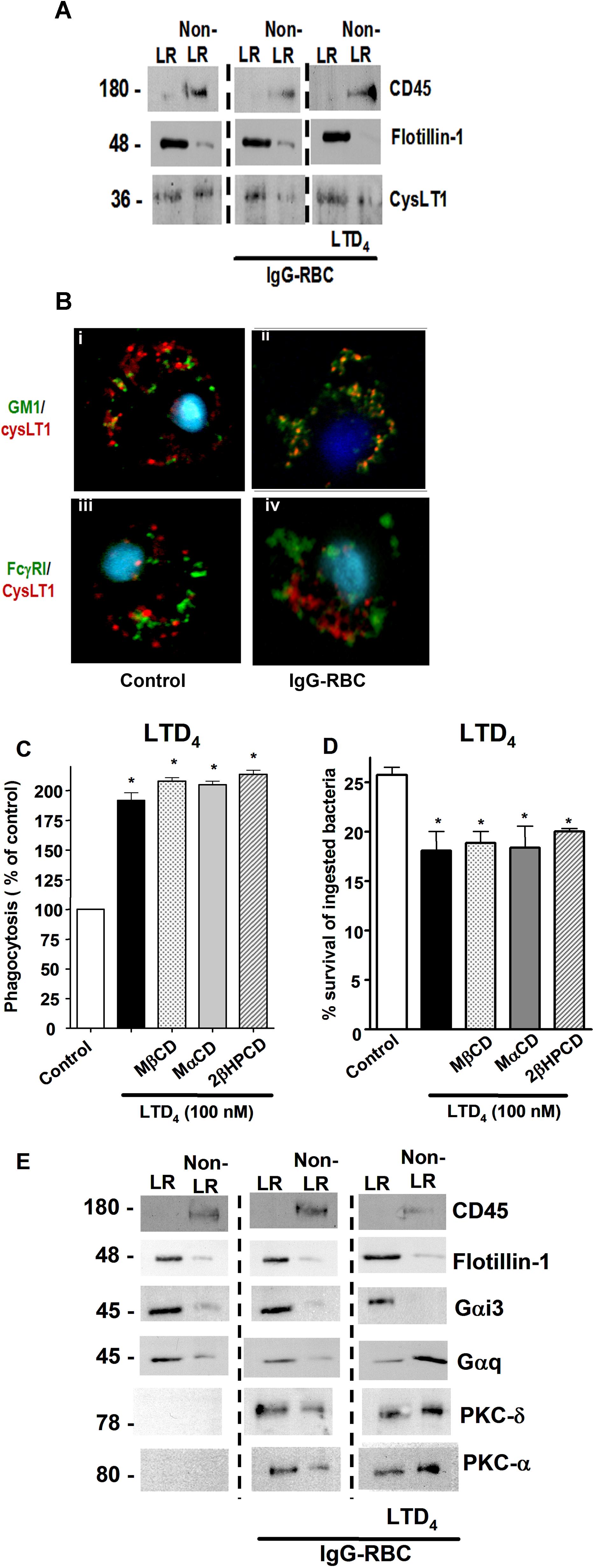
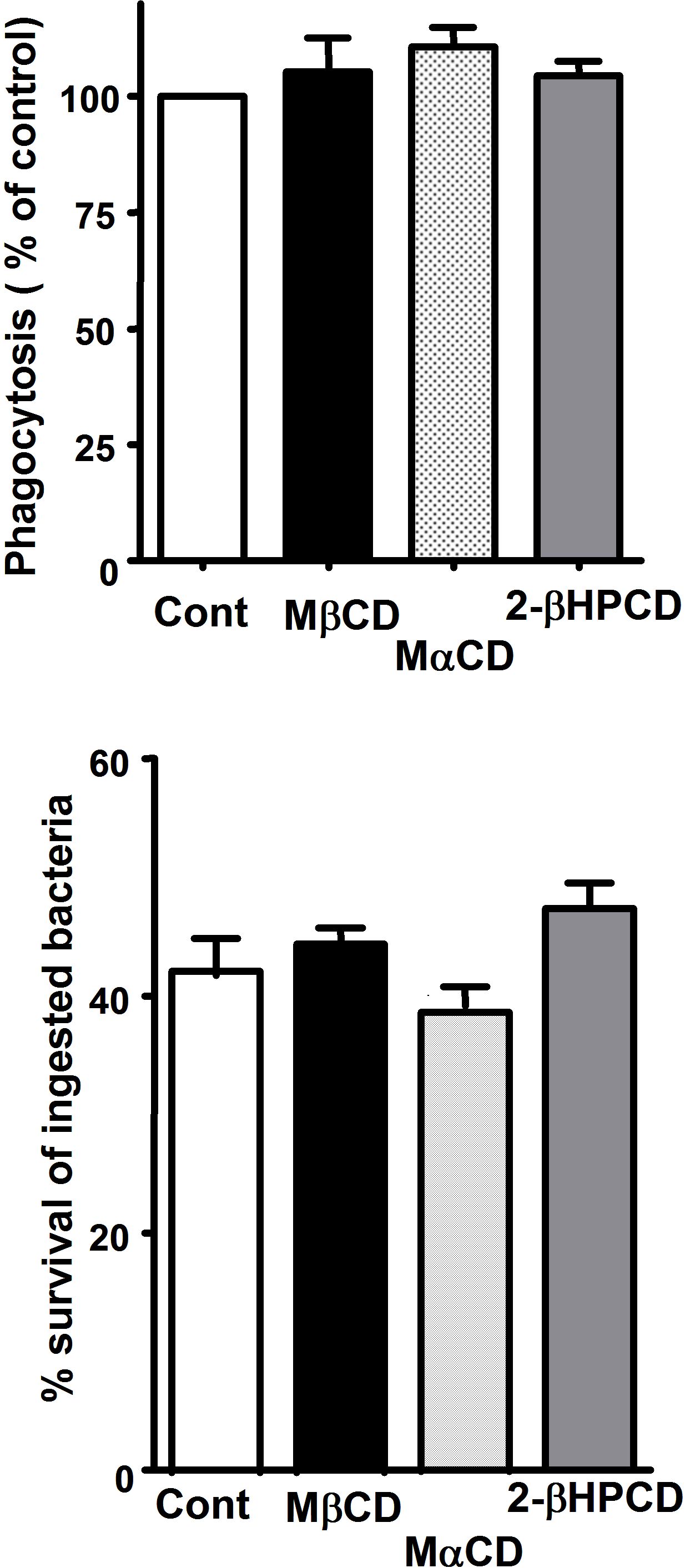
Because both BLT1 and FcγRI were found to redistribute to LRs upon FcγR engagement, we sought to determine whether these microdomains were necessary for the LTB4-induced enhancement of signaling and AM antimicrobial functions. AMs were pretreated with drugs that deplete cholesterol from plasma membranes and thereby functionally disrupt rafts, namely MβCD, 2βΗPCD, or the negative control congener MαCD. That MβCD in fact disrupted LR integrity was evidenced by the facts that CD45 and flotillin-1 were now distributed among both LR and non-LR fractions (supplemental Figure 3A-B), and that both GM1 and BLT1 were colocalized with CD45 in IgG-RBC–stimulated AMs (data not shown). To investigate the functional consequences of LR disruption, cyclodextrin compounds were added at 5 mM for 5 minutes30 before addition of LTB4 (10 nM), after which phagocytosis of IgG-RBCs was determined. With intact LR integrity, LTB4 (Figure 3A) increased phagocytosis, as expected on the basis of our previous reports.16,18 LR disruptors MβCD and 2βΗPCD substantially attenuated LTB4-enhanced phagocytosis (Figure 3A); the specificity of this phenomenon was supported by the fact that MαCD had no effect on the augmentation of phagocytosis by LTB4. Parallel experiments were performed to determine whether LRs were necessary for LTB4 potentiation of another important AM effector function, namely killing of immune serum-opsonized K pneumoniae.15 AMs were pretreated with the LR disruptors or the negative congener, followed by the addition of LTB4, as described above. MβCD and 2βΗPCD specifically abolished the enhancing effect of LTB4 on AM bacterial killing (Figure 3B). The fact that LR disruption failed to reduce basal phagocytosis or bacterial killing below control (non-LTB4) levels (Figure 3A-B and supplemental Figure 4A-B) suggests that LRs are not essential for basal FcγR-mediated effector functions in AMs. These data indicate that BLT1 localization within LRs is required for LTB4 to enhance antimicrobial activity against IgG-opsonized targets. Because CysLT1 also redistributed to LRs upon FcγR engagement, the functional requirement for the integrity of these microdomains in LTD4-induced enhancement of AM antimicrobial functions was determined. LTD4 (100 nM) treatment elicited the expected increases in both phagocytosis and bacterial killing; however, pretreatment with the LR disruptors MβCD and 2βΗPCD had no impact on these effects of LTD4 (supplemental Figure 4C-D).
Figure 3.

LR integrity is required for LTB4 enhancement of AM signaling and antimicrobial functions. (A) AMs were pretreated for 5 minutes with vehicle control or 5 mM MβCD, 2-βHPCD, or MαCD, followed by 5-minute incubation with or without 10 nM LTB4. They were then incubated with IgG-RBCs (50:1) and phagocytic indices, determined as described in “Phagocytosis and bacterial killing.” Phagocytosis is expressed as the percentage of the control value in which no drugs were added, and each bar represents the mean ± SEM from 3 individual experiments, each performed in quintuplicate. *P < .05 compared with control; #P < .001 vs LTB4-treated cells. (B) AMs were preincubated with cyclodextrin compounds as in panel A before the addition of 50:1 serum-opsonized K pneumoniae. Thirty minutes after infection, cells were incubated with 10 nM LTB4 or vehicle. Microbicidal activity was assessed and expressed as the mean ± SEM percentage of survival of ingested bacteria from 3 individual experiments, each performed in triplicate. *P < .05 compared with control; #P < .001 vs LTB4-treated cells. (C) AMs were pretreated for 5 minutes with vehicle control or cyclodextrins, followed by 5-minute treatment with or without 10 nM LTB4, after which they were incubated with 30:1 IgG-RBCs for 15 minutes. Immunoblots depict phosphorylated forms of PKC-α, PKC-δ, ανδ Syk, as well as β-actin as a loading control. Results shown are representative of 3 independent experiments. (D) Relative PKC-α, PKC-δ, and Syk phosphorylation was determined by densitometric analysis of immunoblots from 3 different experiments, as depicted in panel C, and is expressed as a percentage of control. The intensity of phosphorylation was quantitated as the density of the phosphorylated protein band divided by that of the actin band, and this ratio was then expressed relative to that of the untreated control, which was set at 100%. The dashed line in the figure indicates lanes in the membrane that contained experimental conditions that were not pertinent and were omitted. *P < .05 vs IgG-RBC; #P < .001 vs IgG-RBC plus LTB4.
Next, the importance of raft integrity for intracellular signaling cascades activated by BLT1 ligation in the context of FcγR cross-linking was determined. AMs were pretreated as described in “Cholesterol depletion” with the LR disruptor MβCD or with the inactive congener MαCD, after which they were treated for 5 minutes with LTB4 and subsequently with IgG-RBCs for 15 minutes. FcγR engagement activated (ie, phosphorylated) PKC-α, PKC-δ, and Syk, and pretreatment with exogenous LTB4 potentiated activation of each of these kinases (Figure 3C-D). LR disruption had no effect on basal activation of these 3 kinases during FcγR engagement (data not shown), but did abolish their potentiation by LTB4 (Figure 3C-D), confirming the importance of LR integrity for the molecular events underlying LTB4 enhancement of antimicrobial function.
BLT1 associates with FcγRI within LRs
Because our data indicated that BLT1 and FcγRI are both localized to LR microdomains during FcγR engagement and also elicit convergent signaling and functional responses, we asked whether these 2 receptors physically interact. Confocal microscopy demonstrated that although a fraction of immunodetectable BLT1 colocalized with FcγRγI in resting cells, the degree of colocalization increased markedly upon FcγR engagement (Figure 4Ai-ii). We observed no colocalization between CysLT1 and FcγRI under any condition tested (supplemental Figure 2Biii-iv).
Figure 4.

FcγRI and BLT1 colocalize and associate in AM LR microdomains after FcγR engagement. (A) AMs were incubated with (ii) or without (i) 10:1 IgG-RBCs for 10 minutes, and the cells were fixed. They were then stained with anti-BLT1 and anti-FcγRI primary antibodies, followed by fluorescein isothiocyanate-labeled and rhodamine-labeled secondary antibodies, respectively, and visualized by confocal microcopy. Confocal images were taken with identical settings to allow comparison of staining. Images are from one experiment and are representative of 5 independent experiments. (B,D-E) AMs were incubated with or without 30:1 IgG-RBCs for 10 minutes, and membrane fractions were obtained by Optiprep gradient centrifugation. LR and non-LR membrane fractions were pooled, and pooled LR or non-LR fractions were immunoprecipitated with an antibody against either FcγRI (B), BLT1 (D), or nonspecific rabbit IgG (E), as described in “Phagocytosis and bacterial killing.” The immunoprecipitates were subjected to immunoblot analysis using antibodies against the proteins shown on the right. The data shown are from a single experiment representative of 3 independent experiments. (C) Association of BLT1 and Gαi3 with FcγRI was determined by densitometric analysis of immunoblots of FcγRI immunoprecipitates from 3 different experiments, as depicted in panel B, and expressed as the LR/non-LR ratio of each protein. *P < .05 vs control.
To confirm the microscopic studies, we immunoprecipitated FcγRI from LR and non-LR membrane fractions of AMs that had been challenged or not with IgG-RBCs, and performed immunoblot analysis on the immunoprecipitates using antibodies against FcγRI, BLT1, and the irrelevant GPCR G2A. As presented in Figure 3B-C, a small proportion of BLT1 coimmunoprecipitated with FcγRI in LR fractions of resting cells, but this increased markedly after FcγR engagement. G2A and BLT2 failed to coimmunoprecipitate with FcγRI in either LR or non-LR fractions of resting or stimulated cells (data not shown). The FcγRI immunoprecipitates were also examined for the presence of the small G proteins Gαi3 and Gαq. Like BLT1, Gαi3, but not Gαq, also strongly coimmunoprecipitated with FcγRI within LRs of IgG-challenged AMs (Figure 4B-C). The presence of G2A, BLT2, and Gαq in the membrane fractions was confirmed by their detection using Western blot of the supernatants resulting from precipitation (data not shown). In complementary fashion, reverse immunoprecipitation with anti-BLT1 coimmunoprecipitated FcγRI and flotillin-1 in the LR fraction (Figure 4D). Pretreatment with exogenous LTB4 before addition of IgG-RBCs did not modify the observed colocalization of BLT1 and FcγR in either confocal microscopy or immunoprecipitation experiments (data not shown). Immunoprecipitation with a nonspecific IgG failed to coimmunoprecipitate either FcγRI (Figure 4E) or BLT1 (data not shown). Taken together, these data suggest that the dependence on LR integrity for the antimicrobial actions of BLT1 reflects the association within LRs of BLT1 with both FcγRI as well as its primary coupled Gα protein, Gαi3.
FcγR-induced tyrosine phosphorylation of BLT1 by Src kinase mediates its association with FcγRI in LRs
Although serine35 and threonine36 phosphorylation of BLT1 have been implicated in receptor desensitization, neither its tyrosine phosphorylation nor a potential positive role for its phosphorylation in downstream signaling events has previously been investigated. Therefore, phosphorylation of BLT1 was examined by immunoblot analysis in pooled membrane fractions of BLT1 immunoprecipitates. BLT1 was tyrosine phosphorylated in non-LR fractions in resting cells, but tyrosine-phosphorylated receptor redistributed to LR fractions after IgG-RBC challenge. BLT1 was also phosphorylated on serine residues in both LR and non-LR in resting cells, but serine-phosphorylated receptor was confined to LRs upon FcγR engagement (Figure 5A-B).
Figure 5.

BLT1 phosphorylation by Src is required for IgG-induced FcγRI-BLT1 association in LRs. (A) AMs were incubated with or without 30:1 IgG-RBCs for 10 minutes and subjected to membrane fractionation. LR and non-LR membrane fractions were pooled and immunoprecipitated with an antibody against BLT1, after which immunoprecipitates were subjected to immunoblot analysis for the proteins shown on the right. (B) Relative distribution of total BLT1 as well as tyrosine- and serine-phosphorylated BLT1 was determined by densitometric analysis of immunoblots from 3 different experiments, as depicted in panel A, and is expressed as the LR/non-LR ratio of each. *P < .05 vs control. (C) AMs were pretreated with or without the Src kinase inhibitor PP2 (1 μM) or its negative control congener PP3 (1 μM) for 20 minutes, followed by incubation with or without 30:1 IgG-RBCs for 10 minutes. Pooled LR and non-LR fractions were prepared and subjected to immunoblot analysis for the proteins indicated at the right. (D) Relative distribution of BLT1 and FcγRI was determined by densitometric analysis of immunoblots from 3 different experiments, as depicted in panel C, and is expressed as the LR/non-LR ratio of each. *P < .05 vs control, and #P < .05 vs IgG-RBC. (E) AMs were incubated with or without PP2 or PP3 for 20 minutes, followed by challenge with 30:1 IgG-RBCs for 10 minutes. LR and non-LR membrane fractions were pooled and immunoprecipitated with anti-BLT1 antibody. The immunoprecipitates were subjected to immunoblot, as in panel A. (F) Relative distribution of total BLT1, tyrosine-phosphorylated BLT1, FcγRI, and Src was determined by densitometric analysis of immunoblots from 3 different experiments, as depicted in (E), and expressed as LR/non-LR ratio of each. *P < .05 vs control, and #P < .05 vs IgG-RBC. (G) AMs were pretreated with PP2 or PP3, followed by incubation with (ii-iv) or without (i) 10:1 IgG-RBCs for 10 minutes. Cells were fixed and stained with CTxB-Alexa 488/anti-CTxB, Alexa 647 mouse anti-BLT1, and phycoerythrin mouse anti-FcγRI primary antibodies, and then visualized by confocal microcopy. Yellow patches signify areas of colocalization of FcγRI and GM1; purple patches, colocalization of FcγRI and BLT1; and white/gray patches, colocalized BLT1, FcγRI, and GM1 (the latter indicated by solid arrows). A dashed arrow pointing to a blue dot indicates BLT1 alone. Results are representative of at least 3 independent experiments. (H) AMs were pretreated for 5 minutes with vehicle control or PP2 or PP3 (100 nM), followed by 5-minute incubation with or without 10 nM LTB4. They were then incubated with IgG-RBCs (50:1), and phagocytic indices were determined as described in “Methods.” Phagocytosis is expressed as the percentage of the control value (IgG-RBCs alone) to which no drugs were added, and each bar represents the mean ± SEM from 3 individual experiments, each performed in quintuplicate. *P < .05 vs control, and #P < .05 vs LTB4.
We next explored the role of kinases in the redistribution of BLT1 to LRs upon IgG-RBC challenge, initially by pretreating AMs with the generalized PTK inhibitor genistein (100 μM) or the PKC inhibitor calphostin C (100 nM), followed by IgG-RBC stimulation and LR fractionation. Neither genistein nor calphostin C affected the distribution of the LR marker flotillin-1 or the non-LR marker CD45, suggesting that phosphorylation state does not affect LR integrity. Interestingly, genistein, but not calphostin C treatment inhibited BLT1 redistribution to LRs during IgG-RBC challenge, whereas FcγRI distribution was unaffected by both inhibitors (supplemental Figure 5A-B). This effect of genistein was confirmed by immunoprecipitation experiments, where once again genistein, but not calphostin C, abolished the association of FcγRI with BLT1 in both LR and non-LR fractions (supplemental Figure 5C-D).
According to the program GPS 2.0 (http://gps.biocuckoo.org/down.php),37 the only PTKs for which BLT1 contains consensus phosphorylation sequences are Src family members Fgr and B-cell lymphocyte kinases. To investigate the role of Src family kinases in the BLT1 interaction with FcγRI, AMs were pretreated with the Src kinase inhibitor PP2 (1 μM) or its negative control congener PP3 (1 μM). As shown in Figure 5E and F, PP2 specifically decreased tyrosine phosphorylation of immunoprecipitated BLT1 within LRs. Moreover, as assessed by immunoblot analysis, PP2 (but not PP3) prevented BLT1 redistribution to LRs during IgG-RBC challenge, while not affecting redistribution of FcγRI (Figure 5C-D); this effect of PP2 was confirmed by immunoprecipitation experiments (Figure 5E-F). Interestingly, we also observed that Src kinase itself physically associated with BLT1 within LRs during FcγR engagement, an association that was decreased by PP2 treatment (Figure 5E-F). Thus, Src not only mediates BLT1 phosphorylation and drives redistribution of the GPCR to LRs, where it associates with FcγRI and Gαi3, but is itself a component of this multimolecular complex within rafts.
Confocal microscopy was performed to verify the effects of Src inhibition, using anti-FcγRI conjugated to PE; anti-BLT1 conjugated to Alexa 647, which was pseudocolored as dark blue; and anti-CTxB conjugated with Alexa 488. AMs were pretreated with PP2 or PP3, and then challenged with IgG-RBCs. Resting cells demonstrated no colocalization between GM1 and either BLT1 or FcγRI, but some colocalization between BLT1 and FcγRI, as indicated by the purple dots in the merged image (Figure 5Gi). Upon IgG-RBC challenge, colocalization of GM1, BLT1, and FcγRI was evidenced by the white-gray dots (bold arrow in Figure 5Gii). Although PP2 failed to prevent FcγRI colocalization with GM1, it did impair colocalization between BLT1 and both FcγRI as well as GM1. The scattered blue dots indicated by dashed arrows represent BLT1 in non-LR microdomains resulting from Src inhibitor treatment (Figure 5Giv). As expected, there was no effect of PP3 on the colocalization of BLT1/FcγRI or on the redistribution of the receptors to LRs (Figure 5Giii).
We next investigated whether Src activity is required for LTB4/BLT1 effects on FcγR-mediated phagocytosis by pretreating AMs with PP2 or PP3, followed by LTB4 challenge. PP2 at 100 nM (but not PP3) exerted no effect on basal phagocytosis of IgG-RBCs, but completely inhibited LTB4 enhancement of FcγR-mediated phagocytosis (Figure 5H). At a concentration of 1 μM, PP2 also inhibited basal phagocytosis by approximately 70% (data not shown). This indicates that BLT1 phosphorylation by Src after engagement of FcγRI is also required for the enhancement by LTB4 of AM antimicrobial functions.
Discussion
The studies reported in this work used the AM, a primary macrophage that resides on the pulmonary alveolar surface, to provide novel insights into the membrane organization of antimicrobial signaling cascades elicited by the phagocytic receptor FcγRI as well as by the GPCR BLT1, which mediates the immunostimulatory actions of endogenously generated LTB4. Collectively, our findings demonstrate that during FcγR engagement, Src kinase phosphorylates BLT1, and this mediates the direct association of BLT1 with FcγRI and also enhances the activation of key downstream signaling molecules within LR microdomains (Figure 6). The importance of LR integrity for signaling downstream from BLT1 is evidenced by the complete disruption of both the signaling events and the potent antimicrobial actions of LTB4 after cholesterol depletion. By contrast, such dependency on LR integrity was not observed for the antimicrobial effects mediated by an alternative class of LT receptor, CysLT1. Our findings using both biochemical and imaging approaches represent the first examples of which we are aware of (1) GPCR redistribution to LRs upon engagement of an immunoreceptor, and (2) a direct interaction between a GPCR and an immunoreceptor.
Figure 6.
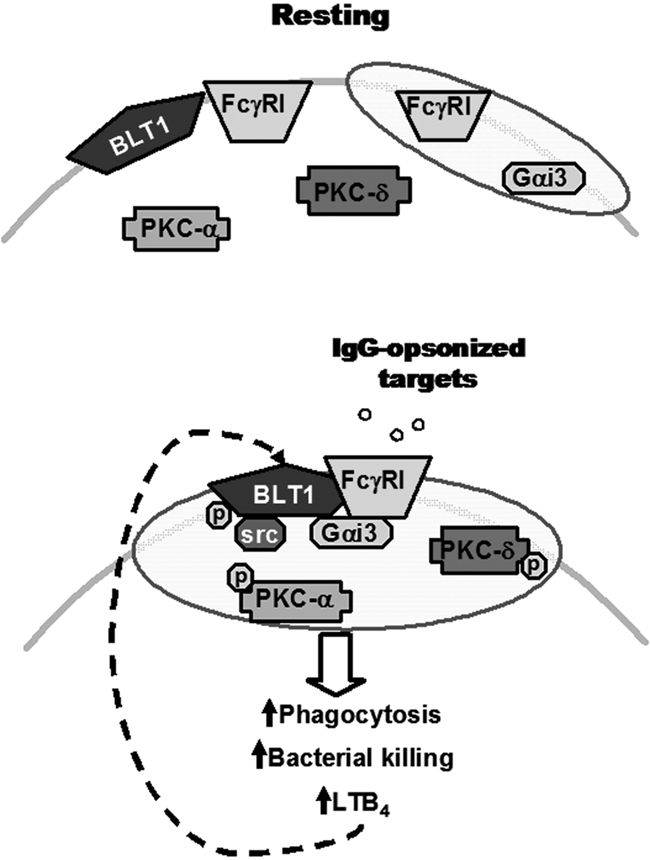
Proposed model for the IgG-induced FcγRI/BLT1/Gαi3/Src complex in LRs that mediates LTB4 enhancement of AM antimicrobial functions and signaling. (Top) In resting cells, BLT1, PKC-α, PKC-δ, and a proportion of FcγRI are located in non-LR domains, whereas Gαi3 and a proportion of FcγRI are constitutively present in LRs (indicated as shaded ovals). (Bottom) Upon FcγRI engagement, Src kinase phosphorylates BLT1, which promotes BLT1 redistribution to LRs as well as the formation of a complex within LRs consisting of this GPCR, its preferentially coupled G protein Gαi, FcγRI, and Src itself. Addition of exogenous LTB4 causes LRs to become further enriched in PKC isoforms, which are required for optimal ingestion of IgG-coated targets. Thus, LRs serve as platforms within which phosphorylated BLT1 interacts with FcγRI and downstream signaling molecules to mediate the augmentation by LTB4 of AM antimicrobial functions, explaining the LR dependence of LTB4/BLT1 actions. Signals from FcγRI also enhance LTB4 generation, which amplifies AM function in an autocrine and paracrine manner. The relative size of the trapezoids represents the relative amount of FcγRI in a given membrane domain. Phosphorylation is indicated by “p.”
Although LTs enhance phagocytosis of microbes via a myriad of opsonic or pathogen recognition receptors,38 our previous studies examining LT-induced signaling in AMs have primarily centered on the context of ingestion via FcγR.14 FcγRI is the most abundantly expressed immunoreceptor isoform in this cell type in humans;4 in this study, we have verified that this is also the case for the rat AM, and have further demonstrated that this isoform mediates basal and LTB4-stimulated IgG-RBC ingestion in these cells. Several reports indicate that FcγRII either is localized to LRs constitutively or is recruited to LRs upon its engagement in human neutrophils,39 U937 cells,20 RAW 264.7 cells,23 and natural killer cells.40 Beekman et al have shown that FcγRI is constitutively present in LRs of human monocytes, U937, and FcγRI/FcRγ chain-transfected IIA1.6 cells,24 but the localization of this FcγR isoform in macrophages was previously unknown. Furthermore, the role of LRs in opsonic phagocytosis is controversial. Kwiatkowska et al showed that LRs are important for the ingestion of IgG-coated targets in U937 cells,20 whereas Lamberti et al41 and Naroeni and Porte42 reported that MβCD failed to interfere with ingestion of IgG-opsonized pathogens in neutrophils and J774.A1 cells, respectively. In our model, cholesterol depletion had no impact on baseline FcγR-mediated phagocytosis or bacterial killing. Our results show that both BLT1 and FcγRI were predominantly localized to non-LR fractions in resting AMs. By contrast, Sitrin et al previously reported that BLT1 was constitutively present in LRs of human neutrophils.30 Differences in BLT1 localization to LRs between these 2 phagocyte populations remain to be explained, but could reflect differences in fractionation protocols, in the activation states of the 2 cell types, or in their LR compositions. That BLT1 translocation to LRs is a specific consequence of FcγR engagement, rather than a universal finding during phagocytosis, was evidenced by the fact that it failed to redistribute to LRs upon AM stimulation with Candida albicans (data not shown), whose ingestion is mediated by pathogen recognition receptors such as the mannose and dectin receptors.43 There is abundant precedent for GPCRs to redistribute to LRs upon binding by their cognate ligands.44 Although LTB4 is generated under conditions of FcγR engagement in AMs,16 the inability of exogenously added LTB4 to itself elicit LR redistribution of its high-affinity GPCR BLT1 argues against the participation of this lipid mediator in this phenomenon. The novel finding that BLT1 instead translocates to LRs upon engagement of this opsonic receptor is consistent with many other lines of evidence indicating a physiologic role for LTB4 in antimicrobial defense.38
Immunoprecipitation studies showed that BLT1 is partially associated with FcγRI primarily in non-LR domains of resting AMs. FcγR engagement results in a much more robust association between BLT1 and FcγRI within LRs, and this complex also includes Gαi3, the G protein we have shown to be most critical for mediating antimicrobial actions of LTB4 via BLT1 in AMs, as well as Src kinase, which mediates the interaction between BLT1 and FcγRI. Although detergent-free preparation of LRs offers several advantages (eg, avoiding differential solubilization of specific LR subsets and preferential solubilization of outer leaflet membrane lipids25,33), it is theoretically possible that immunoprecipitation under these conditions could pull down all raft components and thereby overestimate the physical interaction between partner proteins within LRs. However, confocal microscopic analysis provided independent verification of the interaction between FcγRI and BLT1. Moreover, the fact that several other proteins shown to be associated with LRs (Gαq, the lysophosphatidylcholine receptor G2A, CysLT1, BLT2, and PKC-α and PKC-δ) failed to coimmunoprecipitate with either FcγRI or BLT1 argues strongly for the specificity of the interactions reported in this study. Although Src-dependent phosphorylation of BLT1 is required for BLT1 association with FcγRI and redistribution to LRs, our present studies cannot distinguish between these 2 possible scenarios: (1) phosphorylation promotes BLT1 association with FcγRI in non-LR domains, permitting immunoreceptor to carry GPCR into LRs, and (2) phosphorylation directly promotes BLT1 translocation to LRs, where GPCR associates with immunoreceptor. Even though we have shown that Src phosphorylation is necessary for the interaction between BLT1 and FcγRI in LRs, we cannot exclude the possibility that another molecule(s) may serve as a bridge to mediate such association. Although FcγR has not previously been demonstrated to associate with a GPCR, FcγRIIa has been reported to associate with platelet endothelial cell adhesion molecule–145 and glycoprotein Ib-IX-V complex,46 and FcγRIIIa has been shown to physically associate with the IL-12 receptor in the LR in natural killer cells.40 Another example of an interaction between 2 different classes of receptors includes that of insulin-like growth factor receptor 1 with chemokines CXC chemokine receptor 447 and C-C chemokine receptor 5.48
The signals generated in response to FcγRI engagement that results in physical association between BLT1 and FcγRI within LR microdomains were investigated. Posttranslational modifications such as palmitoylation, myristoylation, and phosphorylation promote the ability of proteins to interact with the unique lipid environment of LRs.49 Among the proteins that are susceptible to acylation and phosphorylation are receptors including GPCRs, G proteins, and Src family kinases.44 We studied the role of phosphorylation in promoting both the association of BLT1 with FcγRI as well as the redistribution of both receptors to LRs. Whereas FcγRI engagement enhanced both serine and tyrosine phosphorylation of BLT1 in LRs, BLT1 redistribution to and physical association with FcγRI in LRs were not abrogated by the protein kinase C inhibitor calphostin C. Although serine/threonine phosphorylation of BLT1 has been implicated in receptor desensitization,35,36 neither its tyrosine phosphorylation nor a role for its phosphorylation in downstream signaling events has been reported. In contrast to calphostin C, the broad spectrum PTK inhibitor genistein did abrogate the tyrosine phosphorylation of BLT1 as well as its redistribution to LRs and its interaction with the immunoreceptor. Because BLT1 contains consensus sequences for phosphorylation by Src kinases and because these kinases are also known to be activated by FcγR ligation and to carry out tyrosine phosphorylation of the FcγR ITAM,5 it was of interest to investigate their role in formation of the BLT1/FcγRI complex as well as in translocation of both receptors to the LR. Like genistein, PP2 attenuated BLT1 phosphorylation and prevented its colocalization with both GM1 and FcγRI, but did not impair FcγRI/GM1 colocalization. Inhibition of another PTK robustly activated by FcγR engagement, Syk, failed to abrogate BLT1 redistribution or interaction with FcγRI, consistent with the lack of consensus sequences in BLT1 for phosphorylation by Syk (data not shown). The functional significance of these actions of Src kinase on BLT1 is indicated by the fact that the Src inhibitor PP2 abolished LTB4 effects on the enhancement of IgG-RBC ingestion. Confirmation by RNA silencing would strengthen our conclusions derived from Src inhibition, but this approach awaits identification of the specific Src family member responsible. Tyrosine phosphorylation of GPCRs after engagement of growth factor receptor tyrosine kinases has been reported,50 and Delcourt et al have shown that Src kinase-dependent phosphorylation of the GPCR pituitary adenylate cyclase–activating polypeptide receptor type I mediates its association with insulin-like growth factor-1.51 Our work extends this type of interaction with a GPCR to a nonreceptor tyrosine kinase immunoreceptor and defines the permissive role of LRs in supporting it. The applicability of our findings to other rat macrophage populations and to AMs of species other than rat remains to be determined.
Our studies thus identify a novel mode of interaction of a GPCR, BLT1, with FcγRI within LRs, which is triggered by immunoreceptor engagement. The ability of FcγRI engagement to affect the recruitment of BLT1 serves to amplify the function of this opsonic receptor. These data provide new insights into the spatial organization and molecular interactions involved in lipid mediator receptor signaling to enhance antimicrobial functions of AMs, responses that are pivotal to immune defenses in the lung.
Supplementary Material
Acknowledgments
We thank Sang-Hoon Kim for performing phagocytosis experiments with PP2, Teresa Marshall and Casey Lewis for technical assistance, Thomas G. Brock and the Microscopy Image Analysis Laboratory Core at University of Michigan for assistance with immunofluorescence microscopy, Sara L. Emery for assistance with lipid raft fractionation, and members of the Peters-Golden laboratory for their input on this project.
This work was supported by National Institutes of Health grants HL058897 (M.P.-G.), HL078727 (D.M.A.), and AI 060983 (R.G.S.). C.H.S. was supported by an American Lung Association Senior Postdoctoral Research Fellowship.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.H.S. designed the research, performed experiments, analyzed data, and wrote the paper; D.M.A. and R.G.S. designed the research and analyzed data; and M.P.-G. designed research, supervised the work, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Marc Peters-Golden, 6301 MSRB III, Box 5642, 1150 West Medical Center Dr, University of Michigan Health System, Ann Arbor, MI 48109-5642; e-mail: petersm@umich.edu.
References
- 1.Gordon S. Macrophage heterogeneity and tissue lipids. J Clin Invest. 2007;117:89–93. doi: 10.1172/JCI30992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox D, Greenberg S. Phagocytic signaling strategies: Fcγ receptor-mediated phagocytosis as a model system. Semin Immunol. 2001;13:339–345. doi: 10.1006/smim.2001.0330. [DOI] [PubMed] [Google Scholar]
- 3.Groves E, Dart AE, Covarelli V, Caron E. Molecular mechanisms of phagocytic uptake in mammalian cells. Cell Mol Life Sci. 2008;65:1957–1976. doi: 10.1007/s00018-008-7578-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger M, Norvell TM, Tosi MF, Emancipator SN, Konstan MW, Schreiber JR. Tissue-specific Fcγ and complement receptor expression by alveolar macrophages determines relative importance of IgG and complement in promoting phagocytosis of Pseudomonas aeruginosa. Pediatr Res. 1994;35:68–77. doi: 10.1203/00006450-199401000-00015. [DOI] [PubMed] [Google Scholar]
- 5.Greenberg S. Diversity in phagocytic signalling. J Cell Sci. 2001;114:1039–1040. doi: 10.1242/jcs.114.6.1039. [DOI] [PubMed] [Google Scholar]
- 6.Pike LJ. Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J Lipid Res. 2006;47:1597–1598. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Greenberg S, Grinstein S. Phagocytosis and innate immunity. Curr Opin Immunol. 2002;14:136–145. doi: 10.1016/s0952-7915(01)00309-0. [DOI] [PubMed] [Google Scholar]
- 8.Peters-Golden M, Henderson WR., Jr Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 9.Yokomizo T, Kato K, Terawaki K, Izumi T, Shimizu T. A second leukotriene B4 receptor, BLT2: a new therapeutic target in inflammation and immunological disorders. J Exp Med. 2000;192:421–432. doi: 10.1084/jem.192.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature. 1997;387:620–624. doi: 10.1038/42506. [DOI] [PubMed] [Google Scholar]
- 11.Lynch KR, O'Neill GP, Liu Q, et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399:789–793. doi: 10.1038/21658. [DOI] [PubMed] [Google Scholar]
- 12.Heise CE, O'Dowd BF, Figueroa DJ, et al. Characterization of the human cysteinyl leukotriene 2 receptor. J Biol Chem. 2000;275:30531–30536. doi: 10.1074/jbc.M003490200. [DOI] [PubMed] [Google Scholar]
- 13.Landry Y, Niederhoffer N, Sick E, Gies JP. Heptahelical and other G-protein-coupled receptors (GPCRs) signaling. Curr Med Chem. 2006;13:51–63. [PubMed] [Google Scholar]
- 14.Mancuso P, Peters-Golden M. Modulation of alveolar macrophage phagocytosis by leukotrienes is Fc receptor-mediated and protein kinase C-dependent. Am J Respir Cell Mol Biol. 2000;23:727–733. doi: 10.1165/ajrcmb.23.6.4246. [DOI] [PubMed] [Google Scholar]
- 15.Serezani CH, Aronoff DM, Jancar S, Mancuso P, Peters-Golden M. Leukotrienes enhance the bactericidal activity of alveolar macrophages against Klebsiella pneumoniae through the activation of NADPH oxidase. Blood. 2005;106:1067–1075. doi: 10.1182/blood-2004-08-3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mancuso P, Standiford TJ, Marshall T, Peters-Golden M. 5-Lipoxygenase reaction products modulate alveolar macrophage phagocytosis of Klebsiella pneumoniae. Infect Immun. 1998;66:5140–5146. doi: 10.1128/iai.66.11.5140-5146.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peres CM, Aronoff DM, Serezani CH, Flamand N, Faccioli LH, Peters-Golden M. Specific leukotriene receptors couple to distinct G proteins to effect stimulation of alveolar macrophage host defense functions. J Immunol. 2007;179:5454–5461. doi: 10.4049/jimmunol.179.8.5454. [DOI] [PubMed] [Google Scholar]
- 18.Canetti C, Hu B, Curtis JL, Peters-Golden M. Syk activation is a leukotriene B4-regulated event involved in macrophage phagocytosis of IgG-coated targets but not apoptotic cells. Blood. 2003;102:1877–1883. doi: 10.1182/blood-2003-02-0534. [DOI] [PubMed] [Google Scholar]
- 19.Campos MR, Serezani CH, Peters-Golden M, Jancar S. Differential kinase requirement for enhancement of FcγR-mediated phagocytosis in alveolar macrophages by leukotriene B4 vs. D4. Mol Immunol. 2009;46:1204–1211. doi: 10.1016/j.molimm.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 20.Kwiatkowska K, Frey J, Sobota A. Phosphorylation of FcγRIIA is required for the receptor-induced actin rearrangement and capping: the role of membrane rafts. J Cell Sci. 2003;116:537–550. doi: 10.1242/jcs.00254. [DOI] [PubMed] [Google Scholar]
- 21.Aman MJ, Tosello-Trampont AC, Ravichandran K. FcγRIIB1/SHIP-mediated inhibitory signaling in B cells involves lipid rafts. J Biol Chem. 2001;276:46371–46378. doi: 10.1074/jbc.M104069200. [DOI] [PubMed] [Google Scholar]
- 22.Barnes NC, Powell MS, Trist HM, Gavin AL, Wines BD, Hogarth PM. Raft localization of FcγRIIa and efficient signaling are dependent on palmitoylation of cysteine 208. Immunol Lett. 2006;104:118–123. doi: 10.1016/j.imlet.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 23.Kono H, Suzuki T, Yamamoto K, Okada M, Yamamoto T, Honda Z. Spatial raft coalescence represents an initial step in FcγR signaling. J Immunol. 2002;169:193–203. doi: 10.4049/jimmunol.169.1.193. [DOI] [PubMed] [Google Scholar]
- 24.Beekman JM, van der Linden JA, van de Winkel JG, Leusen JH. FcγRI (CD64) resides constitutively in lipid rafts. Immunol Lett. 2008;116:149–155. doi: 10.1016/j.imlet.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Pike LJ. Lipid rafts: heterogeneity on the high seas. Biochem J. 2004;378:281–292. doi: 10.1042/BJ20031672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tager AM, Bromley SK, Medoff BD, et al. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol. 2003;4:982–990. doi: 10.1038/ni970. [DOI] [PubMed] [Google Scholar]
- 27.Peters-Golden M, McNish RW, Hyzy R, Shelly C, Toews GB. Alterations in the pattern of arachidonate metabolism accompany rat macrophage differentiation in the lung. J Immunol. 1990;144:263–270. [PubMed] [Google Scholar]
- 28.MacDonald JL, Pike LJ. A simplified method for the preparation of detergent-free lipid rafts. J Lipid Res. 2005;46:1061–1067. doi: 10.1194/jlr.D400041-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Weerth SH, Holtzclaw LA, Russell JT. Signaling proteins in raft-like microdomains are essential for Ca2+ wave propagation in glial cells. Cell Calcium. 2007;41:155–167. doi: 10.1016/j.ceca.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 30.Sitrin RG, Emery SL, Sassanella TM, Blackwood RA, Petty HR. Selective localization of recognition complexes for leukotriene B4 and formyl-Met-Leu-Phe within lipid raft microdomains of human polymorphonuclear neutrophils. J Immunol. 2006;177:8177–8184. doi: 10.4049/jimmunol.177.11.8177. [DOI] [PubMed] [Google Scholar]
- 31.Soehnlein O, Kai-Larsen Y, Frithiof R, et al. Neutrophil primary granule proteins HBP and HNP1-3 boost bacterial phagocytosis by human and murine macrophages. J Clin Invest. 2008;118:3491–3502. doi: 10.1172/JCI35740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaus K, Rodriguez M, Ruberu KR, et al. Domain-specific lipid distribution in macrophage plasma membranes. J Lipid Res. 2005;46:1526–1538. doi: 10.1194/jlr.M500103-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Chamberlain LH. Detergents as tools for the purification and classification of lipid rafts. FEBS Lett. 2004;559:1–5. doi: 10.1016/s0014-5793(04)00050-x. [DOI] [PubMed] [Google Scholar]
- 34.Harder T, Scheiffele P, Verkade P, Simons K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J Cell Biol. 1998;141:929–942. doi: 10.1083/jcb.141.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mollerup J, Eriksen HN, Albertsen J, Wulff T, Lambert IH, Hoffmann EK. Homologous desensitization of the mouse leukotriene B4 receptor involves protein kinase C-mediated phosphorylation of serine 127. Cell Physiol Biochem. 2007;20:143–156. doi: 10.1159/000104162. [DOI] [PubMed] [Google Scholar]
- 36.Gaudreau R, Le Gouill C, Venne MH, Stankova J, Rola-Pleszczynski M. Threonine 308 within a putative casein kinase 2 site of the cytoplasmic tail of leukotriene B4 receptor (BLT1) is crucial for ligand-induced, G-protein-coupled receptor-specific kinase 6-mediated desensitization. J Biol Chem. 2002;277:31567–31576. doi: 10.1074/jbc.M202723200. [DOI] [PubMed] [Google Scholar]
- 37.Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2. 0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol Cell Proteomics. 2008;7:1598–1608. doi: 10.1074/mcp.M700574-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peters-Golden M, Canetti C, Mancuso P, Coffey MJ. Leukotrienes: underappreciated mediators of innate immune responses. J Immunol. 2005;174:589–594. doi: 10.4049/jimmunol.174.2.589. [DOI] [PubMed] [Google Scholar]
- 39.Rollet-Labelle E, Marois S, Barbeau K, Malawista SE, Naccache PH. Recruitment of the cross-linked opsonic receptor CD32A (FcγRIIA) to high-density detergent-resistant membrane domains in human neutrophils. Biochem J. 2004;381:919–928. doi: 10.1042/BJ20031808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kondadasula SV, Roda JM, Parihar R, et al. Colocalization of the IL-12 receptor and FcγRIIIa to natural killer cell lipid rafts leads to activation of ERK and enhanced production of interferon-γ. Blood. 2008;111:4173–4183. doi: 10.1182/blood-2007-01-068908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lamberti Y, Perez Vidakovics ML, van der Pol LW, Rodriguez ME. Cholesterol-rich domains are involved in Bordetella pertussis phagocytosis and intracellular survival in neutrophils. Microb Pathog. 2008;44:501–511. doi: 10.1016/j.micpath.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 42.Naroeni A, Porte F. Role of cholesterol and the ganglioside GM(1) in entry and short-term survival of Brucella suis in murine macrophages. Infect Immun. 2002;70:1640–1644. doi: 10.1128/IAI.70.3.1640-1644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Filler SG. Candida-host cell receptor-ligand interactions. Curr Opin Microbiol. 2006;9:333–339. doi: 10.1016/j.mib.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 44.Chini B, Parenti M. G-protein coupled receptors in lipid rafts and caveolae: how, when and why do they go there? J Mol Endocrinol. 2004;32:325–338. doi: 10.1677/jme.0.0320325. [DOI] [PubMed] [Google Scholar]
- 45.Sullam PM, Hyun WC, Szollosi J, Dong J, Foss WM, Lopez JA. Physical proximity and functional interplay of the glycoprotein Ib-IX-V complex and the Fc receptor FcγRIIA on the platelet plasma membrane. J Biol Chem. 1998;273:5331–5336. doi: 10.1074/jbc.273.9.5331. [DOI] [PubMed] [Google Scholar]
- 46.Thai le M, Ashman LK, Harbour SN, Hogarth PM, Jackson DE. Physical proximity and functional interplay of PECAM-1 with the Fc receptor FcγRIIa on the platelet plasma membrane. Blood. 2003;102:3637–3645. doi: 10.1182/blood-2003-02-0496. [DOI] [PubMed] [Google Scholar]
- 47.Akekawatchai C, Holland JD, Kochetkova M, Wallace JC, McColl SR. Transactivation of CXCR4 by the insulin-like growth factor-1 receptor (IGF-1R) in human MDA-MB-231 breast cancer epithelial cells. J Biol Chem. 2005;280:39701–39708. doi: 10.1074/jbc.M509829200. [DOI] [PubMed] [Google Scholar]
- 48.Mira E, Lacalle RA, Gonzalez MA, et al. A role for chemokine receptor transactivation in growth factor signaling. EMBO Rep. 2001;2:151–156. doi: 10.1093/embo-reports/kve027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Helms JB, Zurzolo C. Lipids as targeting signals: lipid rafts and intracellular trafficking. Traffic. 2004;5:247–254. doi: 10.1111/j.1600-0854.2004.0181.x. [DOI] [PubMed] [Google Scholar]
- 50.Hadcock JR, Port JD, Gelman MS, Malbon CC. Cross-talk between tyrosine kinase and G-protein-linked receptors: phosphorylation of β2-adrenergic receptors in response to insulin. J Biol Chem. 1992;267:26017–26022. [PubMed] [Google Scholar]
- 51.Delcourt N, Thouvenot E, Chanrion B, et al. PACAP type I receptor transactivation is essential for insulin-like growth factor-1 receptor signalling and antiapoptotic activity in neurons. EMBO J. 2007;26:1542–1551. doi: 10.1038/sj.emboj.7601608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}