

Abstract
The exact role of a disintegrin and metalloproteinase with thrombospondin motifs-1 (ADAMTS-1) and the underlying mechanism of its involvement in tumor metastasis have not been established. We have now demonstrated that overexpression of ADAMTS-1 promotes pulmonary metastasis of TA3 mammary carcinoma and Lewis lung carcinoma cells and that a proteinase-dead mutant of ADAMTS-1 (ADAMTS-1E/Q) inhibits their metastasis, indicating that the prometastatic activity of ADAMTS-1 requires its metalloproteinase activity. Overexpression of ADAMTS-1 in these cells promoted tumor angiogenesis and invasion, shedding of the transmembrane precursors of heparin-binding epidermal growth factor (EGF) and amphiregulin (AR), and activation of the EGF receptor and ErbB-2, while overexpression of ADAMTS-1E/Q inhibited these events. Furthermore, we found that ADAMTS-1 undergoes auto-proteolytic cleavage to generate the NH2- and COOH-terminal cleavage fragments containing at least one thrombospondin-type-I-like motif and that overexpression of the NH2-terminal ADAMTS-1 fragment and the COOH-terminal ADAMTS-1 fragment can inhibit pulmonary tumor metastasis. These fragments also inhibited Erk1/2 kinase activation induced by soluble heparin-binding EGF and AR. Taken together, our results suggest that the proteolytic status of ADAMTS-1 determines its effect on tumor metastasis, and that the ADAMTS-1E/Q and the ADAMTS-1 fragments likely inhibit tumor metastasis by negatively regulating the availability and activity of soluble heparin-binding EGF and AR.
Keywords: ADAMTS-1, tumor metastasis, invasion, proteolytic cleavage, heparin-binding EGF, amphiregulin
Introduction
A disintegrin and metalloproteinase with thrombospondin motifs-1 (ADAMTS-1) is a member of the family of multifunctional proteins known as ADAMTS (Porter et al., 2005). ADAMTS-1 has a unique domain organization, being composed of the propeptide, metalloproteinase, disintegrin, and cysteine-rich (Cys-rich) domains, domains containing thrombospondin type I-like (TSP-1) motifs, and a spacer region (Kuno et al., 1997; Vazquez et al., 1999; Cal et al., 2002; Figure 1A). There are at least 19 members of the ADAMTS family, of which ADAMTS-1 was the first to be identified. Disruption of the Adamts-1 gene in mice results in reduced growth, abnormalities in ureteral, adrenal and adipose tissues, and infertility in female mice (Shindo et al., 2000).
Figure 1.
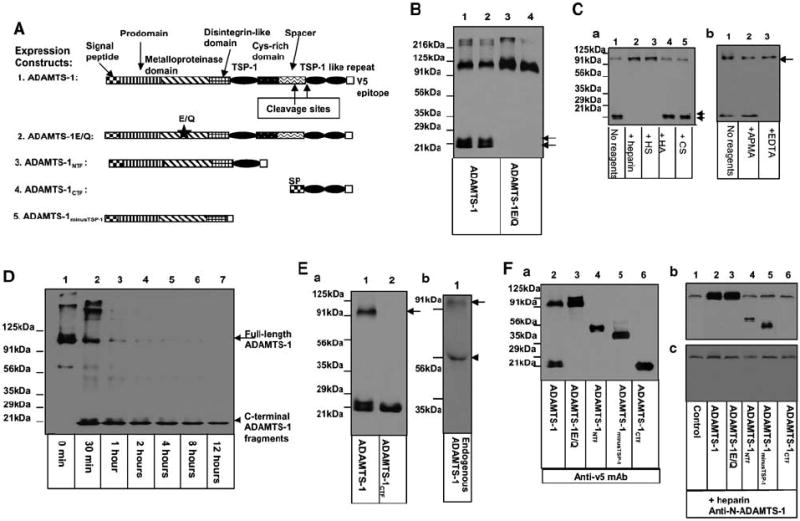
ADAMTS-1 undergoes auto-proteolytic cleavage, and the self-cleavage of ADAMTS-1 is regulated. (A) Diagram of the expression constructs used in the experiments. (B) The proteolytic cleavage of ADAMTS-1 requires its own metalloproteinase activity. The cell culture supernatants derived from Cos-7 cells transfected with the COOH-terminal v5-epitope-tagged ADAMTS-1 (lanes 1 and 2) or ADAMTS-1E/Q (lanes 3 and 4) were analysed by Western blotting with anti-v5 antibody. Arrows indicate the COOH-terminal cleavage fragments of ADAMTS-1. (C) The cleavage of ADAMTS-1 is inhibited by heparin and HS. In (Ca), TA3ADAMTS-1 cells were cultured in the absence (lane 1) or presence of 100 μg/ml of heparin (lane 2), HS (lane 3), hyaluronan (lane 4), or CS (lane 5) for 48 h. The COOH-terminal cleavage fragments are indicated by arrows. In (Cb), TA3ADAMTS-1 cells were cultured in the absence (lane 1) or presence of 200 μm APMA (lane 2) or 500 μm EDTA (lane 3) for 48 h. Cell supernatants were analysed by Western blotting with anti-v5 antibody. (D) Auto-proteolytic cleavage capacity of ADAMTS-1 was assessed by incubating 100 ng of purified ADAMTS-1 in Tris buffer at 37°C for 0 min (lane 1), 30 min (lane 2), or 1 (lane 3), 2 (lane 4), 4 (lane 5), 8 (lane 6), or 12 (lane 7) h. Reaction products were assessed by Western blotting with anti-v5 antibody. (Ea) Western blotting with anti-v5 mAb shows that v5-tagged ADAMTS-1CTF (lane 2) displays a molecular size similar to that of the smaller COOH-terminal cleavage fragment of ADAMTS-1 (lane 1). (Eb) Western blotting of supernatants derived from TA3wt cells using a polyclonal rabbit antibody against an NH2-terminal peptide of mouse ADAMTS-1 (anti-N-ADAMTS-1). (F) Supernatants were derived from TA3wtb (lane 1), TA3ADAMTS-1(lane 2), TA3ADAMTS-1E/Q (lane 3), TA3ADAMTS-1NTF (lane 4), TA3ADAMTS-1minusTSP-1 (lane 5), or TA3ADAMTS-1CTF (lane 6) cells that were cultured in the absence (Fa) or presence (Fb and c) of 100 μg/ml of heparin. Supernatants were analysed directly by Western blotting with anti-v5 antibody (Fa) to assess the expression levels of the v5-tagged exogenous ADAMTS-1 variants or with anti-N-ADAMTS-1 polyclonal antibody (Fb) to detect both endogenous and exogenous ADAMTS-1 and its NH2-terminal variants. One set of supernatants were passed through anti-v5 antibody affinity columns to absorb the v5-tagged exogenous ADAMTS-1 variants. Equal amounts of the flow-through proteins were analysed by Western blotting with anti-N-ADAMTS-1 antibody to detect endogenous ADAMTS-1 (Fc).
ADAMTS-1 is cleaved within its spacer region by matrix metalloproteinase-2, -8, and -15 (MMP-2, -8, and −15; Rodriguez-Manzaneque et al., 2000). There is evidence that other members of the ADAMTS family are proteolytically cleaved as well. ADAMTS-4 is auto-catalytically cleaved at two sites within the spacer/Cys-rich domain (Flannery et al., 2002), and that ADAMTS-2 undergoes autocatalytic processing, with several fragments being generated by cleavage at the end of the spacer domain (Colige et al., 2005).
Several studies have shown that ADAMTS-1 is capable of degrading aggrecan and versican, which are components of extracellular matrix (ECM) barriers (Kuno et al., 2000; Sandy et al., 2001; Russell et al., 2003). However, despite this suggestive evidence of a role for ADAMTS-1 in tumor invasion and metastasis, recent studies, characterizing the expression level of ADAMTS-1 in various cancers and its role in tumor growth and metastasis, have yielded apparently contradictory results.
ADAMTS-1 has been shown to be highly upregulated in breast cancer with elevated metastatic activity (Kang et al., 2003; Minn et al., 2005) and, in pancreatic cancers, a higher ADAMTS-1 mRNA level has been found to correlated with severe lymph node metastasis or retroperitoneal invasion and a worse prognosis, even though the median expression level of ADAMTS-1 in these tumors was lower than that in the noncancerous pancreas (Masui et al., 2001). These data suggest that ADAMTS-1 may promote tumor invasion and metastasis. In contrast, in another study (Porter et al., 2004), ADAMTS-1 mRNA levels were found to be down-regulated in breast carcinoma samples when compared to the non-neoplastic mammary tissues. However, this study did not find a strong link between ADAMTS-1 mRNA levels and the clinicopathological features of these breast cancers. ADAMTS-1 has also been shown to inhibit basic fibroblast growth factor (bFGF)-induced vascularization in the cornea pocket assay, vascular endothelial growth factor (VEGF)-induced angiogenesis in the chorioallantoic membrane assay, and tumor growth in vivo (Vazquez et al., 1999), as likely achieved by sequestering VEGF165 from its receptor through the carboxyl-terminal region containing the two last TSP repeats and at least part of the spacer region (Luque et al., 2003). The metalloproteinase activity of ADAMTS-1 is apparently required for its antiangiogenesis and antitumor growth activity (Iruela-Arispe et al., 2003). In contrast, overexpression of ADAMTS-1 has been found to promote subcutaneous growth of transfected CHO cells, but inhibit the experimental metastasis of the same transfectants (Kuno et al., 2004). However, these studies did not investigate the cleavage status of ADAMTS-1 in their experimental models. One possibility to explain the apparently contradictory results that have been reported thus far is that the requirement of metalloproteinase activity for the antitumor effect of ADAMTS-1 may reflect the fact that the antitumor effect of ADAMTS-1 is actually produced by the cleavage fragments rather than the full-length molecule, and that the metalloproteinase is responsible for generating these fragments.
In the current study, we have investigated the effects of full-length ADAMTS-1 and its fragments on tumor growth and metastasis, and have sought to elucidate the potential mechanism(s) underlying their effects. Our results have shown that overexpression of full-length ADAMTS-1 promotes pulmonary metastasis of TA3 murine mammary carcinoma (TA3) and Lewis lung carcinoma (LLC) cells by enhancing tumor cell invasion and tumor angiogenesis. ADAMTS-1 was found to promote the shedding of two transmembrane precursors of the epidermal growth factor family that bind to heparin, amphiregulin (AR), and heparin-binding epidermal growth factor (HB-EGF), and activation of epidermal growth factor receptor (EGFR). On the contrary, overexpression of a protease-dead ADAMTS-1 mutant (ADAMTS-1E/Q) inhibited these processes, suggesting that the metalloproteinase activity is required for the prometastatic activity of ADAMTS-1. In addition, we have demonstrated that ADAMTS-1 undergoes auto-proteolytic cleavage, generating NH2- and COOH-terminal cleavage fragments (ADAMTS-1NTCF and ADAMTS-1CTCF). In contrast to the results obtained for full-length ADAMTS-1, overexpression of two fragments (the NH2-terminal ADAMTS-1 fragment (ADAMTS-1NTF) and the COOH-terminal ADAMTS-1 fragment (ADAMTS-1CTF)) that mimic the proteolytic cleavage fragments of ADAMTS-1 inhibited pulmonary metastasis of TA3 and LLC cells by inhibiting tumor cell extravasation, proliferation, and survival and by repressing tumor angiogenesis. ADAMTS-1NTF and ADAMTS-1CTF were found to inhibit the activation of Erk1/2 kinase induced by soluble AR and HB-EGF. In addition, we have demonstrated that the auto-proteolytic cleavage of ADAMTS-1 is inhibited by heparin/heparan sulfate (HS), suggesting that the level of HS/heparan sulfate proteoglycans (HSPGs) in the microenvironment likely determines which form of ADAMTS-1 (full-length or cleaved) is predominantly present in the microenvironment and exerting pro- or antitumor activity. The ADAMTS-1 expressed by TA3 and LLC cells is maintained predominantly in the full-length form in vivo and therefore is able to exert its prometastatic activity. Taken together, our results suggest that the proteolytic status of ADAMTS-1 determines its effect on tumor metastasis, and that ADAMTS-1E/Q and the ADAMTS-1 fragments inhibit tumor metastasis, most likely by negatively regulating the availability and activity of soluble HB-EGF and AR.
Results
ADAMTS-1 undergoes auto-proteolytic cleavage, and the self-cleavage of ADAMTS-1 is regulated
As ADAMTS-1 is an active metalloproteinase (Kuno et al., 1999), we investigated the possibility that this molecule can be cleaved through its own metalloproteinase activity as well as by other MMPs. To this end, we generated a protease-dead mutant of ADAMTS-1 (ADAMTS-1E/Q) by substituting Q for E386 in the zinc-binding pocket of the metalloproteinase domain, then transiently transfecting Cos-7 cells with expression constructs containing the COOH-terminal v5-tagged wild-type ADAMTS-1 or ADAMTS-1E/Q. Analysis of cell culture supernatants from the transfected cells indicated that only wild-type ADAMTS-1, and not ADAMTS-1E/Q, was cleaved to generate COOH-terminal cleavage fragments that can be detected with anti-v5 antibody (Figure 1B, arrows), suggesting that the metalloproteinase activity of ADAMTS-1 is required for the cleavage.
We investigated how ADAMTS-1 cleavage is regulated. For this purpose, we added a variety of reagents to a stable TA3 transfectant expressing v5-epitope-tagged ADAMTS-1, and then analysed the cell culture supernatants 48 h later. Our results showed that heparin and HS (100 μg/ml) blocked the proteolytic cleavage of ADAMTS-1, whereas the other glycosaminoglycans (GAGs), chondroitin sulfate and hyaluronan, had no effect (Figure 1Ca). In addition, treatment of the cells with EDTA (0.5 mm) significantly inhibited the cleavage, whereas addition of p-aminophenylmercuric acetate (APMA, 0.2 mm), as expected, had no significant effect, since the majority of the full-length ADAMTS-1 was already in the active form, judging by its molecular size (Figure 1Cb, arrow). This result suggests that ADAMTS-1 cleavage is achieved through metalloproteinase activity and regulated by the rates of synthesis and degradation of HS/HSPGs in the microenvironment, in which ADAMTS-1 is produced.
We were therefore able to obtain full-length ADAMTS-1 protein by culturing transfected Cos-7 cells in the presence of heparin. The culture supernatants of the transfected cells were collected and purified through two affinity columns, as described in Materials and methods. When we tested the purified ADAMTS-1 protein in an in vitro proteolytic cleavage assay, we found that it was auto-proteolytically cleaved to release the v5-epitope-tagged COOH-terminal fragments with the molecular sizes that were similar to those of the fragments generated in cell culture (Figure 1D, arrowhead). A lack of accumulation of the COOH-terminal cleavage fragments suggests that further proteolytic cleavage of ADAMTS-1 occurs under this assay condition. A similar auto-proteolytic cleavage of ADAMTS-4 has already been reported in vitro (Flannery et al., 2002). It is important to consider the possibilities that the loss of the proteolytic cleavage capacity of ADAMTS-1E/Q may have resulted from a conformational change that renders the cleavage site inaccessible, and that the presence of other proteases as the minor contaminants of the purified ADAMTS-1 preparation may explain the additional proteolytic cleavage of ADAMTS-1. Further studies are required to exclude these possibilities.
Full-length ADAMTS-1 promotes pulmonary metastasis of TA3 and Lewis lung carcinoma (LLC) cells, whereas ADAMTS-1E/Q and ADAMTS-1NTF/ADAMTS-1CTF inhibit the process
Although ADAMTS-1 has been found to inhibit angiogenesis in the cornea pocket assay and in the chorioallantoic membrane assay, and tumor growth in vivo (Vazquez et al., 1999), it is unknown whether the molecule was present in full-length form or as the cleavage fragments in these experiments. In addition, several studies have indicated that ADAMTS-1 is highly upregulated in breast cancer with elevated metastatic capacity (Kang et al., 2003; Minn et al., 2005), and increased expression of ADAMTS-1 is correlated to the enhanced metastatic potential and a worse prognosis in pancreatic cancers (Masui et al., 2001), implying that ADAMTS-1 facilitates tumor metastasis. Therefore, to determine the precise roles of ADAMTS-1 and the ADAMTS-1 fragments in tumor metastasis, we investigated how overexpression of full-length ADAMTS-1, ADAMTS-1E/Q, and the fragments of ADAMTS-1 affects the metastasis of TA3 and LLC cells, both of which express endogenous ADAMTS-1 (Figures 1Eb, Fb, and 3Ab).
Figure 3.
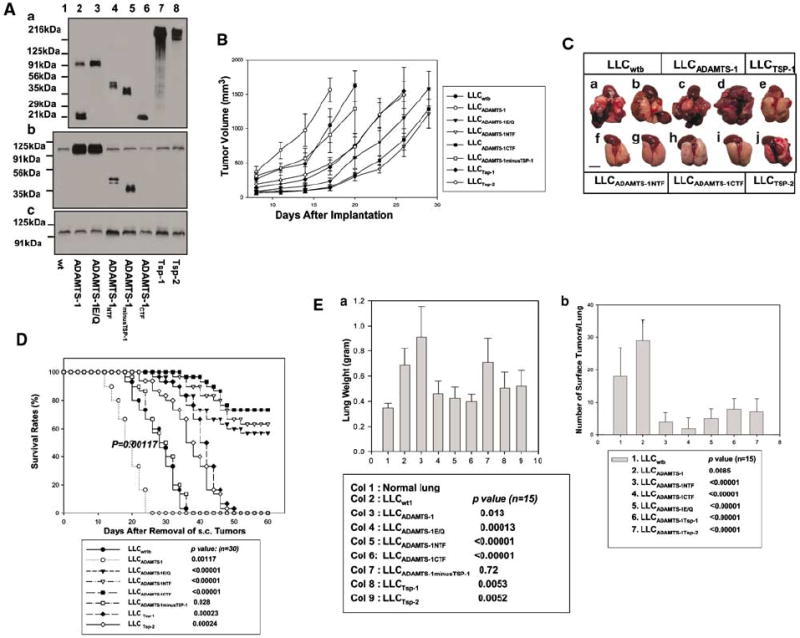
Full-length ADAMTS-1 and ADAMTS-1E/Q/ADAMTS-1NTF/ADAMTS-1CTF have opposite effects on the subcutaneous growth and the spontaneous metastasis of LLC cells. (A) Cell culture supernatants were derived from LLC transfectants transfected with the expression vectors (lane 1), or expressing ADAMTS-1 (lane 2), ADAMTS-1E/Q (lane 3), ADAMTS-1NTF (lane 4), ADAMTS-1minusTSP-1 (lane 5), ADAMTS-1CTF (lane 6), Tsp-1 (lane 7), and Tsp-2 (lane 8). These cells were cultured in the absence (Aa) or presence (Ab and c) of 100 μg/ml of heparin. Supernatants (Aa) were analysed directly by Western blotting with anti-v5 antibody (Aa) to assess the expression levels of the v5-tagged exogenous ADAMTS-1 variants and Tsp-1/Tsp-2 or with anti-N-ADAMTS-1 polyclonal antibody (Ab) to detect both endogenous and exogenous ADAMTS-1 and its NH2-terminal variants. One set of supernatants were passed through the anti-v5 antibody affinity columns to absorb the v5-tagged exogenous ADAMTS-1 variants. Equal amounts of the flow-through proteins were analysed by Western blotting with anti-N-ADAMTS-1 antibody to detect endogenous ADAMTS-1 exclusively (Ac). (B) The growth rates of the subcutaneous tumors derived from the different LLC transfectants are expressed as the mean of tumor volume ±s.d. A total of 12 mice were used for each type of transfectants. (C) Representative mouse lungs 3 weeks after removal of the subcutaneous (s.c.) tumors were derived from the following cells: LLCwt1b (a, b), LLCADAMTS-1(c, d), LLCADAMTS-1NTF (f, g), LLCADAMTS-1CTF (h, i), LLCTsp-1 (e), or LLCTsp-2 (j) cells. (D) Survival rates after removal of the subcutaneous tumors derived from various LLC transfectants (30 mice per type of transfectant). (E) Pulmonary metastatic burden as indicated by the mean weights of mouse lungs 3 weeks after removal of the subcutaneous tumors, and by counting the number of surface pulmonary tumors under a dissection microscope (15 mice per group).
As shown in Figure 1B and C, the COOH-terminal cleavage fragments of ADAMTS-1 are heterogeneous in molecular size, suggesting that, like ADAMTS-4 (Flannery et al., 2002), ADAMTS-1 is likely cleaved at more than one site within the spacer/Cys-rich region (Figure 1A, arrows). This hypothesis was supported by Western blot analysis of the expressed v5-tagged COOH-terminal fragment of ADAMTS-1 that contains the last two TSP-1 type I-like motifs (ADAMTS-1CTF, amino acids 842–951; Figure 1A #4). The molecular size of ADAMTS-1CTF was similar to that of the shortest COOH-terminal cleavage fragment of v5-tagged ADAMTS-1 (Figure 1Ea). This result suggests that, in addition to the previous identified cleavage site within the spacer region (Rodriguez-Manzaneque et al., 2000; Figure 1A, larger arrow), there is at least one additional cleavage site at the junction between the spacer region and the second TSP-1 motif (Figure 1A, the smaller arrow).
In order to identify the NH2-terminal cleavage fragment of ADAMTS-1, we have generated a polyclonal rabbit antibody against an NH2-terminal peptide of mouse ADAMTS-1 according to Russell et al. (2003). Western blotting with this antibody showed that the endogenous ADAMTS-1 expressed by TA3wt cells is cleaved to release an NH2-terminal cleavage fragment (Figure 1Eb, arrowhead).
We found that it was difficult to express the NH2-terminal fragments of ADAMTS-1 containing various parts of the spacer and/or Cys-rich domains (data not shown). Therefore, we used two ADAMTS-1 fragments, ADAMTS-1NTF and ADAMTS-1CTF, to mimic the NH2- and COOH-terminal cleavage fragments of ADAMTS-1 (ADAMTS-1NTCF and ADAMTS-1CTCF), respectively. ADAMTS-1NTF contains the NH2-terminal domains of ADAMTS-1 through the end of the first TSP-1 motif (amino acids 1–596) and ADAMTS-1CTF contains the last two TSP-1 motifs (amino acids 842–951). Another ADAMTS-1 fragment (ADAMTS-1minusTSP-1), which contains the NH2-terminal ADAMTS-1 domains through the end of the disintegrin domain and has no TSP-1 motifs (amino acids 1–545; Figure 1A and Fa), was used to investigate the role of the TSP-1 motif in tumor metastasis.
In order to reliably assess the effects of full-length ADAMTS-1, ADAMTS-1E/Q, and the ADAMTS-1 fragments on tumor metastasis, we first eliminated heterogeneity in the TA3 cells by transfecting them with the empty expression vector containing a neomycin-resistance gene. A clonal TA3 cell (TA3wt1) that underwent pulmonary metastasis after intravenous (i.v.) injection was selected. Our reverse transcriptase–polymerase chain reaction (RT–PCR) and Western blot results showed, that like their parental cells, the selected cells expressed ADAMTS-1 endogenously (Figure 1Fc). We then transfected these TA3wt1 cells with the expression constructs containing the blasticidin-resistance gene and different ADAMTS-1 cDNA inserts (Figure 1A). Five independent clonal TA3 transfectants containing the empty expression vector alone (TA3wtb) or expressing an intermediate-to-high level of the transfected gene products were identified (data not shown) and used as the pooled populations (Figure 1Fa and b) in our pulmonary metastasis experiments. We found that after removal of the exogenous v5-tagged ADAMTS-1 variants with the anti-v5-antibody affinity columns, these pooled TA3 transfectants expressed a similar level of endogenous ADAMTS-1 (Figure 1Fc). These transfectants displayed similar morphology and growth rates under the cell culture conditions (data not shown).
Our results showed that overexpression of ADAMTS-1 but not ADAMTS-1minusTSP-1 significantly accelerated pulmonary metastasis of TA3 transfectants and shortened the survival time of the experimental mice (Figure 2A–C). In contrast, overexpression of ADAMTS-1NTF, ADAMTS-1CTF, and ADAMTS-1E/Q, but not ADAMTS-1minusTSP-1, inhibited the pulmonary metastasis of TA3 transfectants (Figure 2A–C), suggesting that the prometastatic activity of ADAMTS-1 is dependent on the metalloproteinase activity. Theses results also indicated that the inhibitory effect of ADAMTS-1NTF and ADAMTS-1CTF requires the TSP-1 motifs, which are present in ADAMTS-1NTF and ADAMTS-1CTF but not in ADAMTS-1minusTSP-1, and that the antitumor activity derived from the TSP-1 motifs is likely masked in full-length ADAMTS-1.
Figure 2.
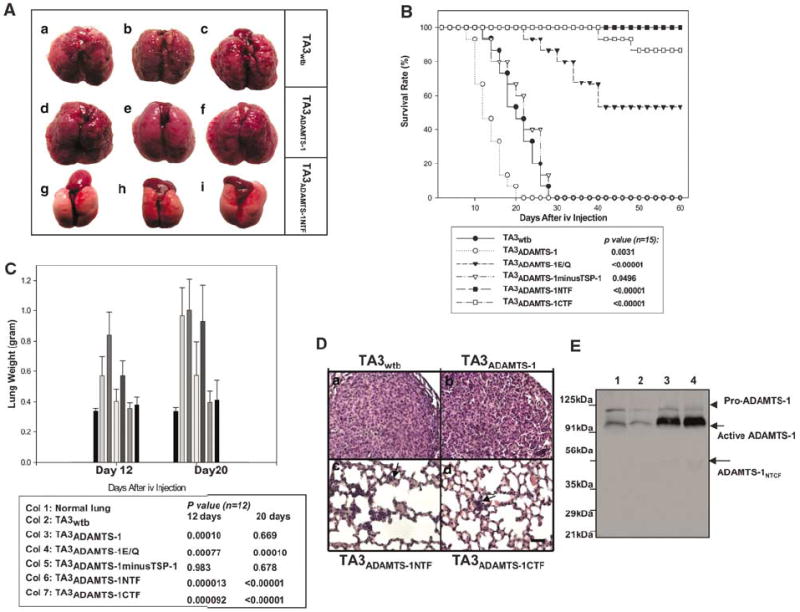
Full-length ADAMTS-1 promotes pulmonary metastasis, whereas ADAMTS-1E/Q and ADAMTS-1NTF/ADAMTS-1CTF inhibit metastasis. (A) Representative mouse lungs 2–3 weeks after i.v. injection of the following cells: TA3wtb (a–c) or TA3ADAMTS-1(d–f), or TA3ADAMTS-1NTF (g–i) cells. (B) Survival rates of mice injected i.v. with various TA3 transfectants (15 mice per group). (C) Pulmonary metastatic burden as indicated by mouse lung weights at 12 and 20 days after i.v. injection of TA3 transfectants (12 mice per group). (D) Representative H&E-stained lung sections from mice injected with the following cells: TA3wtb (a), TA3ADAMTS-1 (b), TA3ADAMTS-1NTF (c), and TA3ADAMTS-1CTF (d). Arrows indicate the micrometastases (Dc and d). Bar, 100 μm. (E) Anti-N-ADAMTS-1 polyclonal antibody was used to detect endogenous and exogenous ADAMTS-1 protein expressed by TA3wtb and TA3ADAMTS-1 cells in vivo. Protein lysates from pulmonary metastases derived from TA3wtb (lanes 1 and 2) and TA3ADAMTS-1(lanes 3 and 4) cells. The arrow indicates mature proteolytically active ADAMST-1, and the arrowhead indicates the pro-ADAMTS-1 protein.
The metastatic tumors derived from TA3wtb, TA3ADAMTS-1, and TA3ADAMTS-1minusTSP-1 cells were invasive and tended to fuse together (Figure 2A, Da, and b, and data not shown), making it impossible to counter the metastatic lesions with any degree of accuracy. Thus, the metastatic burden of the experimental mice was quantified by determining the average weight of the mouse lungs (Figure 2C). As there was a significant difference between the survival time of the experimental and control mice and the mice usually succumbed to pulmonary metastasis when the metastatic burden increased the lung weight to ~1 g, we measured the metastatic burden of the remaining surviving mice at days 12 and 20 after i.v. injection of TA3 transfectants. In all, 12 experimental mouse lungs were weighted for each type of the transfectants at each time point. Our results showed that overexpression of ADAMTS-1 shortened both the time that is required to reach the maximal metastatic burden and the survival time of the mice, whereas overexpression of ADAMTS-1NTF or ADAMTS-1CTF and to a lesser extent of ADAMTS-1E/Q inhibited pulmonary metastasis of TA3 cells and rendered many of the experimental mice free of metastatic disease (Figure 2A–C).
Histological analysis of the lung sections from the experimental mice showed that TA3wtb, TA3ADAMTS-1, and TA3ADAMTS-1minusTSP-1 cells were invasive, with the tumor cells occupying most of the pulmonary space (Figure 2Da and b, and data not shown). In contrast, only micrometastases were detected in the lungs receiving TA3ADAMTS-1NTF, TA3ADAMTS-1CTF, or TA3ADAMTS-1E/Q cells (Figure 2Dc and d, arrows; data not shown). To assess whether endogenous and exogenous ADAMTS-1 expressed by TA3 transfectants are cleaved in vivo, the pulmonary tumors derived from TA3wtb and TA3ADAMTS-1 cells were lysed, and the proteins were analysed by Western blotting with anti-N-ADAMTS-1 polyclonal antibody, which recognizes the NH2-terminus of mouse ADAMTS-1 (Figure 1Eb). Our result indicated that endogenous and exogenous ADAMTS-1 were predominantly maintained in full-length form in vivo and no significant NH2-terminal cleavage fragments were detected (Figure 2E). This result suggests that ADAMTS-1 is maintained in the full-length form in the metastases derived from TA3ADAMTS-1 cells to exert prometastatic activity and the proteolytic cleavage status of ADAMTS-1 determines its effect (stimulatory or inhibitory) on tumor metastasis.
We then used a subline of LLC that is capable of spontaneous pulmonary metastasis after removal of the primary subcutaneous tumors (Xu et al., 2004a) to confirm the effects of full-length ADAMTS-1, ADAMTS-1E/Q, and the ADAMTS-1 fragments on tumor metastasis and subcutaneous tumor growth, comparing these effects to those obtained with thrombospondin-1 (Tsp-1) and thrombospondin-2 (Tsp-2). For this purpose, we established LLC transfectants containing empty expression vectors (LLCwtb) or expressing v5-epitope-tagged full-length ADAMTS-1, ADAMTS-1E/Q, ADAMTS-1NTF, ADAMTS-1CTF, Tsp-1, or Tsp-2 (Figure 3Aa). ADAMTS-1, Tsp-1, and Tsp-2 are members of thrombospondin type I repeat superfamily (TRS; Tucker, 2004). Tsp-1 is a 450-kilodalton (kDa) homotrimeric ECM protein and is considered as a potent antitumor molecule. Systemic injection or overexpression of Tsp-1 inhibits the in vivo growth of several tumor cells, including LLC cells (Volpert et al., 1998; Streit et al., 2000; Miao et al., 2001). Five independent clonal LLC transfectants expressing an intermediate-to-high level of each transfected gene product were identified and used as pooled populations (Figure 3Aa) in subcutaneous tumor growth and spontaneous pulmonary metastasis experiments following established protocols (Xu et al., 2004a). After removal of the exogenous v5-tagged ADAMTS-1 variants with the anti-v5-antibody affinity columns, these pooled LLC transfectants were found to express similar levels of endogenous ADAMTS-1 (Figure 3Ab and c).
We found that expression of full-length ADAMTS-1 promoted subcutaneous tumor growth and spontaneous pulmonary metastasis of LLC transfectants; in contrast, ADAMTS-1NTF, ADAMTS-1CTF, or to a lesser extent ADAMTS-1E/Q had inhibitory effects (Figure 3B–E). Even though LLC transfectants expressed higher levels of Tsp-1 and Tsp-2 than the levels of ADAMTS-1NTF or ADAMTS-1CTF, the inhibitory effect of these ADAMTS-1 fragments was greater than that of Tsp-1 or Tsp-2 (Figure 3B–E).
ADAMTS-1NTF/ADAMTS-1CTF and ADAMTS-1E/Q inhibit pulmonary tumor metastasis by inhibiting proliferation and survival of the tumor cells and by repressing tumor angiogenesis
To determine the mechanism underlying the prometastatic effect of full-length ADAMTS-1 and the antimetastatic effect of ADAMTS-1NTF/ADAMTS-1CTF and ADAMTS-1E/Q, we analysed the rates of proliferation and apoptosis of TA3 transfectants and examined their effect on tumor angiogenesis in vivo. We observed that expression of ADAMTS-1NTF, ADAMTS-1CTF, or ADAMTS-1E/Q, but not of ADAMTS-1minusTSP-1, inhibited tumor cell proliferation, promoted apoptosis of the tumor cells, and inhibited tumor angiogenesis. However, increased expression of full-length ADAMTS-1, supplementing the effect of endogenous ADAMTS-1, had a weakly positive effect on tumor cell proliferation and an inhibitory effect on tumor cell apoptosis and also promoted tumor angiogenesis (Figure 4B).
Figure 4.
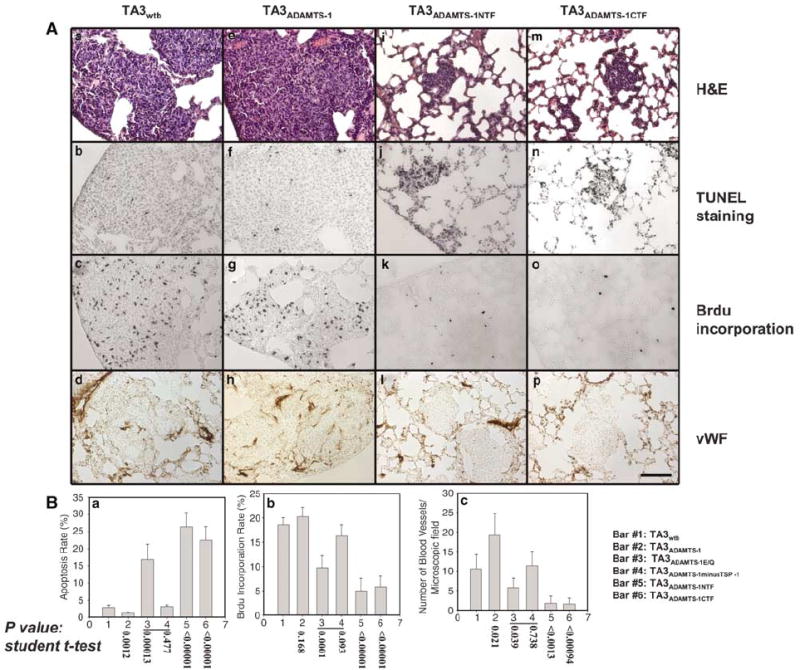
ADAMTS-1NTF/ADAMTS-1CTF and ADAMTS-1E/Q inhibit pulmonary metastasis of TA3 cells by inhibiting proliferation and survival of tumor cells and by repressing tumor angiogenesis. Mouse lungs were sectioned 6 days after i.v. injection of the following cells: TA3wtb (Aa–d), TA3ADAMTS-1 (Ae–h), TA3ADAMTS-1NTF (Ai–l), and TA3ADAMTS-1CTF (Am–p). Sections were stained with H&E (Aa, e, i, m) or reacted with Apoptag to detect apoptotic tumor cells in lung parenchyma (Ab, f, j, n), with anti-BrdU antibody to detect proliferating tumor cells (Ac, g, k, o), or with anti-vWF antibody to reveal blood vessels within macro- or micro-metastases (Ad, h, l, p). Bar: 120 μm. Quantitative data demonstrating the effects of ADAMTS-1, ADAMTS-1E/Q, and the fragments of ADAMTS-1 on tumor cell apoptosis and proliferation and on tumor angiogenesis are shown in (Ba–c).
ADAMTS-1 promotes activation of EGFR and ErbB-2 and shedding of AR and HB-EGF transmembrane precursors
Activation of EGFR and ErbB-2 is known to promote proliferation and survival of breast carcinoma cells and is essential for the progression of breast cancers (Stern, 2000; Yarden and Sliwkowski, 2001). ErbB-2 has no known direct ligands; however, it is activated by forming heterodimers with other ErbB receptors (Burgess et al., 2003). To determine whether ADAMTS-1 promotes the activation of EGFR and/or ErbB-2 in vivo, we assessed the activity of EGFR and ErbB-2 in the lungs of mice that had received i.v. 24 h earlier with the following cells: TA3wtb, TA3ADAMTS-1, TA3ADAMTS-1NTF, TA3ADAMTS-1CTF, or TA3ADAMTS-1E/Q. We have previously shown that extravasation of TA3 cells into the lung parenchyma occurs less than 24 h after i.v. injection (Yu et al., 1997). In order to normalize the number of TA3 transfectants to be included in the protein lysates used in the immunoprecipitation experiments, we first performed a tumor cells tracking assay to analyse extravasation of various TA3 transfectants into mouse lung parenchyma at 24 h after i.v. injection. Our results indicated that ADAMTS-1 promoted TA3 cell extravasation, whereas ADAMTS-1E/Q and to a lesser extent ADAMTS-1NTF/ADAMTS-1CTF inhibited the process (Figure 5A and B).
Figure 5.
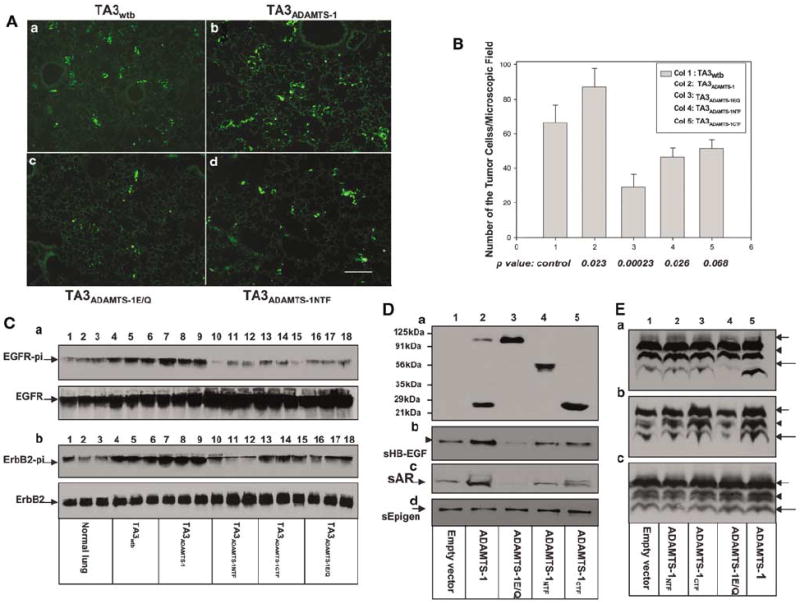
ADAMTS-1NTF/ADAMTS-1CTF and ADAMTS-1E/Q inhibit activation of EGFR and ErbB-2, whereas ADAMTS-1 promotes activation of these receptors and shedding of AR and HB-EGF precursors. (A) Tumor cell tracking assay. Mouse lungs were fixed and sectioned 24 h after i.v. injection of green fluorescein-labeled pooled populations of the TA3 transfectants. TA3wtb (Aa), TA3ADAMTS-1 (Ab), ADAMTS-1E/Q, (Ac), and TA3ADAMTS-1NTF (Ad) residing in the lung parenchyma are shown. (B) Pulmonary extravasation rates of TA3 transfectants are expressed as average number of the tumor cells residing in the lung parenchyma per microscopic field. (C) Activities of EGFR and ErbB-2 in vivo were assessed by immunoprecipitation with anti-EGFR (Ca) or anti-ErbB-2 (Cb) antibody and of lysates derived normal mouse lungs (lanes 1–3) or the lungs implanted, 24 h earlier, with pooled populations of the following cells: TA3wtb (lanes 4–6), TA3ADAMTS-1 (lanes 7–9), TA3ADAMTS-1NTF (lanes 10–12), TA3ADAMTS-1CTF (lanes 13–15), or TA3ADAMTS-1E/Q (lanes 16–18). Each lane represents the result from one mouse lung. To normalize the number of tumor cells in the lysates, based on tumor cell extravasation rates (B), we used 100 μg of protein lysate derived from normal lungs or the lungs implanted with TA3wtb cells, 71, 143, 130, or 230 μg of protein lysates from the lungs implanted with the following cells, respectively: TA3ADAMTS-1, TA3ADAMTS-1NTF, TA3ADAMTS-1CTF, or TA3ADAMTS-1E/Q. Precipitated proteins were analysed by Western blotting with antiphosphotyrosine antibody to detect phosphor-EGFR (Ca, upper panel) or phosphor-ErbB-2 (Cb, upper panel), or with anti-EGFR (Ca, bottom panel) and anti-ErbB-2 (Cb, bottom panel) antibody to detect total EGFR and ErbB-2, respectively. (D, E) ADAMTS-1 promotes shedding of the transmembrane precursors of HB-EGF (lane 2, Db, and lane 5, Ea) and AR (lane 2, Dc, and lane 5, Eb) but not epigen (lane 2, Dd) or TGF-α (lane 5, Ec) precursors, and the shedding is inhibited by ADAMTS-1E/Q (lane 3, Db and c; and lane 4, Ea and b). 293 cells were cotransfected with expression constructs containing cDNA inserts encoding the v-5 epitope tagged HB-EGF (Db and Ea), AR (Dc and Eb), or epigen (Dd)/TGF-fN (Ec) precursors and the empty expression vector (lane 1), ADAMTS-1 (lane 2, D, and lane 5, E), ADAMTS-1E/Q (lane 3, D, and lane 4, E), ADAMTS-1NTF (lane 4, D, and lane 2, E), or ADAMTS-1CTF (lane 5, D, and lane 3, E). Concentrated serum-free cell culture supernatants derived from these cells were analysed by Western blotting using anti-v5 antibody to detect the expression level of the transfected gene products (Da), and anti-HB-EGF (Db), -AR (Dc), or -epigen (Dd) antibody to detect soluble AR, HB-EGF, or epigen. These cotransfected 293 cells were then lysed and equal amounts of proteins were analysed by Western blotting with anti-v5 antibody to detect the fully glycosylated HB-EFG (Ea, arrow), AR (Eb, arrow), or TGFα (Ec, arrow) precursors, the smaller immature glycoprotein of these precursors (E, arrowheads), or the COOH-terminal cleavage fragments (E, longer arrows).
Normal mouse lungs and the lungs from the experimental mice that had been injected with TA3 transfectants i.v. 24 h earlier were lysed and used in immunoprecipitation experiments to pull down EGFR or ErbB-2. Antiphosphotyrosine antibody was used to detect phosphor-EGFR or phosphor-ErbB-2 in the precipitates. These experiments demonstrated that overexpression of ADAMTS-1 promotes activation of EGFR and ErbB-2 (Figure 5C), whereas expression of ADAMTS-1NTF, ADAMTS-1CTF, or ADAMTS-1E/Q inhibited their activation (Figure 5C).
TA3 cells express several GFs belonging to the EGF family, including HB-EGF, AR, and epigen (data not shown). We next investigated the possibility that the increased activation of EGFR and ErbB-2 induced by ADAMTS-1 is achieved through shedding/activating the transmembrane precursors of these GFs, the ligands of ErbB receptor tyrosine kinases. The EGF family GFs include EGF, transforming growth factor-α (TGF-α), HB-EGF, AR, betacellulin, epiregulin, neuregulin, and epigen (Massagué and Pandiella, 1993; Harris et al., 2003); all of these are shed from the cell surface (Peschon et al., 1998; Lee et al., 2003; Sahin et al., 2004). Increasing evidence suggests that shedding of EGF family GF precursors regulates the availability and bioactivity of these factors and is essential for the activation of the ErbB-signaling pathways (Iwamoto et al., 2003; Jackson et al., 2003; Lee et al., 2003; Yamazaki et al., 2003). Members of the ADAM family, particularly ADAM17, have been shown to play an essential but not sole role in the shedding of these factors (Merlos-Suarez et al., 2001; Sahin et al., 2004).
Like HB-EGF and AR, ADAMTS-1 is known to bind to HSPGs. Thus, we focused our attention on determining whether ADAMTS-1 plays a role in the constitutive shedding of these two precursors. To achieve this, we cotransfected 293 cells with AR/HB-EGF/epigen or TGF-α precursors that had been tagged with a v5 epitope at the COOH-terminal end, with empty expression vector, or with the expression constructs containing full-length ADAMTS-1, ADAMTS-1E/Q, or the ADAMTS-1 fragments. Serum-free cell culture supernatants derived from the cotransfected cells were collected, concentrated, and analysed (Figure 5D). The transfected 293 cells were lysed, and equal amounts of the proteins were analysed by Western blotting with anti-v5 antibody, which detects the fully glycosylated full-length EGF family precursors (Figure 5E, arrow), the smaller immature precursors (Figure 5E, arrowheads), and the COOH-terminal cleavage fragments (Figure 5E, longer arrows). 293 cells express ADAMTS-1 endogenously (data not shown). We found that overexpression of ADAMTS-1 promoted shedding of the precursors of AR and HB-EGF but not of epigen and TGF-α (Figure 5D and E), whereas overexpression of ADAMTS-1E/Q inhibited the shedding, most likely in a dominant-negative fashion, by inhibiting the activity of endogenous ADAMTS-1. The ADAMTS-1 fragments had no significant effect on the shedding (Figure 5D and E).
It is generally accepted that endogenous soluble EGF family ligands are not reliably detectable (Lee et al., 2003), perhaps because these soluble/active ligands are immediately used by adjacent cells in vivo. Thus, most of the shedding experiments have been carried out in cell cultures using embryonic fibroblasts from ADAM-knockout mice or 293/Cos-7/CHO cells overexpressing the tagged precursors (Merlos-Suarez et al., 2001; Gschwind et al., 2003; Lee et al., 2003; Sahin et al., 2004). Our results obtained using 293 cells overexpressing tagged HB-EGF/AR and different variants of ADAMTS-1 suggest that ADAMTS-1 promotes shedding/activation of HB-EGF and AR precursors in vitro, and the results demonstrating the enhanced EGFR/ErbB-2 activation by ADAMTS-1 in vivo (Figure 5C) provide indirect support evidence for this possibility.
ADAMTS-1 promotes invasion of TA3 cells through Matrigel
Pericellular proteolysis by MMPs is essential for tumor cell invasion (Yu and Stamenkovic, 1999, 2000; Sternlicht and Werb, 2001), and ADAMTS-1 has been shown to play an important role in degrading versican (Sandy et al., 2001), an important component of the ECM and blood vessel wall. We have now shown that ADAMTS-1 promotes extravasation of TA3 cells into the lung parenchyma (Figure 5A and B). To determine whether this effect is achieved through the promotion of tumor cell invasion through the ECM, we investigated how full-length ADAMTS-1, ADAMTS-1E/Q, and the fragments of ADAMTS-1 affect the invasion of TA3 cells through Matrigel, which mimics the basement membrane as a barrier to tumor cell invasion. TA3ADAMTS-1 displayed two- and four-fold higher invasion indices than did TA3wt1/TA3ADAMTS-1minusTSP-1 cells and TA3ADAMTS-1NTF/TA3ADAMTS-1CTF/TA3ADAMTS-1E/Q cells, respectively (Figure 6). This result confirms that ADAMTS-1 promotes tumor cell invasion, while ADAMTS-1E/Q and, to a lesser extent, ADAMTS-1NTF, or ADAMTS-1CTF inhibits the process.
Figure 6.
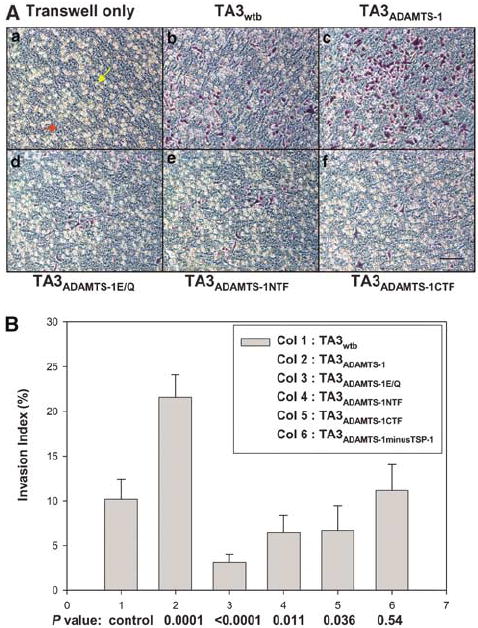
ADAMTS-1 promotes invasion of TA3 transfectants through Matrigel. (A) Representative pictures of TA3 transfectants that have invaded through the coated Matrigel. (Aa) Transwell coated with Matrigel only. The yellow and red arrows point to the 8-μm pore and coated Matrigel, respectively; (Ab) TA3wtb; (Ac) TA3ADAMTS-1; (Ad) TA3ADAMTS-1E/Q; (Ae) TA3ADAMTS-1NTF; or (Af) TA3ADAMTS-1CTF cells. (B) The invasion indexes of the various TA3 transfectants.
ADAMTS-1NTF and ADAMTS-1CTF inhibit activation of Erk1/2 kinases induced by soluble HB-EGF and AR
Since ADAMTS-1NTF and ADAMTS-1CTF had no significant inhibitory effect on the shedding of AR and HB-EGF precursors (Figure 5D and E), we investigated the possibility that instead of inhibiting the availability of soluble HB-EGF and AR (shedding), they affect the activity of these soluble factors. We assessed the effect of the ADAMTS-1 fragments on activation of Erk1/2 kinases induced by soluble HB-EGF and AR. In these experiments, soluble AR, HB-EGF, or bFGF was added to serum-starved MCF-10A mammary epithelial cells in the presence and absence of purified ADAMTS-1NTF, ADAMTS-1CTF, ADAMTS-1E/Q, or full-length ADAMTS-1. Our result indicated that ADAMTS-1NTF and ADAMTS-1CTF, but not full-length ADAMTS-1 or ADAMTS-1E/Q, inhibited Erk1/2 kinase activation induced by soluble AR and HB-EGF, but not by bFGF (Figure 7).
Figure 7.
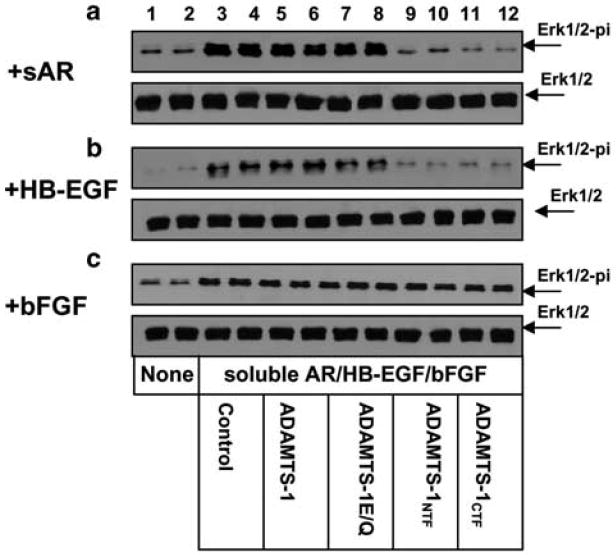
ADAMTS-1NTF and ADAMTS-1CTF inhibit activation of Erk1/2 kinases induced by soluble AR and HB-EGF. Soluble AR (5 ng/ml), HB-EGF (4 ng/ml), or bFGF (15 ng/ml) was applied to serum-starved MCF-10A cells for 25 min in the absence (lanes 3 and 4) or presence of 400 ng of purified full-length ADAMTS-1 (lanes 5 and 6), ADAMTS-1E/Q (lanes 7 and 8), ADAMTS-1NTF (lanes 9 and 10), or ADAMTS-1CTF (lanes 11 and 12). Serum-free medium alone was applied to cells in lanes 1 and 2. The cells were lysed and equal amounts of protein were analysed by Western blotting with anti-phospho-Erk1/2 antibody to detect phosphor-Erk1/2 (upper panels, a–c) or with anti-Erk antibody to detect the total amount of Erk1/2 (bottom panels, a–c).
ADAMTS-1NTF contains a middle TSP-1 motif (ADAMTS-1-mTSP-1; Figure 8) that is very similar (but not identical) to the second and third TSP-1 repeats (WXXWXXW; Figure 8) in Tsp-1, which exhibit antitumor and antiangiogenic activity (Tolsma et al., 1993; Iruela-Arispe et al., 1999; Adams, 2001; Yee et al., 2004). On the other hand, ADAMTS-1CTF contains two COOH-terminal TSP-1 modules (ADAMTS-1-cTSP-1-1 and ADAMTS-1-cTSP-1-2; Figure 8) that have a lower degree of homology to the TSP-1 repeats in Tsp-1. Even though the concentration of ADAMTS-1NTF used in these experiments in Figure 7 was lower than that of ADAMTS-1CTF, similar inhibitory effects were obtained, suggesting that the typical mTSP-1 motif in ADAMTS-1NTF has a more potent inhibitory effect than do the atypical TSP-1 motifs in ADAMTS-1CTF.
Figure 8.

The multiple amino-acid sequence alignment of the second and third TSP-1 repeats of Tsp-1 and the middle and the COOH-terminal TSP-1 motifs of ADAMTS-1. ADAMTS-1NTF contains a middle TSP-1 motif (ADAMTS-1−mTSP-1), whereas ADAMTS-1CTF contains two COOH-terminal TSP-1 modules (ADAMTS-1-cTSP-1-1 and ADAMTS-1-cTSP-1-2).
To determine the molecular mechanism underlying the antiangiogenic activity of ADAMTS-1NTF and ADAMTS-1CTF, we investigated how these fragments affect the bioactivities of several important angiogenic factors in endothelial cells. In the presence or absence of purified ADAMTS-1 or the ADAMTS-1 fragments, we assessed the bioactivities of VEGF165, bFGF, HB-EGF, and AR by determining their ability to induce activation of Erk1/2 kinases, the downstream factors of their corresponding receptors (VEGFR, FGFR, and ErbB). In these experiments, ADAMTS-1NTF and ADAMTS-1CTF, but not full-length ADAMTS-1 or ADAMTS-1minusTSP-1, inhibited the activation of Erk1/2 kinases in human umbilical vein endothelial cells (HUVECs) induced by VEGF165, soluble HB-EGF, and soluble AR, but not by bFGF (data not shown). These results suggest that ADAMTS-1NTF and ADAMTS-1CTF inhibit tumor angiogenesis by inhibiting the activities of several soluble heparin-binding factors that are essential for endothelial cell proliferation and survival.
Discussion
In the present study, we have demonstrated that ADAMTS-1 undergoes auto-proteolytic cleavage and that full-length ADAMTS-1 and the ADAMTS-1 fragments, ADAMTS-1NTF and ADAMTS-1CTF, display pro- and antitumor activity, respectively. We have also shown that the metalloproteinase activity of ADAMTS-1 is required for its protumor activity. In addition, we have demonstrated that the antitumor activity of the ADAMTS-1 fragments requires the TSP-1 motif, which is likely masked in the full-length molecule. Our results imply that the inhibitory effects of ADAMTS-1NTF and ADAMTS-1CTF on the bioactivity of soluble HB-EGF and AR contributes to their antitumor activity; in contrast, ADAMTS-1E/Q likely acts in a dominant-negative manner by inhibiting the shedding of HB-EGF and AR precursors and thus the availability of soluble HB-EGF and AR. Furthermore, we have shown that auto-proteolytic cleavage of ADAMTS-1 can be inhibited by HS, suggesting that the level of HSPGs in the microenvironment likely regulates which form of ADAMTS-1 (full-length or cleaved) is available in the microenvironment to exert its pro- or antitumor activity, respectively.
The antitumor activity displayed by the ADAMTS-1 fragments is likely masked in the full-length molecule
We have established that full-length ADAMTS-1 displays protumor activity and is therefore a potential attractive target for cancer therapy; conversely, the fragments of ADAMTS-1, ADAMTS-1NTF, and ADAMTS-1CTF display potent antitumor activity and have potentials as anticancer agents. These results are consistent with recent studies in cancer research that have demonstrated the pro- and antitumor activity, respectively, of particular intact proteins and their proteolytic fragments (O’Reilly et al., 1999; Pfeifer et al., 2000; Yi and Ruoslahti, 2001; Maeshima et al., 2002). How can auto-proteolytic cleavage convert a protumor factor into an antitumor agent? We have shown that an NH2-terminal fragment of ADAMTS-1 (ADAMTS-1minusTSP-1) that does not contain any TSP-1 motifs also has no antitumor activity, suggesting that the antitumor activity of ADAMTS-1NTF and ADAMTS-1CTF resides in the TSP-1 motif. Even though full-length ADAMTS-1 contains all three TSP-1 motifs, it displays protumor activity, suggesting that the antitumor activity in the TSP-1 motif is masked in the full-length molecule. Auto-proteolytic cleavage of ADAMTS-1 within the spacer/Cys-rich region likely destroys the substrate-binding domain(s) and renders the NH2-terminal cleavage fragments that contain the metalloproteinase domain incapable of binding to and causing the shedding of AR and HB-EGF precursors. This cleavage probably releases the cryptic antitumor activity in the TSP-1 motifs as well (Figure 9).
Figure 9.
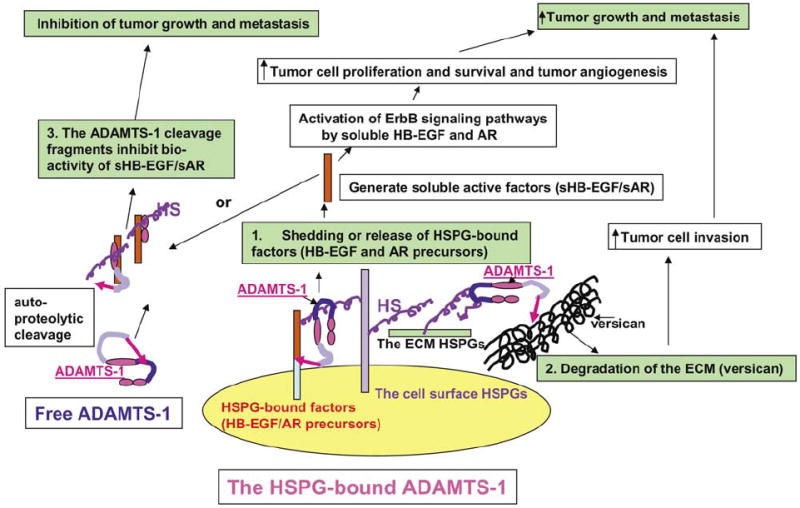
Model showing the potential mechanisms underlying the opposite effects of ADAMTS-1 and the ADAMTS-1 fragments on tumor metastasis. We propose that full-length ADAMTS-1 promotes tumor metastasis by stimulating tumor cell proliferation/survival/invasion and tumor angiogenesis through shedding/activation of the HSPG-bound factors, including HB-EGF and AR transmembrane precursors. We further postulate that this process requires the metalloproteinase activity of ADAMTS-1, and that full-length ADAMTS-1 binds to its substrates through its spacer/Cys-rich domain. In addition, we propose that the antitumor activity in the ADAMTS-1 fragments, ADAMTS-1NTF and ADAMTS-1CTF, resides in the TSP-1 domains, which exert the antitumor activity by inhibiting the bioactivity of several soluble heparin-binding growth/angiogenic factors, including AR, HB-EGF, and VEGF.
ADAMTS-1 may mediate the shedding of heparin-binding EGF family GFs
Although the functional differences between mature soluble EGF family GFs and their transmembrane precursors have not been well-established, the phenotypic similarities between TGF-α- and ADAM17-null mice and between HB-EGF-null and HB-EGF cleavage-resistant mice clearly suggest that shedding of these precursors is essential to the availability and activity of these GFs (Peschon et al., 1998; Iwamoto et al., 2003; Yamazaki et al., 2003). Several members of ADAM family, including ADAM 9, 10, 12, and 17, have been implicated in the shedding of HB-EGF and AR precursors (Black et al., 1997; Blobel, 2000; Herren 2002; Sunnarborg et al., 2002; Gschwind et al., 2003; Lee et al., 2003). Studies using cells from ADAM 9-, 10-, 12-, 15-, and/or 17-null mice have shown that ADAM 17 is the major but not only sheddase for the EGF family GF precursors, suggesting that other member(s) of ADAM and/or ADAMTS family likely play(s) a role as well, especially in the non-12-O-tetradecanoyl-phorbol-13-acetate (TPA)-induced, constitutive shedding of the transmembrane precursors of these factors (Merlos-Suarez et al., 2001; Sahin et al., 2004).
Even though it is well established that activation of EGFR and ErbB-2 promotes carcinoma formation and progression, it is largely unknown how ligands of these receptors contribute to the processes. AR and HB-EGF are upregulated in breast (LeJeune et al., 1993; Normanno et al., 1995; Salomon et al., 1995; Panico et al., 1996; Visscher et al., 1997), prostate (Bostwick et al., 2004), and gastric cancers (Cook et al., 1992; Kitadai et al., 1993; Ebert et al., 1994; Normanno et al., 1995) and keratinocytic tumors (Billings et al., 2003). Shedding of HB-EGF is a critical step in tumor formation (Miyamoto et al., 2004), and overexpression of AR promotes growth of keratinocytic tumors (Billings et al., 2003).
Like HB-EGF and AR, ADAMTS-1 binds to HS through its spacer region and the TSP-1 motifs (Kuno and Matsushima, 1998). HS/HSPG likely brings the metalloproteinase domain of ADAMTS-1 close to the HSPG-bound factors, making ADAMTS-1 an ideal sheddase for these HSPG-bound factors. The activities of ADAMs are regulated through their cytoplasmic tails through intracellular signaling pathways that include the activation of protein kinase C (PKC; Blobel, 2000; Herren, 2002). It is conceivable that ADAM 17 plays an essential role in the TPA-stimulated shedding of AR and HB-EGF precursors (Sahin et al., 2004), whereas ADAMTS-1 plays an important role in the constitutive, non-PKC-dependent shedding of AR and HB-EGF precursors. Furthermore, it is highly likely that the soluble AR and HB-EGF shed by ADAMTS-1 promote tumor cell survival and proliferation and tumor angiogenesis in vivo. Additional studies are required to confirm this hypothesis and to determine whether ADAMTS-1 directly promotes the shedding in vivo. We have also shown that full-length ADAMTS-1, but not ADAMTS-1NTF, promotes shedding of AR and HB-EGF, suggesting that the intact spacer/Cys-rich domain is required for the shedding and that this domain likely contains the substrate recognition/binding site(s), which is(are) destroyed by auto-proteolytic cleavage within the region.
We have shown that ADAMTS-1E/Q, but not ADAMTS-1NTF/ADAMTS-1CTF, inhibits the shedding of HB-EGF and AR precursors (Figure 5D–E), whereas ADAMTS-1NTF and ADAMTS-1CTF, but not ADAMTS-1E/Q, inhibit the activation of Erk1/2 kinase induced by soluble AR and HB-EGF (Figure 7). These results suggest that these two functional domains of ADAMTS-1, the spacer/Cys-rich domain and TSP-1 motifs, are involved in regulating the availability and activity of soluble HB-EGF and AR, respectively. A recent study has shown that ADAMTS-1 inhibits VEGF activity by blocking the interaction between VEGF and its receptor (Luque et al., 2003). Additional study is required to determine whether ADAMTS-1NTF and ADAMTS-1CTF inhibits the bioactivity of soluble HB-EGF and AR by sequestering these soluble factors through the unmasked TSP-1 motifs by directly binding to these factors or by indirectly forming the complexes with these factors through HS (Figure 9).
The function of ADAMTS-1 is regulated by HS/HSPGs
We have shown that heparin/HS inhibits the proteolytic cleavage of ADAMTS-1, and that full-length ADAMTS-1 and the ADAMTS-1 fragments (ADAMTS-1NTF and ADAMTS-1CTF) have opposite effects on tumor metastasis, indicating that the effect of ADAMTS-1 on tumor metastasis depends on its cleavage status (described in the model in Figure 9). In a microenvironment that is highly enriched in HS/HSPGs, ADAMTS-1 binds to HSPGs through its spacer/Cys-rich region, thereby protecting this region from proteolytic cleavage and preserving ADAMTS-1 in full-length form, allowing it to bind and cleave its substrates, including AR and HB-EGF precursors. In this situation, full-length ADAMTS-1 exerts its protumor activity by releasing/activating proproliferation/survival and -angiogenic factors and by promoting tumor cell invasion, and the antitumor activity residing in the TSP-1 motifs is likely masked. In contrast, in a microenvironment lacking HS/HSPGs, ADAMTS-1 is cleaved within the spacer/Cys-rich region, generating the cleavage fragments that have lost the substrate (AR and HB-EGF)-binding sites and contain unmasked antitumor TSP-1 motifs (Figure 9).
Materials and methods
Cell lines and reagents
HUVECs were obtained from Cambrex (Walkersville, MD, USA). TA3 and LLC transfectants were established and maintained as described (Xu and Yu, 2003; Xu et al., 2004a). Anti-v5 epitope (Invitrogen), -von Willebrand factor (vWF) (Dako), -phosphorylated tyrosine (BD Transduction Lab), -EGFR, -ErbB-2, -Erk1/2, and -phospho-Erk1/2 (Santa Cruz) antibodies, the 5-bromo-2′-deoxy-uridine (BrdU)-cell proliferation kit (Roche), Apoptag kit (Chemicon), and heparin and HS (Sigma) were used in the experiments. A rabbit polyclonal antibody recognizing an NH2-terminal peptide (317CNWQKQHNSPSDR DPEHYD335 according to Russell et al., 2003) of mouse ADAMTS-1 was generated to detect endogenous ADAMTS-1.
RT–PCR, mutagenesis, and preparation of expression constructs
Full-length mouse ADAMTS-1 was obtained by RT–PCR using a pair of primers consisting of 24 nucleotides of the 3′ and 5′ extremities of the coding sequence of ADAMTS-1 (accession number NM_009621). The stop codon was omitted from the reverse primers to allow us to fuse ADAMTS-1 to the COOH-terminal v5 epitope tag existing in the expression vector (pEF6/v5-HisTOPO, Invitrogen). The various mutations and deletions of ADAMTS-1 were generated as detailed in Figure 1A using the QuikChange™ and ExSite PCR-based site-directed mutagenesis kits (Stratagene).
Transfection
Lipofectamine (Invitrogen) was used to transfect TA3 and LLC cells with the empty expression vector or the expression constructs containing cDNA inserts encoding ADAMTS-1, ADAMTS-1E/Q, or various fragments of ADAMTS-1 (Figure 1A). TA3 and LLC transfectants were selected on the basis of their resistance to blasticidin (Invitrogen). Expression of v5-epitope-tagged full-length ADAMTS-1, ADAMTS-1E/Q, and the ADAMTS-1 fragments was determined by Western blotting with anti-v5 antibody (Invitrogen).
ADAMTS-1 production and purification, proteolytic cleavage assay, and Western blot analysis
The cell culture supernatants derived from Cos-7 and TA3 transfectants expressing v5-epitope- tagged ADAMTS-1, ADAMTS-1E/Q, or various ADAMTS-1 fragments (Figure 1A) were collected and purified through Ni+-Probond (Invitrogen) and anti-v5 antibody-conjugated affinity columns (Sigma) as described (Xu et al., 2004b). The autoproteolytic cleavage capacity of ADAMTS-1 was assessed by in vitro proteolytic cleavage assays using purified ADAMTS-1. In this assay, 100 ng of ADAMTS-1 was incubated in 50 mm Tris-acetate buffer (pH 7.3) containing 5 mm CaCl2 and 0.1 m NaCl at 37°C for 30 min, or 1, 2, 4, 8, or 12 h. The reactions were stopped by the addition of 8 × SDS sample buffer, and the reaction products were analysed by Western blotting with anti-v5 mAb.
To assess ADAMTS-1 cleavage in the cellular context and determine how the cleavage is regulated, Cos-7 and TA3 transfectants expressing ADAMTS-1 or ADAMTS-1E/Q were cultured for 48 h in the absence or presence of various reagents as detailed in Figure 1, and the cell culture supernatants were collected and analysed by Western blotting with anti-v5 antibody.
Tumor cell tracking, subcutaneous tumor growth, and pulmonary metastasis experiments
To examine the pulmonary extravasation of TA3 transfectants, cells were labeled with green 5-chloromethyl-fluorescein diacetate (CMFDA, Molecular Probes, Inc.) as described (Yu et al., 1997), and injected intravenously into A/Jax syngenic mice at 1 × 106 cells/mouse (the Jackson Lab). The mice were killed 24 h after the injection, and their lungs were removed, fixed, and sectioned. The localization of tumor cells inside the lung parenchyma was revealed by fluorescence microscopy, and the extent of tumor cell extravasation was determined by counting the number of tumor cells residing in the lung parenchyma in five randomly selected × 100 microscopic fields. Experimental (TA3) and spontaneous (LLC) pulmonary metastasis was carried out as detailed previously (Yu et al., 1997; Xu et al., 2004a). Pooled populations of TA3 or LLC transfectants that express an intermediate-to-high level of ADAMTS-1, ADAMTS-1E/Q, ADAMTS-1CTF, ADAMTS-1NTF, ADAMTS-1minusTSP-1, Tsp-1, or Tsp-2, or containing the empty expression vector, were used in these experiments. In all, 15 or 30 mice were injected intravenously (TA3) or subcutaneously (LLC) with each type of the transfectants.
In the spontaneous pulmonary metastasis experiments, the pooled LLC transfectants were injected subcutaneously into C57BL/6 mice following an established protocol (Xu et al., 2004a). When the subcutaneous tumors became easily visible, the longest and shortest diameters of the solid tumors were measured with a caliper in every third day for the next 3 weeks. The tumor volume, expressed as the mean±s.d., was calculated by using the following formula: tumor volume = 1/2 × (shortest diameter)2 × longest diameter (mm3; Hamid et al., 2000; Nokihara et al., 2000). The mice were killed when the tumor size reached ~1.5 cm in the longest diameter or at the end of the experiments.
For spontaneous pulmonary metastasis experiments, the subcutaneous tumors derived from different LLC transfectants were surgically removed when the tumors reach ~1.5 cm in the longest diameter (Xu et al., 2004a). The experimental mice were observed daily after i.v. injection or removal of the subcutaneous tumors. Those that showed signs of severe morbidity were euthanized and considered as if they had died on that day, and the number of surviving mice was recorded. The survival rates were calculated as follows: survival rate (%) = (number of mice still alive/total number of the experimental mice) × 100%. For the experimental metastasis of TA3 transfectants, pulmonary metastatic burden was assessed at 12 and 20 days after i.v. injection by weighting the mouse lungs, since the nature of the pulmonary metastasis made it impossible to count individual tumor nodules. The pulmonary metastatic burden of the LLC transfectants was assessed by counting the number of surface pulmonary tumors under a dissecting microscope and measuring the weights of the lungs. Mice that were free of symptom at 60 days after i.v. injection or removal of the primary tumors were killed, and their lungs were examined.
Transwell tumor cell invasion assay
Tumor cell invasion assays were performed using Transwell cell culture chambers with 8-μm pores (Costar) that were coated with a layer of Matrigel (Collaborative Biomedical). Dulbecco’s modified Eagle’s medium (DMEM) containing 2% fetal bovine serum (FBS) was added to the lower chambers of the Transwells. TA3 transfectants (2 × 105/well) were seeded on the top of the Transwell in triplicate and incubated for 24 h. The bottom filters were fixed and stained. The cells in the top chambers were removed by wiping with cotton swabs, and the stained cells that had migrated through the Matrigel were counted under a microscope. Six randomly selected × 100 microscopic fields were countered. The invasion index of the various TA3 transfectants was calculated according to the following formula:
Histology and immunohistochemistry
To determine the rate of tumor cell proliferation rate in vivo, 5-bromo-2′-deoxyuridine (BrdU) was injected into the mice 4 h prior to killing. The mouse lungs were fixed, sectioned, and stained with hematoxylin and eosin (H&E) as described (Yu and Stamenkovic, 1999; Xu et al., 2004a). In addition, the sections were reacted with anti-von Willebrand factor (vWF) antibody to assess tumor angiogenesis, with anti-BrdU antibody to detect proliferating cells, or with an Apoptag kit to detect apoptotic cells in situ. The total number of tumor cells and the number of tumor cells that were positive for anti-BrdU antibody or TUNEL-staining were determined in five randomly selected × 400 microscopic fields within the pulmonary macro- and micro-metastases. Altogether, >500 cells were counted for each type of the transfectant. The proliferation and the apoptotic rates were calculated as follows: proliferation/apoptosis rate = (number of anti-BrdU or TUNEL-positive cells per microscopic field/total number of tumor cells per microscopic field) × 100%. To assess the extent of tumor angiogenesis, the vWF-positive blood vessels were countered in six randomly selected × 200 microscopic fields within macro- or micro-metastases. The number of blood vessels per microscopic field was expressed as the mean±s.d.
EGFR and ErbB-2 phosphorylation
RIPA buffer (50 mm Tris-HCl, pH 7.4, with 50 mm NaCl, 1% Triton-X100, 0.1% SDS, 2 mm EDTA, 2 mm sodium orthovanadate, 2 mm sodium fluoride, 2 mm phenylmethyl-sulfonyl fluoride, 1 mm leupeptin, 1 mm pepstatin A, and 10 μg/ml aprotinin) was used to extract the lung tissues derived from the mice that had been injected i.v. 24 h earlier with or without different TA3 transfectants (1 × 106/mouse). The proteins were immunoprecipitated by incubation with agarose beads conjugated with anti-EGFR or anti-ErbB-2 antibody (Santa Cruz). The precipitated proteins were analysed by Western blotting with antiphosphotyrosine antibody (BD Bioscience) to detect phosphor-EGFR and phosphor-ErbB-2 or with anti-EGFR or anti-ErbB-2 antibody (Santa Cruz) to detect the total amount of EGFR or ErbB-2.
Shedding of the EGF family GFs and activation of Erk1/2 kinases
Shedding of the transmembrane precursors of AR, HB-EGF, or epigen/TGF-α by ADAMTS-1, ADAMTS-1E/Q, or the ADAMTS-1 fragments was assessed by cotransfecting 293 cells with the expression constructs containing cDNA inserts that encode these EGF family precursors and the ADAMTS-1 constructs detailed in Figure 5. The concentrated cell culture supernatants from the cotransfected 293 cells and the lysates of the transfected cells were analysed by Western blotting to detect the shed soluble GFs or the COOH-terminal cleavage fragments using the corresponding antibodies (R&D Systems, Santa Cruz, and Invitrogen).
The effects of ADAMTS-1, ADAMTS-1E/Q, and the ADAMTS-1 fragments on Erk1/2 kinase activation induced by soluble AR (5 ng/ml), HB-EGF (4 ng/ml), or bFGF (15 ng/ml) were assessed by treating the serum-starved MCF-10A cells with purified soluble AR, HB-EGF, or bFGF in the absence or presence of purified ADAMTS-1, ADAMTS-1E/Q, or the ADAMTS-1 fragments (400 ng). The MCF-10A cells were then lysed, and equal amounts of proteins were analysed by Western blotting with anti-phospho-Erk1/2 to detect phosphor-Erk1/2 or with anti-Erk antibody to detect the total amount of Erk1/2 protein.
The effects of ADAMTS-1, ADAMTS-1E/Q, and the ADAMTS-1 fragments on Erk1/2 activation in HUVECs were assessed by adding VEGF165 (15 ng), bFGF (15 ng), AR (5 ng), or HB-EGF (4 ng) to the serum-starved HUVECs in the absence or presence of 400 ng of ADAMTS-1, ADAMTS-1−TSP-1, ADAMTS-1NTF, or ADAMTS-1CTF for 20 min. The HUVECs were then lysed, and equal amounts of proteins were subjected to Western blotting with anti-phospho-Erk1/2 and anti-Erk antibodies (Santa Cruz) to detect phosphor-Erk1/2 and the total amount of Erk1/2, respectively.
Statistics
Student’s t-test was used to analyse statistical differences between the control and experimental groups. Differences were considered statistically significant at P <0.05.
Acknowledgments
We thank Dr Deborah McClellan for excellent editorial assistance. This work was supported by a fund from NIH (RO1HL074117).
Abbreviations
- ADAMTS-1
a disintegrin and metalloproteinase with thrombospondin motifs-1
- ADAMTS-1E/Q
a protease-dead ADAMTS-1 mutant
- ADAMTS-1NTF
the NH2-terminal ADAMTS-1 fragment
- ADAMTS-1CTF
the COOH-terminal ADAMTS-1 fragment
- ADAMTSNTCF
the NH2-terminal cleavage fragment of ADAMTS-1
- ADAMTSCTCF
the COOH-terminal cleavage of ADAMTS-1
- APMA
p-aminophenylmercuric acetate
- AR
amphiregulin
- bFGF
basic fibroblast growth factor
- BrdU
5-bromo-2′-deoxy-uridine
- CMFDA
Green 5-chloromethyl-fluorescein diacetate
- Cys-rich
cysteine-rich
- DMEM
Dulbecco’s modified Eagle’s medium
- ECM
extracellular matrix
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- GAG
glycosaminoglycans
- GF
growth factors
- HS
heparan sulfate
- HSPGs
heparan sulfate proteoglycans
- HB-EGF
heparin-binding epidermal growth factor
- H&E
hematoxylin and eosin
- HUVECs
human umbilical vein endothelial cells
- kDa
kilodalton
- LLC
Lewis lung carcinoma
- MMP
matrix metalloproteinase
- RT–PCR
reverse transcriptase–polymerase chain reaction
- PKC
protein kinase C
- TA3
TA3 murine mammary carcinoma
- TPA
12-O-tetradecanoylphorbol-13-acetate
- TSP-1
thrombospondin type I-like
- Tsp-1
thrombospondin-1
- Tsp-2
thrombospondin-2
- VEGF
vascular endothelial growth factor
- vWF
von Willebrand factor
References
- Adams JC. Annu Rev Cell Dev Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- Billings SD, Southall MD, Li T, Cook PW, Baldridge L, Moores WB, et al. Am J Pathol. 2003;163:2451–2458. doi: 10.1016/S0002-9440(10)63600-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Blobel CP. Curr Opin Cell Biol. 2000;12:606–612. doi: 10.1016/s0955-0674(00)00139-3. [DOI] [PubMed] [Google Scholar]
- Bostwick DG, Qian J, Maihle NJ. Prostate. 2004;58:164–168. doi: 10.1002/pros.10322. [DOI] [PubMed] [Google Scholar]
- Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett PT, Leahy DJ, et al. Mol Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- Cal S, Obaya AJ, Llamazares M, Garabaya C, Quesada V, Lopez-Otin C. Gene. 2002;283:49–62. doi: 10.1016/s0378-1119(01)00861-7. [DOI] [PubMed] [Google Scholar]
- Colige A, Ruggiero F, Vandenberghe I, Dubail J, Kesteloot F, Van Beeumen J, et al. J Biol Chem. 2005;280:34397–34408. doi: 10.1074/jbc.M506458200. [DOI] [PubMed] [Google Scholar]
- Cook PW, Pittelkow MR, Keeble WW, Graves-Deal R, Coffey RJ, Jr, Shipley GD. Cancer Res. 1992;52:3224–3227. [PubMed] [Google Scholar]
- Ebert M, Yokoyama M, Kobrin MS, Friess H, Lopez ME, Buchler MW, et al. Cancer Res. 1994;54:3959–3962. [PubMed] [Google Scholar]
- Flannery CR, Zeng W, Corcoran C, Collins-Racie LA, Chockalingam PS, Hebert T, et al. J Biol Chem. 2002;277:42775–42780. doi: 10.1074/jbc.M205309200. [DOI] [PubMed] [Google Scholar]
- Gschwind A, Hart S, Fischer OM, Ullrich A. EMBO J. 2003;22:2411–2421. doi: 10.1093/emboj/cdg231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid AS, O’Donnell AL, Balu D, Pohl MB, Seyler MJ, Mohamed S, et al. Cancer Res. 2000;60:7094–7098. [PubMed] [Google Scholar]
- Harris RC, Chung E, Coffey RJ. Exp Cell Res. 2003;284:2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- Herren B. News Physiol Sci. 2002;17:73–76. doi: 10.1152/nips.01373.2001. [DOI] [PubMed] [Google Scholar]
- Iruela-Arispe ML, Carpizo D, Luque A. Ann NY Acad Sci. 2003;995:183–190. doi: 10.1111/j.1749-6632.2003.tb03221.x. [DOI] [PubMed] [Google Scholar]
- Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Circulation. 1999;100:1423–1431. doi: 10.1161/01.cir.100.13.1423. [DOI] [PubMed] [Google Scholar]
- Iwamoto R, Yamazaki S, Asakura M, Takashima S, Hasuwa H, Miyado K, et al. Proc Natl Acad Sci USA. 2003;100:3221–3226. doi: 10.1073/pnas.0537588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson LF, Qiu TH, Sunnarborg SW, Chang A, Zhang C, Patterson C, et al. EMBO J. 2003;22:2704–2716. doi: 10.1093/emboj/cdg264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Kitadai Y, Yasui W, Yokozaki H, Kuniyasu H, Ayhan A, Haruma K, et al. Jpn J Cancer Res. 1993;84:879–884. doi: 10.1111/j.1349-7006.1993.tb02061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno K, Bannai K, Hakozaki M, Matsushima K, Hirose K. Biochem Biophys Res Commun. 2004;319:1327–1333. doi: 10.1016/j.bbrc.2004.05.105. [DOI] [PubMed] [Google Scholar]
- Kuno K, Kanada N, Nakashima E, Fujiki F, Ichimura F, Matsushima K. J Biol Chem. 1997;272:556–562. doi: 10.1074/jbc.272.1.556. [DOI] [PubMed] [Google Scholar]
- Kuno K, Matsushima K. J Biol Chem. 1998;273:13912–13917. doi: 10.1074/jbc.273.22.13912. [DOI] [PubMed] [Google Scholar]
- Kuno K, Okada Y, Kawashima H, Nakamura H, Miyasaka M, Ohno H, et al. FEBS Lett. 2000;478:241–245. doi: 10.1016/s0014-5793(00)01854-8. [DOI] [PubMed] [Google Scholar]
- Kuno K, Terashima Y, Matsushima K. J Biol Chem. 1999;274:18821–18826. doi: 10.1074/jbc.274.26.18821. [DOI] [PubMed] [Google Scholar]
- Lee DC, Sunnarborg SW, Hinkle CL, Myers TJ, Stevenson MY, Russell WE, et al. Ann NY Acad Sci. 2003;995:22–38. doi: 10.1111/j.1749-6632.2003.tb03207.x. [DOI] [PubMed] [Google Scholar]
- LeJeune S, Leek R, Horak E, Plowman G, Greenall M, Harris AL. Cancer Res. 1993;53:3597–3602. [PubMed] [Google Scholar]
- Luque A, Carpizo DR, Iruela-Arispe ML. J Biol Chem. 2003;278:23656–23665. doi: 10.1074/jbc.M212964200. [DOI] [PubMed] [Google Scholar]
- Maeshima Y, Sudhakar A, Lively JC, Ueki K, Kharbanda S, Kahn CR, et al. Science. 2002;295:140–143. doi: 10.1126/science.1065298. [DOI] [PubMed] [Google Scholar]
- Massagué J, Pandiella A. Annu Rev Biochem. 1993;62:515–541. doi: 10.1146/annurev.bi.62.070193.002503. [DOI] [PubMed] [Google Scholar]
- Masui T, Hosotani R, Tsuji S, Miyamoto Y, Yasuda S, Ida J, et al. Clin Cancer Res. 2001;7:3437–3443. [PubMed] [Google Scholar]
- Merlos-Suarez A, Ruiz-Paz S, Baselga J, Arribas J. J Biol Chem. 2001;276:48510–48517. doi: 10.1074/jbc.M103488200. [DOI] [PubMed] [Google Scholar]
- Miao WM, Seng WL, Duquette M, Lawler P, Laus C, Lawler J. Cancer Res. 2001;61:7830–7839. [PubMed] [Google Scholar]
- Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, et al. J Clin Invest. 2005;115:44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Hirata M, Yamazaki A, Kageyama T, Hasuwa H, Mizushima H, et al. Cancer Res. 2004;64:5720–5727. doi: 10.1158/0008-5472.CAN-04-0811. [DOI] [PubMed] [Google Scholar]
- Nokihara H, Yanagawa H, Nishioka Y, Yano S, Mukaida N, Matsushima K, et al. Cancer Res. 2000;60:7002–7007. [PubMed] [Google Scholar]
- Normanno N, Kim N, Wen D, Smith K, Harris AL, Plowman G, et al. Breast Cancer Res Treat. 1995;35:293–297. doi: 10.1007/BF00665981. [DOI] [PubMed] [Google Scholar]
- O’Reilly MS, Pirie-Shepherd S, Lane WS, Folkman J. Science. 1999;285:1926–1928. doi: 10.1126/science.285.5435.1926. [DOI] [PubMed] [Google Scholar]
- Panico L, D’Antonio A, Salvatore G, Mezza E, Tortora G, De Laurentiis M, et al. Int J Cancer. 1996;65:51–56. doi: 10.1002/(SICI)1097-0215(19960103)65:1<51::AID-IJC9>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, et al. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Kessler T, Silletti S, Cheresh DA, Verma IM. Proc Natl Acad Sci USA. 2000;97:12227–12232. doi: 10.1073/pnas.220399597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter S, Clark IM, Kevorkian L, Edwards DR. Biochem J. 2005;386:15–27. doi: 10.1042/BJ20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter S, Scott SD, Sassoon EM, Williams MR, Jones JL, Girling AC, et al. Clin Cancer Res. 2004;10:2429–2440. doi: 10.1158/1078-0432.ccr-0398-3. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Manzaneque JC, Milchanowski AB, Dufour EK, Leduc R, Iruela-Arispe ML. J Biol Chem. 2000;275:33471–33479. doi: 10.1074/jbc.M002599200. [DOI] [PubMed] [Google Scholar]
- Russell DL, Doyle KM, Ochsner SA, Sandy JD, Richards JS. J Biol Chem. 2003;278:42330–42339. doi: 10.1074/jbc.M300519200. [DOI] [PubMed] [Google Scholar]
- Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, et al. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon DS, Normanno N, Ciardiello F, Brandt R, Shoyab M, Todaro GJ. Breast Cancer Res Treat. 1995;33:103–114. doi: 10.1007/BF00682718. [DOI] [PubMed] [Google Scholar]
- Sandy JD, Westling J, Kenagy RD, Iruela-Arispe ML, Verscharen C, Rodriguez-Mazaneque JC, et al. J Biol Chem. 2001;276:13372–13378. doi: 10.1074/jbc.M009737200. [DOI] [PubMed] [Google Scholar]
- Shindo T, Kurihara H, Kuno K, Yokoyama H, Wada T, Kurihara Y, et al. J Clin Invest. 2000;105:1345–1352. doi: 10.1172/JCI8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DF. Breast Cancer Res. 2000;2:176–183. doi: 10.1186/bcr51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit M, Velasco P, Riccardi L, Spencer L, Brown LF, Janes L, et al. EMBO J. 2000;19:3272–3282. doi: 10.1093/emboj/19.13.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunnarborg SW, Hinkle CL, Stevenson M, Russell WE, Raska CS, Peschon JJ, et al. J Biol Chem. 2002;277:12838–12845. doi: 10.1074/jbc.M112050200. [DOI] [PubMed] [Google Scholar]
- Tolsma SS, Volpert OV, Good DJ, Frazier WA, Polverini PJ, Bouck N. J Cell Biol. 1993;122:497–511. doi: 10.1083/jcb.122.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker RP. Int J Biochem Cell Biol. 2004;36:969–974. doi: 10.1016/j.biocel.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Vazquez F, Hastings G, Ortega MA, Lane TF, Oikemus S, Lombardo M, et al. J Biol Chem. 1999;274:23349–23357. doi: 10.1074/jbc.274.33.23349. [DOI] [PubMed] [Google Scholar]
- Visscher DW, Sarkar FH, Kasunic TC, Reddy KB. Breast Cancer Res Treat. 1997;45:75–80. doi: 10.1023/a:1005845512804. [DOI] [PubMed] [Google Scholar]
- Volpert OV, Lawler J, Bouck NP. Proc Natl Acad Sci USA. 1998;95:6343–6348. doi: 10.1073/pnas.95.11.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Liu YJ, Yu Q. Cancer Res. 2004a;64:6119–6126. doi: 10.1158/0008-5472.CAN-04-1054. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liu YJ, Yu Q. J Biol Chem. 2004b;279:41179–41188. doi: 10.1074/jbc.M400292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Yu Q. J Biol Chem. 2003;278:8661–8668. doi: 10.1074/jbc.M208181200. [DOI] [PubMed] [Google Scholar]
- Yamazaki S, Iwamoto R, Saeki K, Asakura M, Takashima S, Yamazaki A, et al. J Cell Biol. 2003;163:469–475. doi: 10.1083/jcb.200307035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Yee KO, Streit M, Hawighorst T, Detmar M, Lawler J. Am J Pathol. 2004;165:541–552. doi: 10.1016/s0002-9440(10)63319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Ruoslahti E. Proc Natl Acad Sci USA. 2001;98:620–624. doi: 10.1073/pnas.98.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Genes and Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Genes and Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Toole BP, Stamenkovic I. J Exp Med. 1997;186:1985–1996. doi: 10.1084/jem.186.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]