

The FOXP2 gene has been implicated in the etiology of severe speech and language disorders (Lai, Fisher, Hurst, Vargha-Khadem, & Monaco, 2001) and was subsequently proposed as an important gene in the evolution of speech in humans (Enard, Przeworski, Fisher, Lai, Wiebe, Kitano, Monaco, & Paabo, 2002; Zhang, Webb, & Podlaha, 2002). Much of what is known about FOXP21 has come from a three generation family that has come to be known as the KE family consisting of 37 members, 15 of whom have a FOXP2 missense mutation and severe speech and language impairment (Lai et al., 2001). Identification of this gene was aided by an independent case with a speech disorder who had a balanced translocation involving the FOXP2 gene. (Lai, Fisher, Hurst, Levy, Hodgson, Fox, Jeremiah, Povey, Jamison, Green, Vargha-Khadem, & Monaco, 2000; Lai et al., 2001). Sequencing of the FOXP2 gene showed that it contains a forkhead box binding domain and the mutation found in the KE family (denoted as R553H) resulted in the protein containing an arginine-to-histidine amino acid substitution within the DNA binding domain (Lai et al., 2001). FOXP2 is now known to be a transcriptional regulatory gene (Li, Weidenfeld, & Morrisey, 2004; Shu, Yang, Zhang, Lu, & Morrisey, 2001) that is expressed in multiple organ systems including the brain. Functional studies of the R553H mutation have revealed that this mutation impedes the transport of the FOXP2 protein across the nuclear membrane and also limits binding of this protein to DNA target sequences (Vernes, Nicod, Elahi, Coventry, Kenny, Coupe, Bird, Davies, & Fisher, 2006). Studies of FOXP2 expression in the brain of mouse, rat, and humans show it is expressed in the cortical plate, thalamus, inferior olives, cerebellum and basal ganglia including the striatum. (Lai, Gerrelli, Monaco, Fisher, & Copp, 2003; Takahashi, Liu, Hirokawa, & Takahashi, 2003).
The first published description of affected individuals in the KE family described them as having apraxia2 of speech and expressive and receptive language problems (Hurst, Baraitser, Auger, Graham, & Norell, 1990). Soon afterward Gopnik and colleagues (Crago & Gopnik, 1994; Gopnik & Crago, 1991; Gopnik, 1990; Ullman & Gopnik, 1999) also described them as having a very specific grammatical deficit involving rule-based morphological paradigms. Vargha-Khadem and colleagues (Vargha-Khadem, Watkins, Price, Ashburner, Alcock, Connelly, Frackowiak, Friston, Pembrey, Mishkin, Gadian, & Passingham, 1998; Vargha-Khadem, Watkins, Alcock, Fletcher, & Passingham, 1995) subsequently described these affected individuals as having prominent orofacial apraxia. More recently Vargha-Khadem's group (Watkins, Dronkers, & Vargha-Khadem, 2002) compared the affected family members with the unaffected family members as well as an acquired aphasia group with regard to intelligence, language, and limb and oral facial praxis. The affected family members performed more poorly than the unaffected family members in all but limb praxis. A statistical analysis indicated that nonword repetition of complex words was the strongest predictor of group membership. Structural brain imaging by this same group (Watkins, Vargha-Khadem, Ashburner, Passingham, Connelly, Friston, Frackowiak, Mishkin, & Gadian, 2002) has indicated abnormalities of either elevated or reduced grey matter density in the caudate nucleus, putamen, cerebellum, temporal cortex, motor cortex, and the inferior frontal gyrus. Further, several of these regions, particularly the caudate, were shown to be abnormal bilaterally (Belton, Salmond, Watkins, Vargha-Khadem, & Gadian, 2003). Functional imaging demonstrated under activation of the left (Broca's area) and right inferior frontal gyri and in the putamen (Liegeois, Baldeweg, Connelly, Gadian, Mishkin, & Vargha-Khadem, 2003). Over activation during language tasks was also observed in the posterior parietal, occipital and postcentral regions that are not normally associated with language. Liegeois and colleagues concluded that FOXP2 may have an important role in the development of the frontostriatal network that supports procedural learning.
Vargha-Khadem and colleagues (Vargha-Khadem, Gadian, Copp, & Mishkin, 2005) have summarized their work concerning the core phenotype associated with this mutation in FOXP2 as “a higher order orofacial motor impairment or dyspraxia that is best exemplified in speech, because speech requires the precise selection, coordination and timing of sequences of rapid orofacial movements” (p. 132). These authors acknowledged that associated with this praxis impairment are language and even nonverbal cognitive impairments; however, they favored the view that these are secondary to the putative core motor impairment. This position stands in contrast with the position initially taken by Gopnik (1990) that the core deficit was a language impairment. Ullman and Gopnik (1999) later provided a cognitive account of the KE phenotype that became associated with the KE family. Based upon Ullman's model of declarative and procedural learning mechanisms in language learning, they proposed that the morphological rule problems of the KE family were rooted in procedural learning deficits. Procedural learning is generally believed to be rooted in the corticostriatal system found to be functionally aberrant in the KE family (Liegeois et al., 2003). A motor speech disorder such as apraxia is also compatible with a procedural learning/memory deficit because this system is closely tied to the learning and performance of complex sequential motor patterns. At this time, the specific nature of the cognitive, speech and language phenotype associated with the FOXP2 mutation found in the KE family remains somewhat unclear.
The mutation of FOXP2 involved a change in a single DNA base pair and thus comprised a point mutation. As was noted earlier, this R553H mutation was a missense mutation that resulted in a change in one amino acid of the resulting FOXP2 protein. Only one other study has reported the speech and language characteristics of individuals with point mutations in FOXP2. MacDermot and colleagues (MacDermot, Bonora, Sykes, Coupe, Lai, Vernes, Vargha-Khadem, McKenzie, Smith, Monaco, & Fisher, 2005) recruited 49 patients worldwide who had speech articulation problems in the absence of other developmental or sensory problems. Phenotyping in this study was performed via physician and parent questionnaire. A mutation screen of FOXP2 in these cases yielded three mutations that resulted in likely changes in the FOXP2 protein. One mutation (R328X) was found in two affected siblings and their mother who by history was also affected. This mutation consisted of a point mutation creating a stop codon. Unlike the R553H mutation of the KE family this mutation caused early termination of translation resulting in the FOXP2 protein being shortened. Both children with the R328X mutation were described as having significant deficits in receptive and expressive language in addition to difficulties with speech sound production. Their mother was described as having poor speech clarity and very simple grammatical constructions, but satisfactory vocabulary. The second form of FOXP2 mutation (Q17L) in one affected sibling consisted of a point mutation that changed the amino acid composition of FOXP2, but not its functional properties (Vernes et al, 2006). This child also had an affected sibling without the substitution. Finally one proband had a region in FOXP2 containing an expansion in a polyglutamine encoding region within exon 5. Additional phenotypic information was not provided on these two cases.
Several other studies have reported individuals with chromosomal abnormalities involving chromosomal rearrangements with breakpoints within FOXP2 or deletions spanning FOXP2. The effects of these types of genetic alterations on genetic function may be different from the point mutations described above. The functional consequences of these chromosomal abnormalities are also more difficult to determine. The first case of a chromosomal rearrangement involving FOXP2 was the case of CS who was found to have a translocation involving a breakpoint in FOXP2 and this finding aided in the identification of FOXP2. CS was found to have a de novo balanced translocation [46, XY t(5;7)(q22; q31.2)] that was later found to transect exons 3b and 4 of FOXP2 (Lai et al., 2001). CS was described as having a severe oral facial apraxia (Lai et al., 2001) that also involved speech and language impairment (Lai et al., 2000). Little other phenotypic information has been provided for this patient. An adult (AA) with a deletion covering the entire region of 7q31 and thus including FOXP2 as well as other genes in this region was described by Liegeois and colleagues (Liegeois, Lai, Baldeweg, Fisher, Monaco, Connelly, & Vargha-Khadem, 2001). AA was described as having severe verbal and orofacial dyspraxia. Additionally, expressive vocabulary and grammar was impaired. Abnormal fMRI and ERP results were found in verbal tasks as well. Zeesman and colleagues (Zeesman, Nowaczyk, Teshima, Roberts, Cardy, Brian, Senman, Feuk, Osborne, & Scherer, 2006) recently reported one child with a deletion in the 7q31 region including FOXP2. This child was described as having praxis deficit that included her not being able to spontaneously cough, sneeze or laugh. Additionally she was found to have dyspmorphic features and a mild developmental delay. Lennon and colleagues (Lennon, Cooper, Peiffer, Gunderson, Patel, Peters, Cheung, & Bacino, 2007) have also recently reported a case of a child with a deletion in the 7q31 region including FOXP2 but also including WNT – a gene that has been associated with autism. This child demonstrated dysmorphic features and mild to moderate mental retardation. Some signs of oral facial motor impairment were noted. These features along with the language impairment were interpreted to be evidence of apraxia.
Recently a series of patients were recruited because of either abnormal FOXP2 or developmental verbal dyspraxia (DVA; Feuk, Kalervo, Lipsanen-Nyman, Skaug, Nakabayashi, Finucane, Hartung, Innes, Kerem, Nowaczyk, Rivlin, Roberts, Senman, Summers, Szatmari, Wong, Vincent, Zeesman, Osborne, Cardy, Kere, Scherer, & Hannula-Jouppi, 2006). These authors described 22 of these individuals, 13 of whom the authors describe as having DVD. The phenotypic characterizations of the speech and language of these individuals with DVD consisted of general descriptions of speech and language delays. The authors did not provide their standards for the determination of DVD, but based on information provided it appears to be founded on a history of late talking and articulation disorder.
One individual reported to have DVD had a translocation of 7q31 with a breakpoint falling within FOXP2. Five of the cases with reported DVD had paternal deletions of the 7q31-32 region that included FOXP2. An additional 7 DVD cases had maternal uniparental disomy of chromosome 7 (two chromosomes from the mother and none from the father) and Silver-Russell Syndrome – a growth retardation syndrome. An expression study showed that these cases with two maternal copies of chromosome 7 had lower levels of FOXP2 expression than normal. Likewise, the five cases of paternal deletions, and the one translocation would have a reduced level of FOXP2 protein production. All four cases with chromosomal abnormalities in the 7q31-32 region were not found to have DVD. In two of these cases without DVD the deletions did not include FOXP2. In another two cases without DVD the abnormality was a paternal uniparental disomy (two chromosomes from the father and none from the mother) of chromosome 7, in which FOXP2 expression levels were found to be normal.
The addition of these new cases with various mutations, deletions and translocations involving FOXP2, support the findings from the KE family that disruption in FOXP2 is associated with speech, language, and often cognitive impairments. These recent new cases also place some form of developmental apraxia of speech in the forefront as the core phenotype. Recently, we have described the speech characteristics of a mother (B) and daughter (T) with a balanced 7;13 translocation involving FOXP2 (Shriberg, Ballard, Tomblin, Duffy, Odell, & Williams, 2006). The findings of the speech analysis for these individuals supported the existing findings of a motor speech impairment in both individuals. The nature of this motor speech impairment was most consistent with spastic dysarthria, but quantitative support for both dysarthria and apraxia of speech were documented. The past studies of affected individuals from the KE family also presented evidence of language and cognitive deficits, particularly impairments in grammar. Thus, if FOXP2 is involved in more than speech, we should expect to see language impairment in these individuals. The focus of this report is to consider whether the phenotype found with abnormalities of FOXP2 is associated with language as well as speech impairment. We will begin with a report of the findings from cytogenetic and molecular genetic studies that have determined the specific locus of the break points on chromosome 7 and 13 for T and B. Of particular interest is whether location of the breakpoint in FOXP2 is likely to have functional consequences that are similar to those found in the point mutation of the KE family. We will then provide a description of the cognitive and language features of these two individuals and will compare these descriptions to cognitive and language descriptions of the affected members of the KE family from prior publications.
Study One: Breakpoint Localization
A chromosomal translocation occurs when two different chromosomes each break apart and the resulting pieces from one chromosome join with a piece from the alternate chromosome. This results in two derived chromosomes that are both combinations of the two originals. When there is no loss or gain of DNA in this process, the translocation is considered to be balanced. The location of the break may or may not reside within a gene. If the translocation on either (or both) chromosome occurs within the elements defining a gene, the function of that gene may be disrupted. Also, unbalanced translocations can often occur in which DNA is lost. Translocations can be identified by means of cytogenetic studies. Such studies had been performed on both T and B indicated that the translocations involved chromosomes 7 and 13. It was not known the exact location of the breakpoints or whether there had been loss of DNA. Therefore, it was necessary to identify the specific locations of these breakpoints on both chromosomes 7 and 13 and determine if DNA had been lost in this translocation.
Method
Participants
The mother (B) and daughter (T) were first referred to our team by a geneticist because they were known to have a translocation involving the 7q31 region containing FOXP2 and both had long-term developmental speech and language problems. Prior cytogenetic studies had shown that B's parents had normal chromosomes. Therefore the chromosomal rearrangement was determined to have originated with B and thus T received both derived 7 and 13 chromosomes from B. T and B have been found by history and examination by physicians of this research group to be healthy individuals with no clinically significant dysmorphology or neurological signs other than marked speech sound impairment.
Since the initial referral, B and T have been examined on three occasions. During this time, B was between 50 and 52 years of age and T was between 18 and 20 years of age. During the second examination period, blood samples from B and T were obtained and used as the source of DNA for cytogenetic and molecular genetic analyses. All research with these individuals was approved by the University of Iowa Institutional Review Board.
Procedures
Fluorescent In Situ Hybridization Analysis
The first step in localizing the breakpoint requires the use of cytogenetic methods that allow the region of the breakpoint to be visualized. Fluorescent in situ hybridization (FISH) involves creating a strand of DNA (clone) that is labeled with fluorescent dye and this strand is then allowed to hybridize with a specific complementary sequence of DNA in the region of interest on the chromosome. Specifically we used G-banded metaphase spreads of the patient cell lines. Clones for FISH were selected using the genome map provided by the University of California Santa Cruz (UCSC) Genomics Bioinformatics Group (http://genome.ucsc.edu). Bacterial Artificial Chromosome (BAC) clones, from RP11 libraries, and fosmid clones were obtained (Children's Hospital of Oakland Research Institute) and directly labeled with SpectrumOrange-dUTP (Vysis, Inc, Downers Grove, IL, USA). The labeled probes were hybridized to metaphase cells and the chromosomes were analyzed using an Olympus BX60 fluorescent microscope. The Applied Imaging System's Cytovision computer program was used to digitize the images to visualize the signals.
Long-range PCR Amplification Across Breakpoints
FISH methods provide a means to narrow the likely breakpoint to a small region, but the resolution of this method does not allow the exact location and DNA sequence to be determined. To accomplish the latter, molecular genetic methods must be employed. Polymerase Chain Reaction (PCR) methods allow copies of DNA sequences to be created beginning with sequence specific primers that determine the endpoints. PCR reactions exponentially amplify this particular section of the chromosome, allowing for its isolation and eventual sequencing.
Primers were designed to amplify a PCR product that contained the breakpoint, primers were designed using sequence obtained from the University of California Santa Cruz (UCSC) genome browser within the fosmids used for cytogenetic studies shown to have spanned the breakpoint for each respective chromosome. Primers were picked with a length of 27 bp and optimal melting temperature (Tm) of 64°C. Specific primer information is available upon request.
In order to amplify a product on the mother and daughter's derivative 7 and derivative 13 chromosomes, forward primers on chromosome 7 were paired with reverse primers on chromosome 13, and vice versa, until a product was amplified. This was continued until a ∼800bp product containing the break was amplified for both derivative chromosomes. Normal chromosome 7 and chromosome 13 primer pairs were also used to amplify the region surrounding the break on the normal chromosomes. These products served as controls against which the derivative chromosome sequences were compared. All reactions were performed in 25 μl final volume using the TaKaRa LA PCR kit, version 2.1. Denaturation steps were performed at 94°C, and annealing/extension took place at 68°C. Extension times began at 10 minutes for the first 10 cycles, and were then increased to 15 minutes + 15 seconds/cycle for the remaining 20 cycles. Extension times were initially calculated using 1 minute/kb as a general guideline.
Sequence Verification
Sequencing was performed on the derivative 7, derivative 13, normal 7 and normal 13 PCR products using the primers previously described as well as new primers designed from within the final long-range PCR products. This sequence was then analyzed in order to find the exact location of the translocation in each derivative chromosome. This was done by comparing the derivative chromosome sequence to the UCSC sequence data as well as to normal chromosome sequence obtained from the patient.
Results
The cytogenetic and molecular genetic methods above resulted in our locating the breakpoint on chromosome 7 of the FOXP2 gene (7q31.1) between 114064876 and 114064883, within intron 6. The breakpoint on chromosome 13 was found in intron 7 of the replication factor C subunit 3 gene (RFC3) (13q13.2), between 33306028 and 33306035. RFC3 is a gene involved in elongation of primed DNA templates. A 6bp homologous sequence was found to comprise the locus of the break point. This sequence was common to the breakpoint region on both 7 and 13. Figure 1 shows the DNA sequences for the normal and derived chromosomes and identifies the specific location of the breakpoint. Neither of these break points involved deletion or transposition of DNA material. The resulting balanced translocation produced two novel coding sequences (FOXP2-RFC3 and RFC3-FOXP2) comprised of combinations of exons3 of FOXP2 and RFC3. These sequences that could potentially produce a fusion protein product containing amino acid sequences from both FOXP2 and RFC3. The fusion protein coding sequence for each of these fusion genes are provided in the Appendix.
Figure 1.
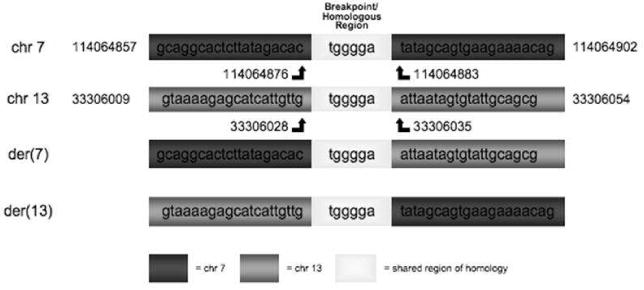
Results of sequencing at breakpoints on chromosomes 7 and 13 for TB translocation. The chromosomes involved in the translocation are identified as derived chromosomes (der) and are the two lower sequences whereas the normal sequence for each chromosome are at the top. A region of 6 bases common to both chromosomes is identified as a homologous region. The upper portion of the figure shows the location of these breakpoints within chromosomes 7 and 13. Within FOXP2, boxes are exons (sequences transcribed into protein) and lines are introns (nontranscribed sequences).
Discussion
The cytogenetic and molecular genetic study of the characteristics of this translocation demonstrated that there was no loss of genetic material involved in this balanced translocation. The breakpoint on Chromosome 7 involved FOXP2. Due to the location of this breakpoint in intron 6, and its union with a portion of chromosome 13, we can expect disruption of the normal FOXP2 protein. An inspection of the fusion protein coding sequence of the FOXP2-RFC3 fusion gene (See Appendix) indicated that the breakpoint induced a frame shift at the union of exon 6 of FOXP2 and exon 8 of RFC3. This frame shift results in a change in the amino acids in the RFC3 product and the occurrence of a stop codon 12 amino acids from the breakpoint. Thus in fusion FOXP2-RFC3 sequence is comprised of a truncated FOXP2 sequence and very little of the RFC3 sequence. Whether this fusion protein product would result in a functional protein can only be determined by expression studies that are not in the scope of this paper. However, it is likely that the mRNA produced by this fusion gene would be degraded by nonsense mediated RNA decay (Chang, Imam, & Wilkinson, 2007) and therefore no translation into protein would occur.
This chromosomal translocation differs from the missense point mutation of the KE family in exon 14 of FOXP2 on chromosome 7, but both changes may influence the function of the protein. Recently it has been shown that the missense mutation of the KE family results in abnormalities of the R553H binding domain of the FOXP2 protein. This mutation results in limited movement of the R553H protein into the nucleus. Furthermore this mutation limits the localization of FOXP2 to DNA binding sites in the nucleus Additionally, this FOXP2 protein variant is less effective in inhibiting expression of downstream targets FOXP2 (Vernes et al., 2006). Vernes et al. also described the functional properties of a severe truncation of FOXP2 resulting in a protein product R328X that was found in two affected siblings and their mother originally reported by (MacDermot et al., 2005). This mutation is also a point mutation that results in an abnormal stop codon in the middle of FOXP2 which results in a severely truncated protein in much the same place as the break point found in the der7 chromosome in this study. The cell based-based studies of Vernes et al. suggested that this protein was non-functional and is compatible with the notion that those with such mutation will present with a reduced dose of FOXP2. As noted earlier, it is likely that the FOXP2-RFC3 fusion product resulting from the translocation would be similar to this R328X product in producing a reduced dose of functional FOXP2. Thus, the findings from T and B are consistent with all other findings of individuals with FOXP2 mutations in that they are likely to have a reduced dose, but not a complete absence of FOXP2 protein. In this case the basis for the FOXP2 reduction comes via a large scale translocation rather than a point mutation in the case of the R328X. Unlike the R553H mutation, these nonfunctional products will result in a reduced dose of FOXP2, but no active mutant protein variants such as occurs with mutation found within the KE family.
In addition to a breakage of FOXP2, this analysis also revealed that RFC3 on chromosome 13 was affected by this translocation. The protein product from the RFC3-FOXP2 fusion gene on der13 chromosome also contained a frameshift resulting in a premature stop codon (See Appendix). Therefore, the fate of this fusion gene is likely to be the same as that of the FOXP2-RFC3 fusion gene on der7. That is, it will be degraded by nonsense mediated decay and thus not be translated. RFC3 is a gene that contributes the p38 subunit of the RFC complex comprised of 5 subunits. The RFC protein complex plays a role in DNA replication by contributing to the formation of a scaffold to support DNA elongation. The deletion of critical regions of the p38 component in RFC3 have been shown to result in impaired RFC formation and function (Uhlmann, Gibbs, Cai, ODonnell, & Hurwitz, 1997). An important role for RFC involves the cell-cycle checkpoint pathways that control sequences of activity during cell division, one part of which is to recognize and respond to damaged DNA - a function called DNA-integrity checkpoint activation. A loss of this function can result in cancer and neurodegenerative disease (Pearce & Humphrey, 2001). The phenotype consisting of a developmental speech and language disorder shown in T and B is not consistent with acquired disorders associated with somatic cell damage as appears to be found with mutations of RFC3. RFC3 is 1Mb centromeric to the neurobeachin gene (NBEA). NBEA is largely expressed in the brain and an individual with autism and a balanced translocation involving NBEA has been reported (Castermans, Wilquet, Parthoens, Huysmans, Steyaert, Swinnen, Fryns, Van de Ven, & Devriendt, 2003). NBEA is believed to be important for normal functioning of neurotransmitter release from vesicles at pre-synaptic terminals. Additionally, RFC3 and NBEA fall within a region that has been linked to autism and in particular individuals with autism who have family histories of language impairment.(Collaborative Linkage Study of Autism, 2000). Whether this linkage can be attributed to NBEA remains unknown. The break in RFC3 could have an impact on the function of nearby genes such as NBEA via a position effect that alters NBEA expression due to changes in regulatory sequences for NBEA function. We should note that position effects on nearby genes could also be involved in the breakpoint within FOXP2. Thus, we cannot rule out a role for the chromosome 13 break in the speech, language and cognitive phenotype of T and B. It does seem unlikely that RFC3 itself contributes to this particular phenotype. In contrast, the involvement of FOXP2 in the chromosome 7 breakpoint is a very strong candidate in contributing to the speech, language, and cognitive phenotype of these individuals.
Study Two: Language Characteristics
We noted in Study One that the functional nature of the FOXP2 impairment in the TB family should be similar to that of the KE family albeit not identical. In both cases, the downstream regulation of gene expression will be affected. Because this is not the same kind of disruption of FOXP2, it is likely that these functional effects could be different in kind or magnitude. Therefore, it is important to carefully describe the phenotype of T and B to further understand the possible cognitive and linguistic systems affected by FOXP2 and downstream genes it affects.
As we noted earlier, recent reports of individuals with FOXP2 abnormalities have emphasized speech as the principal phenotype. We have previously reported the speech characteristics of T and B (Shriberg et al., 2006). In this study we will complement that report by emphasizing the language and cognitive abilities of T and B. Two questions serve as the focus of this investigation. First, we wish to determine whether language impairment is likely to be a part of the phenotypic profile of FOXP2 abnormalities. Second, if language impairment is found, do we find that the features of this impairment are similar to that reported in the KE family. Therefore, this study will focus on language measures obtained from T and B that are either identical or similar to those that have been reported for the KE family members and therefore allow for a comparison of the KE phenotype with T and B. Thus, this study aimed to characterize the language status of T and B and to descriptively compare this profile with the KE family.
Methods
Participants
The participants were T and B as described in Study One.
Language Measures
Vocabulary
Measures of receptive and expressive vocabulary have been provided for the KE family via British Vocabulary Test (BVT: Dunn, Dunn, Whetton, & Pintillie, 1982) and the Wechsler Adult Intelligence Scale-III (WAIS-III; Wechsler, 1997). To parallel the BVT, the Peabody Picture Vocabulary Test:3 (PPVT-3; Dunn, 1997) was used in conjunction with the Test of Expressive Vocabulary (EVT; Williams, 1997). Likewise the vocabulary subtest of the Wechsler Adult Intelligence Scale-III (WAIS-III; Wechsler, 1997) was administered to mirror the data from the KE family.
Grammar
Several measures of sentence use were also administered. The Test of Receptive Grammar (TROG; Bishop, 1982) has been used with the KE family by (Watkins et al., 2002). The TROG is a standardized test of sentence comprehension in which the participant listens to sentences and points to pictures depicting the critical meaning of the utterance. The content of the TROG consists of sentences that vary in grammatical (syntax and morphology) form. The sentences are single clauses and thus mastery of this test by 8 year-olds is common. Sentence comprehension was also examined using the Clinical Evaluation of Language Fundamentals: Concepts and Directions (CELF:III C&D; Semel, Wiig, & Secord, 1995). The CELF:III-C & D test evaluates sentence comprehension in contexts where lexical content is reduced to color, shape, and a limited set of actions (touch, point). Utterance complexity is increased by adding clauses and more complex adverbial relations among clauses. Thus, this test provides a mix of greater working memory demands in conjunction with more complex syntax. Production of sentences within elicited contexts was measured using sentence repetition and closure tasks. The Clinical Evaluation of Language Fundamentals: Recalling Sentences (CELF:III-RS; Semel et al., 1995) requires the repetition of sentences of a range of different grammatical forms and lengths. Performance on the CELF:III-RS has been found to be particularly sensitive to specific language impairment (Conti-Ramsden, Botting, & Faragher, 2001).
Tense marking on regular and irregular verbs has been reported for members of the KE family by Ullman and Gopnik using the Ulman Grammatic Completion Task (UGCT; Ullman & Gopnik, 1999). Therefore, this test was administered to the TB family. The test includes 16 regular forms with real and 12 regular forms with nonsense forms. It also tests irregular forms in 16 real words and 14 novel words. The items are presented orally using a standard frame “Every day I (make) my lunch. Just like every day, yesterday I made my lunch.” The participant hears the verb set off above in parentheses in its present tense and then is expected to complete the utterance “yesterday I” with the appropriate past tense inflected verb as shown by italic. For this study these sentences were randomized and as in Ullman and Gopnik arranged so that similar sounding verbs did not occur in sequence. These sentences were audio recorded and were presented via headphones. Preceding the test items were 4 practice items. Ullman and Gopnik used this test with some affected members of the KE family. For some individuals, the sentences were read by the family members and for other individuals the sentences were read to them.
The responses on the UGCT were coded according to a set of categories described by Ullman and Gopnik (1999). These categories are described in Table 1. Two of these categories – past tense marking and over-regularization are classified together for the purposes of this paper as conventional forms of past tense marking. An additional set of response categories consisted of responses that did not reflect past tense use or made it difficult to evaluate whether the person could mark tense on the particular verb being tested. For this paper these categories will be merged into one category that is named unconventional or unmarked. The coding of these responses was performed by separate coders to evaluate coding reliability. These coding decisions were in very good agreement (total percent agreement = 95%; Kappa=.89).
Table 1. Categories of past tense marking with the Ullman Past Tense Inflection Task.
| Conventional Tense Marked Forms | ||
| Past Marked | Verbs inflected appropriately (cross-crossed, make-made) or irregular novel words marked either with irregular change (criv, crove). Or regular inflections (crive-crived). | |
| Over-regularized | Real irregular verbs were coded as over regularized if the regular past affix was used. | |
| Unmarked or Unconventional Responses | ||
| Unmarked Alternative marking | Verbs with bare stems (look-look, dig-dig). Verbs with inflections that were not past tense | |
| forms (ing, en, -s). | ||
| Conceptually plausible | Use of a different verb that was semantically similar to the stimulus (hit-pound). | |
| Phonological proximate | Use of a different real verb that was phonologically similar to the stimulus (slip-slap) | |
| Unclear or no response | Responses that did not fall into the categories above |
Finally, language use was observed in a standard narrative production task using a method developed by Saffran et al. (Saffran, Berndt, & Schwartz, 1989) Narrative samples were obtained by asking T and B to tell the story of Cinderella. In each case they were shown a wordless picture book to aid them in remembering the story and then asked to retell the story. Their narratives were then transcribed and coded for use of obligatory morphemes, clausal structure, and grammaticality of sentences using Systematic Analysis of Language Transcripts (SALT: Miller & Chapman, 1993) conventions.
Several of the language measures described above provided norms. In those cases where norms were available for T and B's chronological age group, those norms were used to report scores of relative standing. To aid comparisons across measures, these standard scores were transformed to have a mean of 100 and standard deviation of 15 or to have a mean of 10 and a standard deviation of 3. Some of the normative tables included scores for older children and young adults, but did not extend to B's age level. In such cases the raw scores could be norm referenced with regard to age equivalence scores. In this case, the age equivalence for a given raw score was taken as the age of the normative group that was associated with a 50th percentile score. Age equivalence scores are often viewed as being problematic for clinical decision making because they do not have a known and fixed variance. However, these scores can be validly used to represent relative ability level and were used for this purpose.
Results
Since there were only two individuals in the TB family and the individual data from the affected members of the KE family were not available, comparisons between these two families will be descriptive. Of particular interest in these comparisons are the profiles shown across tasks for the two families. Absolute levels of performance will also be examined but are not viewed as significant as the profiles of relative strengths and weaknesses.
Intelligence
The results of the WAIS-III testing of the TB family are provided in Table 2 along with average scores for each subtest reported by Wakins et al. (2002) for the affected family members (N=13) in the KE family. To facilitate comparison of these data the average scores for the TB family and the affected members of the KE family are shown in Figure 2. The average score for the normative sample for the subscales is 10 with a standard deviation of 3. In both families considerable variation can be seen across subtests. Overall, both families show below average scores and both families show a pattern where the average verbal score is 10 points below the average performance IQ. The TB family members show an overall better level of performance than the affected members of the KE family and in particular performed much better on two of the verbal subtests – information and similarities and the KE family performed better than the TB family members on the Picture Completion scale. With the exception of these two subtests, there was a remarkably similar pattern of relative strengths and weaknesses. Within the performance scales, Block Design and Object Assembly were both relative strengths for both families whereas, Picture Arrangement and Digit Symbol Coding were relative weaknesses. Within the verbal scales, Vocabulary, Arithmetic, and Digit Repetition were relative weaknesses for affected members of both families.
Table 2. Results of the Wechsler Adult Intelligence Scale-III for T (daughter) and B (mother) and the average scores for the affected members of the KE family (N=13).
| WAIS-III Scale | TB | Affected KE | |||
|---|---|---|---|---|---|
| B | T | Mean | Std. Dev. | ||
| Non-verbal | Picture Completion | 9 | 6 | 8 | 2.4 |
| Digit Symbol-Coding | 4 | 5 | 5.4 | 2. | |
| Block Design | 8 | 8 | 9.1 | 2.7 | |
| Object Assembly | 10 | 9 | 8.5 | 2.1 | |
| Picture Arrangement | 9 | 6 | 6 | 2.4 | |
| Performance IQ | 95 | 87 | 83.2 | 10.6 | |
| Verbal | Vocabulary | 6 | 5 | 5 | 1.8 |
| Similarities | 8 | 9 | 5.9 | 2.4 | |
| Arithmetic | 4 | 7 | 5.7 | 1.7 | |
| Digit Span | 5 | 8 | 6.3 | 2.4 | |
| Information | 11 | 5 | 5.9 | 1.8 | |
| Verbal IQ | 81 | 81 | 74.1 | 8.8 | |
| Full Scale IQ | 88 | 81 | na* | na | |
na = not available
Figure 2.
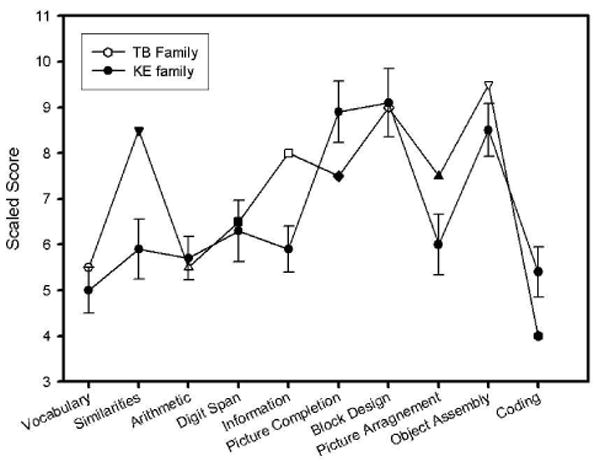
Mean scale scores (10=average, 1 SD=3) for subtests of the Wechsler Adult Intelligence Scale-III for T (daughter) and B (mother) in comparison with mean (bars represent standard error of the mean) scores for the 13 affected members of the KE family (Closed circles; Watkins et al., 2002).
Vocabulary
As shown above on the WAIS-III vocabulary test (Figure 2), scores for both TB family members were similar to the affected members of the KE family were within the range of scores reported for affected members of the KE family. T and B received standard scores of 76 (z-score = -1.66; scaled score=5) and 80 (z-score=-1.33, scaled score=6) respectively. T and B obtained the same standard scores on both the PPVT-3 and the EVT where they scores of 79 (z-score=-1.4) and 83 (z score=-1.13). The affected members of the KE family were reported by Vargha-Khadem (1995) to have a mean BVT of 65.38 (SD=11.37; z-score=-2.31). Thus, on this measure of vocabulary the affected members of the KE family was about 1 SD lower than the TB family; however, it must be acknowledged that the norms for the two tests are not the same and therefore comparison of absolute scores is problematic.
Sentence Development and Use
The raw scores on the TROG for T and B were 72 and 73 respectively where the maximum raw score was 80. In comparison, the average TROG score for the affected members of the affected members of the KE family was 71 (SD=4.82). The norms for the TROG do not extend to adults and therefore standard score interpretation is not possible; however, both T and B had 15 of 20 item blocks correct which according to the test manual yield an age equivalent score of 8 years for normal hearing children. The CELF:III C&D also assesses sentence comprehension. T received a raw score of 24. The CELF-III provided norms for individuals of Ts age and her performance yielded a standard score of 5. B received a raw score of 16 and no norms were available for adults of her age; however, this raw score represents average performance for 6 year olds.
Sentence imitation skills were reflected on the CELF:III RS test. T received a raw score of 30 which yields a standard score of 3 and an age equivalence of 7 years. B received a raw score of 16 on this test and again because of her age no standard score was available. A raw score of 16 is 1 SD below the average performance levels of children 6 to 6-11; the youngest group for which norms were provided on this test.
The pattern of responses provided by T and B on the Past Tense Inflection task used in Ullman and Gopnik (1999) are shown in Table 3 along with data from affected members from the KE family and response rates for normal age mates as reported by Ullman and Gopnik. The data show that T and B were more likely to mark past tense than the affected members of the KE family but were also less likely to doso than the normal controls. Similar to the affected members of the KE family, rates of past-marking was lower with the younger family member, T, than the older family member, B. Ullman and Gopnik provided guidelines for comparison of individuals' performance levels with the control group. In the case of the past-marked responses these represent values that fall below a cut-off established as being 1.5 times the interquartile range of the normal controls. These values are shown in parentheses. Both B and T's performance levels fell below these cut-off values for marking past for both real regular forms and B's performance also fell below this criterion irregular real words (norms were not available for irregular novel words). Unlike B, T also showed a greater tendency to mark past on the novel regulars using an irregular form than the regular form, whereas B rarely used an irregular and instead used the regular form.
Table 3. Percentage of past tense use in T (daughter) and B (mother) and KE family members and controls.
| Young Adults | Middle Age Adults | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| T | AW | RO | Controls | B | PA | JO | VA | Controls | |
| Real Regulars | |||||||||
| Past-Marked | 88* | 69 | 0 | 99 (97) | 94* | 100 | 100 | 31 | 100 (99) |
| Unconventional or Unmarked | 12 | 32 | 100 | 0 | 6 | 0 | 0 | 68 | 0 |
| Real Irregulars | |||||||||
| Past-Marked | 100 | 36 | 64 | 96 (94) | 100 | 79 | 93 | 0 | 95 (85) |
| Over-regular | 0 | 21 | 7 | 0 | 0 | 0 | 0 | 0 | na |
| Unconventional or Unmarked | 0 | 43 | 28 | 2 (0) | 0 | 0 | 0 | 100 | 1 (0) |
| Novel Regular | |||||||||
| Past-Marked | 33* | 8 | 0 | 96 (84) | 92 | 83 | 75 | 8 | 94 (75) |
| Unconventional and Unmarked | 50* | 91 | 100 | 1 (0) | 8* | 17 | 25 | 92 | 0 (0) |
| Novel Irregular | |||||||||
| Regularized | 21 | 7 | 0 | na | 71 | 36 | 36 | 0 | na |
| Irregularized | 36 | 7 | 7 | na | 0 | 7 | 7 | 7 | na |
| Unmarked and Unconventional | 43 | 86 | 93 | na | 29 | 56 | 57 | 93 | |
Note. Affected KE family members are identified as AW, RO, PA, JO, VA. Data from KE family are from Ullman and Gopnik (Ullman & Gopnik, 1999). na = not available.
Response rates for T or B that fell outside of 1.5 times the interquartile range for typical controls from Ullman and Gopnik.
Both T and B retold the Cinderella story. Their narratives were analyzed with regard to measures of productivity, sentence length, grammaticality, and sentence complexity. The results of this analysis are shown in Table 4 along with normative data for adults in this same task reported by Thompson et al. (Thompson & Shapiro, 1995). These data show that T was much more expressive than B producing nearly twice as many words in her story. In fact, T's story was more than 1 SD above the mean length for normal adults. B's story was somewhat shorter than the mean for normal adults, but was still within the normal range. T's sentence length as reflected in MLU was quite similar to the average performance of the normal group; whereas B's was 1.5 SD below the mean. The use of obligatory bound morphemes in normal adults is expected to be nearly 100%. In contrast, B produced obligated morphemes on 84% of the opportunities and T produced these on 91% of opportunities. This rate of acceptable grammar was even lower when all forms of grammatical use were coded. In contrast to the near 90% use by the normal group, T and B's performance approached 57% and 62% respectively. The use of complex sentences by T and B reflects the rate of embedding and proportion of complex sentences. In each case, T and B were substantially below the rates shown for the normal adults. Finally, speaking rate was computed and compared with norms from Goldman-Eisler (1968) where it can be seen that T and B were substantially below the expected range of speaking rate for adults.
Table 4. Descriptive data regarding language usage from a narrative generation sample for T (daughter) and B (mother) in comparison with normative performance levels.
| Language Variable | T | B | Normative Data | |
|---|---|---|---|---|
| Mean | SD | |||
| Total utterances | 39 | 28 | 23.6 | 7.1 |
| Total words produced | 523 | 271 | 323.7 | 113.6 |
| MLU (morphemes) | 14.3 | 11.1 | 14.5 | 2.2 |
| Obligatory bound morphemes produced | 39/43 | 21/25 | -- | -- |
| % grammatical sentences | 56.8 | 61.9 | 89.8 | 8.0 |
| Mean number of embeddings per utterance | 0.51 | 0.62 | 1.0 | 0.2 |
| % complex sentences | 43.2 | 42.9 | 57.5 | 16.9 |
| Words / min | 90.6 | 80.84 | Range | 150-250 |
Note. Normative data obtained from Thompson and Shapiro (1995)
Language: Discussion
Point mutations and chromosomal rearrangements of FOXP2 have been consistently associated with both speech motor impairments and impairments of language development (Feuk et al., 2006; Gopnik, 1990; Gopnik & Crago, 1991; Hurst et al., 1990; Lai et al., 2000; 2001; MacDermot et al., 2005; Ullman & Gopnik, 1999; Vargha-Khadem et al., 1995; Vargha-Khadem et al., 1998; Watkins et al., 2002; Zeesman et al., 2006). Despite the acknowledgement of language deficits, many of these reports emphasize the speech motor impairment and more specifically consider the primary phenotype associated with mutations of FOXP2 to be developmental verbal apraxia of speech. In fact, the only data provided with regard to the language features associated with FOXP2 variations come from studies of the KE family who have one particular mutation of FOXP2. Two research groups have reported on the KE family, one led by Gopnik and the other by Vargha-Khadem. Gopnik's reports focused solely on the language impairments of the affected family members arguing that the deficit displayed by these individuals was rooted in a morphosyntactically specific deficit. Vargha-Khadem's reports have argued against these claims by showing a broader deficit in cognition, speech and language. Vargha-Khadem's group has acknowledged that the affected members of the KE family do have language deficits. However, this group has emphasized the speech production deficits. Watkins and colleagues (2002) appeared to favor an explanation wherein developmental apraxia affects phonological memory that in turn results in poor language learning. They do acknowledge that other relationships could also exist (Watkins et al., 2002).
This present study describes the language and nonverbal skills of a mother and daughter (T and B) with a balanced translocation that involves FOXP2 on chromosome 7 and RFC3 on chromosome 13. The speech skills of these two individuals have been described previously (Shriberg et al., 2006). Consistent with the reports of other individuals with FOXP2 abnormalities, T and B also demonstrated significant impairments of speech. The characteristics of the speech deficit in both these individuals were consistent with both a spastic dysarthria and apraxia of speech.
The profile of language and nonverbal skills in the TB family when compared with the affected members of the KE family was quite similar. Because we didn't have access to the individual data from the KE family and the TB family only contained two individuals, this comparison can only be made by visual inspection of the data. As shown in Figure 2, the profile across the WAIS-3 subtests is quite similar. The exception was B's very good performance on the information subtest. This is a task that although verbal, emphasizes declarative and executive skills, that is, knowledge one is taught and then employed in a strategic fashion. Despite B's language problems, she is very curious and is an avid reader and user of the internet. The depressed performance of the affected members of the KE family on this subtest, may reflect their educational experiences and cultural background. This conjecture is supported by the fact that even the unaffected members of the affected members of the KE family also scored low on this subtest. Another contrast between the TB family and the affected members of the affected members of the KE family concerns vocabulary. The TB family showed depressed expressive and receptive vocabulary abilities. Both T and B were approximately 1.3 SD below the mean for their age group on the WAIS-3 vocabulary subtest, the PPVT-3 and the EVT. The average WAIS-3 vocabulary subtest score for the KE family was also at this level. In contrast, the affected KE family members were about 2.3 SDs below the mean on the BPVT which is considered to be a parallel test to the PPVT-3. This discrepancy may be due to differences in the normative samples of the two tests. Hick et al. (Hick, Joseph, Conti-Ramsden, & Serratrice, 2002) have reported that 2 of 3 children scored substantially lower on the BPVT than EVT which would be consistent with this interpretation.
The finding that depressed vocabulary development is found in individuals with FOXP2 abnormalities would be particularly expected within the framework of Vargha-Khadem and colleague's proposal that these abnormalities cause speech-motor impairments that result in phonological memory impairments. Gathercole and Baddeley (1989) propose that phonological memory is necessary for word learning. In their theory, phonological memory requires subvocal rehearsal to refresh information in the phonological loop. According to Baddeley and Wilson (1985) apraxia compromises this rehearsal and thus should restrict word learning. Baddeley has not advanced as similar stance with regard to grammatical development and phonological memory and therefore we might predict that lexical deficits should be the most prominent form of language deficit associated with apraxia of speech and thus mutations of FOXP2. We should also note that Baddeley and Wilson have shown that dysarthria does not result in a disruption of phonological memory (Baddeley & Wilson, 1985) and therefore should the motor deficit associated with FOXP2 mutations be solely a subtype of dysarthria, the coherence of Vargha-Khadem's account would be compromised.
Earlier we noted the difficulties the affected members of the KE family have had with sentence use, in particular. The language data from the TB family are again consistent with the general patterns described for the KE family by both Vargha-Kahdem (1995) and Ullman and Gopnik (1999). The TROG was given to both families and the results were very similar for the TB and KE affected family members. Moreover, the age equivalence scores obtained on the CELF-3 concepts and directions subtest which wasn't given to the KE family were very similar to age equivalence scores on to the TROG obtained by the KE family. Additionally, the CELF-3 sentence imitation subtest revealed substantial limitation in sentence use. On all these tasks, age equivalence scores were in the range of below 6 years to 8 years of age. Standard scores could be computed for T on the two CELF-3 subtests and these placed her between 1.66 and 2 SD below the mean for her age. These performance levels were near or at the floor of these tests for her age group. Very similar findings of significantly depressed sentence production were found in the narrative production task where high levels of ungrammatical utterances and simple sentences placed both T and B substantially below expectations for adults on this task. The fact that this poor performance with sentence use extends across both comprehension and production suggests that this difficulty with grammar and sentence use is not likely to be directly attributable to their speech production difficulties constraining their output.
Abnormality in grammar was also shown with regard to past tense marking on the UGCT. Interestingly, the patterns of performance were different for the two individuals. B displayed a strong tendency to employ regular tense marking paradigms for both regular and irregular words whereas T showed a preference for irregular tense marking. B's use of regular forms could be seen quite well on the novel irregular words which could be inflected with either an irregular form or a regular form. B regularized these forms 64% of the time and used an irregular form only 7% of the time. She also used regular forms for the novel regular words at a level similar to the normative sample. Within the Words-and-Rules (WR) model that employs either a rule-based or lexical-based solution to tense marking (Pinker and Ullman, 2002), this performance might be interpreted as evidence of a productive morphological rule system for regular tense marking. Ullman (1998; 2001) has expanded the WR model by proposing that words are acquired and stored in a declarative system and rules are learned and stored in a procedural system. If so, B's performance would be evidence of both an intact morphological rule system and procedural system which is contrary to Ullman and Gopnik's (1999) account for the affected KE family members. T's performance on the other hand would be very consistent with an impairment of a rule-based procedural learning system. As such, the same FOXP2 chromosomal rearrangement in T and B would be implicated in two very different cognitive and linguistic patterns of behavior.
Recently, Hartshorne and Ullman (2006) have found a similar pattern of over-regularization in young girls as compared with boys. These data were explained within the WR model by allowing regular forms to be generated via the stored lexical association mechanism as well as rule based means. In this model, regular forms can be stored in memory and from this store associative processes generate regularization to novel forms based on phonological similarity. This account would allow, in the case of B, for the over-regularization of tense on novel words that could take an irregular form despite an impaired procedural learning system. Thus, a tendency to over-regularize and under-regularize as shown by B and T respectively can be explained by the WR model. Harshorne and Ullman noted that this latter revised form of the WR model brings it much closer to a connectionist approach to past tense generation (McClelland & Patterson, 2002; Rumelhart & McClelland, 1986). In this case a single association mechanism that is sensitive to the phonological properties of the root drives the generalizations toward either a regular or quasi-regular (irregular) form. A key feature of an association learning system is that it would allow for a protracted learning process since individual lexical items could be learned and incorporated into the lexicon throughout life. The fact that both B and the older affected members of the KE family showed better use of the regular form than T and the younger KE family members is consistent with a slower association learning process.
General Discussion
Several reports regarding individuals with point mutations, deletions and translocation breakponts of FOXP2 have recently added to the evidence provided by the original point mutation reported for the KE family. These reports have focused on the speech sound impairments of these individuals although language impairment has also been reported. In this study, we have focused on the language phenotype in two individuals with a translocation breakpoint involving FOXP2 and the RFC3 gene on chromosome 13. By identifying the nature of the breakpoint within FOXP2, we can conclude that these individuals will have a haploinsufficiency of the FOXP2 protein product. The functional consequences of this translocation with regard to FOXP2, therefore, will be similar although not identical to the point mutation found in the KE family.
The measures of language obtained for T and B were intended to provide comparable as well as complementary language data to that reported for the KE family. In many ways T and B's overall cognitive and language profiles were similar to profiles reported for the affected family members in the KE family. In addition to the presence of a motor speech disorder as already described by us in an earlier paper (Shriberg et al., 2006), both family sets demonstrated noticeable problems in language particularly with regard to their grammatical abilities. Vocabulary abilities in both families were a relative strength. These similarities of the language profiles in the two family sets provide support for a role for FOXP2 in the neurodevelopmental pathways of the substrates of language as well as speech. It remains unclear whether this role is mediated by motor impairments affecting speech and phonological memory or whether there are systems such as the procedural system that affect learning of aspects of language directly. In recent years, the procedural system has been found to serve a range of cognitive functions, such as perceptual pattern learning that does not involve motor actions (Schultz, Apicella, Romo, & Scarnati, 1998). Thus, the language deficits found may not be by products of apraxia of speech, but rather both the poor motor performance and poor language could arise from a common impairment involving the procedural system. This view point has also been voiced by Watkins and colleagues (2002) and Lai and colleagues (Lai et al., 2003).
Although this study provides additional support for a role of FOXP2 in the neurodevelopmental pathways for language, we also have to acknowledge that to date, common population allelic variations in coding regions of FOXP2 have not been found to be associated with variations in language or speech development. At this time the coding regions of FOXP2 appear to be quite invariant. Recently, we have reported that an association of allelic variation in a regulatory (noncoding) region of FOXP2 is associated with language status as well as learning performance on a serial response task (Tomblin, Bjork, & Christiansen, 2007). These findings do support the possible role for FOXP2 in language and procedural learning. Furthermore, FOXP2 is known to be a regulatory gene and therefore it interacts with other downstream genes to influence their expression. Thus, the result of FOXP2 mutations in the KE, and TB families as well as in other individuals is a product of the altered actions of the abnormal FOXP2 protein on these downstream genes. It is very possible that population wide allelic variations in these downstream genes could be associated with differences in speech and language function. Thus, a critical question at this time has to do with the identity of these downstream genes. In fact, recently, the identity of many of these downstream genes has been determined (Spiteri, Konopka, Coppola, Bomar, Oldham, Ou, Vernes, Fisher, Ren, & Geschwind, 2007; Vernes, Spiteri, Nicod, Groszer, Taylor, Davies, Geschwind, & Fisher, 2007). This work has shown that FOXP2 can either lower or elevate expression of these downstream genes and that many of these genes play a role in neural development, neural plasticity and neural transmission.
The similarities of the cognitive, speech and language profiles of the TB and KE families are consistent with their shared abnormality of FOXP2. However, with regard to the TB family we cannot completely discount the role of RFC3 in their speech and language phenotype. As noted earlier, RFC3 itself does not have an obvious function that would be related to cognitive, speech and language skills alone because this gene is involved in more general DNA replication processes. Although a direct effect of RFC3 is not obvious, studies indicate that genetic effects often involve complex interactive pathways with other genes and environmental effects. Thus the RFC3 gene should remain as a possible candidate for further studies to determine if it is associated with speech and/or language in the general population. Additionally, as was noted earlier, the neurobeachin gene (NBEA) is nearby on chromosome 13. NBEA does have a plausible role for cognition, speech and language due to its role in neural function and one case of an individual with autism who had an NBEA deletion. Thus a breakpoint in this region could influence the functioning of this gene by altering the DNA environment of this gene.
The findings from this study are consistent with a mounting literature concerning the speech and with regard to this study the language manifestations of individuals with abnormal FOXP2. It is reasonable to ask, however, whether these features of language impairment, couldn't be attributed to non-genetic sources. In this regard, there is little doubt that the mother's (B) language difficulties could and likely did influence the daughter's (T) via environmental pathways. However B, in whom this mutation originated, did have siblings who had normal speech and language by report of B's mother and by B's report. These siblings also did not have this translocation. Additionally, the literature has provided remarkably consistent evidence of an association between abnormalities of FOXP2 that result in limitations in the dose of gene product from this gene. Thus, the current findings are intended to add to the weight of evidence in support of a role for FOXP2 in the pathways that are important for speech and language development. These findings, however, should not be taken as evidence that environmental factors do not also play a role in these pathways.
Conclusions
The genetic and language findings described in this report support the hypothesis that disruptions in FOXP2 affect mechanisms involved with both speech and language development. More generally, this report demonstrates the opportunities that exist for the use of genetics in the study of speech and language disorders. Specifically, advances in understanding are likely to come from the hypotheses that can be generated about the distal and proximal causal pathways of associated with the genetic substrates of speech and language development and use. These hypotheses arise from the recognition that genes are only one element in a complex interactive causal system that emerges from neurobiology and multiple types of environments. The identification of a gene that serves a role in this complex system will rarely provide a simple deterministic explanation for atypical rather than typical speech and language development. Rather, by identifying such genes we have the opportunity to constrain the hypothesis space within which this complexity operates and from which the elegance of speech and language arises.
Acknowledgments
This research was supported by National Institute on Deafness and Other Communicative Disorders Grant R01DC007643. We thank Connie Ferguson for her able assistance in data collection as well as the insightful suggestions provided by an anonymous reviewer.
Glossary
- Coding region
the portion of a gene that contains codons that are translated into amino acids in a protein
- Codon
a sequence of three nucleotide bases (triplet) that code for either an amino acid during translation or signal the start point or the stop point during transcription
- Derivative (der) chromosome
a chromosome that is formed by the union of two different chromosomes via a translocation or a chromosome that has been structurally altered by deletions or inversions of chromosomal material
- Frame shift
a mutation involving either the addition or deletion of one or two nucleotides that results in a change in the composition of a codon triplet
- Mutation
a permanent change in the composition of DNA in an organism
- Nonsense mutation
a point mutation that introduces a premature stop codon that results in a shortened protein product
- Point mutation
a change in one nucleotide consisting of a loss, addition, or substitution of a nucleotide base
- Stop codon
a codon that signals a termination of the translation of a protein
- Transcription
the process by which messenger RNA sequence (mRNA) is copied from a DNA (gene) sequence
- Translation
the process by which a sequence of amino acids are joined in accord with the codon sequence of mRNA
- Translocation
a transfer of a segment of DNA to a different chromosome
- Uniparental disomy
a genotype in which an individual receives both chromosomes from one parent and no copy of this chromosome from the other parent. This results in a normal dose of genetic material, however, imprinting can result in abnormal gene expression
Appendix
RFC3-FOXP2 Fusion Protein Coding Sequence
 .AGC.CTC.TGG.GTG.GAC.AAG.TAT.CGG.CCC.TGC.TCC.TTG.GGA.CGG.CTG.GAC.TAT.CAC.AAG.GAG.CAG.GCG.GCC.CAG.CTG.CGG.AAC.CTG.GTG.CAG.TGT.GGT.GAC.TTT.CCT.CAT.CTG.TTA.GTG.TAC.GGA.CCA.TCA.GGT.GCT.GGA.AAA.AAG.ACA.AGA.ATT..TGT.ATT.CTA.CGT.GAA.CTT.TAT.GGT.GTT.GGA.GTG.GAA.AAA.TTG.AGA.ATT.GAA.CAT.CAG.ACC.ATC.ACA.ACT.CCA.TCT.AAA.AAA.AAA.ATT.GAA.ATT.AGC.ACC.ATT.GCA.AGT.AAC.TAC.CAC.CTT.GAA.GTT.AAT.CCT.AGT.GAT.GCT.GGA.AAT.AGT.GAC.CGA.GTA.GTC.ATT.CAG.GAG..TTG.AAA.ACA.GTG.GCA.CAA.TCA.CAA.CAA.CTT.GAA.ACA.AAC.TCT.CAA.AGG.GAT.TTT.AAA.GTG.GTA.TTA.TTG.ACA.GAA.GTT.GAC.AAA.CTC.ACC.AAA.GAT.GCT.CAG.CAT.GCC.TTG.CGA.AGA.ACC..GAA.AAA.TAT..TCT.ACC.TGC.AGA.TTG.ATC.TTG.TGC.TGC.AAT.TCT.ACA.TCT.AAA.GTG.ATC.CCA.CCT.ATT.CGT.AGT.AGG.TGC.TTG.GCG.GTT.CGT.GTG.CCT.GCT.CCC.AGC.ATT.GAA.GAT.ATT.TGC.CAC.GTG.TTA.TCT.ACT.GTG.TGT.AAG.AAG.GAA.GGT.CTG.AAT.CTT.CCT.TCA.CAA.CTG.GCT.CAT.AGA.CTT.GCA.GAG.AAG.TCT.TGT.AGA.AAT.CTC.AGA.AAA.GCC.CTG.CTT..TGT.GAA.GCC.TGC.AGA.GTG.CAA.CAA.TAT.CCT.TTT.ACT.GCA.GAT.CAA.GAA.ATC.CCT.GAG.ACA.GAT.TGG.GAG.GTG.TAT.CTG.AGG.GAG.ACT.GCA.AAT.GCT.ATT.GTC.AGT.CAG.CAA.ACT.CCA.CAA.AG|||C.TGG.CTT.AAG.TCC.TGC.TGA.GAT.TCA.GCA.GTT..GAA.AGA.AGT.GAC.TGG.AGT.TCA.CAG.TAT.GGA.AGA.CAA.TGG.CAT.TAA.ACA.TGG.AGG.GCT.AGA.CCT.CAC.TAC.TAA.CAA.TTC.CTC.CTC.GAC.TAC.CTC.CTC.CAA.CAC.TTC.CAA.AGC.ATC.ACC.ACC.AAT.AAC.TCA.TCA.TTC.CAT.AGT.GAA.TGG.ACA.GTC.TTC.AGT.TCT.AAG.TGC.AAG.ACG.AGA.CAG.CTC.GTC.ACA.TGA.GGA.GAC.TGG.GGC.CTC.TCA.CAC.TCT.CTA.TGG.CCA.TGG.AGT.TTG.CAA.
.AGC.CTC.TGG.GTG.GAC.AAG.TAT.CGG.CCC.TGC.TCC.TTG.GGA.CGG.CTG.GAC.TAT.CAC.AAG.GAG.CAG.GCG.GCC.CAG.CTG.CGG.AAC.CTG.GTG.CAG.TGT.GGT.GAC.TTT.CCT.CAT.CTG.TTA.GTG.TAC.GGA.CCA.TCA.GGT.GCT.GGA.AAA.AAG.ACA.AGA.ATT..TGT.ATT.CTA.CGT.GAA.CTT.TAT.GGT.GTT.GGA.GTG.GAA.AAA.TTG.AGA.ATT.GAA.CAT.CAG.ACC.ATC.ACA.ACT.CCA.TCT.AAA.AAA.AAA.ATT.GAA.ATT.AGC.ACC.ATT.GCA.AGT.AAC.TAC.CAC.CTT.GAA.GTT.AAT.CCT.AGT.GAT.GCT.GGA.AAT.AGT.GAC.CGA.GTA.GTC.ATT.CAG.GAG..TTG.AAA.ACA.GTG.GCA.CAA.TCA.CAA.CAA.CTT.GAA.ACA.AAC.TCT.CAA.AGG.GAT.TTT.AAA.GTG.GTA.TTA.TTG.ACA.GAA.GTT.GAC.AAA.CTC.ACC.AAA.GAT.GCT.CAG.CAT.GCC.TTG.CGA.AGA.ACC..GAA.AAA.TAT..TCT.ACC.TGC.AGA.TTG.ATC.TTG.TGC.TGC.AAT.TCT.ACA.TCT.AAA.GTG.ATC.CCA.CCT.ATT.CGT.AGT.AGG.TGC.TTG.GCG.GTT.CGT.GTG.CCT.GCT.CCC.AGC.ATT.GAA.GAT.ATT.TGC.CAC.GTG.TTA.TCT.ACT.GTG.TGT.AAG.AAG.GAA.GGT.CTG.AAT.CTT.CCT.TCA.CAA.CTG.GCT.CAT.AGA.CTT.GCA.GAG.AAG.TCT.TGT.AGA.AAT.CTC.AGA.AAA.GCC.CTG.CTT..TGT.GAA.GCC.TGC.AGA.GTG.CAA.CAA.TAT.CCT.TTT.ACT.GCA.GAT.CAA.GAA.ATC.CCT.GAG.ACA.GAT.TGG.GAG.GTG.TAT.CTG.AGG.GAG.ACT.GCA.AAT.GCT.ATT.GTC.AGT.CAG.CAA.ACT.CCA.CAA.AG|||C.TGG.CTT.AAG.TCC.TGC.TGA.GAT.TCA.GCA.GTT..GAA.AGA.AGT.GAC.TGG.AGT.TCA.CAG.TAT.GGA.AGA.CAA.TGG.CAT.TAA.ACA.TGG.AGG.GCT.AGA.CCT.CAC.TAC.TAA.CAA.TTC.CTC.CTC.GAC.TAC.CTC.CTC.CAA.CAC.TTC.CAA.AGC.ATC.ACC.ACC.AAT.AAC.TCA.TCA.TTC.CAT.AGT.GAA.TGG.ACA.GTC.TTC.AGT.TCT.AAG.TGC.AAG.ACG.AGA.CAG.CTC.GTC.ACA.TGA.GGA.GAC.TGG.GGC.CTC.TCA.CAC.TCT.CTA.TGG.CCA.TGG.AGT.TTG.CAA. .GCC.AGG.CTG.TGA.AAG.CAT.TTG.TGA.AGA.TTT.TGG.ACA.GTT.TTT.AAA.GCA.CCT.TAA.CAA.TGA.ACA.CGC.ATT.GGA.TGA.CCG.AAG.CAC.TGC.TCA.GTG.TCG.AGT.GCA.AAT.GCA.GGT.GGT.GCA.ACA.GTT.AGA.AAT.ACA.GCT.TTC.TAA.AGA.ACG.CGA.ACG.TCT.TCA.AGC.AAT.GAT.GAC.CCA.CTT.GCA.CAT.GCG.ACC.CTC.AGA.GCC.CAA.ACC.ATC.TCC.CAA.ACC.TCT.AAA.TCT.GGT.GTC.TAG.TGT.CAC.CAT.GTC.GAA.GAA.TAT.GTT.GGA.GAC.ATC.CCC.ACA.GAG.CTT.ACC.TCA.AAC.CCC.TAC.CAC.ACC.AAC.GGC.CCC.AGT.CAC.CCC.GAT.TAC.CCA.GGG.ACC.CTC.AGT.AAT.CAC.CCC.AGC.CAG.TGT.GCC.CAA.TGT.GGG.AGC.CAT.ACG.AAG.GCG.ACA.TTC.AGA.CAA.ATA.CAA.CAT.TCC.CAT.GTC.ATC.AGA.AAT.TGC.CCC.AAA.CTA.TGA.ATT.TTA.TAA.AAA.TGC.AGA.TGT.CAG.ACC.TCC.ATT.TAC.TTA.TGC.AAC.TCT.CAT.AAG.GCA.GGC.TAT.CAT.GGA.GTC.ATC.TGA.CAG.GCA.GTT.AAC.ACT.TAA.TGA.AAT.TTA.CAG.CTG.GTT.TAC.ACG.GAC.ATT.TGC.TTA.CTT.CAG.GCG.TAA.TGC.AGC.AAC.TTG.GAA.GAA.TGC.AGT.ACG.TCA.TAA.TCT.TAG.CCT.GCA.CAA.GTG.TTT.TGT.TCG.AGT.AGA.AAA.TGT.TAA.AGG.AGC.AGT..GAC.TGT.GGA.TGA.AGT.AGA.ATA.CCA.GAA.GCG.AAG.GTC.ACA.AAA.GAT.AAC.AGG.AAG.TCC.AAC.CTT.AGT.AAA.AAA.TAT.ACC.TAC.CAG.TTT.AGG.CTA.TGG.AGC.AGC.TCT.TAA.TGC.CAG.TTT.GCA.GGC.TGC.CTT.GGC.AGA.GAG.CAG.TTT.ACC.TTT.GCT.AAG.TAA.TCC.TGG.ACT.GAT.AAA.TAA.TGC.ATC.CAG.TGG.CCT.ACT.GCA.GGC.CGT.CCA.CGA.AGA.CCT.CAA.TGG.TTC.TCT.GGA.TCA.CAT.TGA.CAG.CAA.TGG.AAA.CAG.TAG.TCC.GGG.CTG.CTC.ACC.TCA.GCC.GCA.CAT.ACA.TTC.AAT.CCA.CGT.CAA.GGA.AGA.GCC.AGT.GAT.TGC.AGA.GGA.TGA.AGA.CTG.CCC.AAT.GTC.CTT.AGT.GAC.AAC.AGC.TAA.TCA.CAG.TCC.AGA.ATT.AGA.AGA.CGA.CAG.AGA.GAT.TGA.AGA.AGA.GCC.TTT.ATC.TGA.AGA.TCT.GGA..A
.GCC.AGG.CTG.TGA.AAG.CAT.TTG.TGA.AGA.TTT.TGG.ACA.GTT.TTT.AAA.GCA.CCT.TAA.CAA.TGA.ACA.CGC.ATT.GGA.TGA.CCG.AAG.CAC.TGC.TCA.GTG.TCG.AGT.GCA.AAT.GCA.GGT.GGT.GCA.ACA.GTT.AGA.AAT.ACA.GCT.TTC.TAA.AGA.ACG.CGA.ACG.TCT.TCA.AGC.AAT.GAT.GAC.CCA.CTT.GCA.CAT.GCG.ACC.CTC.AGA.GCC.CAA.ACC.ATC.TCC.CAA.ACC.TCT.AAA.TCT.GGT.GTC.TAG.TGT.CAC.CAT.GTC.GAA.GAA.TAT.GTT.GGA.GAC.ATC.CCC.ACA.GAG.CTT.ACC.TCA.AAC.CCC.TAC.CAC.ACC.AAC.GGC.CCC.AGT.CAC.CCC.GAT.TAC.CCA.GGG.ACC.CTC.AGT.AAT.CAC.CCC.AGC.CAG.TGT.GCC.CAA.TGT.GGG.AGC.CAT.ACG.AAG.GCG.ACA.TTC.AGA.CAA.ATA.CAA.CAT.TCC.CAT.GTC.ATC.AGA.AAT.TGC.CCC.AAA.CTA.TGA.ATT.TTA.TAA.AAA.TGC.AGA.TGT.CAG.ACC.TCC.ATT.TAC.TTA.TGC.AAC.TCT.CAT.AAG.GCA.GGC.TAT.CAT.GGA.GTC.ATC.TGA.CAG.GCA.GTT.AAC.ACT.TAA.TGA.AAT.TTA.CAG.CTG.GTT.TAC.ACG.GAC.ATT.TGC.TTA.CTT.CAG.GCG.TAA.TGC.AGC.AAC.TTG.GAA.GAA.TGC.AGT.ACG.TCA.TAA.TCT.TAG.CCT.GCA.CAA.GTG.TTT.TGT.TCG.AGT.AGA.AAA.TGT.TAA.AGG.AGC.AGT..GAC.TGT.GGA.TGA.AGT.AGA.ATA.CCA.GAA.GCG.AAG.GTC.ACA.AAA.GAT.AAC.AGG.AAG.TCC.AAC.CTT.AGT.AAA.AAA.TAT.ACC.TAC.CAG.TTT.AGG.CTA.TGG.AGC.AGC.TCT.TAA.TGC.CAG.TTT.GCA.GGC.TGC.CTT.GGC.AGA.GAG.CAG.TTT.ACC.TTT.GCT.AAG.TAA.TCC.TGG.ACT.GAT.AAA.TAA.TGC.ATC.CAG.TGG.CCT.ACT.GCA.GGC.CGT.CCA.CGA.AGA.CCT.CAA.TGG.TTC.TCT.GGA.TCA.CAT.TGA.CAG.CAA.TGG.AAA.CAG.TAG.TCC.GGG.CTG.CTC.ACC.TCA.GCC.GCA.CAT.ACA.TTC.AAT.CCA.CGT.CAA.GGA.AGA.GCC.AGT.GAT.TGC.AGA.GGA.TGA.AGA.CTG.CCC.AAT.GTC.CTT.AGT.GAC.AAC.AGC.TAA.TCA.CAG.TCC.AGA.ATT.AGA.AGA.CGA.CAG.AGA.GAT.TGA.AGA.AGA.GCC.TTT.ATC.TGA.AGA.TCT.GGA..A
FOXP2-RFC3 Fusion Protein Coding Sequence
..CAG.GAA.TCT.GCG.ACA.GAG.ACA.ATA.AGC.AAC.AGT.TCA..AAT.CAA.AAT.GGA..AGC.ACT.CTA.AGC.AGC.CAA.TTA.GAT.GCT.GGC.AGC.AGA.GAT.GGA.AGA.TCA.AGT.GGT.GAC.ACC.AGC.TCT.GAA.GTA.AGC.ACA.GTA.GAA.CTG.CTG.CAT.CTG.CAA.CAA.CAG.CAG.GCT.CTC.CAG.GCA.GCA.AGA.CAA.CTT.CTT.TTA.CAG.CAG.CAA.ACA.AGT.GGA.TTG.AAA.TCT.CCT.AAG.AGC.AGT.GAT.AAA.CAG.AGA.CCA.CTG.CAG.GTG.CCT.GTG.TCA.GTG.GCC...ACT.CCC.CAG.GTG.ATC.ACC.CCT.CAG.CAA..CAG.CAG.ATC.CTT.CAG.CAA.CAA.GTC.CTG.TCT.CCT.CAG.CAG.CTA.CAA.GCC.CTT.CTC.CAA.CAA.CAG.CAG.GCT.GTC..CTG.CAG.CAG.CAA.CAA.CTA.CAA.GAG.TTT.TAC.AAG.AAA.CAG.CAA.GAG.CAG.TTA.CAT.CTT.CAG.CTT.TTG.CAG.CAG.CAG.CAG.CAA.CAG.CAG.CAG.CAG.CAA.CAA.CAG.CAG.CAA.CAA.CAG.CAG.CAG.CAA.CAA.CAA.CAA.CAA.CAG.CAG.CAA.CAA.CAG.CAG.CAG.CAG.CAG.CAA.CAG.CAG.CAG.CAG.CAG.CAA.CAG.CAT.CCT.GGA.AAG.CAA.GCG.AAA.GAG.CAG.CAG.CAG.CAG.CAG.CAG.CAG.CAA.CAG.CAA.TTG.GCA.GCC.CAG.CAG.CTT.GTC.TTC.CAG.CAG.CAG.CTT.CTC.CAG..CAA.CAA.CTC.CAG.CAG.CAG.CAG.CAT.CTG.CTC.AGC.CTT.CAG.CGT.CAG.GGA.CTC.ATC.TCC.ATT.CCA.CCT.GGC.CAG.GCA.GCA.CTT.CCT.GTC.CAA.TCG.CTG.CCT.CAA.G|||GC.TCC.TTG.AAG.TTC.GTG.GAA.GGC.TGT..AGC.TTC.TAA.CTC.ATT.GTA.TTC.CTC.CTG.AGA.TAA.TAA.TGA.AGG.GCC.TTC.TCT.CAG.AAC.TGT.TAC.ATA.ATT.GTG..GAC.AAC.TGA.AAG.GGG.AGG.TGG.CAC.AAA.TGG.CAG.CTT.ACT..AGC.ATC.GTC.TAC.AGC.TGG.GTA.GCA.AAG.CCA.TTT.ATC.ACT.TGG.AAG.CGT.TTG.TGG.CCA.AAT.TCA.TGG.CAC.TTT.ATA.AGA.AGT.TCA.TGG.AGG..GAT.TGG.AAG.GCA.TGA.TGT.TCT.GA
 (Shaded)
(Shaded)
Stop codon (Bold)
RFC3 portion of fusion sequence (Italic)
||| denotes breakpoint
Footnotes
Genetic nomenclature identifies human genes by capitalized italic font and the protein product in capitalized standard font. Non-human genes are in italic with lower case.
Apraxia and dyspraxia are treated as synonyms in this paper. We will use apraxia except in cases where the authors of a specific paper have used dyspraxia.
An exon is a portein coding sequence of DNA.
Contributor Information
J. Bruce Tomblin, Department of Speech Pathology and Audiology, University of Iowa, Iowa City, Iowa.
Marlea O'Brien, Department of Speech Pathology and Audiology, University of Iowa, Iowa City, Iowa.
Larry Shriberg, Waisman Center, University of Wisconsin, Madison, Wisconsin.
Charles Williams, Department of Pediatrics, University of Florida, Gainesville, Florida.
Jeff Murray, Department of Pediatrics, University of Iowa, Iowa City, Iowa.
Shivanand Patil, Department of Pediatrics, University of Iowa, Iowa City, Iowa.
Jonathan Bjork, Department of Pediatrics, University of Iowa, Iowa City, Iowa.
Steve Anderson, Department of Neurology, University of Iowa, Iowa City, Iowa.
Kirrie Ballard, Department of Speech Pathology, University of Sidney, Sidney, Australia.
Reference List
- Baddeley A, Wilson B. Phonological Coding and Short-Term-Memory in Patients Without Speech. Journal of Memory and Language. 1985;24:490–502. [Google Scholar]
- Belton E, Salmond CH, Watkins KE, Vargha-Khadem F, Gadian DG. Bilateral brain abnormalities associated with dominantly inherited verbal and orofacial dyspraxia. Human Brain Mapping. 2003;18:194–200. doi: 10.1002/hbm.10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop D. TROG: Test for reception of grammar. Manchester, U. K.: Age and Cognitive Performance Research Centre; 1982. [Google Scholar]
- Castermans D, Wilquet V, Parthoens E, Huysmans C, Steyaert J, Swinnen L, et al. The neurobeachin gene is disrupted by a translocation in a patient with idiopathic autism. Journal of Medical Genetics. 2003;40:352–356. doi: 10.1136/jmg.40.5.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YF, Imam JS, Wilkinson MF. The Nonsense-Mediated Decay RNA Surveillance Pathway. Annual Review of Biochemistry. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- Collaborative Linkage Study of Autism. American Journal of Human Genetics. 2000. An autosomal genomic screen for autism; pp. 278–281. [DOI] [PubMed] [Google Scholar]
- Conti-Ramsden G, Botting N, Faragher B. Psycholinguistic markers for specific language impairment (SLI) Journal of Child Psychology and Psychiatry and Allied Disciplines. 2001;42:741–748. doi: 10.1111/1469-7610.00770. [DOI] [PubMed] [Google Scholar]
- Crago MB, Gopnik M. From families to phenotypes: Theoretical and clinical implications of research into the genetic basis of specific language impairment. In: Watkins RV, Rice ML, editors. Specific language impairments in children. Baltimore; MD, US: Paul H. Brookes Publishing Co.; 1994. pp. 35–51. [Google Scholar]
- Dunn LM. Peabody Picture Vocabulary Test - Third Edition. Circle Pines, MN: Ameerican Guidance Service; 1997. [Google Scholar]
- Dunn LM, Dunn L, Whetton C, Pintillie D. British Picture Vocabulary Scale. Windsor, UK: NFER-Nelson; 1982. [Google Scholar]
- Enard W, Przeworski M, Fisher SE, Lai CSL, Wiebe V, Kitano T, et al. Molecular evolution of FOXP2, a gene involved in speech and language. Nature. 2002;418:869–872. doi: 10.1038/nature01025. [DOI] [PubMed] [Google Scholar]
- Feuk L, Kalervo A, Lipsanen-Nyman M, Skaug J, Nakabayashi K, Finucane B, et al. Absence of a paternally inherited FOXP2 gene in developmental verbal dyspraxia. American Journal of Human Genetics. 2006;79:965–972. doi: 10.1086/508902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gathercole SE, Baddeley AD. Evaluation of the role of phonological STM in the development of vocabulary in children: a longitudinal study. Journal of Memory and Language. 1989;28:200–213. [Google Scholar]
- Goldman-Eisler F. Psycholinguistics: Experiments in Spontaneous Speech. London: Academic Press; 1968. [Google Scholar]
- Gopnik M. Feature-blind grammar and dysphasia. Nature. 1990;344:715. doi: 10.1038/344715a0. [DOI] [PubMed] [Google Scholar]
- Gopnik M, Crago MB. Familial aggregation of a developmental language disorder. Cognition. 1991;39:1–50. doi: 10.1016/0010-0277(91)90058-c. [DOI] [PubMed] [Google Scholar]
- Hartshorne JK, Ullman MT. Why girls say ‘holded’ more than boys. Developmental Science. 2006;9:21–32. doi: 10.1111/j.1467-7687.2005.00459.x. [DOI] [PubMed] [Google Scholar]
- Hick R, Joseph K, Conti-Ramsden G, Serratrice L. Vocabulary profiles of children with specific language impairment. Child Language Teaching and Therapy. 2002;18:165–180. [Google Scholar]
- Hurst JA, Baraitser M, Auger E, Graham F, Norell S. An extended family with a dominantly inherited speech disorder. Developmental Medicine and Child Neurology. 1990;32:704–717. doi: 10.1111/j.1469-8749.1990.tb16948.x. [DOI] [PubMed] [Google Scholar]
- Lai CS, Fisher SE, Hurst JA, Levy ER, Hodgson S, Fox M, et al. The SPCH1 region on human 7q31: genomic characterization of the critical interval and localization of translocations associated with speech and language disorder [see comments] American Journal of Human Genetics. 2000;67:357–368. doi: 10.1086/303011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519–523. doi: 10.1038/35097076. [DOI] [PubMed] [Google Scholar]
- Lai CS, Gerrelli D, Monaco AP, Fisher SE, Copp AJ. FOXP2 expression during brain development coincides with adult sites of pathology in a severe speech and language disorder. Brain. 2003;126:2455–2462. doi: 10.1093/brain/awg247. [DOI] [PubMed] [Google Scholar]
- Lennon PA, Cooper ML, Peiffer DA, Gunderson KL, Patel A, Peters S, et al. Deletion of 7q31.1 supports involvement of FOXP2 in language impairment: clinical report and review. American Journal of Medical Genetics. 2007;143:791–798. doi: 10.1002/ajmg.a.31632. [DOI] [PubMed] [Google Scholar]
- Li SR, Weidenfeld J, Morrisey EE. Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Molecular and Cellular Biology. 2004;24:809–822. doi: 10.1128/MCB.24.2.809-822.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liegeois F, Baldeweg T, Connelly A, Gadian DG, Mishkin M, Vargha-Khadem F. Language fMRI abnormalities associated with FOXP2 gene mutation. Nature Neuroscience. 2003;6:1230–1237. doi: 10.1038/nn1138. [DOI] [PubMed] [Google Scholar]
- Liegeois F, Lai CSL, Baldeweg T, Fisher S, Monaco AP, Connelly A, et al. Behavioural and neuroimaging correlates of a chromosome 7q31 deletion containing the SPCH1 gene. Society of Neuroscience Abstracts; 2001. p. 27. Program No 529.17. [Google Scholar]
- MacDermot KD, Bonora E, Sykes N, Coupe AM, Lai CSL, Vernes SC, et al. Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. American Journal of Human Genetics. 2005;76:1074–1080. doi: 10.1086/430841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland JL, Patterson K. ‘Words or Rules’ cannot exploit the regularity in exceptions. Trends in Cognitive Science. 2002;6:464–465. doi: 10.1016/s1364-6613(02)02012-0. [DOI] [PubMed] [Google Scholar]
- Miller J, Chapman R. SALT: Systematic analysis of language transcripts [Computer software] Madison, WI: Language Analsysis Laboratory; 1993. [Google Scholar]
- Pearce AK, Humphrey TC. Integrating stress-response and cell-cycle checkpoint pathways. Trends in Cell Biology. 2001;11:426–433. doi: 10.1016/s0962-8924(01)02119-5. [DOI] [PubMed] [Google Scholar]
- Rumelhart DE, McClelland JL. In: Parallel distributed processing: Explorations in the microstrucres of cognition. Mcclelland JL, Rumelhart DE, editors. Cambridge, MA: MIT Press; 1986. pp. 216–271. [Google Scholar]
- Saffran EM, Berndt RS, Schwartz MF. The quantitative-analysis of agrammatic production - procedure and data. Brain and Language. 1989;37:440–479. doi: 10.1016/0093-934x(89)90030-8. [DOI] [PubMed] [Google Scholar]
- Schultz W, Apicella P, Romo R, Scarnati E. Contex-dependent activity in primate striatum reflecting past and future behavioral events. In: Houk J, Davis J, Beiser D, editors. Models of information processing in the basal ganglia. Cambridge, MA: MIT Press; 1998. pp. 11–28. [Google Scholar]
- Semel E, Wiig E, Secord W. Clinical Evaluation of Language Fundamentals-3. San Antonio, TX: The Psychological Corp.; 1995. [Google Scholar]
- Shriberg LD, Ballard KJ, Tomblin JB, Duffy JR, Odell KH, Williams CA. Speech, prosody, and voice characteristics of a mother and daughter with a 7;13 translocation affecting FOXP2. Journal of Speech Language Hearing Research. 2006;49:500–525. doi: 10.1044/1092-4388(2006/038). [DOI] [PubMed] [Google Scholar]
- Shu WG, Yang HH, Zhang LL, Lu MM, Morrisey EE. Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. Journal of Biological Chemistry. 2001;276:27488–27497. doi: 10.1074/jbc.M100636200. [DOI] [PubMed] [Google Scholar]
- Spiteri E, Konopka G, Coppola G, Bomar J, Oldham M, Ou J, et al. Identification of the transcriptional targets of FOXP2, a gene linked to speech and language, in developing human brain. American Journal of Human Genetics. 2007;81:1144–1157. doi: 10.1086/522237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Liu FC, Hirokawa K, Takahashi H. Expression of Foxp2, a gene involved in speech and language, in the developing and adult striatum. Journal of Neuroscience Research. 2003;73:61–72. doi: 10.1002/jnr.10638. [DOI] [PubMed] [Google Scholar]
- Thompson CK, Shapiro LP. Training sentence production in agrammatism: implications for normal and disordered language. Brain and Language. 1995;50:201–224. doi: 10.1006/brln.1995.1045. [DOI] [PubMed] [Google Scholar]
- Tomblin JB, Bjork J, Christiansen MH. Association of FOXP2 genetic markers with procedural learning and language. Paper presented at the Boston University Child Language Development Conference; Boston, MA. 2007. [Google Scholar]
- Uhlmann F, Gibbs E, Cai JS, ODonnell M, Hurwitz J. Identification of regions within the four small subunits of human replication factor C required for complex formation and DNA replication. Journal of Biological Chemistry. 1997;272:10065–10071. doi: 10.1074/jbc.272.15.10065. [DOI] [PubMed] [Google Scholar]
- Ullman M. A role for declarative and procedural memory in language. Brain and Cognition. 1998;37:142–143. [Google Scholar]
- Ullman MT. A neurocognitive perspective on language: The declarative/procedural model. Nature Reviews Neuroscience. 2001;2:717–726. doi: 10.1038/35094573. [DOI] [PubMed] [Google Scholar]
- Ullman MT, Gopnik M. Inflectional morphology in a family with inherited specific language impairment. Applied Psycholinguistics. 1999;20:51–117. [Google Scholar]
- Vargha-Khadem F, Gadian DG, Copp A, Mishkin M. FOXP2 and the neuroanatomy of speech and language. Nature Reviews Neuroscience. 2005;6:131–138. doi: 10.1038/nrn1605. [DOI] [PubMed] [Google Scholar]
- Vargha-Khadem F, Watkins K, Alcock K, Fletcher P, Passingham R. Praxic and nonverbal cognitive deficits in a large family with a genetically transmitted speech and language disorder. Proceedings of the National Academy of Sciences. 1995;92:930–933. doi: 10.1073/pnas.92.3.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargha-Khadem F, Watkins KE, Price CJ, Ashburner J, Alcock KJ, Connelly A, et al. Neural basis of an inherited speech and language disorder. Proceedings of the National Academy of Sciences. 1998;95:12695–12700. doi: 10.1073/pnas.95.21.12695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernes SC, Nicod J, Elahi FM, Coventry JA, Kenny N, Coupe AM, et al. Functional genetic analysis of mutations implicated in a human speech and language disorder. Human Molecular Genetics. 2006;15:3154–3167. doi: 10.1093/hmg/ddl392. [DOI] [PubMed] [Google Scholar]
- Vernes SC, Spiteri E, Nicod J, Groszer M, Taylor JM, Davies KE, et al. High-throughput analysis of promoter occupancy reveals direct neural targets of FOXP2, a gene mutated in speech and language disorders. American Journal of Human Genetics. 2007;81:1232–1250. doi: 10.1086/522238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins KE, Dronkers NF, Vargha-Khadem F. Behavioural analysis of an inherited speech and language disorder: comparison with acquired aphasia. Brain. 2002;125:452–464. doi: 10.1093/brain/awf058. [DOI] [PubMed] [Google Scholar]
- Watkins KE, Vargha-Khadem F, Ashburner J, Passingham RE, Connelly A, Friston KJ, et al. MRI analysis of an inherited speech and language disorder: structural brain abnormalities. Brain. 2002;125:465–478. doi: 10.1093/brain/awf057. [DOI] [PubMed] [Google Scholar]
- Wechsler D. Wechsler Adult Intelligence Scale-Third Edition. San Antonio, TX: The Psychological Corporation; 1997. [Google Scholar]
- Williams K. Expressive Vocabulary Test. Circle Pines, MN: American Guidence Service; 1997. [Google Scholar]
- Zeesman S, Nowaczyk MJ, Teshima I, Roberts W, Cardy JO, Brian J, et al. Speech and language impairment and oromotor dyspraxia due to deletion of 7q31 that involves FOXP2. American Journal of Medical Genetics. 2006;140:509–514. doi: 10.1002/ajmg.a.31110. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Webb DM, Podlaha O. Accelerated protein evolution and origins of human-specific features: FOXP2 as an example. Genetics. 2002;162:1825–1835. doi: 10.1093/genetics/162.4.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]