

Abstract
Intrinsically disordered proteins are predicted to be highly abundant and play broad biological roles in eukaryotic cells. In particular, by virtue of their structural malleability and propensity to interact with multiple binding partners, disordered proteins are thought to be specialized for roles in signaling and regulation. However, these concepts are based on in silico analyses of translated whole genome sequences, not on large-scale analyses of proteins expressed in living cells. Therefore, whether these concepts broadly apply to expressed proteins is currently unknown. Previous studies have shown that heat-treatment of cell extracts lead to partial enrichment of soluble, disordered proteins. Based on this observation, we sought to address the current dearth of knowledge about expressed, disordered proteins by performing a large-scale proteomics study of thermo-stable proteins isolated from mouse fibroblast cells. Using novel multidimensional chromatography methods and mass spectrometry, we identified a total of 1,320 thermo-stable proteins from these cells. Further, we used a variety of bioinformatics methods to analyze the structural and biological properties of these proteins. Interestingly, more than 900 of these expressed proteins were predicted to be substantially disordered. These were divided into two categories, with 514 predicted to be predominantly disordered and 395 predicted to exhibit both disordered and ordered/folded features. In addition, 411 of the thermo-stable proteins were predicted to be folded. Despite the use of heat treatment (60 min. at 98 °C) to partially enrich for disordered proteins, which might have been expected to select for small proteins, the sequences of these proteins exhibited a wide range of lengths (622 ± 555 residues (average length ± standard deviation) for disordered proteins and 569 ± 598 residues for folded proteins). Computational structural analyses revealed several unexpected features of the thermo-stable proteins: 1) disordered domains and coiled-coil domains occurred together in a large number of disordered proteins, suggesting functional interplay between these domains, and 2) more than 170 proteins contained lengthy domains (>300 residues) known to be folded. Reference to Gene Ontology Consortium functional annotations revealed that, while disordered proteins play diverse biological roles in mouse fibroblasts, they do exhibit heightened involvement in several functional categories, including, cytoskeletal structure and cell movement, metabolic and biosynthetic processes, organelle structure, cell division, gene transcription, and ribonucleoprotein complexes. We believe that these results reflect the general properties of the mouse intrinsically disordered proteome (IDP-ome) although they also reflect the specialized physiology of fibroblast cells. Large-scale identification of expressed, thermo-stable proteins from other cell types in the future, grown under varied physiological conditions, will dramatically expand our understanding of the structural and biological properties of disordered eukaryotic proteins.
Keywords: intrinsically disordered proteins, intrinsically unstructured proteins, proteomics, mammalian proteome, thermo-stable proteins
Introduction
Based on theoretical translations of whole genome sequences, approximately 30-40% of all eukaryotic proteins are predicted to be either entirely disordered or contain long disordered regions1, 2. Further, bioinformatics analyses have strongly suggested that these theoretical, intrinsically disordered proteins (DPs) play broad roles in biological systems, especially in molecular signaling and regulation 3-14, and that many DPs are involved in the pathogenesis of a wide range of human diseases, including cancer, malaria, AIDS, and amyloid diseases 12, 14-17. However, despite their predicted high abundance and broad biological roles in eukaryotes, few studies have focused on large-scale analysis of the subset of DPs that are actually expressed in eukaryotic cells at a given time and under specific environmental conditions. It is important to understand not only the theoretical upper limit of the number of all DPs encoded by genomes, but also to understand which DPs are actually expressed under certain physiological conditions and how cells vary their expressed DP repertoire in response to changing conditions and external stimuli. Because it is not currently possible to predict protein expression patterns on the basis of genome sequence information alone, experimental methods are required for large-scale detection of expressed DPs.
We addressed this issue by developing proteomics techniques to study a large fraction of the DPs that are expressed in mouse fibroblast cells. Previously, we and others reported that heat-treatment of soluble cellular extracts afforded modest selectivity for DPs and selectivity against highly abundant, folded proteins (FPs)18-20. This method, combined with two-dimensional polyacrylamide gel electrophoresis (2D PAGE), allowed identification of 114 cytosolic and nuclear DPs from mouse fibroblast cells, many of which are involved in cellular signaling and regulation18. However, due to the inherent low dynamic range of this protein identification method, the majority of these were high abundance proteins. While some are highly abundant, many other proteins involved in signaling and regulation are present at low levels in cells. Thus, it was necessary to use techniques capable of greater proteome penetration to identify a larger fraction of proteins in the intrinsically disordered proteome (referred to as the “IDP-ome” here and the “unfoldome” by others21) of mouse fibroblast cells.
Improved penetrance of the IDP-ome in the current study was achieved using a two step procedure. In the first step, we used multi-dimensional protein identification technology (MudPIT)22, to identify 1,320 thermo-stable (TS) proteins in a heat-treated extract of mouse fibroblast cells. Our past IDP-ome study showed that a large fraction of the proteins detected in the heat-treated, soluble extract from mouse fibroblast cells were DPs18. Therefore, we reasoned that the same selection procedure, combined with highly sensitive MudPIT, would allow identification of a large number of additional, lower abundance, DPs. In the second step of our procedure, the experimentally identified TS proteins were structurally analyzed using bioinformatics methods. While MudPIT was capable of identifying more than 1,300 individual proteins amongst the many thousands that were present in the heat-treated cell extract, it was not possible to structurally characterize each of the identified proteins within the cell extract using mass spectrometry or other analytical methods. Therefore, it was necessary to use sequence analysis algorithms to computationally analyze the structural properties of the identified TS proteins. Using several, well-validated disorder prediction programs, including NORSnet23, IUPred24, and DISOPRED225, we demonstrated that proteins exhibiting significant disorder were over-represented in the TS dataset with respect to the entire mouse proteome, with up to 69% identified as being fully or partially disordered by these prediction methods. In addition, we used the disorder prediction program, PONDR26, to analyze the overall structural properties of each of the TS proteins and classified them as being predominantly disordered (termed “disordered proteins”, DPs), predominantly folded (termed “folded proteins”, FPs), or of mixed character (termed “mixed proteins”, MXPs). Using this classification system, more than 900 proteins were predicted to contain disordered domains and classified as DPs or MXPs. Interestingly, of these >900 proteins, only 53 have previously been experimentally characterized as being either partially or wholly disordered, illustrating the limitations of our current knowledge of disordered proteins that are expressed in living cells.
Proteins in the TS dataset exhibited diverse and novel structural features. First, despite exposure to an extreme temperature, the primary structures of these proteins spanned a wide range of lengths (627 ± 646 residues; average length ± standard deviation), with 50 exceeding 2,000 residues. This range of lengths is generally representative of that for proteins in the entire mouse and human proteomes. Second, a large number of disorder-containing proteins classified as DPs and MXPs (21% and 14%, respectively) also contained segments predicted to be coiled-coils. Since both disordered and coiled-coil domains are known to mediate protein-protein interactions, this observation suggests that these independent domains may cooperate to mediate biological function. Third, almost 200 proteins in the TS dataset were predicted to contain lengthy (>300 residues in length), folded regions while 65 others were predicted to contain trans-membrane (TM) domains. Many of these regions and domains occur in proteins that are predicted to otherwise be extensively disordered, a factor which may mitigate the tendency of folded, hydrophobic polypeptide segments (soluble and globular, and membrane-spanning) to denature and precipitate upon heating. This survey of the unusual structural characteristics of proteins with both disordered and ordered features within the TS dataset highlights how little is currently known about the physical properties of the thousands of proteins expressed in living mouse cells and emphasizes the need for large-scale studies of expressed proteins.
Relationships between disorder (and order) and biological function were analyzed by evaluating the sub-cellular localizations, biological processes and molecular functions associated with all 1,320 proteins in the TS dataset using the Gene Ontology (GO) Consortium database (www.geneontology.org). Importantly, this analysis revealed that DPs and MXPs are involved not only in signaling and regulation, as often noted, but also in a wide range of other, previously uncharacterized biological functions and processes. Relationships between protein disorder and biological function were further probed by analyzing the occurrence of post-translational modifications and alternative splicing for the proteins in the TS dataset.
These novel insights into the structural and functional properties of proteins in the TS dataset were gained by applying state-of-the-art methods to detect a very large number of expressed mouse proteins. Bioinformatics analysis of the sequences of these expressed proteins revealed that the majority were significantly disordered (514 DPs plus 395 MXPs), far exceeding the number reported in our past proteomics study18. Significantly, we estimate that this represents up to ∼75% penetrance of the mouse IDP-ome. This pattern of disordered protein expression reflects the specialized physiology of fibroblasts and is likely to vary with cell type and physiological state. These results provide motivation to apply similar protein detection and analysis methods to other cell types in the future in order to further expand our understanding of the relationships between disorder and biological function for proteins expressed in eukaryotic cells.
Experimental Section
Cell Culture
Arf-null mouse NIH 3T3 fibroblast cells were maintained in Dulbeccos modified Eagles media (DMEM) supplemented with 10% fetal bovine serum and 2 mM glutamine. Cells were grown at 37 °C in a humidified incubator with a 5% CO2 atmosphere. For large-scale experiments cells were grown on 20 cm × 20 cm plates that yielded approximately 1 × 107 cells at 80% confluence.
Thermo-stable Protein Enrichment
Thermo-stable proteins were isolated from mouse fibroblasts as described previously18. Briefly, mouse fibroblasts (8 × 107) were washed with cold PBS buffer, harvested with a cell scraper and resuspended in 1 ml of Buffer A (10 mM sodium phosphate, pH 7.0, 50 mM NaCl, 50 mM DTT, 1 × protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) and 0.1 mM sodium orthovanadate). The cells were lysed and then centrifuged at 16,000 × g for 30 min at 4 °C. The supernatant was transferred to a fresh tube, diluted to a protein concentration of approximately 1 mg/ml with Buffer A and heated at 98 °C for 1 h. Following heating the protein mixture was placed on ice for 15 min and then spun at 16,000 × g for 15 min at room temperature to pellet aggregated and precipitated proteins. Soluble proteins in the supernatant were precipitated with 20% TCA at -20 °C, washed three times with cold (-20 °C) acetone and the pellet was stored at -80 °C for further analysis.
Trypsin Digest of Thermo-stable Proteins
Proteins (330 μg) were dissolved in a solution containing 50 mM Tris pH 8.0 and 8.0 M urea and reduced with 10 mM DTT at 37 °C for 1 hour. Following carboxyamidomethylation by adding iodoacetamide to a final concentration of 50 mM and incubating at room temperature for 1 hour, the protein mixture was digested with 5 μg of endopeptidase lys-C (Sigma Aldrich, St. Louis, MO) at 37 °C for 15 hours. The mixture was diluted 4 fold with a solution containing 10 mM ammonium bicarbonate, pH 8.0, 4 mM CaCl2 and then digested with 10 μg of trypsin (Promega, Madison, WI) at 37 °C for 3 hours. The pH was adjusted to 10.0 by adding 200 mM ammonium formate, pH 10.0, immediately prior to loading onto the reversed-phase HPLC column.
Reversed-phase Chromatography of Tryptic Peptides at High pH
The first dimension of the 2D-LC separation of tryptic peptides was performed off-line on a reversed-phase column at high pH according to published protocols27, 28. Briefly, reversed-phase experiments at high pH were performed on a Xterra MS C18 column (2.1 × 150 mm, 3.5 μm particle) (Waters Corporation, Milford, MA). Mobile phase A was water, B was acetonitrile and C was 200 mM ammonium formate buffer at pH 10. Pump C was used to isocratically deliver 10% of the solvent so that the chromatography solvent always contained 20 mM NH4CO2H. Aliquots (50 μl) of trypsin digested, heat-treated, mouse fibroblast extract (200 μl) were loaded onto a column equilibrated at 30 °C at a flow rate of 200 μl/min. Tryptic peptides were eluted using a gradient of 0 – 50% B buffer (60 min) at a flow rate of 200 μl/min. Fractions (30 s) were collected into tubes containing 10 μl 2% formic acid, evaporated to dryness in a Savant SC110 speedvac and then resuspended in 40 μl of 0.2 formic acid.
LC-MS/MS Analysis and Database Searching
LC-MS/MS analyses were carried out using a Finnigan LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific, Inc., Waltham, MA) in line with a nanoAcquity ultra performance LC system (Waters Corporation, Milford, MA). Peptides were loaded onto a “precolumn” (Symmetry C18, 180 μm i.d × 2 0mm, 5 μm particle) (Waters Corporation) which was connected through a zero dead volume union to the analytical column (BEH C18, 75 μm i.d × 100 mm, 1.7 μm particle) (Waters Corporation) equilibrated with solvent D (0.2% formic acid / 98% water / 2% acetonitrile). The peptides were eluted using a gradient (0-70% E in 60 min, 70-100% E in 10 min, where solvent E was 70% acetonitrile, 0.2% formic acid in water) at a flow rate of 250 nL/min and introduced online into the linear ion trap mass spectrometer using electrospray ionization (ESI). Following acquisition of each full-scan mass spectrum, 10 precursor ions were chosen for collision-activated dissociation (CAD) in a data-dependent manner (one microscan per MS2 spectrum; precursor isolation window m/z ± 1.5 Da, 35% collision energy, 30 ms ion activation, 35 s dynamic exclusion, repeat count 2).
Product ions generated by CAD were searched against the Mus. musculus subset (11,747 sequences) of the SwissProt non-redundant protein sequence database (Version 50.9; 235,673 sequences; 86,495,188 residues) using the MASCOT search engine (Matrix Science Inc., London, U.K.). The following residue modifications were allowed: fixed, cysteine (carbamidomethylation) and variable, methionine (oxidation). The following parameters were used: enzyme, trypsin; mass values, monoisotopic; protein mass, unrestricted; peptide mass tolerance, ± 1.5 Da; fragment mass tolerance, ± 1.5 Da; maximum number of missed cleavages, 2; instrument type, ESI-TRAP; and number of queries searched, 531,134. For display purposes, the significance threshold of p < 0.05, an ions score cut-off of 35, and the requirement of bold red were used. Identifications from the automated search were further validated through manual inspection; this process yielded 1,320 validated protein identifications which is termed the TS protein dataset (Suppl. Table 1).
Bioinformatics Analysis of Protein Disorder
Proteins in the TS dataset were analyzed with regard to order/disorder using several different disorder prediction programs and different criteria for structural classification. First, we used three complementary disorder predictors, NORSnet23, IUPred24, and DISOPRED229, to predict the number of proteins in the TS protein dataset which contained at least one disordered region ≥30 residues in length. We used these three predictors because they use complementary sequence analysis methods and are known to give complementary results23, 30. For example, NORSnet23 uses feed-forward neural networks trained on polypeptide regions predicted to lack secondary structure to predict the location of disordered regions within proteins. In contrast, IUPred24 uses an empirically-derived energy function based on the statistics of amino acid contacts in proteins to predict the location of disordered regions. Finally, DISOPRED229 uses a support vector machine-based algorithm trained on residues that are disordered in high-resolution X-ray crystal structures to predict the location of disordered regions. In order to define a residue as disordered we used three different parameter sets to establish prediction thresholds that were determined through independent studies on proteins listed in the DisProt database [www.disprot.org]. The stringency levels associated with these parameter sets were: 1) 10% false positive rate on a per-residue basis (termed “Stringent”), 2) 1% false positive rate on a per-protein basis (termed “Intermediate”), and 3) 5% false positive rate on a per-protein basis (termed “Permissive”). Different protein training sets and/or empirical data were used to develop the NORSnet, IUPred, and DISOPRED2 predictors. As noted, the three levels of prediction stringency were achieved by empirical adjustment of prediction parameters using experimentally verified disordered proteins in the DisProt database. The prediction results for the TS protein dataset are largely independent of the methods used for establishing prediction stringency because only a small fraction of the proteins in the TS dataset (4.9%) exhibited sequence similarity to proteins in DisProt.
Second, we used the VL-XT disorder predictor and the charge-hydropathy analysis tool within the PONDR suite of programs26, 31, 32 to classify the average structural features of each protein in the TS dataset. The VL-XT algorithm predicts the likelihood that each residue in a protein exists in an ordered or disordered conformation using 1) a feed-forward neural network trained using the physical attributes of disordered regions from a small set of proteins (calcineurin sequences from 13 species) and ordered regions from structured proteins in the NRL-3D database32, and 2) two feed-forward networks trained on the sequences of 115 N-terminal and 84 C-terminal disordered regions, respectively, from proteins in the PDB-select-25 database31. Individual residue prediction scores ranged from 0 (order) to 1 (disorder) and these values for each residue were averaged over all residues to give the average PONDR order/disorder score. As was the case for the three predictors described above, we argue that the prediction results for the TS protein dataset based on use of PONDR are largely independent of the methods used to develop this predictor because 1) only two proteins in the TS dataset are related to the calcineurin sequences used for training and 2) a relatively small number of disordered terminal segments of structured proteins from many organisms were used for training, making significant overlap with proteins in the TS dataset unlikely. In addition, PONDR was used to compute the average charge (C) and hydropathy (H) score for each protein according to Uversky, et al.33. Individual C and H values were related to a line defined by C = 2.785 × H − 1.151 in a two-dimensional coordinate system; the (C, H) values for individual proteins occurred either on the left-hand side or right-hand side of this line. Proteins were classified as follows18: DPs exhibited an average disorder/order score > 0.5, or an average disorder/order score ≤ 0.5 and > 0.32 and (C, H) scores which occurred to the left of the boundary line; FPs exhibited an average disorder/order score < 0.32, or an average disorder/order score ≤ 0.5 and ≥ 0.32 and (C, H) scores which occurred to the right of the boundary line; and MXPs did not satisfy the previous criteria. Our classification system, while developed independently, has relevance to an earlier report on computational methodologies used to identify “mostly disordered” proteins2. This report, which also used PONDR disorder/order and (C, H) scores to evaluate order and disorder within proteins although in a quantitatively different manner than presented herein, noted that proteins that were predicted by both scores to be disordered, and others which were predicted by the PONDR score to be disordered and the (C, H) score to be ordered, were likely to constitute distinct structural classes, the former corresponding to highly extended, disordered proteins and the latter corresponding to proteins with collapsed but disordered polypeptide chains (e.g. molten globules). These observations suggest that consideration of both PONDR and (C, H) scores allows different types of disordered proteins to be discriminated, justifying our use of three structural categories (DP, MXP, and FP) to classify the TS proteins detected in our study. The ability of these two structural parameters to discriminate between different types of disordered proteins may arise because they detect different structural features of polypeptide chains, as suggested by the observation of only a weak linear correlation between the PONDR and (C, H) scores for proteins in the TS dataset (R = 0.69). Average hydropathy scores (Suppl. Table 3) exhibited a similar poor linear correlation with PONDR scores (R = 0.69) and average charge scores were even more weakly correlated with PONDR scores (R = 0.33). As noted earlier, we view our PONDR-based disorder/order prediction results and structural classifications for mouse TS proteins to be largely independent of the manner in which the various algorithms which comprise PONDR were trained because many different proteins from many different organisms were used for training. We are not aware that any of the training datasets were enriched in thermostable proteins so as to introduce bias in our disorder/order prediction results.
The results of these analyses were stored in a MySQL database and accessed through a web interface written in PHP. The web interface displayed protein identifications and PONDR analysis results. Data could be sorted according to charge, hydropathy, average PONDR score, and other parameters to facilitate manual analysis. The following information derived from these structural analyses is included in Suppl. Table 3: protein length (number of residues), average PONDR VL-XT score, average charge score, average hydropathy score, the distance of these values from the boundary line between disordered and ordered proteins (as define above), and structural classification. In addition, proteins in the TS dataset were searched against the DisProt database (http://www.disprot.org/) 34 using BLAST to identify matches with >20% identity. When matches were found, the ID number, source organism name and percentage identity (with respect to the mouse TS dataset entry) for the DisProt entries were included in Suppl. Table 3.
Analysis of GO terms and other bioinformatics analyses
The biological properties of proteins within the TS protein dataset were analyzed by reference to the classification system of the Gene Ontology (GO) Consortium35. For these analyses, the TS proteins were divided into two groups: disordered proteins (DPs + MXPs; 909 proteins) and folded proteins (FPs; 411 proteins). For each group, the proteins were functionally classified using GO terms for three categories (level-0 terms): cellular component, biological process, and molecular function. The mouse gene and GO term association file available from Mouse Genome Informatics (MGI, ftp://ftp.informatics.jax.org/pub/reports/index.html#go) was used and all the mouse protein or gene identifiers were converted to Swiss-Prot primary accession numbers for the downstream analyses. Fisher's exact test was used to determine the over-represented or under-represented GO terms for the three ontology categories noted above and the P values were corrected for multiple testing using the false discovery rate (FDR) controlling procedure of Benjamini and Hochberg36. A cutoff of FDR < 0.01 was used to score significantly over- or under-represented GO terms, corresponding to a 1% false positive rate. The results of these analyses for level-2 terms are summarized in Fig. 3 and the results for terms at all levels are given in Suppl. Table 10. In Fig. 3, only the results for over-represented or under-represented level-2 GO terms associated with ≥10 disordered or folded/ordered proteins are presented. We have focused our functional analysis of proteins in the TS dataset on level-2 terms in three level-0 categories (cellular component, biological process, and molecular function) because, at level-2, a modest number of GO terms were shown to be over- or under-represented, allowing the overall results to be discussed in the text. Further, level-2 term names often provide insights into specific biological function of proteins with which they are associated. We report all over- or under-represented GO terms in Suppl. Table 10 to provide more detailed insights into the biological functions of proteins in the TS dataset.
Figure 3.

Biological functions associated with disordered (DPs + MXPs) and folded/ordered proteins (FPs) in TS dataset. Graphical representation of over- and under-representation of GO terms for disordered proteins (DPs + MXPs) and folded ordered proteins (FPs) for three functional categories, (A) cell component, (B) biological process, and (C) molecular function. Results are shown only for over- and under-represent GO terms with false discover rate (FDR) values <0.01 and with ≥10 associated proteins. The column labeled Background indicates the percentage of all theoretical mouse proteins that exhibited a particular GO term using a gray scale. The columns labeled (DPs + MXPs) and FPs indicate the extent of over- (red scale) or under-representation (green scale) of a particular GO term, given as [(LH/LT) – (BH/BT)]/(BH/BT) × 100; where: LH is the number of disordered or folded proteins associated with a particular GO term, LT is the total number of disordered or folded proteins with any GO term, BH is the number of theoretical mouse proteins associated with a particular GO term, and BT is the number of theoretical mouse proteins associated with any GO term. The color and gray scales are defined in the lower right. Gray boxes in the columns labeled (DPs + MXPs) and FPs indicate that the noted GO term was not over- or under-represented for the indicated structural class and have the same shade as the box labeled Background; asterisks in the columns labeled (DPs + MXPs) and FPs indicate that zero proteins in the indicated structural class were associated with the noted GO term.
In addition, information on the occurrence of known sites of post-translational modification and alternative splicing for proteins in the TS dataset was obtained using the proteomics software suite ProteinCenter (Proxeon Biosystems A/S, Odense Denmark).
In the course of these proteomics studies, the SwissProt identifications for 37 proteins in the TS dataset were updated; the original names for these appear in Suppl. Table 1 and the new names, with synonyms indicated in brackets, appear in Suppl. Table 3.
Protein-Protein Interactions
The OPHID database (http://128.100.65.8/ophidv2.201/index.jsp) was queried to identify proteins having known or predicted protein-protein interactions. This database is comprised of 295,131 interactions of which 162,054 are known and 133,885 are predicted. The Protein Information Resource (PIR; http://pir.georgetown.edu/) was used to extract information regarding protein three-dimensional structures (RSCB database). The disordered protein database DisProt (http://www.disprot.org/) was searched to identify proteins having experimentally characterized disordered regions.
Prediction of Protein Transmembrane Helices
We used two methods to predict integral transmembrane helices: TMHMM2 37 and PHDhtm 38. These two methods were among the best such predictors in recent assessments 39, 40. TMHMM2 is based on a hidden Markov model while PHDhtm utilizes a neural network. We ran the two methods with default parameters and reported the number of proteins predicted to have at least one transmembrane helix. Overall TMHMM2 and PHDhtm predicted 65 such proteins, 54 of them in common (Suppl. Table 8).
Prediction of Coiled-Coil Regions
In order to predict coil-coiled regions we used the program MARCOIL 41, a hidden Markov model-based method that was evaluated as the best performing such predictor by a recent assessment 42. We ran MARCOIL with default parameters on several datasets: the entire mouse genome, the TS protein dataset, and individually on the DP, MXP, and FP protein subsets of the TS dataset.
Identification of Folded Domains
The sequences of all proteins in the TS protein dataset were compared to all sequences in the Protein Data Bank (PDB) using the program BLAST43. The list of PDB sequences was retrieved from the Research Collaboratory for Structural Bioinformatics FTP site (ftp://ftp.rcsb.org) and formatted as a searchable database for BLAST using the NCBI program “formatdb”. The BLAST analyses were performed twice, once saving all sequences in which domains of ≥ 60 residues exhibited sequence identities of ≥25% with respect to at least one sequence in the PDB, and a second time saving all sequences in which domains of ≥ 300 residues exhibited sequence identities of ≥25% with respect to at least one sequence in the PDB. In cases where more than one structure matched the query protein, only the structure with the highest bit score was retained.
Results
Large-scale Identification of Thermo-stable Proteins from Mouse Fibroblast Cells
Thermo-stable (TS) mouse proteins were obtained by heating the soluble extract from fibroblast cells at 98 °C for 1 hour, followed by centrifugation to remove precipitates. Proteins were digested with endoproteinase Lys-C and trypsin. The resulting peptides were fractionated by two-dimensional ultra-high performance liquid chromatography, and subjected to tandem mass spectrometry to identify the proteins from which they were derived. For this purpose, the eluent stream from the second chromatographic separation was introduced into a linear ion-trap mass spectrometer and subjected to electrospray ionization. From the ions detected in full-scan spectra, precursors were selected in a data-dependent manner for collision-activated dissociation. The resulting product ion spectra were assigned to peptide sequences, and these sequences were compiled to form a protein list, by using the MASCOT search engine. A total of 1,320 non-redundant TS proteins were identified (Suppl. Table 1A). All proteins were identified with two or more peptides and 1,289 proteins (97.7%) were identified by 5 or more peptides (Suppl. Table 1B). This is approximately 5-fold and 10-fold higher than the number of proteins previously identified by 2D polyacrylamide gel electrophoresis (2D PAGE) analysis of untreated and heat-treated extracts, respectively 18. Additional details of the configuration and performance of the instruments used in the MudPIT procedure employed to identify these soluble, heat-stable proteins will be provided in a separate manuscript (submitted).
Structural Analysis of Thermo-stable Mouse Proteins
We used two different approaches to computationally analyze the occurrence of disorder in proteins in the TS dataset. In a first approach, we used three complementary disorder predictors, NORSnet23, IUPred24 and DISOPRED225, to estimate the frequency with which disordered segments of ≥30 residues occurred within these proteins. For each predictor, three different, empirically-derived levels of stringency were applied for these predictions corresponding to different false positive rates (Suppl. Table 2). At the intermediate stringency level corresponding to a 1% false positive rate per protein, 488 (836) proteins (37% (63%) of all TS proteins) were predicted to contain at least one disordered segment of ≥30 residues by all three (at least one) of the predictors. The percentage of all theoretical proteins in the mouse proteome predicted by all three predictors to contain at least one disordered segment of ≥30 residues was 40% and the percentage predicted by at least one of the three predictors was 46%. The former percentage is similar to that obtained for proteins in the TS dataset while the latter is significantly smaller, suggesting that proteins with at least one disordered segment of ≥30 residues are over-represented in the TS dataset. These analyses indicate that the TS protein dataset is a rich source of expressed, disordered proteins.
In a second computational approach, we used the program PONDR26 to predict the average structural properties of and to structurally classify each protein in the TS dataset (Suppl. Table 3). Based on this analysis, proteins were classified as being predominantly disordered (termed “disordered proteins”, DPs), predominantly folded/ordered (termed “folded proteins”, FPs), or of mixed disordered and folded character (termed “mixed proteins”, MXPs). While the computational analysis approach discussed above accurately predicted the occurrence of short disordered segments within TS proteins, the probability of occurrence of these segments increased with protein size. Since the proteins in the TS dataset exhibited a remarkably wide range of lengths (627 ± 646 residues), we also used the second analysis approach, which classified proteins on the basis of average disorder/order and charge-hydropathy scores, to normalize for protein length. The details of our structural classification system are given under Materials and Methods. For clarity, proteins classified as DPs or FPs were predicted to be predominantly disordered or folded, respectively. Proteins classified as having mixed character often exhibited both disordered segments and folded domains. However, proteins in this class may also exhibit structural features which fall between disorder and order; for example proteins in this class may exhibit collapsed but disordered structures (e.g. molten globules), as was previously suggested2. Interestingly, the proportions of DPs, MXPs and FPs in the current TS dataset (39%, 30% and 31%, respectively) were similar to those reported previously for proteins identified by 2D PAGE (Figure 1 and Suppl. Table 4)18. Proteins in each structural category exhibited a wide range of sequence lengths: DPs, 622 ± 555 residues; MXPs, 693 ± 784 residues; and FPs, 569 ± 598 residues. These values are slightly larger than the average value for mouse proteins in SwissProt (average length, 485 residues)44 and all predicted human proteins (510 ± 604 residues)45 and indicate that the length distribution of proteins in the TS dataset is generally representative of that observed in the entire mouse and human proteomes. We note, however, that the MudPIT methods that were used to detect TS proteins may introduce bias toward the detection of proteins with long sequences since these proteins are more likely to yield multiple, detectable tryptic peptides. However, because the length distribution of the TS proteins is in accord with that observed for other proteomes, we believe that this potential bias was a minor factor in our study.
Figure 1. Percentage of proteins identified as DPs, MXPs and FPs in control and heat-treated samples from mouse fibroblasts.

The percentages of proteins classified as DPs (blue bars), MXPs (open bars), and FPs (orange bars) in datasets derived from 2D PAGE analyses of untreated (left) and heat-treated (center) mouse fibroblast cell extracts are compared to those determined through MudPIT analysis (right) of a similar heat-treated extract. The total number of proteins detected in each experiment is shown at the top in parentheses.
Coexistence of Disordered and Coiled-coil Domains in Thermo-stable Mouse Proteins
We previously noted that a significant number of TS proteins from mouse fibroblasts exhibited segments predicted to fold into oligomeric coiled-coil structures46. We believe that proteins containing coiled-coil domains survive our heat-treatment procedure because these domains are comprised predominantly of charged and polar residues and, therefore, are highly soluble, even under conditions of thermal denaturation. For example, the leucine-zipper heptad motif, which comprises coiled-coil polypeptide segments, consists of two hydrophobic residues47, 48 separated by several charged and hydrophilic residues which confer high solubility under conditions of heat-treatment. Therefore, proteins which contain coiled-coil domains, possibly in addition to other disordered and/or folded domains, may remain soluble at 98° C. In addition, while coiled-coil domains are known in hundreds of cases to adopt folded structures49, the chemical nature of residues in this motif (five of seven are either charged or small and polar47, 48) causes many coiled-coil segments to be predicted to be disordered by PONDR18. Therefore, because we identified coiled-coil proteins in the past in heat-treated mouse fibroblast extracts18 and because these segments are likely to be folded but are predicted by PONDR to be disordered18, we used several approaches to analyze the occurrence of coiled-coil segments within the proteins in our TS dataset. Initially, all TS protein sequences were analyzed using the coiled-coil prediction program MARCOIL41. In total, 13% (166) of the TS proteins were predicted to contain a least one coiled-coil segment ≥30 residues in length (99% confidence limit per residue). Most of these coiled-coil proteins were structurally classified as DPs (108, 21% of all DPs) or MXPs (48, 12% of all MXPs) (Suppl. Figure 1) and relatively few as FPs (10, 2% of all FPs). We tested our hypothesis that heat-treatment may enrich for coiled-coil domain-containing proteins by comparing coiled-coil predictions for proteins in the TS dataset and the entire mouse proteome. Using a more stringent cutoff for prediction of coiled-coil segments by MARCOIL (90% confidence limit per protein), we determined that coiled-coil segments were over-represented for proteins in the TS dataset (7.6% of the proteins identified contained coiled-coil segments) in comparison with all proteins in the mouse proteome (3% contained coiled-coil segments). These results suggested that heat-treatment is selective for coiled-coil domain-containing proteins but that, overall, these proteins constitute very small factions of the TS protein dataset and theoretical mouse proteome, respectively.
The observation that coiled-coil segments were predicted to primarily occur in DPs and MXPs was a concern because it was possible that inaccurate prediction of these segments as being disordered influenced the structural classification of the proteins in which they occur. However, it was also possible that inaccurate disorder predictions of coiled-coil domain-containing proteins did not lead to structural misclassification and that disordered and coiled-coil segments coexist within these proteins. To distinguish between the two possibilities, we determined whether disordered and coiled-coil segments occurred separately, or coincidently, within protein sequences. To address this issue, for all proteins in the TS dataset predicted to contain a coiled-coil domain (and all theoretical mouse proteins), we determined the number of residues that were predicted to exhibit disordered character, coiled-coil character, and both structural features, and then determined the percentage of disordered and coiled-coil residues that exhibited both structural characteristics (Suppl. Table 5). These analyses were performed individually for coiled-coil domain-containing DPs, MXPs, and FPs, as well as for all of these proteins together. Further, these analyses were performed using three disorder predictors (NORSnet, DISOPRED2, and IUPred) that are independent of PONDR. The results indicate that, using either NORSnet or DISOPRED2, the extent of overlap between disordered and coiled-coil character in coiled-coil domain-containing proteins is very small (<5% as a percentage of the number of disordered residues and <4% as a percentage of the number of coiled-coil residues). The results using IUPred suggest extensive overlap of disordered and coiled-coil character in the proteins under study; however, this is an artifact of the algorithm used by IUPred, which bases its predictions of disorder on the likelihood of pair-wise contact between amino acids. Due to the infrequent occurrence of hydrophobic residues in coiled-coil segments, which have a high likelihood for pair-wise contacts in folded proteins, coiled-coils are predicted to be disordered (data not shown). In summary, these computational sequence analysis results strongly suggest that a significant fraction of disordered proteins within the TS protein dataset (21% of the DPs and ∼12% of the MXPs) contain at least one coiled-coil segments of ≥30 residues. Further, results from two disorder predictors (NORSnet and DISOPRED2) indicate that coiled-coil and disordered domains overlap to only a very small extent. Considering the prevalence of coiled-coil segments in the disordered proteins identified in this study, we suggest that new disorder predictors be developed, that detect the heptad repeat pattern of coiled-coil segments in addition to disordered polypeptide segments, to determine the generality of our findings regarding the coexistence of disordered and coiled-coil segments within proteins.
Validation of Protein Structural Classifications by Reference to the DisProt Database
The availability of the DisProt database of experimentally characterized, disordered proteins (http://www.disprot.org/)34 provided the opportunity to validate our PONDR-based structural classification system. We note that while some of the proteins that are now in the DisProt database were used in the training of the various PONDR algorithms, these algorithms were developed well before DisProt was established. Therefore, our PONDR-based predictions of protein disorder/order are largely independent of the current content of DisProt. Unfortunately, we observed that less than 5% of the mouse TS proteins exhibited sequence similarity to proteins archived in DisProt: 36 DPs, 17 MXPs, and 12 FPs (Suppl. Table 3). It must be emphasized that proteins deposited in the DisProt database exhibit a wide range of structural features and are disordered to widely varied extents; for example, some protein entries have been shown experimentally to be entirely disordered while others may exhibit only one short disordered segment. Therefore, it was necessary to evaluate the primary structural data for proteins in DisProt that exhibited sequence matches to proteins in the TS protein dataset in order to evaluate the validity of our structural classifications. Such a review confirmed that the proteins that we classified as DPs have been shown experimentally be extensively disordered, including but not limited to 4E-BP1, calpastatin, CREB, p21Cip1, p27Kip1, Sp1, stathmin, and WASP (Suppl. Table 3). Further, similar review of information regarding the 17 MXPs noted above indicated that the “mixed” structural classification was appropriate. For example, the 500 residue long N-terminal domain of one MXP, glucocorticoid receptor, was predicted and has been experimentally shown to be disordered50 while the C-terminal, ligand binding domain (∼280 residues long) was predicted to be folded and its structure has been previously determined51 (Suppl. Figure 2). In another case, the N-terminal domain of nucleoplasmin-3 was predicted to be ordered and the Xenopus ortholog has been shown experimentally to fold into a pentameric β-propeller structure52 while the shorter C-terminal domain of both the Xenopus and mouse proteins was predicted and experimentally demonstrated to be disordered53 (Suppl. Figure 3). In these two examples, the term “mixed” applies in the sense that the proteins exhibit both disordered and structured/ordered features. An example of an MXP which exhibits a different “mixed” structural profile is 60S acidic ribosomal protein P1 (Suppl. Figure 4). The N-terminus of this 108 residue long protein was predicted by PONDR to be ordered and the C-terminus, disordered; these features led to our classification as an MXP. However, experimental studies showed that the foldedness of the P1 protein depended on pH, being folded below pH 3.9 and disordered above54. While PONDR was not developed to predict the pH dependence of structural properties, the algorithm is sensitive to the sequence features that give rise to this pleomorphic behavior. Finally, virtually all of the proteins classified by us as FPs that also appear in the DisProt database possess one or more folded domains which comprise a large portion of the polypeptide sequence but which also exhibit one or more experimentally characterized disordered segments, often at the N- and/or C-termini. An exception is cystatin B (Suppl. Figure 5), a small protein which was predicted to and is known to be almost entirely folded55. This protein appears in the DisProt database because a disease-associated truncation mutant, that interrupts the globular fold, is unstructured in solution56; thus, our assignment of full-length mouse cystatin B as an FP is appropriate. This critical review of structural information for DPs, MXPs and FPs that appear in the TS dataset as well as in the DisProt database independently validates our method of structural classification by documenting a strong correlation between predicted and experimentally observed protein structural features. In addition, it serves to strengthen our view that assignment of the term “intrinsically disordered” to a particular protein must be qualified with information about the fraction of residues within a given protein that are disordered. We have strived for this by creating three structural classifications which differentiate between proteins that are predominantly disordered (DPs), ordered (FPs) and of mixed character (MXPs). Finally, this review, showing that <5% of the TS proteins we identified have been experimentally characterized as being disordered, underscores the need for broader experimental characterization of disordered proteins expressed in eukaryotic cells.
The Occurrence of Both Small and Large Folded Regions within Thermo-stable Mouse Proteins
As an additional means to validate our structural classification system and to determine the extent to which regions of known three-dimensional (3D) structure occurred within proteins in the TS dataset, we used BLAST43 to search for matches between the sequences of all TS proteins and those deposited in the protein data bank (PDB; http://www.rcsb.org/pdb). As was true for our predictions of disorder, we believe that our predictions of folded proteins are largely independent of the protein sets used to train the PONDR algorithms. For example, a reduced, non-redundant form of the PDB was used in the training of PONDR in 199732; since that time, the total PDB has grown approximately 8-fold (from 6,570 entries in 1997 to 52,821 in 2008)57. Therefore, it is unlikely that a significant fraction of the proteins or domains in the TS dataset that were predicted to be folded using PONDR were used in training the PONDR algorithms. Remarkably, we found that structural information was available for one or more regions of ≥60 residues for most DPs, MXPs and FPs in the TS dataset based on BLAST analysis against the PDB using 25% identity as the cut-off for sequence similarity (Suppl. Table 6A-C). We used these criteria because the minimal size for folded protein domains is approximately 60 residues and 25% identity is an approximate lower limit for domains with similar folds. Only 116 of the 1,320 TS proteins we identified did not exhibit sequence similarity according to the above criteria to proteins in the PDB (Suppl. Table 7). As would be expected based upon their reduced propensities to exist in folded/ordered states, the vast majority of these were classified as either DPs (80 proteins) or MXPs (19 proteins). It must be noted, however, that this method of sequence analysis is not an absolute indicator that a particular ≥60 residue region of a TS protein exists in a folded conformation. La Gall, et al.58, showed that between 5% and 21% of residues in a non-redundant sub-set of PDB entries also listed in Swiss-Prot were predicted to be disordered by various disorder predictors. This observation is consistent with conformational restriction of residues due to the influence of crystal packing of segments at the N- and C-termini of folded regions, and/or within loops, that would otherwise be flexible in solution.
Each of the proteins detected in our study necessarily remained soluble after heat-treatment at 98 °C for 1 hour. Therefore, it is remarkable that such a large number of short, predominantly folded regions (≥60 residues), often subject to thermal denaturation, non-specific aggregation and precipitation upon heating, were identified in the TS protein dataset. However, it must be remembered that these domains exist in the context of very long proteins (627 ± 646 residues) and portions of these proteins outside the putative folded regions may confer thermo-stability. To further explore the ordered/folded features of proteins in the TS dataset, we performed an additional BLAST analysis to identify proteins which contained large regions of known structure. For this, we increased the region length that was searched from ≥60 to ≥300 residues. Remarkably, 17 DPs, 57 MXPs and 100 FPs exhibited long regions (≥300 residues) of known 3D structure. Together, these results indicate that a large fraction of all proteins in the TS dataset are likely to contain at least one small (≥60 residues in length), folded regions. A much smaller fraction of proteins contain large (≥300 residues in length), folded regions, with those classified as MXPs and FPs most likely to exhibit such a region. While some proteins in the TS dataset are predicted to be exclusively disordered, these results show that disordered polypeptide regions most often occur in proteins which exhibit at least one short, folded region. Similarly, most folded proteins we detected exhibit some segments which are disordered, either at the N- or C-termini, or within loops. Thus, the expressed TS proteins we detected in mouse fibroblasts exhibit a wide range of structural features which fall along a continuum from complete disorder to complete order59. Most proteins, however, exhibit some aspects of disorder and order rather than falling at the extremes of this structural continuum. Analysis of sequences and structures of the folded regions within these proteins in the future may provide insights into their apparent and remarkable thermo-stability.
Occurrence of Transmembrane Domains (TMs) within Thermo-stable Mouse Proteins
Disordered polypeptide segments play important biological roles not only in soluble proteins, but also in proteins localized to membranes. For example, a large fraction (∼40%) of human plasma membrane proteins were previously predicted to possess intrinsically disordered domains of ≥30 residues, with most of these domains predicted to be exposed to the cytoplasm60. Therefore, we investigated the occurrence of TM domains within proteins in the TS dataset using TM domain prediction programs, TMHMM237 and PHDhtm38. This analysis showed that 65 proteins contained TM domains (Suppl. Table 8); 11 of these were predicted to be DPs, 9 were predicted to be MXPs, and 45 were predicted to be FPs (Suppl. Table 9, Figure 2). The 45 TM domain-containing FPs exhibited a wide range of sequence lengths (1004 ± 1066 residues) and numbers of TM helices (5.4 ± 4.6 TM helices), as did the 9 MXPs (884 ± 673 residues in length, 2.3 ± 2.1 TM helices). The 11 TM domain-containing proteins classified as DPs exhibited a similar, wide range of sequence lengths (1058 ± 543 residues) but on average contained between 1 and 2 TM domains (1.6 ± 1.8 TM helices). Overall, 55% of the TM domain-containing proteins exhibited 1 or 2 TM helices, with the remainder exhibiting between 4 and 16 TM helices. In summary, while present in the TS protein dataset, TM domain-containing proteins constitute a minor portion of all proteins identified.
Figure 2. Percentage of proteins classified as DPs, MXPs and FPs in the mouse TS protein dataset that contain known sites of post-translational modification (PTM) and phosphorylation, alternate splice variants or transmembrane domains.

Key: blue bars, PTMs; red bars, phosphorylation; black bars, alternative splice variants; and white bars, transmembrane domains. The total number of proteins in each structural class is shown at the top in parentheses.
Biological Classification of Thermo-stable Mouse Proteins
We investigated relationships between the biological characteristics of proteins in the TS dataset and their structural classification in order to understand the biological roles of both disordered and folded/ordered proteins expressed in mouse fibroblasts. Specifically, to perform this analysis in an unbiased manner, we determined the Gene Ontology (GO) Consortium database (http://www.geneontology.org/)35 terms in three categories, cellular component (CC), biological process (BP), and molecular function (MF), associated with the TS proteins that are over- or under-represented relative to results for the entire theoretical mouse proteome. For these analyses, DPs and MXPs were grouped together to represent disordered proteins and FPs were used to represent folded/ordered proteins. Fisher's exact test was used to identify GO terms that were over- or under-represented in the (DP + MXP) and FP data subsets relative to their occurrence in the mouse proteome and only those terms characterized by a false discovery rate (FDR) values less than 0.01 are discussed. Figure 3 graphically summarizes these results for level-2 terms while Suppl. Tables 10A-F lists all GO terms in the three categories that were significantly over- or under-represented for disordered and folded/ordered proteins. In total, 152, 278, and 173, terms for the level-0 GO categories, cellular component, biological process, and molecular function, were analyzed. In the following section, we focused our functional analyses on over- or under-represented level-2 GO terms because theirs numbers were manageable and their names in many cases offered specific insights into biological function.
Cellular Component
Of 152 level-2 GO terms describing cellular components, only 19 were over- or under-represented amongst proteins in the TS dataset relative to all proteins in the mouse proteome (Figure 3A). Further, a cellular component GO term was found for 756 of 909 total disordered proteins and for 336 of 411 total folded proteins. For disordered proteins, the GO terms for cellular component that are most highly populated (e.g. GO terms that are associated with the largest numbers of proteins considering the whole mouse proteome) and that were over-represented include, “non-membrane-bounded organelle” (220 proteins), “intracellular organelle part” (696 proteins), “organelle part” (218 proteins), “membrane-bounded organelle” (434 proteins), and “intracellular organelle” (527 proteins). Additional, over-represented terms for disordered proteins included, “leading edge” (18 proteins), “cell projection” (52 proteins), “cell projection part” (11 proteins), and “ribonucleoprotein complex” (86 proteins). In addition, both disordered and folded/ordered (FPs) proteins exhibited significant over-representation of several terms, including “protein complex” (106 (DPs + MXPs); 72 FPs), “intracellular” (705 (DPs + MXPs); 275 FPs), and “intracellular part” (696 (DPs + MXPs); 267 FPs), indicating that thermo-stable proteins, in general, exhibit these localization features. Finally, both disordered and folded proteins exhibited significant under-representation of two highly populated GO terms, “membrane part” and “membrane”. Detailed information regarding these analyses is provided in Suppl. Table 10A and 10B for (DPs and MXPs) and FPs, respectively, including all over- and under-represented level-2 and lower level cellular component GO terms, statistics of over- or under-representation relative to all mouse proteins, and the Swiss-Prot names of the over- and under-represented proteins.
Biological Process
Of 278 level-2 GO terms describing biological process, only 28 were over- or under-represented amongst proteins in the TS dataset relative to all proteins in the mouse proteome (Figure 3B). Further, a biological process GO term was found for 705 of 909 total disordered proteins and for 336 of 411 total folded proteins. Eleven significantly over-represented terms were associated only with TS disordered proteins, including “macromolecular complex disassembly” (13 proteins), “chromosome segregation” (10 proteins), “cell division” (29 proteins), “cell cycle” (73 proteins), “cell cycle process” (47 proteins), “macromolecule metabolic process” (370 proteins), “biosynthetic process” (212 proteins), “gene expression” (236 proteins), “establishment of protein localization” (58 proteins), “macromolecule localization” (68 proteins), and “cellular component organization and biogenesis” (161 proteins). Both disordered and folded/ordered proteins and, thus, TS proteins in general, were over-represented in several categories, several of which are highly populated, including “primary metabolic process” (384 (DPs + MXPs); 194 FPs), “cellular metabolic process” (385 (DPs + MXPs); 200 FPs), “cellular localization” (61 (DPs + MXPs); 31 FPs), “establishment of localization in cell” (58 (DPs + MXPs); 30 FPs), “cellular macromolecular complex subunit organization” (45 (DPs + MXPs); 20 FPs) and “macromolecular complex assembly” (36 (DPs + MXPs); 21 FPs). Both disordered and folded/ordered proteins were under-represented in two categories, “system process” and “cell communication”. Folded/ordered proteins alone were under-represented in several additional, highly populated categories, including “regulation of metabolic process”, “regulation of biological process”, and “regulation of cellular process”. Finally, disordered proteins were significantly under-represented in the following categories, “immune response”, “response to chemical stimulus”, and “response to external stimulus”.
Molecular Function
Of 173 level-2 GO terms describing molecular function, only 23 were over- or under-represented amongst proteins in the TS dataset relative to all proteins in the mouse proteome (Figure 3C). Further, a molecular function GO term was found for 758 of 909 total disordered proteins and for 361 of 411 total folded proteins. Twelve significantly over-represented terms were associated with TS disordered proteins, including, “structural constituent of cytoskeleton” (10 proteins), “structural constituent of ribosome” (36 proteins), “microtubule motor activity” (10 proteins), “translation factor activity, nucleic acid binding” (22 proteins), “transcription activator activity” (22 proteins), “transcription cofactor activity” (20 proteins), “nucleic acid binding” (216 proteins), “protein binding” (404 proteins), and “nucleotide binding” (216 proteins). Folded/ordered proteins were also over-represented for “nucleotide binding” (93 proteins) but were under-represented for “nucleic acid binding”. Amongst these molecular function GO terms, only “nucleic acid binding”, “protein binding”, and “nucleotide binding” are highly populated considering all mouse proteins while each of the other terms are populated to the extent of 1.3% or less. Several molecular function GO terms are under-represented amongst disordered proteins, including “substrate-specific transporter activity”, “transmembrane transporter activity”, “signal transducer activity”, “hydrolase activity”, and “transferase activity”. Amongst folded/ordered proteins, several terms associated with catalytic activity are over-represented, including, “isomerase activity” (15 proteins), “oxidoreductase activity” (36 proteins), “cofactor binding” (18 proteins), “vitamin binding” (10 proteins), “ligase activity” (31 proteins), and “hydrolase activity” (85 proteins). Finally, folded/ordered proteins are under-represented in two highly populated categories, including “signal transducer activity” and “nucleic acid binding”.
Overall, these results indicate that the two structural classes of proteins under investigation, disordered and folded/ordered proteins, exhibit distinct functional characteristics when compared using GO terminology, including GO terms for three functional categories, cell component, biological process and molecular function. These comparisons have been performed to reveal GO terms that are over- or under-represented relative to their occurrence in the background of all proteins encoded by the mouse genome. A relatively small fraction (10-13%) of the level-2 GO terms in these three functional categories exhibited over- or under-representation amongst disordered and folded/ordered TS proteins. Further, in the majority of cases, either disordered or folded/ordered proteins, but not both structural types, were over- or under-represented, suggesting that TS proteins with these different structural features perform distinct, specialized biological functions. In contrast to previous analyses which have relied upon the analysis of disordered proteins within theoretical whole proteomes, the results presented herein represent the first large-scale analysis of disordered proteins that are expressed in a particular eukaryotic cell type, in this case mouse fibroblast cells. While the heat-treatment procedure used was a significant factor in determining which mouse proteins were detected in our study, correlations of protein disorder with over-represented functional categories is meaningful in clarifying the actual roles performed by disordered proteins in fibroblast cells. In contrast, under-representation of certain functional classes in disordered proteins cannot be meaningfully interpreted due to the possibility that under-representation stems from heat sensitivity.
Post-translational Modifications of Thermo-stable Mouse Proteins
Proteins in all structural classes, including intrinsically disordered proteins, experience post-translational modifications (PTMs). However, because their sequences are generally enriched in amino acids that are subject to post-translational modification (e.g. Ser, Thr, Lys, and Arg)61 and because disordered polypeptide segments are accessible to enzymes that catalyze modifications, it has been proposed that disordered proteins experience PTMs to a greater extent than do rigid, folded proteins12. We used the ProteinCenter software package, which searches the UniProt database, to identify proteins in the TS dataset that were previously shown to experience post-translational modifications (Suppl. Table 11A and B). More than half of the DPs (66%) contained previously characterized PTM sites (Figure 2, blue bars), with 95% of these corresponding to phosphorylation sites (Figure 2, red bars). Similarly, 53% of MXPs contained PTM sites, with >90% of these corresponding to phosphorylation sites. A somewhat smaller percentage of FPs (43%) contained known sites of PTM while 68% of these were due to phosphorylation. These data support the view that expressed mouse proteins containing disordered segments experience extensive post-translational modification, especially phosphorylation.
Alternative Splicing of Thermo-stable Mouse Proteins
Analysis using ProteinCenter software indicated that 347 of the 1,320 TS proteins (26%) are known to experience alternative splicing (Figure 2, black bars). The percentage of DPs which experience alternative splicing (34%) was more than 2-fold greater than that for FPs (15%). These observations are consistent with a previous report which showed that alternative splicing occurs most frequently within RNA regions which encode disordered protein segments62.
Protein-protein Interactions Involving Thermo-stable Mouse Proteins
Many disordered polypeptides exhibit multiple, short motifs that are either known or predicted to mediate protein-protein interactions. Moreover, these motifs have the potential to interact with multiple binding partners by adopting different conformations when bound to different targets. These observations have led to the suggestion that disordered proteins may serve as hubs in protein-protein interaction networks (24-26, 42). Since the TS dataset contained many proteins with disordered segments, we queried the OPHID protein-protein interaction database63 to determine the number of interaction partners for each as a measure of their hub-like qualities (Suppl. Tables 12-13). The results show that most proteins in each structural class interact with fewer than 50 other proteins and that the decrease in the percentage of proteins with a certain number of interaction partners as the number of partners increases is similar for DPs, MXPs and FPs (Figure 4). This trend is maintained for proteins with both small numbers and large numbers of interaction partners (Figure 4, inset), indicating that the proteins in the different structural classes exhibit similar and widely ranging promiscuity toward interactions. Based on this, we conclude that DPs, MXPs and FPs in the TS dataset exhibit similar, rather than differing, hub-like characteristics. Protein-protein interactions are mediated by both short and long domains, and proteins with long sequences are likely to exhibit the largest number of interaction partners because they are most likely to contain these interaction domains. The interaction profiles for proteins in the TS dataset in the different structural classes may be similar because the average protein length in these classes, and the standard deviation of length, are very similar. These results do not support the suggestions of others noted above. Interestingly, in agreement with our observations, Schnell, et al.64, failed to observe a correlation between protein topological connectivity (hub-like character) and disorder for proteins in whole proteome interaction networks from humans and several other species. However, since our analysis was based upon the information from the OPHID protein-protein interaction database and that of Schnell, et al.64, on information from the Biomolecular Interaction Network Database 65, any biases and limitations in the information in these databases will have influenced the conclusions reached. For example, hub-like DPs may bind to their partners through as yet unknown interaction domains. Protein-protein interactions mediated by such unknown domains are not represented in interaction databases; therefore, the analyses described above may underestimate the number of interactions any protein can experience. As greater numbers of disordered interaction domains are identified and cataloged, the completeness of large-scale interaction databases will improve. Despite these limitations, our analysis suggests strongly that DPs, MXPs and FPs in the TS dataset participate in protein-protein interactions to approximately similar extents.
Figure 4. Analysis of the number of protein interaction partners for proteins in the different structural classes in the TS protein dataset.

The percentage of DPs (blue diamonds), MXPs (black squares) and FPs (orange circles) which interact with up to the given numbers of interaction partners is plotted versus the number of interaction partners. The boxed region is expanded in the upper right. The data represent totals over bins incremented by 5 interaction partners (e.g., 0-5 partners, 6-10 partners, etc.).
Discussion
Bioinformatics analyses have predicted that intrinsically disordered proteins constitute a large proportion (30-40%) of proteins which comprise eukaryotic proteomes and that these proteins are extensively involved in cellular processes such as signaling and regulation. However, despite the significance of the roles played by DPs in normal biological processes and in disease (>75% of human cancer-associated proteins are predicted to be intrinsically disordered14), relationships between their physical properties and biological functions are understood in detail for relatively few and few large-scale proteomics studies have been performed. To begin to address these deficiencies, we previously developed a method for partial enrichment and detection of DPs from mammalian cells18. We showed that heat-treatment of the soluble extract from mouse fibroblast cells resulted in modest enrichment of cytosolic and nuclear DPs involved in cell signaling and regulation. However, a relatively small number of DPs, in comparison with that predicted by bioinformatics studies, were identified primarily due to the low dynamic range of gel-based proteomic analysis. In the present study, we used a novel MudPIT scheme involving both alkaline and acidic reversed phase ultra-high performance liquid chromatography to mine deeper into the mammalian IDP-ome. Using these procedures, we identified a total of 1,320 TS proteins in a mouse fibroblast extract; of these proteins, >900 were predicted to be significantly disordered, about 15-fold more than we had reported previously18. Using three different disorder predictors, we estimate that between 12.4% and 23.4% of the approximately 25,000 proteins in the mouse proteome contain one or more disordered segment(s) of ≥ 30 residues (data not shown). Based upon this, we estimate that the mouse IDP-ome theoretically is comprised of between ∼3,000 and ∼6,000 disordered proteins. However, it is generally accepted that only ∼10,000 mouse proteins (∼40% of the total predicted open reading frames) are expressed in any one cell type at any given time. Therefore, we estimate that on the order of between 1,200 and 2,400 disordered proteins are actually expressed in mouse fibroblasts. Of the 1,320 proteins identified in the TS dataset, ∼900 were predicted to be significantly disordered (514 DPs and 395 MXPs). Based on these figures, we estimate that we have achieved ∼38-75% penetrance of the mouse IDP-ome.
Based on the analysis given above, this work constitutes the largest scale proteomics study of experimentally detected, significantly disordered proteins from mammalian cells reported to date. It should be noted that our structural classification system relied on the use of well-established bioinformatics tools to analyze the sequences of the more than 1,300 TS proteins that were identified using MudPIT. At present, it is not possible to experimentally determine the structural properties of individual proteins within such a large dataset. While proteins with a wide range of predicted structural features were detected, heat-treatment of the soluble extract from mouse fibroblast cells afforded modest selectivity for proteins predicted to be DPs and MXPs. While our study did rely on the use of bioinformatics methods for structural analysis, it differs from past in silico, whole proteome analyses in that our results reflect the protein expression pattern associated with a particular biological state of mouse fibroblast cells; in this case, cells which had reached 80% confluence in culture. Knowledge of the proteins which are actually expressed under these conditions, and thus could be detected using MudPIT, has provided the opportunity to study on a large scale the structural (using bioinformatics tools) and biological (by reference to the GO database) properties of TS proteins expressed in living cells, a large fraction of which were predicted to be intrinsically disordered.
We made several remarkable and unexpected observations in the course of this IDP-omics study. First, while the range of protein lengths comprising the heat-treated TS dataset is generally representative of the lengths of all proteins predicted to exist in the mouse proteome, it is remarkable that many proteins with lengths >1,000 residues survive our harsh heat-treatment procedure. Of course, many of these “thermo-survivors” are DPs, which are known in general to be thermo-stable66. However, many others are MXP or FPs which possess folded domains, with a large number containing large (>300 residues), folded domains. These proteins may be inherently thermo-stable, either in isolation or within multi-protein assemblies. For example, multi-protein assemblies often contain both DPs and FPs and, in some cases, are known to be highly thermo-stable67. Alternatively, some of the thermo-survivors may thermally denature at 98 °C and refold upon cooling prior to processing for MudPIT analysis. Some of the proteins present in the complex fibroblast extract, possibly MXPs or DPs, may serve as chaperones for other proteins, promoting refolding and conferring thermo-stability. An additional explanation is that proteins comprised of both disordered and ordered domains may have been subject to partial digestion by endogenous proteases prior to heat treatment and trypsin digestion, which may have enhanced their ability to survive heat treatment. A key point is that disordered polypeptide segments occur within large proteins which are additionally comprised of many other disordered and folded/ordered domains. The fact that many functional, disordered domains are relatively short in length68 suggests that the, on average, rather large, extensively disordered proteins we have detected in our study may individually perform diverse and complex biological functions. The concept of “one (folded) protein = one biological function” from the earliest days of protein structure/function analysis is passé in light of the rich diversity of disordered and ordered/folded polypeptide segments detected here in proteins expressed in mouse fibroblast cells.
A second unexpected observation was coexistence of disordered and coiled-coil domains within a large fraction of proteins structurally classified as either DPs (21%) or MXPs (12%). While disordered protein domains are known to have the potential to interact with many partners, coiled-coil domains generally mediate homo-meric or hetero-meric interactions amongst coiled-coil domains. This observation suggests a mechanism by which disordered proteins mediate the assembly of protein complexes by coordinating several modes of interaction: 1) homo- or hetero-meric oligomerization mediated by coiled-coil segments, and 2) folding-upon-binding mediated by disordered segments. Precedent for this concept is found in studies of the intrinsically disordered transporter protein, dynein intermediate chain (IC), and its interactions with the folded and dimeric hub protein, LC8 (reviewed in 69). The sequence of IC is predicted to contain both disordered and coiled-coil segments; however, in isolation, IC is intrinsically disordered. Interestingly, in the presence of dimeric LC8, disordered segments—termed interaction motifs (IMs)—from two molecules of IC fold upon binding in hydrophobic grooves on opposite surfaces of the LC8 dimer, which further promotes dimerization via one of the coiled-coil segments of IC (Figure 5). This coupled folding-upon-binding of a disordered IM segment of IC to LC8 and dimerization of a separate coiled-coil segment of IC, may be a general mechanism of cooperation between disordered binding domains and coiled-coil polypeptide segments in disordered proteins. In the case of IC/LC8 interactions, the assembly which forms has a highly extended structure and plays a role in the transport of cargo along microtubules. The identification of coiled-coil segments within a large number of DPs and MXPs in this proteomics study provides the opportunity to test this hypothesis in the future through protein structural studies. Such studies would be aided by the development of disorder predictors that can reliably identify both short interaction motifs and coiled-coil segments.
Figure 5. Cooperation amongst intrinsically disordered and coiled-coil segments within IC promotes binding to LC8.
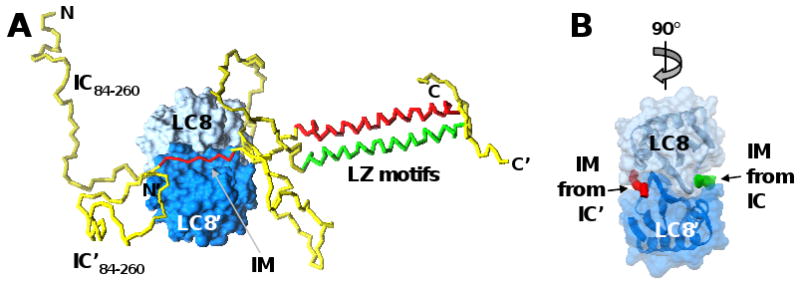
(A) A short interaction motif (IM) from two molecules of the intrinsically disordered protein, IC (residues 84-260 illustrated), adopt rigid, extended structure when bound on opposite faces of the folded, dimeric protein, LC8. While not directly involved in binding to LC8, two leucine-zipper (LZ) motif-containing segments of IC, that are unfolded and monomeric in the absence of LC8, form a coiled-coil dimer when the IM segments of IC bind to LC8. IC is illustrated as a yellow tube, with the IM segments and LZ motifs colored red or green, respectively, in the two molecules. The two subunits of the LC8 dimer are shown in surface representation in dark and light blue, respectively. (B) The LC8 dimer was rotated 90° relative to (A) and only the IM segments of the two IC molecules are illustrated as red and green tubes, respectively. [Modeled after Figure 2 in ref. 69, with permission from the author.]
Additional unexpected observations were made regarding the functional properties of the disordered proteins we detected in our study. Here we provide a brief review of these observations considering the three GO functional categories that were analyzed, cellular component, biological process and molecular function. First, under cellular component, Figure 3A shows over-representation of three level-2 GO terms associated with cell movement (“leading edge”, 18 proteins; “cell projection”, 52 proteins; and “cell projection part”, 11 proteins). In addition to these three level-2 GO terms, many hierarchically related, lower level GO terms are over-represented (according to the same criteria used to analyze level-2 terms, data not shown), including terms such as “myosin complex”, “stress fiber”, “actin filament bundle”, “actin cytoskeleton”, “cell cortex”, and “cortical cytoskeleton”. These observations suggest that disordered proteins play a specialized role in fibroblasts in cytoskeletal structure and cell movement. Another notable observation regarding subcellular localization (Figure 3A) is the association of disordered proteins with level-2 GO terms that include the term “organelle” (5 terms in total, several hundred proteins associated with each term). This indicates that disordered proteins play roles in the organization of biomolecules into organelles, an observation which is likely to be general rather than specific to fibroblast cells. Finally, the association of disordered proteins with GO terms associated with ribonucleoprotein complexes (“ribonucleoprotein complex”, 86 proteins) is noteworthy but not unexpected. Significantly over-represented, lower-level GO terms in this branch of the ontology include “nucleolus”, “ribosome”, and “spliceosome”.
Under biological process (Figure 3B), an unexpected observation was the extensive association of disordered proteins with level-2 GO terms containing the descriptors, “metabolic” (“macromolecule metabolic process”, 370 proteins; “primary metabolic process”, 384 proteins; “cellular metabolic process”, 385 proteins; and “regulation of metabolic process”, 151 proteins) or “biosynthetic” (“biosynthetic process”, 212 proteins). We are not aware of previous studies showing that disordered proteins play extensive roles in the fundamental cellular processes of metabolism and biosynthesis. It is extremely unlikely that extensively disordered proteins play direct roles in these processes, for example as catalysts; however, our results suggest that they play diverse, indirect roles which influence the roles played by folded/ordered catalysts. Another unexpected observation was the association of disordered proteins with processes related to the structural organization of cells. For example, six level-2 GO terms including the descriptors “localization” or “organization” (“cellular localization”, 61 proteins; “establishment of localization in cell”, 58 proteins; “establishment of protein localization”, 56 proteins; “macromolecule localization”, 68 proteins; “cellular component organization and biogenesis”, 161 proteins; and “cellular macromolecular complex subunit organization”, 45 proteins) were shown to be over-represented amongst disordered proteins. These functional associations amongst disordered proteins may be relevant to the associations with cell component GO terms pertaining to organelle structure and organization that were discussed above. Other over-represented level-2 biological process terms associated with disordered proteins were those involved in cell division (“chromosome segregation”, 10 proteins; “cell division”, 29 proteins; “cell cycle”, 73 proteins; “cell cycle process”, 47 proteins) and gene expression (“gene expression”, 236 proteins). These functional associations of disordered proteins, however, were not unexpected; it is well known that disordered proteins are involved in the regulation of cell division59 and gene expression70.
Finally, our analysis of over-represented level-2 molecular function GO terms (Figure 3C) confirmed the observations noted above made on the basis of GO terms for cellular component and biological process. For example, over-represented GO terms associated with disordered proteins include descriptors such as “cytoskeleton”, “ribosome”, or “microtubule” (“structural constituent of cytoskeleton”, 10 proteins; “structural constituent of ribosome”, 36 proteins; and “microtubule motor activity under molecular function”, 10 proteins), or “translation” or “transcription” (“translation factor activity, nucleic acid binding”, 22 proteins; “transcription activator activity”, 22 proteins; and “transcription cofactor activity”, 20 proteins). Further, several highly populated, over-represented GO terms include the descriptor, “binding” (“nucleic acid binding”, 216 proteins; “protein binding”, 404 proteins; and “nucleotide binding”, 216 proteins). These observations are consistent with the general concept that disordered proteins function by folding upon binding their biomolecular targets4, 66.
In conclusion, the expressed proteins we detected in the heat-treated TS dataset that exhibit a significant extent of disorder, classified here as DPs and MXPs, play diverse biological roles in mouse fibroblasts. However, our functional analysis reveals heightened involvement of disordered proteins in several functional categories, including, cytoskeletal structure and cell movement, metabolic and biosynthetic processes, organelle structure, cell division, gene transcription, and ribonucleoprotein complexes. This disordered protein/function expression pattern reflects the specialized biology of mouse fibroblasts at 80% confluence in culture. It is likely that disordered proteins are specialized to perform many of these biological functions in other cell types and in other organisms; however, some of these disordered protein functional classes may be specifically upregulated in fibroblast cells, for example, cytoskeletal structure and cell movement. In addition to exhibiting diverse biological features, the expressed disordered proteins we identified exhibited diverse structural features. We propose that the structure of proteins be considered in the context of a continuum which extends from complete disorder to complete order. Our results show that the majority of the disordered proteins we detected, while dominated by disordered domains, also exhibited ordered features. Similarly, the majority of the ordered proteins we detected, while dominated by ordered/folded domains, also exhibited disordered features. Thus, we believe that most proteins fall within central region of proposed structural continuum, rather than exhibiting features corresponding to either extreme. The complex biological functions of proteins arise from partnerships between disordered and folded domains, which have evolved to perform distinct aspects of biological function. While many folded proteins were detected, our results confirm the predominance of disorder in mammalian proteomes pointed out previously based on studies of theoretical whole proteomes by others1, 2. Because we have substantially penetrated the mouse IDP-ome (∼38-75%), the disordered proteins we identified can serve in the future as quantitative probes of the biological pathways and processes in which they participate. Application of the MudPIT procedures we developed will allow the state of the IDP-ome to be broadly monitored, allowing its role in cell physiology to be more completely understood.
Supplementary Material
Acknowledgments
The authors thank Charles Ross (Department of Structural Biology, St. Jude Children's Research Hospital) for computer support, Perdeep Mehta (Hartwell Center for Bioinformatics and Biotechnology, St. Jude Children's Research Hospital) for assistance in assembling the table of peptide IDs (Suppl. Table 1), members of the Kriwacki laboratory for stimulating discussion, and Elisar Barbar (Oregon State University, Corvallis, Oregon) for providing the pdb file used to prepare Figure 5. This work was supported by the American Lebanese Syrian Associated Charities (ALSAC), National Cancer Institute (5R21CA104568 and 2R01CA082491, RWK), and a Cancer Center (CORE) Support Grant (5P30CA021765, SJCRH).
Abbreviations
- 2D PAGE
two-dimensional polyacrylamide gel electrophoresis
- DPs
disordered proteins
- FPs
folded proteins
- IDP-ome
intrinsically disordered proteome
- MXPs
proteins having mixed order/disorder character
- MudPIT
multi-dimensional protein identification technology
- PDB
protein databank
- PTM
post-translational modification
- TM
transmembrane
- TS
thermo-stable
Footnotes
Supporting Information Available: Supplementary figures showing the occurrence of residues with coiled-coil character in proteins classified as DPs and MXPs; and a comparison of PONDR predictions and experimentally determined disorder for glucocorticoid receptor, nucleoplasmin-3, 60S acidic ribosomal protein P1, and Cystatin-B. In addition, supplementary tables are provided showing results from mass spectrometry analysis of tryptic peptides of mouse TS proteins; the comparison of results of disorder predictions for the mouse TS protein dataset using several protein disorder predictors; Swiss-Prot IDs, descriptions, GO annotations and results of structural analysis using PONDR for proteins in the mouse TS dataset; the numbers of proteins structurally classified as DPs, MXPs and FPs in the past using 2D polyacrylamide gel electrophoresis (PAGE) and in the current TS protein dataset using MudPIT; the influence of coiled-coil segments on the fraction of long disordered segments identified for proteins in the mouse fibroblast TS protein dataset; an analysis of per-residue overlap between disordered and coiled-coil residues for proteins in the mouse fibroblast TS protein dataset and the theoretical mouse proteome; lists of proteins in the TS protein dataset which do and do not exhibit small (≥ 60 residues) folded domains; a comparison of the number of proteins in the mouse TS protein dataset predicted to contain transmembrane domains; lists of proteins in the mouse TS protein dataset predicted to contain one or more transmembrane domains; a list of GO annotation terms for subcellular localization, biological process and molecular function for all proteins in the mouse TS protein dataset; a list of proteins in the mouse TS protein dataset identified to contain known sites of post-translational modification and known sites of phosphorylation; and lists of interaction partners and their numbers identified in the OPHID protein-protein interaction database for proteins in the TS protein dataset.
References
- 1.Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–71. [PubMed] [Google Scholar]
- 2.Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44:1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- 3.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–82. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 4.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 5.Minezaki Y, Homma K, Kinjo AR, Nishikawa K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J Mol Biol. 2006;359:1137–49. doi: 10.1016/j.jmb.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 6.Namba K. Roles of partly unfolded conformations in macromolecular self-assembly. Genes Cells. 2001;6:1–12. doi: 10.1046/j.1365-2443.2001.00384.x. [DOI] [PubMed] [Google Scholar]
- 7.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 8.Tompa P, Csermely P. The role of structural disorder in the function of RNA and protein chaperones. Faseb J. 2004;18:1169–75. doi: 10.1096/fj.04-1584rev. [DOI] [PubMed] [Google Scholar]
- 9.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–56. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6:1899–916. doi: 10.1021/pr060393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 12.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–32. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–98. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 15.Bertoncini CW, Rasia RM, Lamberto GR, Binolfi A, Zweckstetter M, Griesinger C, Fernandez CO. Structural Characterization of the Intrinsically Unfolded Protein beta-Synuclein, a Natural Negative Regulator of alpha-Synuclein Aggregation. J Mol Biol. 2007;17:17. doi: 10.1016/j.jmb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 16.Cheng Y, LeGall T, Oldfield CJ, Dunker AK, Uversky VN. Abundance of intrinsic disorder in protein associated with cardiovascular disease. Biochemistry. 2006;45:10448–60. doi: 10.1021/bi060981d. [DOI] [PubMed] [Google Scholar]
- 17.Feng ZP, Zhang X, Han P, Arora N, Anders RF, Norton RS. Abundance of intrinsically unstructured proteins in P. falciparum and other apicomplexan parasite proteomes. Mol Biochem Parasitol. 2006;150:256–67. doi: 10.1016/j.molbiopara.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 18.Galea CA, Pagala VR, Obenauer JC, Park CG, Slaughter CA, Kriwacki RW. Proteomic studies of the intrinsically unstructured mammalian proteome. J Proteome Res. 2006;5:2839–48. doi: 10.1021/pr060328c. [DOI] [PubMed] [Google Scholar]
- 19.Csizmok V, Szollosi E, Friedrich P, Tompa P. A novel two-dimensional electrophoresis technique for the identification of intrinsically unstructured proteins. Mol Cell Proteomics. 2006;5:265–73. doi: 10.1074/mcp.M500181-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Irar S, Oliveira E, Pages M, Goday A. Towards the identification of late-embryogenic-abundant phosphoproteome in Arabidopsis by 2-DE and MS. Proteomics. 2006;6 1:S175–85. doi: 10.1002/pmic.200500387. [DOI] [PubMed] [Google Scholar]
- 21.Cortese MS, Baird JP, Uversky VN, Dunker AK. Uncovering the unfoldome: enriching cell extracts for unstructured proteins by acid treatment. J Proteome Res. 2005;4:1610–8. doi: 10.1021/pr050119c. [DOI] [PubMed] [Google Scholar]
- 22.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 23.Schlessinger A, Liu J, Rost B. Natively Unstructured Loops Differ from Other Loops. PLoS Comput Biol. 2007;3:e140. doi: 10.1371/journal.pcbi.0030140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–4. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 25.Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20:2138–9. doi: 10.1093/bioinformatics/bth195. [DOI] [PubMed] [Google Scholar]
- 26.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 27.Gilar M, Olivova P, Daly AE, Gebler JC. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J Sep Sci. 2005;28:1694–703. doi: 10.1002/jssc.200500116. [DOI] [PubMed] [Google Scholar]
- 28.Gilar M, Olivova P, Daly AE, Gebler JC. Orthogonality of separation in two-dimensional liquid chromatography. Anal Chem. 2005;77:6426–34. doi: 10.1021/ac050923i. [DOI] [PubMed] [Google Scholar]
- 29.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Schlessinger A, Punta M, Rost B. Natively unstructured regions in proteins identified from contact predictions. Bioinformatics. 2007;23:2376–84. doi: 10.1093/bioinformatics/btm349. [DOI] [PubMed] [Google Scholar]
- 31.Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Predicting Protein Disorder for N-, C-, and Internal Regions. Genome Inform Ser Workshop Genome Inform. 1999;10:30–40. [PubMed] [Google Scholar]
- 32.Romero P, Obradovic Z, Dunker AK. Sequence data analysis for long disordered regions prediction in the calcineurin family. Genome Informatics. 1997;8:110–124. [PubMed] [Google Scholar]
- 33.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–27. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 34.Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, Obradovic Z, Dunker AK. DisProt: the Database of Disordered Proteins. Nucleic Acids Res. 2007;35:D786–93. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300. [Google Scholar]
- 37.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 38.Rost B, Fariselli P, Casadio R. Topology prediction for helical transmembrane proteins at 86% accuracy. Protein Sci. 1996;5:1704–18. doi: 10.1002/pro.5560050824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen CP, Kernytsky A, Rost B. Transmembrane helix predictions revisited. Protein Sci. 2002;11:2774–91. doi: 10.1110/ps.0214502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cuthbertson JM, Doyle DA, Sansom MS. Transmembrane helix prediction: a comparative evaluation and analysis. Protein Eng Des Sel. 2005;18:295–308. doi: 10.1093/protein/gzi032. [DOI] [PubMed] [Google Scholar]
- 41.Delorenzi M, Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18:617–25. doi: 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- 42.Gruber M, Soding J, Lupas AN. Comparative analysis of coiled-coil prediction methods. J Struct Biol. 2006;155:140–5. doi: 10.1016/j.jsb.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 43.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 44.Zhuang Y, Ma F, Li-Ling J, Xu X, Li Y. Comparative analysis of amino acid usage and protein length distribution between alternatively and non-alternatively spliced genes across six eukaryotic genomes. Mol Biol Evol. 2003;20:1978–85. doi: 10.1093/molbev/msg203. [DOI] [PubMed] [Google Scholar]
- 45.Sakharkar MK, Kangueane P, Sakharkar KR, Zhong Z. Huge proteins in the human proteome and their participation in hereditary diseases. In Silico Biol. 2006;6:275–9. [PubMed] [Google Scholar]
- 46.Galea C, Bowman P, Kriwacki RW. Disruption of an intermonomer salt bridge in the p53 tetramerization domain results in an increased propensity to form amyloid fibrils. Protein Sci. 2005;14:2993–3003. doi: 10.1110/ps.051622005. Epub 2005 Oct 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Landschulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–64. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- 48.O'Shea EK, Rutkowski R, Kim PS. Evidence that the leucine zipper is a coiled coil. Science. 1989;243:538–42. doi: 10.1126/science.2911757. [DOI] [PubMed] [Google Scholar]
- 49.Lupas AN, Gruber M. The structure of alpha-helical coiled coils. Adv Protein Chem. 2005;70:37–78. doi: 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- 50.Baskakov IV, Kumar R, Srinivasan G, Ji YS, Bolen DW, Thompson EB. Trimethylamine N-oxide-induced cooperative folding of an intrinsically unfolded transcription-activating fragment of human glucocorticoid receptor. J Biol Chem. 1999;274:10693–6. doi: 10.1074/jbc.274.16.10693. [DOI] [PubMed] [Google Scholar]
- 51.Kauppi B, Jakob C, Farnegardh M, Yang J, Ahola H, Alarcon M, Calles K, Engstrom O, Harlan J, Muchmore S, Ramqvist AK, Thorell S, Ohman L, Greer J, Gustafsson JA, Carlstedt-Duke J, Carlquist M. The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J Biol Chem. 2003;278:22748–54. doi: 10.1074/jbc.M212711200. [DOI] [PubMed] [Google Scholar]
- 52.Dutta S, Akey IV, Dingwall C, Hartman KL, Laue T, Nolte RT, Head JF, Akey CW. The crystal structure of nucleoplasmin-core: implications for histone binding and nucleosome assembly. Mol Cell. 2001;8:841–53. doi: 10.1016/s1097-2765(01)00354-9. [DOI] [PubMed] [Google Scholar]
- 53.Hierro A, Arizmendi JM, De Las Rivas J, Urbaneja MA, Prado A, Muga A. Structural and functional properties of Escherichia coli-derived nucleoplasmin. A comparative study of recombinant and natural proteins. Eur J Biochem. 2001;268:1739–48. [PubMed] [Google Scholar]
- 54.Zurdo J, Gonzalez C, Sanz JM, Rico M, Remacha M, Ballesta JP. Structural differences between Saccharomyces cerevisiae ribosomal stalk proteins P1 and P2 support their functional diversity. Biochemistry. 2000;39:8935–43. doi: 10.1021/bi000363b. [DOI] [PubMed] [Google Scholar]
- 55.Martin JR, Craven CJ, Jerala R, Kroon-Zitko L, Zerovnik E, Turk V, Waltho JP. The three-dimensional solution structure of human stefin A. J Mol Biol. 1995;246:331–43. doi: 10.1006/jmbi.1994.0088. [DOI] [PubMed] [Google Scholar]
- 56.Rabzelj S, Turk V, Zerovnik E. In vitro study of stability and amyloid-fibril formation of two mutants of human stefin B (cystatin B) occurring in patients with EPM1. Protein Sci. 2005;14:2713–22. doi: 10.1110/ps.051609705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.(RCSB), R. C. f. S. B. http://www.rcsb.org/pdb/statistics/contentGrowthChart.do?content=total&seqid=100.
- 58.Le Gall T, Romero PR, Cortese MS, Uversky VN, Dunker AK. Intrinsic disorder in the Protein Data Bank. J Biomol Struct Dyn. 2007;24:325–42. doi: 10.1080/07391102.2007.10507123. [DOI] [PubMed] [Google Scholar]
- 59.Galea CA, Wang Y, Sivakolundu SG, Kriwacki RW. Regulation of cell division by intrinsically unstructured proteins: intrinsic flexibility, modularity, and signaling conduits. Biochemistry. 2008;47:7598–609. doi: 10.1021/bi8006803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Minezaki Y, Homma K, Nishikawa K. Intrinsically disordered regions of human plasma membrane proteins preferentially occur in the cytoplasmic segment. J Mol Biol. 2007;368:902–13. doi: 10.1016/j.jmb.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 61.Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–49. doi: 10.1093/nar/gkh253. Print 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, Cortese MS, Sickmeier M, LeGall T, Obradovic Z, Dunker AK. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci U S A. 2006;103:8390–5. doi: 10.1073/pnas.0507916103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown KR, Jurisica I. Online predicted human interaction database. Bioinformatics. 2005;21:2076–82. doi: 10.1093/bioinformatics/bti273. [DOI] [PubMed] [Google Scholar]
- 64.Schnell S, Fortunato S, Roy S. Is the intrinsic disorder of proteins the cause of the scale-free architecture of protein-protein interaction networks? Proteomics. 2007;7:961–4. doi: 10.1002/pmic.200600455. [DOI] [PubMed] [Google Scholar]
- 65.Bader GD, Donaldson I, Wolting C, Ouellette BF, Pawson T, Hogue CW. BIND--The Biomolecular Interaction Network Database. Nucleic Acids Res. 2001;29:242–5. doi: 10.1093/nar/29.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21(waf1/cip1/sdi1) in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc Natl Acad Sci USA. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bowman P, Galea CA, Lacy E, Kriwacki RW. Thermodynamic characterization of interactions between p27(Kip1) and activated and non-activated Cdk2: intrinsically unstructured proteins as thermodynamic tethers. Biochim Biophys Acta. 2006;1764:182–9. doi: 10.1016/j.bbapap.2005.12.016. Epub 2006 Jan 11. [DOI] [PubMed] [Google Scholar]
- 68.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–48. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 69.Barbar E. Dynein light chain LC8 is a dimerization hub essential in diverse protein networks. Biochemistry. 2008;47:503–8. doi: 10.1021/bi701995m. [DOI] [PubMed] [Google Scholar]
- 70.Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, Dunker AK. Intrinsic disorder in transcription factors. Biochemistry. 2006;45:6873–88. doi: 10.1021/bi0602718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.