

Abstract
We previously reported that central nervous system (CNS) inactivation of Nf1 and p53 tumor suppressor genes in mice results in the development of low-grade to high-grade progressive astrocytomas. When the tumors achieve high grade, they are frequently accompanied by Akt activation, reminiscent of the frequent association of PTEN mutations in human high-grade glioma. In the present study, we introduced CNS heterozygosity of Pten into the Nf1/p53 astrocytoma model. Resulting mice had accelerated morbidity, shortened survival, and full penetrance of high-grade astrocytomas. Haploinsufficiency of Pten accelerated formation of grade 3 astrocytomas, whereas loss of Pten heterozygosity and Akt activation coincided with progression into grade 4 tumors. These data suggest that successive loss of each Pten allele may contribute to de novo formation of high-grade astrocytoma and progression into glioblastoma, respectively, thus providing insight into the etiology of primary glioblastoma. The presence of ectopically migrating neural stem/progenitor lineage cells in presymptomatic Pten-deficient mutant brains supports the notion that these tumors may arise from stem/progenitor cells.
Introduction
Gliomas are neuroectodermal tumors with predominantly glial characteristics (1). Malignant gliomas are characterized by diffuse tumor infiltration rendering surgical resection ineffective and resistance to current chemotherapy and radiation protocols. Median survival of patients with glioblastoma multiforme (GBM), the most common malignant glioma, is about 1 year (2, 3). Despite vigorous basic and clinical studies over the past two decades, the median survival for this disease has only improved marginally (4). Frequently mutated or deleted genes in malignant gliomas include CDK4, INK4A, ARF, RB, EGFR, PDGFR, TP53, and PTEN (1). These glioma signature genes are components of signaling pathways that normally control cell cycle, proliferation, survival, or death.
By genetically engineering signature glioma mutations, several groups have developed mouse models for glioma (5–14). Among these, the Nf1- and p53-deficient mice (Mut3 mice, GFAP-cre;cisNf1f/+;p53−/+) represent a genetic model wherein initially healthy mice eventually develop malignant astrocytomas (8). In these mice, somatic heterozygosity of Nf1, a negative regulator of the Ras pathway, is driven in neural cells by GFAP-cre (15) together with a p53 germline heterozygosity. Histopathologic evaluation uncovered presence of tumors that ranged from low-grade astrocytomas (grade 2) to GBMs (grade 4) with evidence of loss of heterozygosity (LOH) at both tumor suppressor genes (8). The full penetrance of the tumor phenotype permitted examination of presymptomatic mice that revealed abnormal proliferation and hyperplasia in the vicinity of the stem/progenitor cell niche, thus supporting the neural stem/progenitor cell origin of glioma hypothesis (8, 16, 17).
Intriguingly, a majority of high-grade, but not low-grade, gliomas from Mut3 mice were accompanied by appearance of Akt activation (8). Similarly, a majority of grade 3 astrocytomas found in a transgenic mouse line with deficiency in the Rb pathway (TgG(ΔZ)T121 mice) also showed increased Akt activation (9). These findings are consistent with frequent PTEN mutations in human high-grade gliomas (18). AKT is a major downstream effector of the phosphatidylinositol 3-kinase (PI3K) pathway, and PTEN antagonizes the PI3K pathway by dephosphorylating phosphatidylinositol 3,4,5 triphosphate that is required for AKT activation (19, 20). Decreased PTEN expression is also a poor prognosis marker for malignant gliomas (21). In addition, virus-mediated deletion of Pten (both alleles) in postnatal or 4-week-old mouse brains induced formation of higher grade astrocytomas in mouse models with activated Ras signaling in neural cells (13, 22).
Pten-null mice die embryonically, but heterozygous mice survive and develop tumors in diverse organs, including the lymphoid system, endometrium, prostate, and thyroid, but not in the nervous system (23–26). With respect to gliomas, these findings would be consistent with the idea derived from the above-mentioned mouse studies that PTEN mutations are important in glioma progression, but not in initiation (1, 3, 8, 10). However, in other mouse models of neonatal Ntv-a mice, constitutive activation of both the Akt and Ras pathways, but not either alone, induced GBM (7). In addition, constitutive activation of Akt increased glioma incidence in Ntv-a or Gtv-a mice with activated KRas and null for Ink4a/Arf (11). Introduction of germline Pten heterozygosity into the TgG(ΔZ)T121 mice decreased the latency for astrocytoma formation, without significantly changing tumor grade (9). Thus, oncogenic activation in neonates or viral oncogene expression coupled with Pten deficiency or Akt activation can cooperate with additional loss of tumor suppressor function to initiate gliomas. Therefore, the picture of the mechanistic contribution of Pten deficiency in mice and the relation to human gliomagenesis remain hazy.
In humans, PTEN mutations are uniquely associated with high-grade astrocytomas, in which PTEN loss seems to occur principally through LOH (18). To examine causal versus consequential relationship of PTEN mutation in genesis and progression of astrocytoma, we introduced a loxP-Pten allele (27) in the context of our Nf1 and p53 astrocytoma model (Mut3 mice; ref. 8). We find that the inclusion of somatic heterozygosity of Pten causes accelerated tumor formation that more closely resembles de novo (primary) GBM. These results coincide with abnormal neural stem/progenitor populations in vivo.
Materials and Methods
Mice and histology
To generate Mut3 to Mut6 mice (see Fig. 1A for genetic configurations), we crossed Mut3 (8) or Mut4 males with wild-type (wt), loxP-Pten (Ptenf hereafter; ref. 27), p53f (28), or p53f;Ptenf females. Littermate controls used for this study were with a genotype of wt for all alleles, cre only, or Nf1f/+;p53−/+;Ptenf/+ without cre. We maintained the mice in mixed genetic background of C57/BL6, Sv129, and B6/CBA. We observed the mice at least 6 d/wk. For BrdUrd chasing, we injected mice with 50 mg/kg (in PBS) of BrdUrd (Sigma) five times with a 2-h interval and sacrificed the mice 1 d or 1 wk later. We dissected out, processed, and sectioned brains as described (29). To evaluate brain anatomy, we stained every fifth slide with H&E. Mut3 to Mut6 astrocytomas were independently graded by using WHO histopathologic criteria, nuclear atypia, mitotic index, necrosis, and microvascular proliferation (C-H.K. and D.K.B.; Fig. 1C; ref. 2). Whereas grade 2 tumors show nuclear atypia only, grade 3 tumors contain two of the criteria, usually nuclear atypia and mitotic index, and grade 4 GBM additionally harbor necrosis, endothelial proliferation, or both. D.K. Burns, a neuropathologist, was blinded to genotype during the tumor grading. All mouse protocols were approved by the Institutional Animal Care and Research Advisory Committee at University of Texas Southwestern Medical Center.
Figure 1.
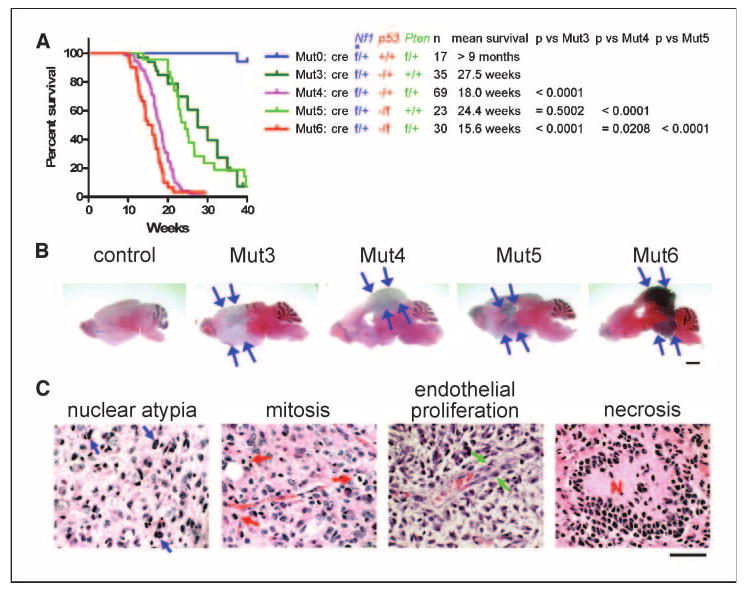
Somatic heterozygosity of Pten significantly shortens survival of brain tumor-forming Mut3 mice. A, comparative analysis on Kaplan-Meier survival curves of five mouse genotypes shows that mortalities of Mut4 or Mut6, but not of Mut5, mice were significantly earlier than that of Mut3 mice. Genetic configuration, number of mice, and mean survival of each genotype and P values between genotypes are shown next to the curves. *, flanked by two loxP sites. B, all symptomatic Mut3 to Mut6 mice analyzed (n = 11, 10, 6, and 8, respectively) harbored brain tumors (arrows), as shown in representative H&E-stained sections. Scale bar, 2 mm. C, brain tumors found in Mut3 to Mut6 mice exhibit morphologic features characteristic of diffusely infiltrating astrocytomas, including nuclear hyperchromasia and pleomorphism. Left, atypical astrocytic nuclei (blue arrows, for example), some of which surround normal cortical neurons. Features of high-grade astrocytomas, including mitotic index (red arrows, for example), endothelial proliferation (green arrows, for example), and necrosis (N) were also present. Scale bar, 50 μm.
Immunohistochemistry
We performed all immunohistochemistry on triplicate or more paraffin sections per group. We chose matched sections from control and mutant based on anatomy of the hippocampus and subventricular zone (SVZ). Antibodies used for immunohistochemistry were against Ki67 antigen (Novocastra), glial fibrillary acidic protein (Gfap; Sigma), Pten (NeoMarkers), nestin (BD Bioscience), phosphorylated Akt (p-Akt; Cell Signaling), BrdUrd (DAKO), Olig2 (Chemicon), or doublecortin (Santa Cruz). We used microwave antigen retrieval for all antibodies, except for doublecortin antibody. We amplified and visualized the primary antibodies as described (30). For BrdUrd/doublecortin double labeling, we treated sections with 2 N HCl for 1 h, briefly washed with 1 mol/L NaHCO3 and PBS, then blocked with 10% donkey serum before antibody incubation. We visualized the signals with Cy2-donkey anti-mouse IgG (Jackson ImmunoResearch) and Cy3-donkey anti-goat IgG.
Magnetic resonance imaging and tumor growth measurement
We initiated magnetic resonance imaging (MRI) studies on asymptomatic Mut3 (21–28 wk) and Mut4 (11–18 wk) mice (Supplementary Table S1). We did follow-up scanning weekly over a 1 to 6 wk of period to detect tumor growth. We performed MRI as described (8), except for using slightly different acquisition conditions for T1-weighed (TR = 200 ms; TE = 15 ms) and T2-weighed (TR = 1800 ms; TE = 80 ms) spin echo multislice axial images. We determined tumor volume on T2-weighed images by manually outlining, with a track ball, the enhancing portion of the mass on each image by using standard “browser” software provided by Varian Inova imaging system. The area measurements were automatically calculated and multiplied by the MRI section thickness to calculate a per-section tumor volume. The total tumor volume was obtained by summing the volume calculations for all sections. After MRI, the mouse was sacrificed and a whole mouse brain was dissected and subjected to histologic analysis, as described above.
Neurosphere culture and immunostaining
We established and maintained neurosphere cultures as described (31) with some modifications. Briefly, we dissected out the lateral walls of the lateral ventricle or whole brain tumor and digested by mechanical trituration and treatment with DNase I (250 units/mL; Invitrogen), papain (2.5 units/mL; Sigma), and neutral protease (1 units/mL; Roche). We washed and plated the cells on ultra-low attachment plates (Corning) in DMEM/F12 media (Invitrogen) containing B27 (without vitamin A), epidermal growth factor (20 ng/mL), and basic fibroblast growth factor (20 ng/mL; Sigma). We fed the cells every 2 to 3 d with the same media and passaged weekly by trypsinization with a seeding density of 2.0 × 104 cells/mL. All experiments using neurospheres as below were done between passages 5 and 10. For differentiation, we seeded 3.0 × 104 cells per well of eight-chamber slide coated with Matrigel (1:20; BD Bioscience) and cultured with Neuralcult with differentiation medium (StemCell Technologies) for 7 d. Then, we fixed the cells with 4% paraformaldehyde for 30 min and performed immunostaining for lineage markers as above (see Immunohistochemistry), except for using TO-PRO-3 (Invitrogen) as counter-staining. We obtained images by using confocal microscopy as described (32). Antibodies used were against β3-tubulin (TuJ1; Covance), Gfap (Dako), CNPase (Chemicon), or nestin.
LOH study and Western blotting
For LOH study, we compared genomic DNA isolated from ear, tumor-derived neurospheres, or tumor sections. To obtain genomic DNA from tumor sections, we choose paraffin blocks of well-formed grade 3 or grade 4 astrocytomas. We cut the remaining paraffin blocks after tumor grading into a 25-μm section and immediately isolated tumor region by careful dissection under microscope. We extracted genomic DNA by using QIAamp DNA micro kit (Qiagen) according to manufacturer's instruction. We used genotyping primers for cre, p53, or p53f as described (28, 33, 34). Primers used to detect Nf1f or Ptenf allele are described in Supplementary Table S2. We performed Western blotting on lysates of neurospheres as described (29). Antibodies used were against Pten (Cell Signaling), p-Akt, Akt, or β-actin (Sigma).
Results
Somatic Pten heterozygosity in neural cells shortens survival of astrocytoma-forming Mut3 mice
Mut3 mice have germline heterozygosity of p53 together with cre-mediated somatic heterozygosity of Nf1 in embryonic and adult neural precursors. Both tumor suppressor genes are located on the same chromosome in mice and humans. The Mut3 genotype harbors the mutations in cis and develops malignant astrocytomas in adult mice with complete penetrance. When the tumors reach high grade (grade 3/grade 4), we observed Akt activation (8). To study the role of PI3K signaling in gliomas, we introduced a loxP-Pten allele (27) into the Mut3 background (termed Mut4). In addition, we developed mutant strains (Mut5 and Mut6), in which the trans wt p53 allele in Mut3 and Mut4 genotypes, respectively, was replaced with a loxP-p53 allele (28). Similar to Mut3 mice, the Mut4 to Mut6 mice (see Fig. 1A for genetic configurations) were viable at birth and indistinguishable from littermate controls until adulthood when they developed variable neurologic abnormalities, including generalized motor seizures, ataxic gait, exaggerated startle response, and unidirectional circling behavior. Interestingly, most symptomatic Mut4 and Mut6 mice died as early as 10 weeks of age and usually within a week after showing signs of morbidity. In contrast, symptomatic Mut3 or Mut5 mice (containing wt Pten alleles) usually survived up to 8 weeks beyond initial appearance of symptoms. Comparison of Kaplan-Meier survival curves of Mut3 to Mut6 mice shows that somatic heterozygosity of Pten enhances mortality (Fig. 1A).
Histopathologic analysis revealed brain tumors in all symptomatic Mut3 to Mut6 brains analyzed (Fig. 1B). All abnormal regions had high expression of the astrocytic and neural precursor marker Gfap and also of the proliferation marker Ki67 antigen, suggesting that the tumors are gliomas (Supplementary Fig. S1A). Higher magnification images of H&E-stained sections from Mut3 to Mut6 tumors showed the typical morphology and histopathology of diffusely infiltrating astrocytomas, including nuclear atypia, mitotic figures, endothelial proliferation, and necrosis (Fig. 1C).
Pten deficiency results in precocious formation of high-grade astrocytomas and accelerated tumor growth
To further classify Mut3 to Mut6 astrocytomas, we applied the WHO brain tumor grading (2). All astrocytomas from symptomatic Mut3 to Mut6 mice contained both nuclear atypia and mitosis, categorizing them as at least grade 3 tumors (Fig. 2A). Occasionally, necrosis or endothelial proliferation was also observed in subsets of the tumors, classifying them as grade 4 (GBM). There was no substantial difference in tumor grade between symptomatic Mut3 and Mut4 mice (Fig. 2A and Supplementary Table 3). A greater incidence of GBM tumors appears in symptomatic mice in which LOH of p53 is driven by cre transgene (Mut5 and Mut6), which is consistent with the finding in human astrocytomas that loss of both p53 alleles is more frequent in higher grade forms (35).
Figure 2.
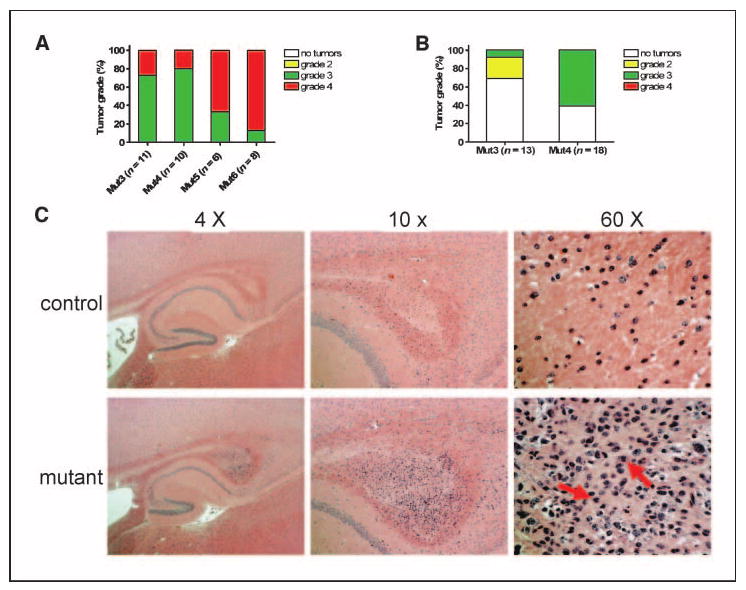
Somatic heterozygosity of Pten accelerates high-grade astrocytoma formation with no evidence of low-grade tumorigenesis. A, all symptomatic Mut3 to Mut6 mice analyzed harbored grade 3 or grade 4 astrocytomas. See Fig. 1A for configuration of genotypes and Materials and Methods for tumor classification. B, of 13 asymptomatic Mut3 mice analyzed (mean, 14.5 wk), three contained grade 2 tumors and one had a grade 3 tumor. In contrast, 11 of 18 asymptomatic Mut4 mice (mean, 14.2 wk) harbored grade 3 tumors. No grade 2 tumors were observed in Mut4 mice. C, a 14-wk-old, asymptomatic Mut4 mouse had a small astrocytoma in the caudal corpus callosum. Higher magnification image reveals the presence of mitotic index (arrows), classifying the tumor as a grade 3 malignancy.
Previously, we reported that astrocytomas from Mut3 mice progress from grade 2 to grade 4 (8). To determine whether similar tumor progression occurs in Pten-deficient Mut4 mice, we compared brains from age-matched, young asymptomatic Mut3 and Mut4 mice. Consistent with the previous report, gliomas from asymptomatic Mut3 mice were primarily classified as grade 2 astrocytomas (Fig. 2B). In age-matched asymptomatic Mut4 mice, tumor incidence was higher (11 of 18) than the Mut3 counterparts (4 of 13). In contrast to Mut3 counterparts, all astrocytomas from asymptomatic Mut4 mice were classified as grade 3 tumors. For example, the smallest astrocytoma found in the caudal corpus callosum of a 14-week-old Mut4 mouse displayed mitoses, indicating a grade 3 malignancy (Fig. 2C). We have yet to classify such small tumors in Mut3 mice as higher than grade 2 (not shown). These data suggest that heterozygosity of Pten in neural cells, in cooperation with deficiency in Nf1 and p53, elevates the incipient tumor to anaplastic astrocytoma (grade 3).
The shortened survival of Mut4 mice, coupled with high-grade classification in the smallest tumors analyzed, suggest an increased growth rate of the Pten-deficient tumors. Thus, we monitored and compared Mut3 and Mut4 tumor growth over time by MRI and confirmed tumor size by H&E staining and Ki67/Gfap immunohistochemistry. To compare tumor growth at similar stages, we examined 21-week-old to 28-week-old asymptomatic Mut3 mice and 11-week-old to 18-week-old asymptomatic Mut4 mice. As indicated in Supplementary Table S1, when observed, most tumors were grade 3. We found a 5-fold higher growth rate of Mut4 astrocytomas compared with Mut3 tumors (Fig. 3A). This was not due to differing tumor stage because the mean tumor size for each group was similar after the second scan (Supplementary Table S1). The accelerated growth rate of Mut4 tumors correlated with increased mitotic index and density of Ki67-positive cells in Mut4 over Mut3 tumors (Fig. 3B). Thus, introduction of somatic heterozygosity at Pten into astrocytoma-forming Mut3 mice resulted in earlier and higher grade tumors with accelerated tumor growth.
Figure 3.
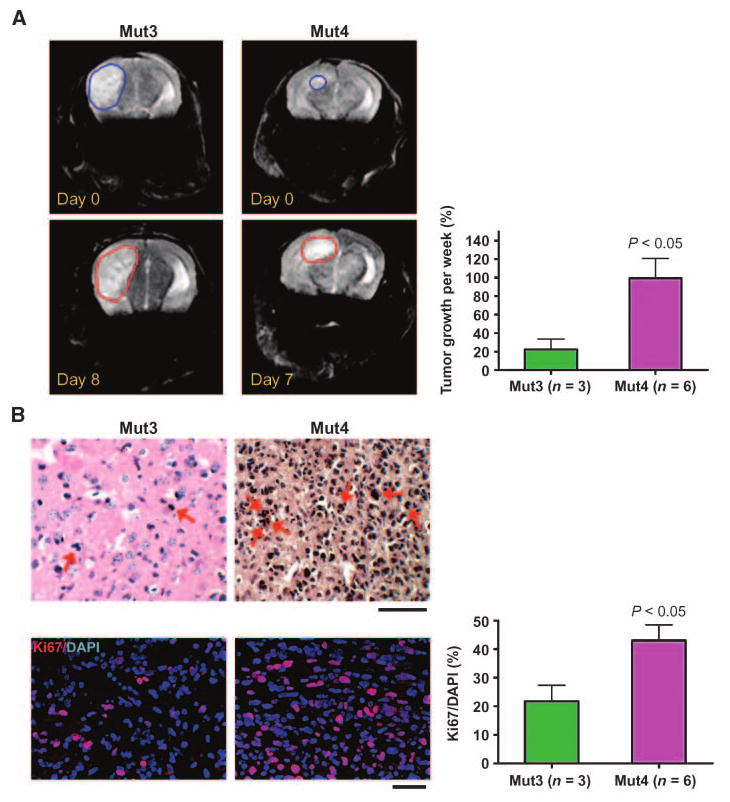
Mut4 tumors grow more rapidly than Mut3 tumors in vivo. A, representative MRI of a Mut3 mouse (n = 3) show tumor boundaries 8 d after (red) the first imaging (blue). The tumor growth was more prominent in Mut4 mice (n = 6). Analysis on tumor size detected by MRI indicates that Mut3 tumors grew 22.43 ± 11.25% (mean ± SE)/wk and Mut4 tumors, 99.45 ± 23.46%, demonstrating significantly faster growth of Mut4 than Mut3 tumors. See Materials and Methods for detailed MRI and tumor size measurement. B, representative images of H&E-stained astrocytoma sections analyzed after final MRI show increased mitotic index (arrows, for example) in Mut4 than in Mut3 tumors (top). Similarly, Ki67 immunoreactivity was higher in Mut4 than in Mut3 tumors (bottom). Statistical analysis of Ki67 density [the highest ratio of Ki67-positive to 4′,6-diamidino-2-phenylindole (DAPI)–positive cells] shows significantly higher value in Mut4 over Mut3 astrocytomas. Scale bars, 50 μm.
Pten inactivation and Akt activation in astrocytomas
To further analyze Mut3 to Mut6 astrocytomas, we established neurosphere cultures from the mouse tumors. When placed in differentiation conditions, the neurosphere cultures showed expression of the three major neural lineage markers (shown as a representative Mut6 culture in Supplementary Fig. S1B), indicating a stem cell–like property in the tumors. Previously, we reported LOH of Nf1 and p53 in astrocytomas from symptomatic Mut3 mice accompanied by Akt phosphorylation in some grade 3 and all grade 4 tumors (8). Similarly, all neurospheres derived from Mut3, Mut4, and Mut6 astrocytomas exhibit LOH at both Nf1 and p53 loci (Fig. 4A). Pten LOH was obvious in one of three Mut3 and all Mut4 and Mut6 neurospheres derived from astrocytomas. Western blotting confirmed the lack of Pten expression and concomitant elevation of p-Akt, a marker for Akt activation, in tumor-derived neurospheres with Pten LOH (Fig. 4B). Interestingly, all Mut3 tumor-derived neurospheres showed reduction or absence of Pten expression, compared with controls (Fig. 4B). Considering that most high-grade, but not low-grade, Mut3 astrocytomas harbor increased p-Akt (8) and that all Mut3 neurospheres in this study have LOH of Nf1 and p53, these data support the notion that high-grade Mut3 tumors undergo loss of Pten after LOH of Nf1 and p53.
Figure 4.
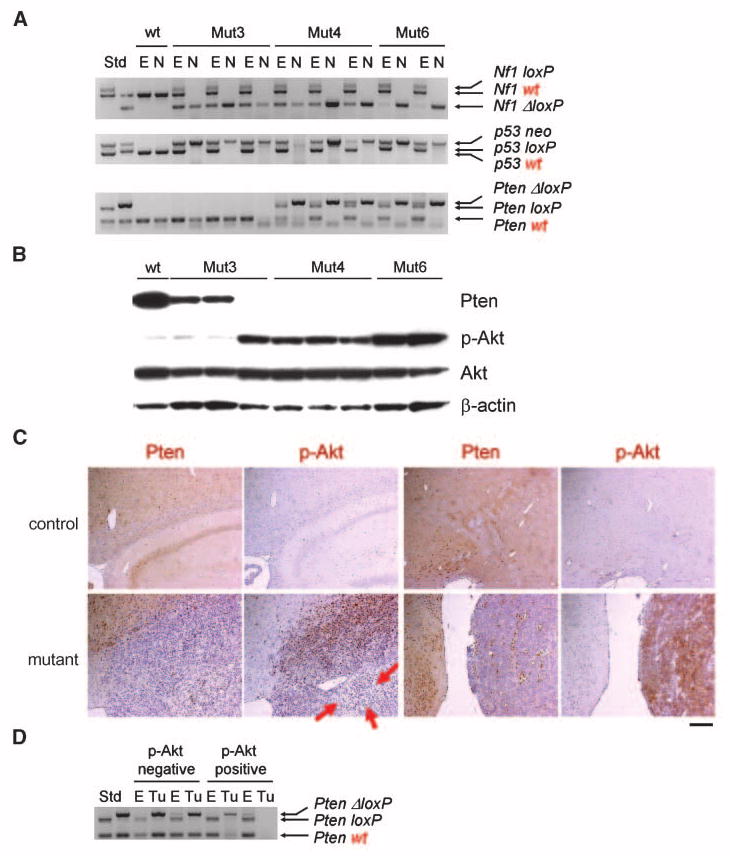
Decreased Pten expression and increased p-Akt levels in astrocytomas. A, Nf1 and p53 LOHs in all tumor-derived neurospheres (see Materials and Methods) accompany frequent Pten LOH. Genomic DNA from the ear (E) or neurospheres (N) derived from Mut3 to Mut6 astrocytomas was subjected to semiquantitative PCR. Size of each allele was compared with those of standard (Std). Whereas wt and mutant (loxP or neo) bands for Nf1 and p53 were found in all Mut3 to Mut6 ears, the wt bands were not detectable in all Mut3 to Mut6 tumor-derived neurospheres, indicating LOH of Nf1 and p53. LOH of Pten was also found in one Mut3 and all Mut4 or Mut6 cases. In most Mut3 to Mut6 cases, recombined loxP (ΔloxP) was also detected in the ear, probably due to partial cre activity. B, Western blotting on the tumor-derived neurospheres in A reveals decreased (two of three Mut3 cases) or lack of Pten expression (all other cases). p-Akt levels were commensurately elevated in all Pten-deficient samples. C, representative adult brain sections immunostained for Pten or p-Akt (brown) show abundant Pten expression and absence of p-Akt in controls. In contrast, representative high-grade astrocytomas from Mut6 mice displayed reduced or undetectable Pten expression and increased levels of p-Akt. There were some tumor regions lacking both Pten and p-Akt signals (arrows, for example). Scale bar, 200 μm. D, in vivo elevation of p-Akt correlates with Pten LOH. Genomic DNA from the ear or tumor sections (Tu) was subjected to semiquantitative PCR. Size of each allele was compared with those of standard. While p-Akt–negative astrocytomas (from Mut4 mice) retained wt and recombined loxP (ΔloxP) alleles, p-Akt–positive tumors showed decrease or absence of wt band.
Next, we measured Pten expression and p-Akt levels in brain tumor sections. Consistent with previous reports (30), normal brains exhibit high Pten expression and no evidence of p-Akt (Fig. 4C). In contrast, Pten expression was low or absent in all astrocytomas examined. All grade 4 and a subset of grade 3 gliomas harbored elevated p-Akt, implying that LOH at Pten plays an important role in grades 3 to 4 progression, as previously suggested (8). Intriguingly, a majority of grade 3 gliomas lacked both Pten and p-Akt expression (i.e., arrows in Fig. 4C). Genotyping of the tumor sections revealed that the p-Akt–negative, Pten-negative astrocytomas retained the wt Pten allele (Fig. 4D). Thus, despite the presence of one intact Pten allele, its protein expression is suppressed without stimulating Akt phosphorylation. Because all astrocytomas analyzed to date from young, asymptomatic Mut4 mice were classified as grade 3 (Fig. 2B), these data suggest haploinsufficiency for Pten in grade 3 tumors in the context of mutations in the Nf1 and p53 genes. In contrast, all p-Akt–positive tumors harbored LOH at the Pten allele (Fig. 4D). Thus, similar to two of three Mut3 tumor-derived neurospheres with decreased Pten expression (Fig. 4B), it seems that Pten heterozygosity does not result in appreciable elevation of p-Akt levels.
Ectopic migration of NSC/progenitor lineage cells precedes tumor initiation
In rodents, NSCs proliferate in the SVZ, the largest germinal center, and transient amplifying progenitors migrate along the rostral migratory stream (RMS) into the olfactory bulb (OB; refs. 36, 37). Previously, we reported evidence that Mut3 astrocytomas arise from the SVZ (8), supporting the NSC/progenitor origin hypothesis for brain tumors (16, 17, 38). A majority of Mut4 to Mut6 astrocytomas (21 of 24) were found in proximity of the SVZ/RMS/OB or nearby regions, such as the striatum and corpus callosum (Supplementary Table S3), and these tumors also express the neural progenitor markers nestin and Sox2 (Supplementary Fig. S1C). To examine the status of neural progenitor populations in these tumor prone mice, we analyzed Mut3 to Mut6 mice during development by immunohistochemistry for Ki67, Gfap, and nestin. Consistent with the early and high-grade tumor appearance seen in Mut4 and Mut6 mice, by 8 weeks of age, a proportion of the mice showed abnormal Ki67, nestin, and Gfap immunoreactivity in the vicinity of the SVZ and RMS, including the striatum and rostral corpus callosum (Fig. 5 and data not shown). To test whether outlying parenchymal abnormal cells result from aberrant migration of NSC/progenitor, we performed a pulse chase experiment to specifically label SVZ stem cells, progenitors, and their descendants. We injected BrdUrd into mice at 6 weeks of age before evidence of cellular abnormality and analyzed the mice 1 day or 1 week later. One day after the BrdUrd pulse, all mice showed BrdUrd reactivity mainly confined to the SVZ (Fig. 6A and data not shown). However, 1 week after injection, Pten-deficient Mut4 (one of three) or Mut6 (three of three) mice exhibited ectopic localization of BrdUrd-containing cells in the vicinity of the SVZ/RMS/OB, including the striatum and corpus callosum (Fig. 6B and Supplementary Table 4). A majority of the BrdUrd-positive ectopic cells also expressed doublecortin, a marker for migrating neuroblasts, or Olig2, a marker for neural progenitors and oligodendroglial precursors (39–41). Mut3 and Mut5 mice did not exhibit such abnormal localization at this early time point consistent with the more retarded appearance of tumors in these mice (Fig. 6A and Supplementary Table S4). Thus, our data indicate that ectopic migration of Nf1;p53;Pten-deficient NSC/progenitor lineage cells temporally precedes the appearance of astrocytomas.
Figure 5.
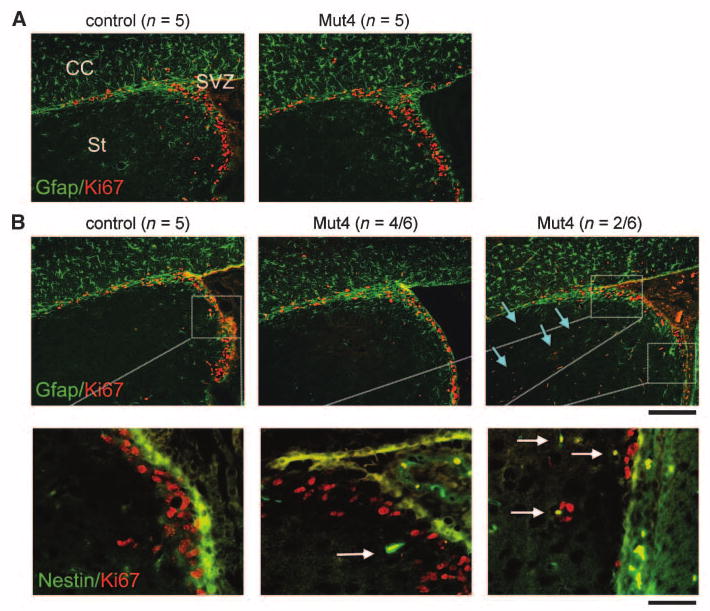
Ectopic positioning of neural stem/progenitor lineage cells precedes the appearance of astrocytomas. Mut3 to Mut6 mice were subjected to detailed histologic and immunocytochemical analysis at various time points preceding any evidence from previous studies that tumors would be present. Brain sections were double-labeled for Ki67, a proliferation marker, and Gfap that is expressed in astrocytes and subsets of neural precursor populations. A, representative images indicate that, at 6 wk of age, there was no substantial difference in Ki67 and Gfap signal between genotypes. CC, corpus callosum; St, striatum. B, by 8 wk of age, two of six Mut4 mice exhibited abnormal Ki67-positive and Gfap-positive cells in the striatum (light blue arrows), which were rare or absent in all control and other Mut4 mice analyzed (top). Higher magnification from the SVZ of adjacent sections stained with Ki67 and nestin, a neural stem/progenitor marker, shows the presence of double-labeled cells (white arrows) in Mut4 sections, which were absent in control (bottom). Scale bar, 200 μm (top and middle) and 50 μm (bottom).
Figure 6.
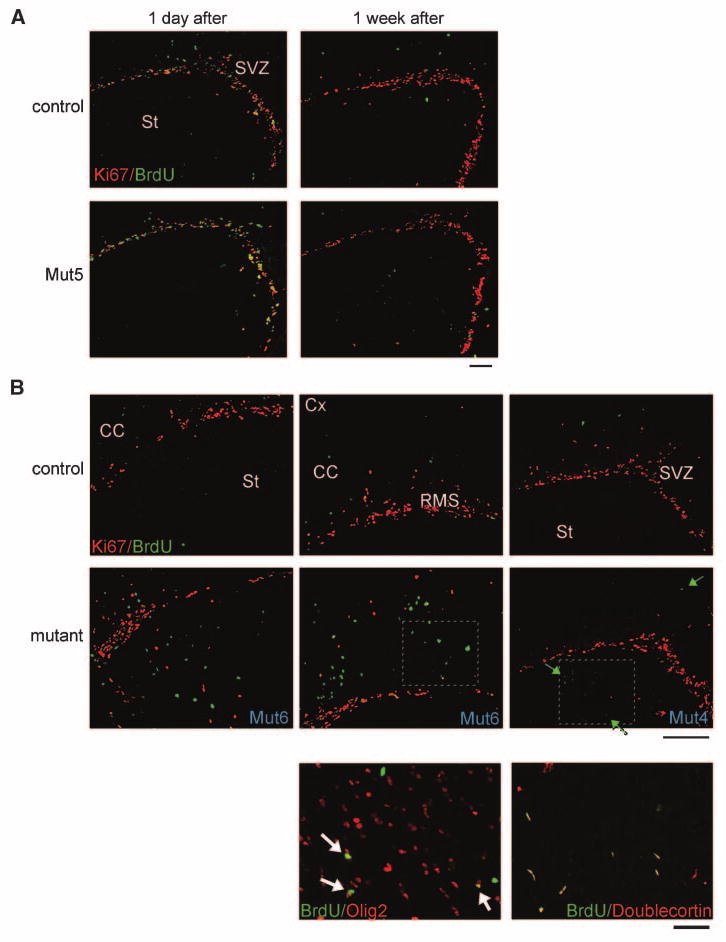
BrdUrd chasing reveals that ectopic migration of neural stem/progenitor lineage cells precedes the appearance of astrocytomas. Mutant and control mice were injected with BrdUrd at 6 wk of age and analyzed 1 d or 1 wk later. A, representative images show that there was no substantial difference between control and Mut5 brains for locations of BrdUrd or Ki67 signal. B, representative images of BrdUrd/Ki67 double-stained brain sections (top and middle) 1 wk after the BrdUrd pulsing exhibit that Mut6 or Mut4 brains harbor ectopically localized BrdUrd-positive cells in the striatum (St), corpus callosum (CC), and/or cortex (Cx), close to SVZ and RMS where neural stem/progenitors are normally present. Some of the BrdUrd-positive ectopic cells were stained by Ki67 (green arrows, for example) and also expressed doublecortin (bottom right). A majority of the BrdUrd-positive ectopic cells expressed Olig2 (bottom left). Such BrdUrd/doublecortin or BrdUrd/Olig2 double-positive cells were fewer or absent in the similar areas of control brains (not shown). Bottom, higher magnification images of immunostained, adjacent sections to the boxed area in the middle. Scale bar, 200 μm (top and middle) and 50 μm (bottom).
Discussion
We have used reverse genetics to explore the etiology and underlying mechanisms of glioma in genetic mouse models designed to test the role and physiologic relevance of somatic mutations identified in human glioma. In extension of previous studies (8), we find that while deficiencies of Nf1 and p53 result in formation of low-grade astrocytomas, Pten deficiency cannot cooperate with Nf1 deficiency alone to initiate gliomas (Mut0 mice in Fig. 1A). Instead, we find that inclusion of Pten deficiency, together with Nf1 and p53 deficiencies, confers de novo GBM features in our tumor models. De novo or primary GBM occurs in relatively older individuals lacking clinical evidence of a prior low-grade lesion, whereas progressive or secondary GBM arises in relatively younger patients through anaplastic progression (1–3). According to a recent survey, de novo GBMs constitute a majority of GBMs (95%) compared with progressive forms (5%; ref. 42). Our current study indicates that, consonant with observations made with human glioma samples (1, 3), Pten deficiency alone does not initiate low-grade tumors but is sufficient to transform low-grade tumors into high-grade tumors (as in progressive GBM) or alternatively to initiate high-grade tumors (de novo GBM).
In our previous glioma models, we observed that a proportion of grade 3 tumors and all grade 4 tumors exhibited activation of Akt and additional markers associated with human glioma progression (8). We reasoned that loss of Pten in the transition from low grade to high grade could be a causal genetic event rather than simply a marker for the transition. In the present study, we show that introduction of somatic heterozygosity of Pten in neural cells causes shortened survival of astrocytoma-forming Mut3 mice. Mice harboring somatic Pten heterozygosity in the context of Nf1 and p53 deficiencies develop precocious tumors with accelerated tumor growth. Histologic and molecular analysis of the incipient tumors in asymptomatic mice failed to reveal features of low-grade tumors, but rather showed consistent evidence of active proliferation, providing compelling evidence that these are de novo high-grade tumors. According to a survey covering >700 patients, p53 mutations are found in 28% of de novo GBMs, although the frequency is higher in progressive GBMs (65%; ref. 43). Although NF1 mutations are rare in human de novo GBMs, we emphasize three relevant features: (a) Individuals with neurofibromatosis 1 have increased incidence of glioma; (b) NF1 deficiency down-regulates cyclic AMP level through adenylate cyclase/PKA signaling (44), which in turn decreases differentiation and death and promotes proliferation of malignant glioma cells (45, 46); (c) NF1 also controls RAS signaling, which is often dysregulated in de novo GBMs (1). Indeed dominant activating RAS mutations are not a feature of glioma. Rather, low-grade persistent activation of the pathway is a feature of common EGFR mutations found in glioma (47). The effect of NF1 inactivation results in a similar passive activation of RAS signaling (48). Thus, although the Pten-deficient Mut4 and Mut6 GBM models described here may not represent precise genocopies of idiopathic human de novo GBMs, we propose that these models provide physiologically relevant phenocopies of these tumors.
A large proportion of human high-grade astrocytomas contain one of two different types of allelic PTEN loss (18). One allelic mutation is either a point or frame-shift mutation within PTEN. The second mutation can be a variable deletion of chromosome 10q23 where PTEN is located. This suggests successive loss of the two PTEN alleles in tumor development. Also frequent is the presence of a wt PTEN allele together with a mutant allele. These observations are consistent with the model that PTEN heterozygosity may have consequences in glioma biology. Previously reported mouse models with deletion of both Pten alleles clearly indicate that loss of Pten function promotes tumor progression (10, 13, 22). However, the mechanistic contribution of Pten heterozygosity in astrocytoma formation remains unclear. Although introduction of germline Pten heterozygosity significantly shortened survival of astrocytoma forming TgG(ΔZ)T121 mice, status of the wt Pten allele or change in Akt activation level has not been reported (9). Our current data support the model that Pten heterozygosity may participate in astrocytoma initiation. We uncovered that a significant proportion of grade 3 tumors retained Pten heterozygosity, which, in cooperation with Nf1 and p53 heterozygosity, produced earlier onset of astrocytomas. The lack of Pten expression in the tumors may be due to epigenetic silencing or limited sensitivity of antibody used for immunohistochemistry. The lack of Akt activation in most Pten-deficient grade 3 astrocytomas suggests involvement of Akt-independent pathway(s), of which mechanisms are under active investigations in other experimental systems (49, 50). Increased p-Akt signal in grade 4 tumors with loss of both Pten alleles indicate that Pten LOH and Akt activation are crucial for grades 3 to 4 progression, resonating with the previously hypothesized role of PTEN inactivation in high-grade gliomas (1, 3, 8, 10, 22).
A continuing puzzle in the study of human glioma is the basis for de novo GBM. The current understanding in this field is confounded by the fact that tumor identification in human subjects, and subsequent histologic and molecular analysis, can only begin once an afflicted individual presents with some form of neurologic deficit that leads to medical attention. Thus, it cannot be ascertained how long the tumor has been present or whether there exists a brief period of low-grade tumorigenic state preceding clinical manifestation, followed by rapid transition to high-grade status. Therefore, the accelerated growth of GBM, in the absence of a reliable mouse model, precludes study and elucidation of tumor initiation or etiology. Although previously reported mouse models using constitutively activated Akt (7, 11), germline Pten heterozygosity (9), or acute deletion of both Pten alleles (10, 13, 22) have provided interesting insights into the involvement of the PI3K/PTEN/AKT pathway in the formation and progression of gliomas, our current models with somatic Nf1 and Pten heterozygosity with p53 deficiency in neural cells allow for stochastic LOH and subsequent tumor development. This in turn provides a clearer picture of how allelic loss of each Pten allele contributes to de novo GBMs. Additionally, immunohistochemical and BrdUrd pulse-chase experiments revealed ectopic migration of neural stem/progenitor lineage cells in young Mut4 and Mut6 mice at an age where no evidence of tumors could be found. Thus, in our model, cells originating in the stem/progenitor niche are the first and only cells to exhibit abnormal features preceding tumor formation. These data add further support to the notion that neural stem/progenitor lineage can be the glioma cell of origin (8, 16, 17, 38). While our studies identify neural stem/progenitor lineage cells as potential sources for glioma formation, these neither address nor preclude alternative mechanisms that could lead to glioma formation from nonneurogenic niche precursors or more differentiated cell types.
We note that our mouse tumor samples isolated distant from the SVZ grow well under neurosphere culture conditions. In addition, preliminary studies indicate the ability of tumor-derived neurospheres to seed orthotopic brains and generate infiltrative tumors.6 Thus, as has been described for human glioma by Dirks and colleagues (17), it seems that Mut3-, Mut4-, or Mut6-derived gliomas contain cells with “cancer stem cell” properties. Continued analysis and comparison to human glioma tumors and cancer stem cells should further refine the properties of these tumors as they relate to human glioma.
A caveat in cre/loxP experiments that include multiple conditional alleles is the possibility of interchromosomal recombination or confounding effects caused by the cre recombinase. We consider it unlikely that cre-mediated interchromosomal recombination affected the survival of Mut4 to Mut6 mice, because neither Mut0 mice in this study (Fig. 1A) nor additional previous controls (8) developed brain tumors.
In summary, we have developed genetic mouse models for de novo GBM that support the idea that in the presence of Nf1, p53, and Pten heterozygosity, de novo high-grade gliomas appear without requisite transition through low-grade status, undergoing LOH at Nf1 and p53, yet retaining Pten heterozygosity. Our data show that Pten heterozygosity confers haploinsufficiency for de novo high-grade tumor formation. The ability to study these mice in great detail may provide insight into mechanism and provide novel therapeutic targets to attack this intractable disease.
Supplementary Material
Acknowledgments
Grant support: Basic Research Fellowships from American Brain Tumor Association (C.-H. Kwon, in memory of Daniel J. Martinelli and Geoffrey J. Cunningham), Cancer Imaging Program grant SAIRP U24CA126608, Advanced Imaging Research Center NMR Center grant NIH BTRP #P41-RR02584, NIH grant R37NS33199, and DOD grants DAMD17-02-1-0638 and DAMD17-03-1-0216 (L.F. Parada). L.F. Parada is an ACS Research Professor.
We thank Albee Messing for GFAP-cre mice; Caroline Gregorian, Zohre German, Steve McKinnon, Randy Wolfe, Shawna Kennedy, Tzong-Shiue Yu, and Brett Whittemore for technical information and assistance; and Sharon Matheny, Steve Kernie, Jon Graff, and Robert Bachoo for suggestions and helpful discussion.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
D. Zhao, J. Chen, and S. Alcantara contributed equally to this work.
S. Alcantara, C.-H. Kwon, and L.F. Parada, unpublished observation.
References
- 1.Zhu Y, Parada LF. The molecular and genetic basis of neurological tumours. Nat Rev Cancer. 2002;2:616–26. doi: 10.1038/nrc866. [DOI] [PubMed] [Google Scholar]
- 2.Kleihues P, Cavenee WK, editors. Pathology and genetics of tumours of the nervous system. Lyon: IARC Press; 2000. [Google Scholar]
- 3.Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–33. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Ding H, Roncari L, Shannon P, et al. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001;61:3826–36. [PubMed] [Google Scholar]
- 6.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109–13. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 7.Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–7. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Y, Guignard F, Zhao D, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–30. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao A, Wu H, Pandolfi PP, Louis DN, Van Dyke T. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell. 2002;1:157–68. doi: 10.1016/s1535-6108(02)00029-6. [DOI] [PubMed] [Google Scholar]
- 10.Xiao A, Yin C, Yang C, Di Cristofano A, Pandolfi PP, Van Dyke T. Somatic induction of Pten loss in a preclinical astrocytoma model reveals major roles in disease progression and avenues for target discovery and validation. Cancer Res. 2005;65:5172–80. doi: 10.1158/0008-5472.CAN-04-3902. [DOI] [PubMed] [Google Scholar]
- 11.Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC. Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res. 2002;62:5551–8. [PubMed] [Google Scholar]
- 12.Uhrbom L, Kastemar M, Johansson FK, Westermark B, Holland EC. Cell type-specific tumor suppression by Ink4a and Arf in Kras-induced mouse gliomagenesis. Cancer Res. 2005;65:2065–9. doi: 10.1158/0008-5472.CAN-04-3588. [DOI] [PubMed] [Google Scholar]
- 13.Wei Q, Clarke L, Scheidenhelm DK, et al. High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Res. 2006;66:7429–37. doi: 10.1158/0008-5472.CAN-06-0712. [DOI] [PubMed] [Google Scholar]
- 14.Charest A, Wilker EW, McLaughlin ME, et al. ROS fusion tyrosine kinase activates a SH2 domain-containing phosphatase-2/phosphatidylinositol 3-kinase/mammalian target of rapamycin signaling axis to form glioblastoma in mice. Cancer Res. 2006;66:7473–81. doi: 10.1158/0008-5472.CAN-06-1193. [DOI] [PubMed] [Google Scholar]
- 15.Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 16.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–83. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 18.Ali IU, Schriml LM, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst. 1999;91:1922–32. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- 19.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 20.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–5. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 22.Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia. 2005;7:356–68. doi: 10.1593/neo.04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Podsypanina K, Ellenson LH, Nemes A, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563–8. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 25.Stambolic V, Suzuki A, de la Pompa JL, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki A, de la Pompa JL, Stambolic V, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–78. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 27.Groszer M, Erickson R, Scripture-Adams DD, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–9. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- 28.Lin SC, Lee KF, Nikitin AY, et al. Somatic mutation of p53 leads to estrogen receptor α-positive and -negative mouse mammary tumors with high frequency of metastasis. Cancer Res. 2004;64:3525–32. doi: 10.1158/0008-5472.CAN-03-3524. [DOI] [PubMed] [Google Scholar]
- 29.Fraser MM, Zhu X, Kwon CH, Uhlmann EJ, Gutmann DH, Baker SJ. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004;64:7773–9. doi: 10.1158/0008-5472.CAN-04-2487. [DOI] [PubMed] [Google Scholar]
- 30.Kwon CH, Luikart BW, Powell CM, et al. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–88. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–16. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- 32.Luikart BW, Nef S, Virmani T, et al. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–86. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwon CH, Zhou J, Li Y, et al. Neuron-specific enolase-cre mouse line with cre activity in specific neuronal populations. Genesis. 2006;44:130–5. doi: 10.1002/gene.20197. [DOI] [PubMed] [Google Scholar]
- 34.Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 35.del Arco A, Garcia J, Arribas C, et al. Timing of p53 mutations during astrocytoma tumorigenesis. Hum Mol Genet. 1993;2:1687–90. doi: 10.1093/hmg/2.10.1687. [DOI] [PubMed] [Google Scholar]
- 36.Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–8. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- 37.Alvarez-Buylla A, Garcia-Verdugo JM, Tramontin AD. A unified hypothesis on the lineage of neural stem cells. Nat Rev Neurosci. 2001;2:287–93. doi: 10.1038/35067582. [DOI] [PubMed] [Google Scholar]
- 38.Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 39.Lu QR, Sun T, Zhu Z, et al. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- 40.Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–71. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- 41.Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, Alvarez-Buylla A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. 2006;26:7907–18. doi: 10.1523/JNEUROSCI.1299-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005;64:479–89. doi: 10.1093/jnen/64.6.479. [DOI] [PubMed] [Google Scholar]
- 43.Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892–9. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 44.The I, Hannigan GE, Cowley GS, et al. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science. 1997;276:791–4. doi: 10.1126/science.276.5313.791. [DOI] [PubMed] [Google Scholar]
- 45.Yoshimura S, Sakai H, Nakashima S, et al. Differential expression of Rho family GTP-binding proteins and protein kinase C isozymes during C6 glial cell differentiation. Brain Res Mol Brain Res. 1997;45:90–8. doi: 10.1016/s0169-328x(96)00239-2. [DOI] [PubMed] [Google Scholar]
- 46.Chen TC, Hinton DR, Zidovetzki R, Hofman FM. Up-regulation of the cAMP/PKA pathway inhibits proliferation, induces differentiation, and leads to apoptosis in malignant gliomas. Lab Invest. 1998;78:165–74. [PubMed] [Google Scholar]
- 47.Huang HS, Nagane M, Klingbeil CK, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem. 1997;272:2927–35. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- 48.Zhu Y, Parada LF. Neurofibromin, a tumor suppressor in the nervous system. Exp Cell Res. 2001;264:19–28. doi: 10.1006/excr.2000.5138. [DOI] [PubMed] [Google Scholar]
- 49.Freeman DJ, Li AG, Wei G, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–30. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 50.Yoo LI, Liu DW, Le Vu S, Bronson RT, Wu H, Yuan J. Pten deficiency activates distinct downstream signaling pathways in a tissue-specific manner. Cancer Res. 2006;66:1929–39. doi: 10.1158/0008-5472.CAN-05-1986. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.