

Abstract
Cardiac IKr is a critical repolarizing current in the heart and a target for inherited and acquired long QT syndrome (LQTS). Biochemical and functional studies have demonstrated that IKr channels are heteromers composed of both hERG 1a and 1b subunits, yet our current understanding of IKr functional properties derives primarily from studies of homo-oligomers of the original hERG 1a isolate. Here we examine currents produced by hERG 1a and 1a/1b channels expressed in HEK-293 cells at near-physiological temperatures. We find that heteromeric hERG 1a/1b currents are much larger than hERG 1a currents and conduct 80% more charge during an action potential. This surprising difference corresponds to a two-fold increase in the apparent rates of activation and recovery from inactivation, thus reducing rectification and facilitating current rebound during repolarization. Kinetic modeling shows these gating differences account quantitatively for the differences in current amplitude between the two channel types. Drug sensitivity was also different. Compared to homomeric 1a channels, heteromeric 1a/1b channels were inhibited by E-4031 with a slower time course and a corresponding four-fold shift in the IC50. The importance of hERG 1b in vivo is supported by the identification of a 1b-specific A8V missense mutation in 1/269 unrelated genotype-negative LQTS patients and absent in 400 control alleles. Mutant 1bA8V expressed alone or with hERG 1a in HEK-293 cells dramatically reduced 1b protein levels. Thus, mutations specifically disrupting hERG 1b function are expected to reduce cardiac IKr and enhance drug sensitivity, and represent a potential mechanism underlying inherited or acquired LQTS.
Keywords: hERG 1a/1b, Kv11.1, KCNH2, ether-à-go-go, arrhythmia, long QT syndrome, potassium channels, hERG 1b, hERG1b
Introduction
Cardiac IKr is a potassium current contributing to ventricular repolarization in mammalian heart.1, 2 The molecular basis of cardiac IKr was first elucidated when its unique biophysical and pharmacological properties were largely reproduced by heterologous expression of the human ether-a-go-go-related gene (hERG1, or KCNH2).3, 4 Together with the discovery of KCNH2 mutations as the pathogenic substrate in families with type 2 long QT syndrome (LQTS) 5, these studies explained the underlying cause of disease as a loss of cardiac IKr. They also identified hERG1 channels as a molecular target for acquired LQTS, a much more prevalent form of the disease arising from IKr block primarily by drugs intended for other therapeutic targets.6 In either manifestation, LQTS is characterized by prolonged ventricular action potentials and a susceptibility to potentially life-threatening arrhythmias known as torsades de pointes (TdP).7
Our understanding of how IKr contributes to ventricular repolarization is based primarily on heterologous expression of the originally-identified hERG 1a subunit. 3, 4, 8, 9 Like other voltage-gated potassium channels, hERG1a channels activate and inactivate upon depolarization. However, because inactivation is much faster than activation, current is suppressed at positive potentials but rebounds upon repolarization as channels quickly recover from inactivation and slowly close. During an action potential, these gating transitions produce a resurgent current that peaks during the repolarizing phase. 10 Gating is modulated by the cytoplasmic amino (N) terminus, with different regions affecting deactivation, inactivation and activation. 11-15
More recent studies suggest that native IKr channels are heteromers arising from coassembly of hERG 1a with 1b, another alpha subunit encoded by an alternate transcript of KCNH2. The two transcripts, first characterized in mouse and human, are identical except for alternate 5′ exons encoding unique cytoplasmic N termini of significantly different sizes (Fig. 1A). 16, 17 Expressed in heterologous systems, the 1a and 1b subunits co-assemble to form functional channels, 17 a process that is facilitated by direct, co-translational interactions of the divergent N termini. 18 Such interactions mask an exposed endoplasmic reticulum (ER) retention signal (RXR) in the 1b subunit, which otherwise prevents efficient expression of hERG 1b homomeric channels. 19 Isoform-specific antibodies can co-immunoprecipitate 1a and 1b subunits from rat, canine and human ventricle, suggesting that both proteins indeed associate to form native IKr channels. In addition, both isoforms show extensive T-tubular distribution in canine myocytes. 20 In a selective knock-out of mouse ERG 1b, fetal IKr deactivation rate is decreased, and a small dofetilide-sensitive component of outward current in adult cardiomyocytes is eliminated. 21
Fig 1.
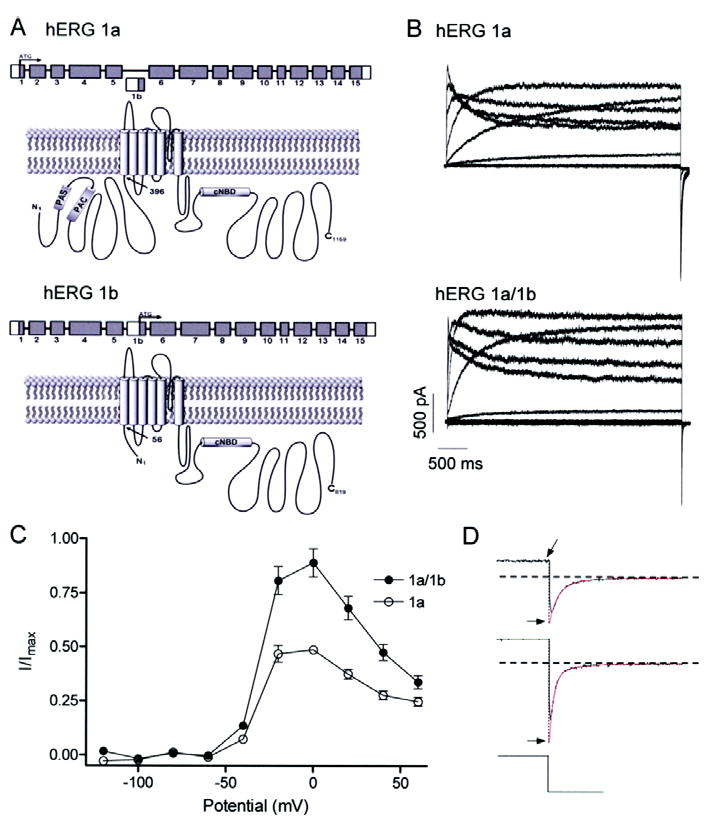
Greater amplitude currents for hERG 1a/1b compared to hERG 1a channels. A, graphic showing N-terminal differences in primary structure between hERG 1a (upper) and hERG 1b (lower). Cytoplasmic N and C termini flank the hydrophobic core. N-terminal differences yield subunits of 1159 and 819 amino acid residues for hERG 1a and 1b, respectively. “PAS”, Per-Arnt-Sim domain; “PAC”, PAS-associated C-terminal domain; “CNBD”, putative cyclic nucleotide binding domain; “TCC”, tetramerization coiled-coil domain. B, currents from stably transfected hERG 1a cells (upper) or hERG 1a stable cells transiently transfected with hERG 1b (lower). All currents were recorded at 34 ± 2°C in response to series of 4-s depolarizing voltage steps ranging from -120 to 60 mV followed by a 5-s repolarizing step to -50 mV. C, steady state current-voltage relations of hERG 1a and hERG 1a/1b channels, both displaying the hallmark negative slope conductance (n = 5 - 6 for both). The currents at the end of each depolarizing pulse were normalized to the absolute value of the extrapolated maximum tail current and plotted as a function of membrane potential. D, double exponential fits (in red) of tail currents evoked following a step to 60 mV extrapolated back to the moment of voltage change to obtain peak current value (Imax; see Methods).
Despite the evidence that heteromeric hERG 1a/1b channels underlie cardiac IKr, we know little about the gating and pharmacological properties of these channels, how hERG 1a/1b channels differ from hERG 1a homomers, or what role the hERG 1b subunit might play in disease. Here we describe for the first time the biophysical properties of hERG 1a/1b heteromeric channels at near-physiological temperatures in mammalian cells. We find that hERG 1a/1b currents are significantly larger than hERG 1a currents attributable to an increased activation rate and a faster rate of recovery from inactivation. In addition, hERG 1a/1b currents peak earlier than 1a currents during the action potential. Despite their increased occupancy of the open state, hERG 1a/1b channels develop E-4031 block with slower kinetics and with four-fold reduced sensitivity compared with hERG 1a channels, suggesting that a loss of the 1b subunit would enhance drug sensitivity and susceptibility to acquired LQTS. Kinetic modeling can account for these differences by introducing “N-mode” gating with alternative transition rates. A role for hERG 1b in LQTS is further supported by the identification of a 1b-specific mutation in a patient with clinically diagnosed LQTS who had remained genotype negative for all known LQTS-susceptibility genes.
Materials and Methods
Cell culture
hERG1a currents were recorded from a stable hERG 1a/HEK-293 cell line. The cells were maintained and passaged using minimum essential medium supplemented with 10% fetal bovine serum, 1% non-essential amino acids, 1% sodium pyruvate and 400 μg/ml geniticin. For recordings of heteromeric 1a/1b channels, 2 micrograms hERG1b cDNA was transiently transfected into a stable hERG1a cell line along with EGFP cDNA, leading to an average ratio of 1a:1b expression of 1:2. Cells were incubated for 36-48 h prior to recording. In preparation for electrophysiological recording, cells were trypsinized, washed in standard MEM media and plated onto cover slips on the day of recording. After a few hours the cover slip was placed in a recording chamber on a Nikon Diaphot inverted microscope. Cells with green fluorescence were chosen for 1a/1b recordings.
Electrophysiological procedures and analysis
Whole-cell patch-clamp recordings were performed using an Axopatch 200B amplifier (Axon Instruments, Inc.). PClamp software (Axon Instruments) was utilized for data acquisition and analysis. The recording chamber was perfused with HEPES-buffered Tyrode solution containing (in mM) 137 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.4 with NaOH). Whole-cell currents were recorded with a fire-polished pipette tip of approximately 1-2 μm with a resistance of 2-4 MΩ when filled with the internal pipette solution. Internal pipette solution contained (in mM) 130 KCl, 1 MgCl2, 5 EGTA, 5 MgATP, 10 HEPES (pH 7.2 with KOH). Currents were digitized at 1-20 kHz unfiltered. Series resistance compensation was typically 60-70% such that voltage errors were less than 5 mV. No leak subtraction was applied; cells exhibiting leak conductance greater than 10% maximal conductance were excluded from the study. Temperature was regulated by an in-line solution heater made locally. All experiments reported here were carried out at 34 ± 2°C to approximate physiological conditions, except for the pharmacology experiments, which were carried out at room temperature (22 ± 2°C) because at higher temperatures the seals were insufficiently stable to endure the long recordings required for measuring drug sensitivity.
Experiments were carried out using both voltage step and simulated action potential protocols to better mimic the native electrical signals in cardiac myocytes. The action potential voltage command was the same as that used previously, a digitized recording from a rabbit ventricular myocyte 10. Differences in rectification between hERG 1a and 1a/1b channels were revealed in I-V plots from currents elicited using both voltage clamp protocols (Fig. 1C and 2B, respectively) and divided by the absolute value of the maximum peak deactivating inward tail current evoked subsequent to a pulse to +60 mV. The peak current, determined using a two-exponential fit to the deactivating current extrapolated back to the moment of the voltage change (Fig. 1D), represents the maximal conductance and is proportional to the number of channels in the cell. This approach requires single channel conductance to be the same for hERG 1a and 1a/1b channels, a reasonable assumption for two channel types identical except for their cytoplasmic N termini; moreover, we know from previous work that removal of the hERG 1a N terminus does not alter single channel conductance.15 In this way the amplitude of outward currents, reflecting the degree of rectification, could be characterized for hERG 1a vs. 1a/1b channels, irrespective of differences in transfection efficiency or the corresponding number of channels expressed in each cell, which would otherwise prohibit comparison.
Fig 2.
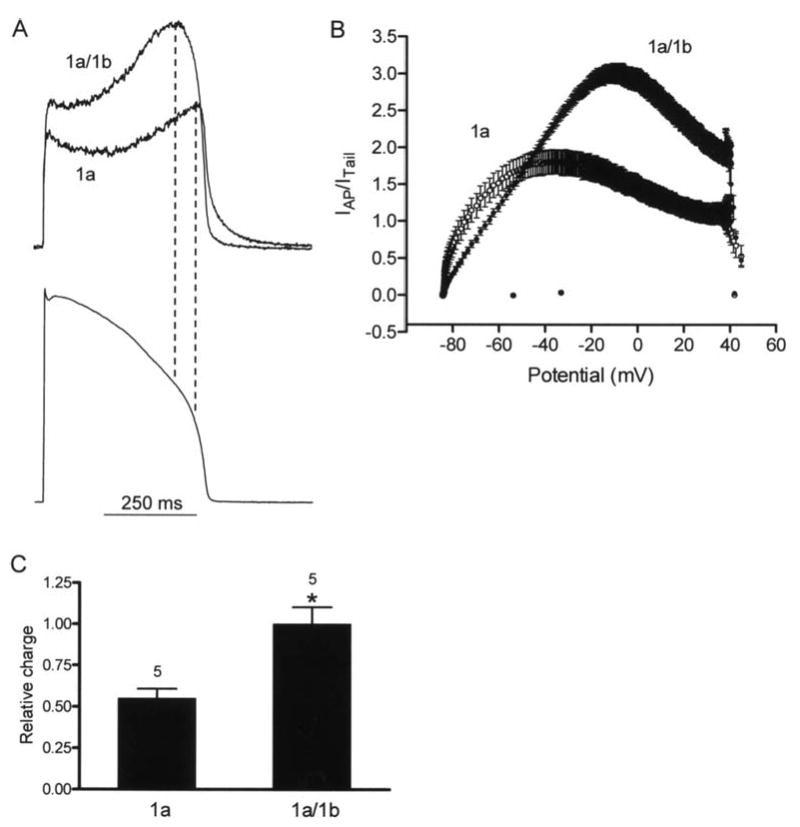
hERG 1a and 1a/1b currents during cardiac action potential clamp. A, upper panel, representative current profiles of hERG 1a/1b and 1a channels recorded at 34 ± 2°C. Currents were normalized to peak tail currents elicited at -105 mV following a prepulse to 60 mV to compare current amplitudes and differences in rectification for a given channel density. Lower panel, voltage command was a digitized rabbit ventricular action potential as previously described 10. B, real-time I-V plots of current for 1a/1b and 1a channels during action potential clamp. Each point is the mean ± s.e.m of five cells. C, relative charge transferred during an action potential command. Values were obtained by integrating the normalized current traces (432.4 ± 45.9 and 786.6 ± 80.7 for 1a and 1a/1b, respectively) and were plotted with an additional normalization to the 1a/1b values. * indicates P < 0.05, Mann Whitney test.
Steady state inactivation was measured as previously published8 but with shorter test potentials to minimize the contamination by the faster deactivation at the higher temperatures used in our study22. Errors due to deactivation were corrected for8 and the resulting normalized currents plotted vs. test voltage and fit with a Boltzmann function. The short pulse causes a positive shift of the V1/2 of the resulting steady-state inactivation curve from what might be considered “true” steady state. Therefore, actual V1/2 values differ based on the method used and at different temperatures, but the observed shift in V1/2 between hERG 1a and 1a/1b channels can provide information about how gating differences result in an alteration of the distribution between the inactivated and activated states.
For drug block measurements, current was activated by a 4-s long depolarizing pulse to 20 mV from a holding potential of -80 mV, and the peak tail current was recorded at -50 mV. After control currents were recorded, the drug was perfused for 2 min and then incubated for 10 min while the cell was held at -80 mV to maintain the channels in a closed state. Following the 10 min incubation, depolarizing steps were applied for 20 times at 15-s interpulse intervals until steady-state block was reached.
GraphPad Prism (GraphPad Software, Inc.) and pClamp were used to analyze data and generate statistical plots. Curves were fit using nonlinear least-squares regression analysis. Conductance and voltage data were fitted to a single Boltzmann function: I/Imax = (Imin) + (Imax −Imin)/{1+exp [(V1/2-V)/k]}, where, V1/2 is the half-activation potential, V is the test voltage and k is the slope factor. The relative charge transfer through 1a/1b and 1a channels was determined by integrating the normalized current traces (area under the curve). IC50 ± s.e.m values were obtained from best-fit results using the following sigmoidal concentration-response function: I = Imax[1+(IC50/X)n] where I denotes relative tail current, Imax is the peak tail current, X is the concentration of test compound in log scale, n is the Hill coefficient reflecting the number of drug binding sites, and IC50 is the concentration at 50% of maximal inhibition. Averaged data are presented as the mean ± standard error of the mean (s.e.m.). Statistical comparisons were made using the Mann Whitney test. P values < 0.05 were considered statistically significant. E-4031 was purchased from Sigma (St. Louis).
Computational Modeling
Detailed computational modeling methods and model equations are provided in the online supplement.
KCNH2 Exon 1b Mutational Analysis
Previously, a comprehensive mutational analysis of all 60 amino acid-encoding exons of the five cardiac channel LQTS-associated genes was completed for 541 consecutive, unrelated patients (358 females; average age at diagnosis = 24 ± 16 years; average QTc = 482 ± 57 ms) who were referred to the Windland Smith Rice Sudden Death Genomics Laboratory at Mayo Clinic, Rochester, MN for LQTS genetic testing between August 1997 and July 200423. This study focused on the 269 unrelated patients in whom no LQTS-associated mutations were identified following this analysis. This cohort is referred to as “genotype-negative” LQTS.
Genomic DNA panels derived from 100 healthy Caucasian and 100 African American subjects was obtained from the NINDS Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds) and served as controls.
Mutational analysis of KCNH2 exon 1b was performed using polymerase chain reaction (PCR), denaturing high performance liquid chromatography (DHPLC), and direct DNA sequencing as previously described24. This alternate exon was not included in the original analysis and KCNH2 exon 1b is not presently included as part of the clinically available genetic test for LQTS. Previously published PCR primers and reaction conditions were used in this study25.
Mutagenesis and western blot analysis
The A8V mutation was introduced into the hERG 1b construct using a PCR-based mutagenesis strategy; sequence analysis confirmed the presence of the mutation and integrity of all other sequence. To generate western blots, HEK-293 cells were transfected with 1.5 ug of each DNA construct using LT1 reagent (Mirus, Madison, WI). Cells were lysed 48 hours post-transfection and processed as described previously 18. Cell lysate (5-10 micrograms) was separated by SDS-PAGE, transferred to Immobilon-P polyvinylide difluoride membranes (Millipore, Bedford, MA) and probed with antibodies raised against the C terminus of hERG26. Membranes were also probed with anti-PDI as a loading control. Standard Western blot procedures were followed for detection of protein signal on X-ray film.
Results
Differences in steady-state current-voltage relations
As a first step in characterizing differences in hERG 1a and 1a/1b channels we recorded whole-cell currents using patch clamp at 34 ± 2°C from HEK-293 cells heterologously expressing the corresponding cDNA (Fig. 1). Currents were activated by 4-sec step commands in 20 mV increments over a range of voltages from -120 mV to 60 mV from a holding potential of -80 mV (Fig. 1B). Tail currents were evoked by a subsequent step to -105 mV. Expression of 1b alone in HEK-293 cells, like Xenopus oocytes17, produced little or no current (data not shown). The currents at the end of each pulse were normalized to the absolute value of the extrapolated maximum tail current obtained for each cell following the step to +60 mV and plotted vs. voltage (Fig. 1C, D; see Methods). This analysis shows that, while both 1a and 1a/1b currents display the hallmark negative-slope conductance characteristic of hERG current-voltage (I-V) relations, hERG 1a/1b currents are much larger and exhibit less rectification.
Currents are larger and peak earlier during the action potential for hERG 1a/1b vs. hERG 1a channels
To understand the physiological ramifications of hERG 1a/1b heteromerization, we evoked currents using voltage clamp commands mimicking ventricular action potentials at 34 ± 2°C (Fig. 2). Normalizing the current evoked using the action potential clamp to the extrapolated peak tail current elicited following a step command in the same cell (see Methods) shows the resulting 1a/1b currents were larger and peaked earlier during the action potential (Fig. 2A). A “dynamic I-V” plot shows normalized currents sampled at 1-ms intervals and averaged over several experiments as a function of the action potential voltage (Fig. 2B). This data transformation highlights the greater amplitude and shift in peak current of 1a/1b relative to 1a. Integrating the normalized current traces indicated hERG 1a/1b currents transfer approximately 80% more charge during the action potential voltage command (Fig. 2C).
Differences in activation and deactivation
To determine the biophysical basis for the reduced rectification of hERG 1a/1b current, we examined gating properties using a variety of paradigms. The steady-state activation curve representing the distribution between the closed and open states over a range of voltages showed no significant shift in voltage of half-maximal activation or slope factor (Fig. 3A). For 1a channels, V1/2 and the slope factor are -26.8 ± 1.3 mV and 6.9 ± 0.2, respectively (n = 6); for 1a/1b channels, the values are -28.6 ± 1.0 mV and 6.2 ± 0.1 (n = 5) (P > 0.05). Thus, the closed-open equilibrium as represented by the Boltzmann distribution is similar for hERG 1a homomeric and 1a/1b heteromeric channels.
Fig 3.
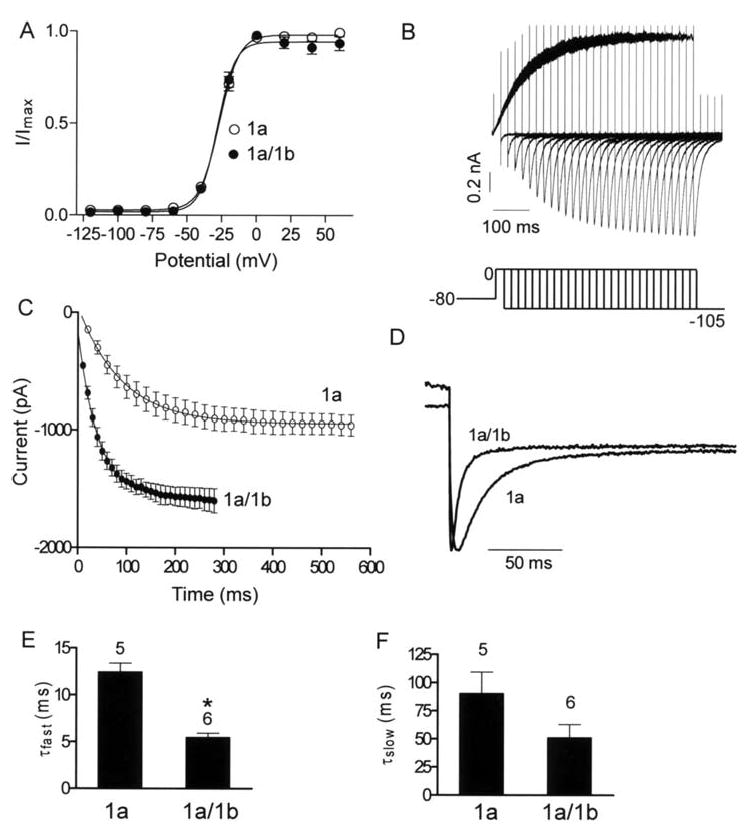
Activation and deactivation properties of hERG 1a and 1a/1b channels. A, Steady-state activation plots. Tail current amplitudes during the -105 mV step were normalized to the maximum tail current amplitude and plotted as a function of the preceding depolarization step to obtain the steady-state activation plots. The line of best fit is a Boltzmann function (Materials and Methods). The V1/2 and the slope factor for 1a channels are -26.8 ± 1.3 mV and 6.9 ± 0.2, (n = 6); and for 1a/1b channels -28.6 ± 1.0 mV and 6.2 ± 0.1, respectively (n = 5; P > 0.05, Mann Whitney test). B, currents evoked using an envelope of tails protocol to determine time course of activation. Peak inward tail currents were evoked by a step to -105 mV following a prepulse of increasing duration to 0 mV. Holding potential was -80 mV. C, apparent activation is faster for hERG 1a/1b compared with hERG 1a currents. The peak amplitudes of the tail currents (B) were plotted against test pulse duration and fit to single exponential function. Time constants of activation were 98.7 ± 17.6 ms and 41.6 ± 6.7 ms for hERG 1a and 1a/1b, respectively. Each point is the mean ± s.e.m of four to six cells. D, deactivation is faster for hERG 1a/1b vs. hERG 1a currents. Scaled tail currents recorded at -105 mV are shown. E & F, time constants of fast and slow components from double exponential fits to deactivating tail currents are plotted for comparison between 1a and 1a/1b channels. * indicates P < 0.05, Mann Whitney test. Temperature is 34 ± 2°C.
Examination of kinetic transitions provides more information regarding the influence of the 1b subunit. Both activation and deactivation transitions between the closed and open states were accelerated. Fig. 3B illustrates the envelope of tails protocol used to measure the activation time constant uncontaminated by the faster inactivation process 4. The tail current, which is proportional to the number of channels having transitioned from closed to open during the preceding step to 0 mV, is plotted as a function of time in Fig. 3C. Pooled data over several experiments were fit with a single exponential function as shown. This analysis reveals a time constant of activation of 98.7 ± 17.6 ms (n = 6) for hERG 1a, and 41.6 ± 6.7 ms (n = 4) for hERG 1a/1b, reflecting an approximately two-fold increase in the apparent rate of transition from the closed to the open state for the heteromeric channels.
We determined deactivation kinetics from tail currents evoked at -105 mV subsequent to a step to 60 mV (Fig. 3D). Fitting the deactivating component of the tail current with the double exponential function y = A0 + Afe-t/τf +Ase-t/τs revealed fast and slow time constants (τ) (Fig. 3E and F, respectively). The fast time constants, which account for approximately 90% of the deactivating component at -105 mV for hERG 1a and 1a/1b, are 12.4 ± 0.9 ms (n = 5) and 5.5 ± 0.4 ms (n = 6), respectively, reflecting a nearly two-fold difference in apparent closing rate (Fig. 3E) (P < 0.05). The time constants of the slow deactivation component for 1a vs. 1a/1b were not significantly different (P > 0.05) (Fig. 3F).
Differences in inactivation and recovery from inactivation
Previous studies have shown that rectification of hERG 1a currents arises because inactivation is much faster than activation, thus minimizing the dwell time in the open state and effectively suppressing outward current in response to positive voltage commands 2-4. We tested the hypothesis that differences in inactivation contribute to the reduced rectification of hERG 1a/1b channels. Using the “three-pulse” protocol that separates inactivation from the temporally overlapping activation process (Fig. 4A), we were surprised to find no differences in the time constant of inactivation (P > 0.05) (Fig. 4B). Instead, there was approximately a two-fold difference in the time constant of recovery from inactivation (P < 0.05) (Fig. 4C,D), consistent with the earlier trajectory of resurgent current and larger current amplitude. There is a corresponding 28-mV shift in the steady-state inactivation curve, with average V1/2's of -14.5 mV and 13.68 mV for hERG 1a and hERG 1a/1b, respectively (Fig. 4E).
Fig 4.
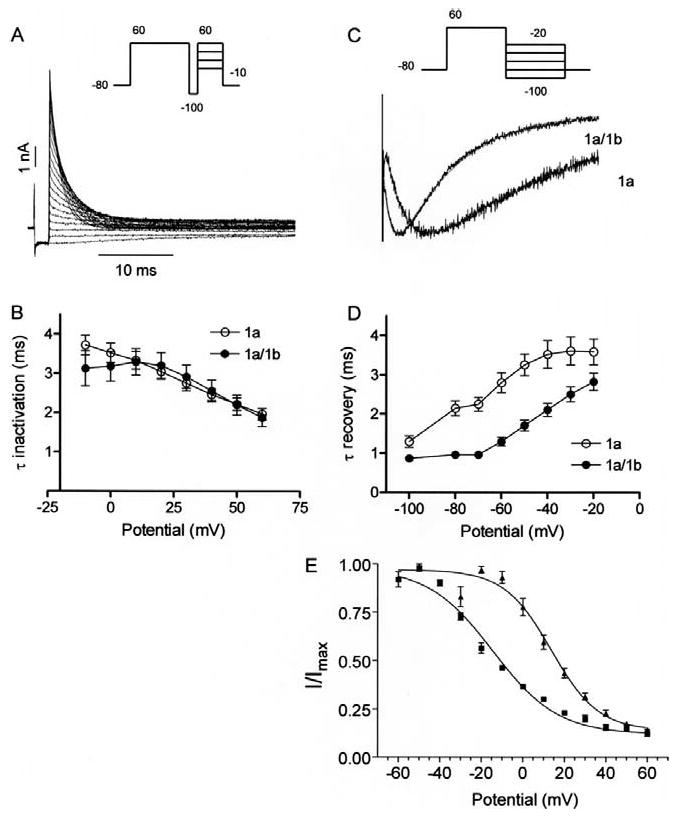
Inactivation properties of hERG 1a and 1a/1b channels. A, hERG currents elicited by the three-pulse protocol to measure the time course of inactivation. A 500-ms pulse to 60 mV to activate and then inactivate hERG is followed by a 2 ms pulse to -100 mV to remove the inactivation. In the third pulse varying the potential between -10 and 60 mV allowed the inactivation time course to be measured as a function of voltage. B, the time constants for onset of inactivation are estimated by fitting the decay of the currents in the third pulse to a single exponential function and plotted as a function of test potential. There were no significant differences in the time constants of inactivation for hERG1a and hERG1a/1b channels. Each point is the mean ± s.e.m of four to six cells (P > 0.05, Mann Whitney test). C, exemplary tail currents with double exponential fits (red) showing faster recovery from inactivation for hERG 1a/1b compared with hERG 1a channels. Tail currents were evoked by a step to -105 mV following a 4-sec, 60 mV pulse. D, plot quantifying data showing recovery from inactivation is faster in hERG 1a/1b compared with hERG 1a channels. Time constants were measured as the single exponential fit to the rising phase (> -80 mV) or as the fast time constant of a double exponential fit (≤ -80 mV) to the tail current. Each point is the mean ± s.e.m of seven to eight cells (P < 0.05, Mann Whitney test). Temperature was 34 ± 2°C. E, steady-state inactivation plot showing shift of V1/2 between hERG 1a and 1a/1b currents. Currents were measured at +40 mV following a series of 2-ms steps to a range of voltages from a holding potential of +40 mV.
Comparison of E-4031 block of hERG 1a and 1a/1b channels
Although hERG 1a/1b and hERG 1a channel pores share the same primary structure, and thus likely the same drug binding sites, we tested the hypothesis that differences in gating kinetics might cause access-related differences in the efficacy of block. Specifically, we predicted the increased open time accounting for larger current amplitudes would enhance drug block in hERG 1a/1b channels. We examined steady-state block by the methanesulfonanilide hERG blocker E-4031 at room temperature. The resulting dose-response curve shows an increase in the IC50 for E-4031 inhibition of hERG 1a/1b vs. 1a currents, from 6.2 ± 1.1 nM to 25.6 ± 4.3 nM, respectively (Fig. 5A). A corresponding slowing of block was observed (Fig. 5B). This reduction in efficacy for hERG 1a/1b channels, although modest, was nonetheless surprising given that E-4031 is an open-channel blocker and 1a/1b channels spend more time in the open state than do 1a channels.
Fig 5.
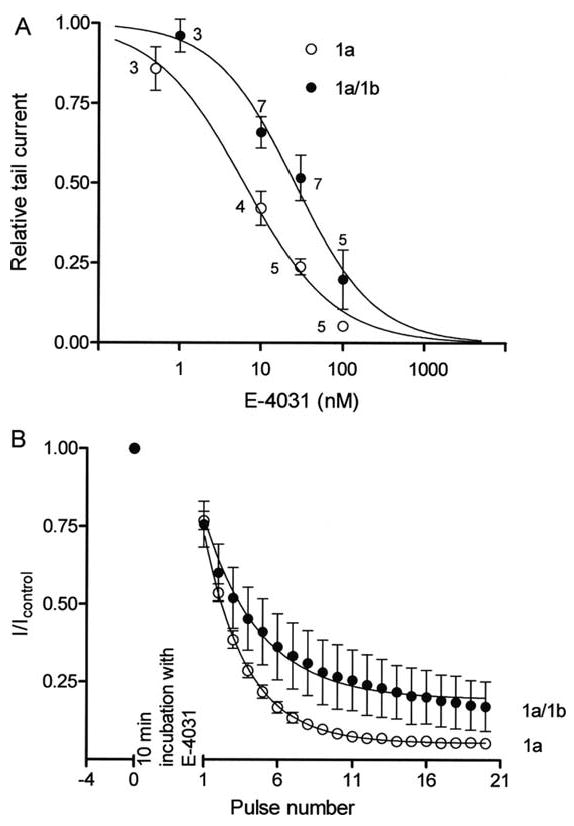
E-4031 block of hERG1a/1b and 1a channels assessed with conventional step protocol at room temperature. Currents were recorded before drug application by stepping from -80 to +20 mV for 4 s and then to -50 mV for 5 s with 15 s interpulse interval. Channels were held closed at -80 mV for 10 min in the presence of E-4031 and then subjected to 20 voltage pulses in the continued presence of the drug. A, steady-state dose-response curves for hERG 1a and 1a/1b channels. The IC50 values for E-4031 drug block for hERG 1a and hERG 1a/1b channels were 6.2 ± 1.1 nM and 25.6 ± 4.3 nM, respectively. B, peak tail current amplitudes plotted as a function of number of pulses and fit to an exponential function to measure the time course of block. Development of E-4031 block is slower for hERG1a/b channels.
A Kinetic Model Explains Increased Current Amplitude and Decreased Drug Sensitivity of hERG 1a/1b Channels
We turned to kinetic modeling to determine whether the faster apparent rates of activation and recovery from inactivation could quantitatively account for the increased current amplitude of hERG 1a/1b channels. To be valid, the model also had to explain the paradoxical reduction in efficacy of drug block. We arrived at our designs for Markov models of hERG 1a/1b and hERG 1a by considering how the known protein structural differences might account for the measured biophysical differences in the currents. Given that hERG 1a is structurally identical to hERG 1b with the exception of its longer and unique N terminus, we propose that the presence of the extended N termini on all four subunits in the hERG 1a channel causes an interaction that introduces an alternative gating mode (“N-mode”) not in effect for hERG 1a/1b channels where the extended N termini are present in three or fewer subunits.
Figure 6 illustrates the state diagrams for hERG 1a/1b and hERG 1a models. In the absence of the hERG blocking drug E-4031, the model for hERG 1a/1b (blue) includes five states connected as in Silva and Rudy27 (c3, c2, c1, o, and i). The model for hERG 1a (red) includes a normal mode of gating with states and transitions identical to hERG 1a/1b. When E-4031 is present, the transition rate ON becomes nonzero allowing movement from o to the E-4031-block mode (green rectangle) in both hERG 1a/1b and hERG 1a models. We allow gating to occur while the drug remains trapped in the pore28-30. The hERG 1a model also includes N-mode gating reflecting the action of the N termini in the hERG 1a homomer previously shown to affect gating12-15 (turquoise rectangle). N-mode access is via the open state along θ/ρ transition rates. N-mode transition rates are altered as described in the upper right corner of the figure. hERG 1a has two equally conducting open states (o and on, circled in black). hERG 1a/1b has one conducting open state (o, circled in black). For hERG 1a, E-4031 block and N-mode gating may exist in isolation or they may overlap (N-mode + E4031-block mode, intersection of green and turquoise rectangles). E-4031 block and N-mode gating are separate and independent processes, but they may occur simultaneously (e.g. along transition path κ). This is achieved by imposing that κ, ν, λ and δ transitions comply with microscopic reversibility. Moreover, simultaneity follows as a natural consequence of our inclusion of the combination N-mode gating together with E4031 block, which implies non-exclusionary binding of the 1a N termini and E-4031.
Fig 6.
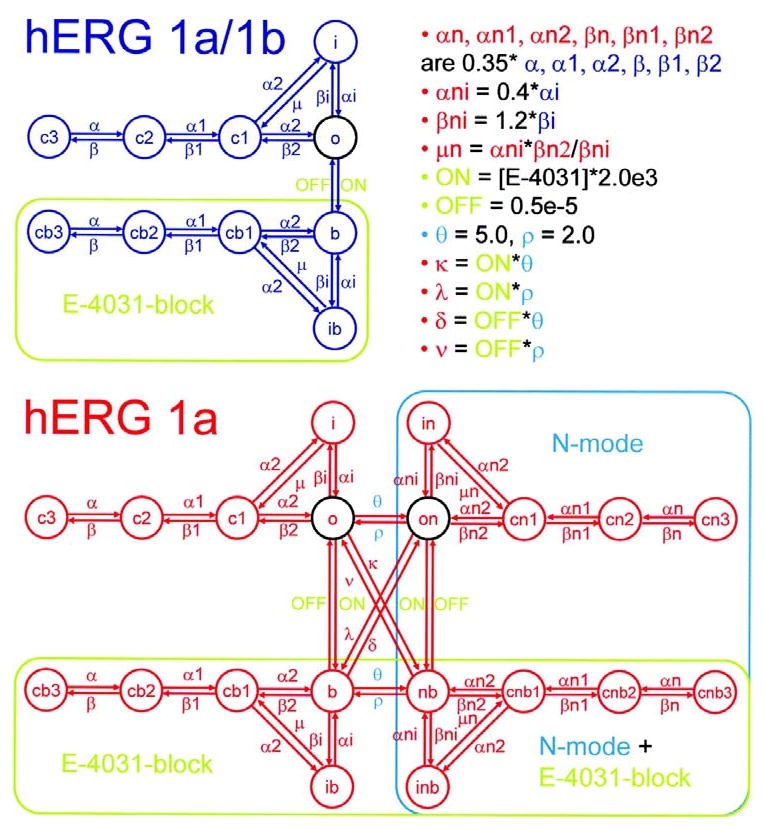
Schematic diagrams for hERG 1a/1b and hERG 1a models. In the absence of E-4031, the model for hERG 1a/1b (blue) operates in normal mode, while the hERG 1a model (red) operates in both normal mode and N-mode (turquoise rectangle). Transition rates for the N-mode are different from those for the normal mode as described in the upper right corner of the figure. Normal mode transition rates are identical for hERG 1a/1b and 1a models. Open states, circled in black, are all equally conducting. The presence of E-4031 allows entry into the E-4031-block mode (green rectangles for both hERG 1a/1b and 1a). E-4031-block mode and N-mode may coincide for hERG 1a (intersection of turquoise and green rectangles).
For model validation, we show simulated reproductions of important experimental results for hERG 1a/1b and hERG 1a in Figs. 7 and 8. We assume that maximum single channel conductance is the same for hERG 1a/1b and hERG 1a, so all differences are attributed to channel kinetic properties. The simulations show general agreement with the experimental observations with respect to normalized steady-state current-voltage relations (Fig. 7A), steady-state conductance-voltage relations (Fig. 7B), and rates of inactivation and recovery from inactivation (Figs. 7C, D). The time constant for activation, and the fast time constant for deactivation were matched by the models (hERG 1a/1b τ activation (ms): experiment 41.6 ± 6.7, model 40.9; hERG 1a τ activation (ms): experiment 98.7 ± 17.6, model 107.1; hERG 1a/1b τ fast deactivation (ms): experiment 5.5 ± 0.4, model 5.8; hERG 1a τ fast deactivation (ms): experiment 12.4 ± 0.9, model 11.0). Thus, the observed alterations in kinetics can quantitatively account for the decrease in rectification and concomitant increase in current amplitude during step or action potential voltage commands (Figs. 7A and 8A, respectively), as well as the increased charge transferred during repolarization (Fig. 8B). The faster rate of deactivation in hERG 1a/1b contributes to the earlier peak and to lower amplitude currents but only at more negative voltages (cf. Fig. 2A). Notably, simulations show E-4031 sensitivity is greater for hERG 1a than hERG 1a/1b, as observed experimentally (Fig. 8C). Also, as observed in the experiments, simulations demonstrate faster block for hERG 1a than for hERG 1a/1b (i.e. maximal degree of block is reached in fewer cycles of the protocol, not shown).
Fig 7.
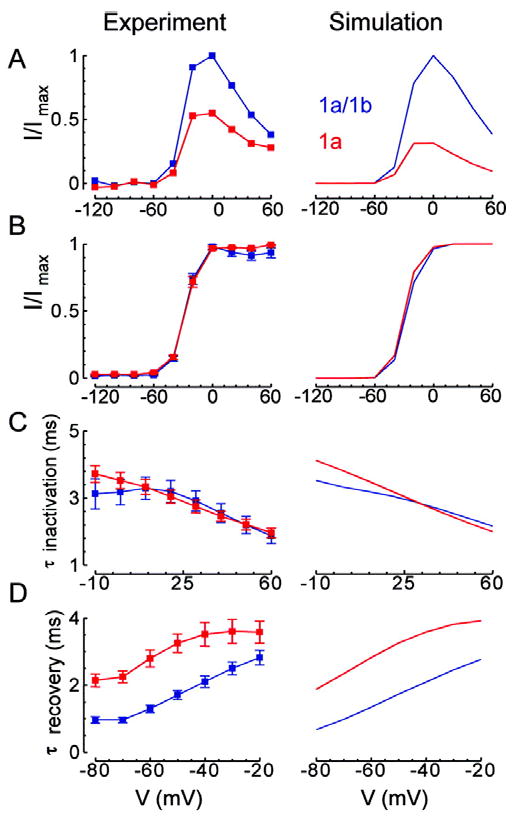
Voltage-dependent behavior. Left column shows experimental results and right column shows corresponding simulation results. hERG 1a/1b is in blue and hERG 1a is in red. A, Step current. Plot shows relative current at the end of 4 seconds at the indicated potentials from a -80 mV holding potential divided by the extrapolated peak tail current at -105 mV. Results are normalized to the maximum for hERG 1a/1b. B, Steady-state activation. Plot shows normalized maximal tail currents at steps to -105 mV following a 4 second step to the indicated potentials from a -80 mV holding potential. For computation of steady state activation, movement from state i to state c1 was not allowed in order to prevent artifactual recovery to c1 instead of o at the most depolarized potentials. C, Time constant for inactivation. We fit decay of the currents elicited by steps to the indicated potentials following a 500 ms step to 60 mV then a 2 ms step to -100 mV from a -80 mV holding potential. D, Time constant for recovery from inactivation. We fit decay of currents elicited by steps to the indicated potentials following a 500 ms step to 60 mV from a -80 mV holding potential.
Fig 8.
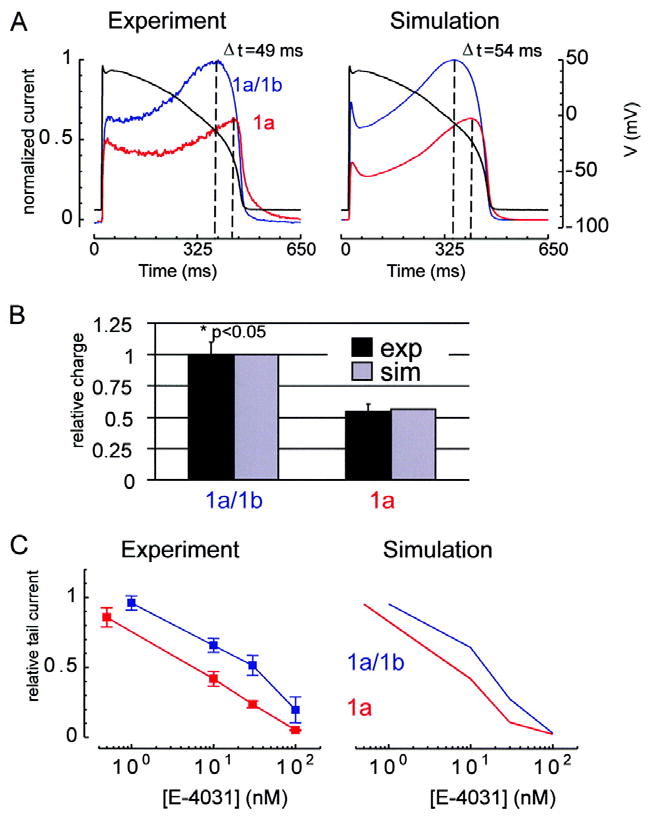
Action potential voltage clamp and E-4031 drug sensitivity. hERG 1a/1b is in blue and hERG 1a is in red. For panels A and C, the left column shows experimental results and the right column shows corresponding simulation results. A, Action potential voltage clamp. Shown are current responses (scale to the left of panel) to the voltage-clamp waveform (black, scale to the right of panel). Results are normalized to the maximum for hERG 1a/1b. The difference in time at which maximum current occurs for hERG 1a/1b versus hERG 1a is reproduced by the simulations (experiment: Δt = 49 ms; simulation: Δt = 54 ms). B, Experiments show that hERG 1a/1b contributes significantly more charge during the action potential than hERG 1a (p<0.05, Mann Whitney test, n=5, s.e.m. error bars). Relative charge is measured by integrating the current and dividing by the value obtained for hERG 1a/1b. Simulations (gray) agree with experiments (black). C, Dose-response curve for E-4031. Tail currents were elicited by 5 second steps to -50 mV following 4 seconds at 20 mV from a 15 second -80 mV holding step. We plot tail currents at the 20th cycle of this protocol divided by that for the 1st cycle for various E-4031 doses.
The model provides some insights into the paradox of why E-4031, a use-dependent channel blocker28, 31-34, is more efficacious in blocking hERG 1a channels, which we show spend less time in the open state than do hERG 1a/1b channels. Although drug binding/unbinding rates (ON/OFF) in the hERG 1a/1b and hERG 1a models are identical, additional E-4031 blocked states are afforded by the combination N-mode + E4031-block mode for hERG 1a channels. This allows for more complete block. Furthermore, blocked states are accessible via additional pathways (i.e. κ and λ). This allows for more rapid block development. Thus, openings, though not as frequent because of different channel kinetics, lead to more complete and rapid E-4031 block for hERG 1a than for hERG 1a/1b even when the same ON/OFF rates are used for both.
Loss of hERG 1b prolongs action potential duration and enhances E-4031 sensitivity in myocyte models
Effects on action potential duration of hERG 1a/1b (blue) and hERG 1a (red) substituted for the native IKr in the human-based Fink modified35 ten Tusscher action potential model36 are shown in Fig. 9A. APD90 (time between activation and 90% repolarization) for hERG 1a is 376 ms. This is 38 ms longer than APD90 for hERG 1a/1b. The hERG 1a versus hERG 1a/1b prolongation is 30 ms when our models are incorporated into the Priebe and Beuckelmann action potential model37 (not shown). Prolongation occurs because N-mode occupancy results in slower activation, and slower closed-state inactivation, effects that are explained in detail in Online Figure I. In the presence of E-4031, APD90 increases for both hERG 1a/1b and hERG 1a (Fig. 9B). Interestingly, ΔAPD90 (defined as APD90 for hERG 1a minus APD90 for hERG 1a/1b) also increases with increasing [E-4031] (Fig. 9C). At 10 mM E-4031, the hERG 1a action potential achieves full repolarization on the odd beats (e.g., 999th beat, shown), but fails to do so on the even beats (e.g., 1000th beat). The alternating pattern for hERG 1a is a consequence of sustained depolarization curtailing deactivation at the end of even beats. This provides immediately available hERG current at the start of odd beats, without the usual delay of slow N-mode activation (channels are still open at t = 0, o + on = 0.11). For [E-4031] ≥ 30 nM, both hERG 1a and hERG 1a/1b action potentials fail to repolarize during the pacing cycle length (not shown). Similar results were found using our hERG models and following the same protocol in the Priebe and Beuckelmann action potential model37 (not shown). Specifically, with increasing doses of E-4031, APD90 becomes longer for both hERG 1a/1b and hERG 1a. Also, ΔAPD90 between hERG 1a and hERG 1a/1b increases with increasing E-4031 concentrations ([E-4031] = 0 nM, ΔAPD90 = 30 ms; [E-4031] = 5 nM, ΔAPD90 = 258 ms; [E-4031] = 10 nM, ΔAPD90 = 1187 ms). Using Priebe and Beuckelmann, both hERG 1a/1b and hERG 1a action potentials fail to repolarize during the pacing cycle length for [E-4031] ≥ 30 nM. In Online Figure II we use the Luo-Rudy model38 to demonstrate that early afterdepolarizations are evoked in a rate-dependent manner selectively for hERG 1a. Taken together with the fact that excessive APD prolongation can cause arrhythmia, these results suggest that hERG blocking drugs are more arrhythmogenic for hERG 1a than they are for hERG 1a/1b.
Fig 9.
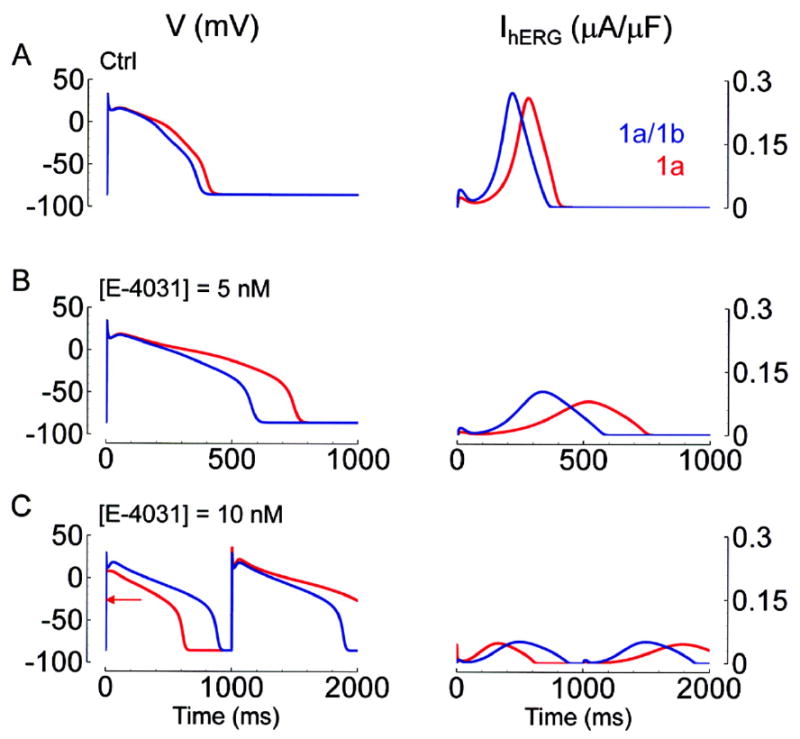
Effect of E-4031 on the action potential. The left column shows action potentials from the Fink modified35 ten Tusscher action potential model36 in which hERG 1a/1b (blue) and hERG 1a (red) were substituted for the native IKr. As [E-4031] increases (0, 5, and 10 nM for panels A, B, and C respectively), APD90 increases, and IhERG decreases for both hERG 1a/1b and 1a. ΔAPD90 (the difference in APD90 between hERG 1a/1b and hERG 1a) also increases with [E-4031] ([E-4031] = 0 nM, ΔAPD90 = 38 ms; [E-4031] = 5 nM, ΔAPD90 = 168 ms; [E-4031] = 10 nM, ΔAPD90 = 728 ms). In all cases, APD90 is longer and peak IhERG is smaller for hERG 1a. For control and for [E-4031] = 5 nM, peak IhERG occurs later for hERG 1a. For hERG 1a at [E-4031] = 10 nM (bottom row, red), repolarization fails to occur within the 1 sec cycle length on odd numbered beats. To mark the takeoff potential of the odd beat for hERG 1a, we place a red arrow. The alternating pattern for hERG 1a is a consequence of sustained depolarization curtailing deactivation at the end of even beats.
Identification of a hERG 1b-Specific Mutation in a Patient with LQTS
Seeking evidence for the importance of hERG 1b in vivo, we performed mutational analysis of the hERG1b-specific alternate exon (i.e. KCNH2 exon 1b) in 269 unrelated LQTS referral patients who remained genotype-negative following a comprehensive mutational analysis of the LQTS genes KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2. This analysis revealed an alanine-to-valine substitution at amino acid position 8 of the hERG1b N terminus (p. A8V, c. 23C>T) in an 8-year-old Caucasian female who had experienced exercise-induced cardiac arrest and demonstrated on serial electrocardiograms a QTc ranging from 450 to 480 ms (Fig. 10). This nonsynonymous variant was absent in 400 references alleles (100 Caucasians and 100 African Americans) and represents the first report of a potential hERG1b-specific disease mutation.
Fig 10.
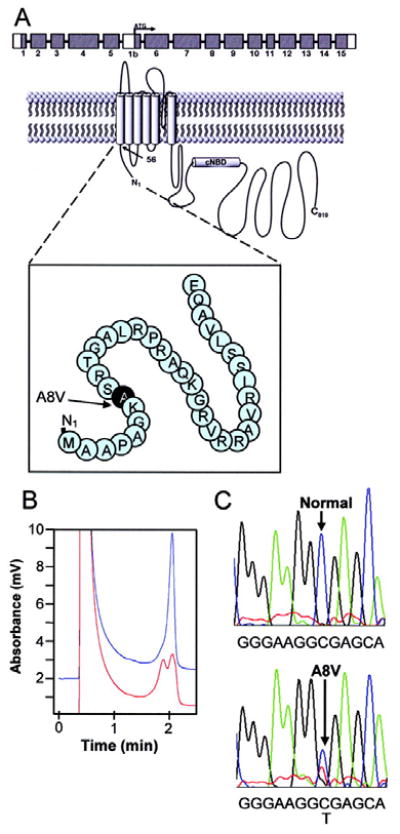
Molecular characterization of mutation hERG 1b 23C>T encoding A8V in patient DNA. A, Schematic of hERG 1b N-terminal sequence encompassing A8V mutation. B, Denaturing high performance liquid chromatography (DHPLC) chromatogram revealing a wild-type (blue peak) and an abnormal elution profile (red peak). C, Corresponding DNA sequencing chromatograms revealing the heterozygote 23C>T missense mutation encoding A8V identified in HERG exon 1b.
Western blot analysis showed the mutation confers a dramatic loss of hERG1b protein which was only slightly mitigated by co-expression with hERG1a (Fig. 11A). In four separate experiments, no 1b signal could be detected when hERG A8V-1b was expressed in isolation. Coexpression with 1a, which stabilizes the 1b protein18, showed the A8V mutation reduced 1b protein levels respectively reduced to 3%, 1%, 6% and 43% in the four experiments. Despite the low protein levels, A8V-1b clearly coassembled with stably expressed wild type 1a subunits to modify the current properties (cf. Figs. 11B,D). Compared with the 1a controls, the mutant reduced total conductance (and therefore surface expression levels) as inferred from tail current amplitudes and variably reduced rectification as reflected in the ratio of the current amplitude at the end of the test pulse relative to the tail current amplitude. A detailed functional analysis of differences between 1a/1b and 1a/A8V-1b channels will be required to fully understand the biophysical effects of the mutation. Independent of the functional properties, such a dramatic reduction in 1b protein levels in native tissues, where hERG maturation is highly efficient20, is expected to decrease the fraction of hERG 1a/1b heteromers and thus reduce the contribution of IKr to ventricular repolarization. These findings suggest the hERG 1b A8V mutation predicts a loss of function in patients.
Fig 11.
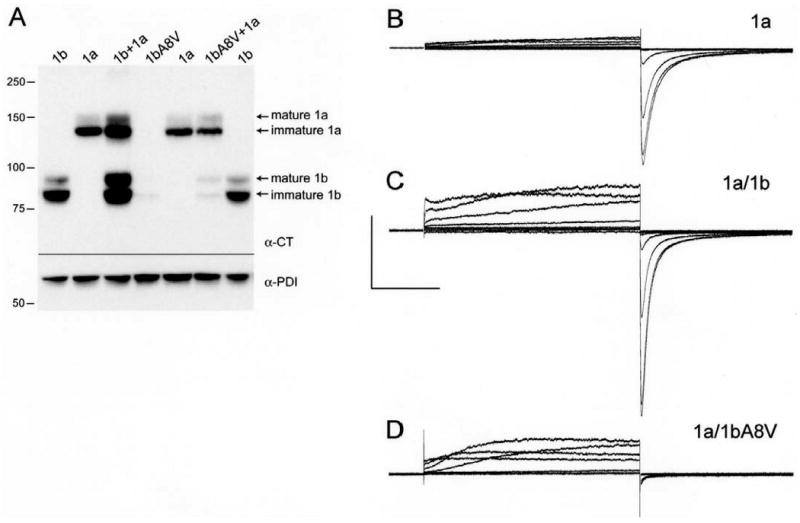
Loss of hERG 1b protein attributed to A8V mutation in HEK-293 cells. A, Western blot analysis of HEK-293 cell lysates probed with a C-terminal pan-hERG antibody (α-CT).20 Protein disulfide isomerase (PDI), an ER-resident protein, was used as a loading control (α-PDI). Lane 1, hERG 1b. Lane 2, hERG 1a. Lane 3, hERG 1b + hERG 1a. Lane 4, mutant hERG 1bA8V. Lane 5, hERG 1a. Lane 6, mutant hERG 1bA8V + hERG 1a. Lane 7, hERG 1b. B, Current traces from hERG 1a stably expressed in HEK-293 cells recorded at room temperature, evoked from a holding potential of -80 mV and stepped from -100 to +60 mV, followed by a step to -105 mV. C, Currents as in B but from stable 1a cells transiently transfected with hERG 1b. D, Currents as in B but from stable 1a cells transiently transfected with A8V-1b. Capacitive transient upon repolarization is deleted in D to make small tail current more visible.
Discussion
In this study we carried out biophysical, pharmacological, computational and genetic analyses to understand the physiological and pathological role of hERG 1b subunits in heteromeric hERG 1a/1b channels contributing to cardiac IKr. We used whole-cell voltage clamp at temperatures approximating those in mammalian systems to observe differences in biophysical properties of hERG 1a/1b heteromers compared with the more commonly studied hERG 1a homomers. We found that hERG 1a/1b currents are significantly larger, transferring approximately 80% more repolarizing charge, due to differences in gating kinetics that reduce rectification and cause the current to peak earlier during the action potential. Apparent activation rates were faster for 1a/1b channels, whereas rates of inactivation over the same voltage range were unchanged. Because of the faster activation at voltages corresponding to the peak of the action potential, there is less rectification and the current amplitude is greater. Upon repolarization, recovery from inactivation is faster for 1a/1b channels, thus giving rise to an earlier peak (and greater current amplitude) before deactivation ensues. Although deactivation was also faster for 1a/1b channels, current amplitude was reduced only when the action potential reached more negative potentials and currents were small due to the diminishing driving force. E-4031 drug block was slower for hERG 1a/1b than 1a channels, and exhibited a corresponding four-fold shift in IC50 to higher concentrations. The differences in amplitude and drug sensitivity could be explained in kinetic models by inclusion of N-liganded states in the 1a homomeric channels that are absent in the 1a/1b heteromers, which possess fewer 1a N termini. The faster activation, deactivation and recovery from inactivation of hERG 1a/1b compared with 1a is reminiscent of the differences between IKr in guinea pig myocytes and hERG 1a currents under similar recording and temperature conditions 39-41, further supporting the hypothesis that heteromers of hERG 1a and 1b subunits emulate the native IKr. In myocyte computational models, replacing native IKr with hERG 1a or hERG 1a/1b showed hERG 1a was associated with longer action potential durations (APD's). Differences in APD were enhanced in the presence of E-4031, consistent with enhanced drug sensitivity in the absence of the hERG 1b subunit. The importance of hERG 1b in normal physiological function in the human heart is supported by the identification of a 1b-specific mutation, A8V, in a patient referred for LQTS testing but exhibiting no mutations in other known LQTS loci.
The larger amplitude of 1a/1b currents suggest that selective loss of the 1b subunit would give rise to significantly reduced IKr in native tissues, and the 1b isoform (and specifically the KCNH2 1b exon encoding the unique N-terminal sequences, and/or its promoter) may be a target for mutations causing long QT syndrome. Indeed, a 1b-selective knockout in mouse reduced IKr and elicited arrhythmia in a subset of affected animals 21, consistent with the predictions of our findings in a heterologous system. Because IKr contributes much more to repolarization in humans than in adult mice 42, even a 50% reduction of the 1b subunit in individuals heterozygous for 1b mutations is expected to be deleterious. Our results suggest the reduction in IKr current amplitude with the loss of mouse ERG 1b may be explained by differences in gating per se, without alterations in ERG 1a trafficking, but we cannot rule out a dominant-negative effect of the hERG 1b mutant on IKr surface density. Our findings suggest that 1b-specific mutations in humans could reduce IKr levels and thus account for previously unmapped cases of inherited, type 2 LQTS or for subclinical reductions of IKr that increase susceptibility to hERG blockers and acquired LQTS.
The observation that hERG 1a/1b channels exhibit lower sensitivity to drug block compared with hERG 1a may have implications for acquired LQTS. If hERG 1a/1b heteromers represent a significant fraction of channels contributing to IKr, down-regulation of 1b would enhance sensitivity to hERG blockers up to four-fold and increase the risk for arrhythmia by having a predominant population of more drug-sensitive hERG 1a channels rather than the relatively less sensitive hERG 1a/1b channels. As an alternate transcript, expression of hERG 1b is under the control of its own promoter43 and thus could be subject to independent regulation; it is interesting to speculate that developmental regulation of the 1b subunit might account for the three-fold decrease in dofetilide affinity observed in mouse IKr between fetal and neonatal mouse cardiomyocytes 42, mirroring the reduced drug sensitivity of hERG 1a/1b compared with hERG 1a channels. A detailed understanding of how hERG 1a and 1b expression is regulated during human development will be important for knowing whether a loss of the 1b subunit could confer susceptibility of the fetus or infant to sudden cardiac death. Indeed, sudden infant death syndrome (SIDS) is increasingly attributed to cardiac ion channelopathy 44, 45, as in a study showing reduction in IKr due to SIDS mutations or polymorphisms common to both hERG isoforms 46.
Differences in drug sensitivity between hERG 1a and 1a/1b channels provide insights into the mechanism of drug block. If inhibition were due only to occlusion upon drug binding to hydrophobic residues lining the pore in the channel's open state 47, 48, block should develop more quickly for channels spending more time in the open state. Accordingly, since the larger hERG 1a/1b currents reflect increased occupancy of the open state, one expects block to develop more rapidly. Moreover, the more rapid deactivation of the heteromers might be expected to enhance drug trapping, which contributes to the efficacy of block.29 Instead, drug sensitivity was reduced in hERG 1a/1b channels vs. hERG 1a channels, and the development of drug block was slower. This finding suggests that differences in N-terminal structure lead to differences in drug block. Our kinetic model suggests that N-mode gating allows for many more states that can also be bound by drug, thus enhancing efficacy. We can also predict from gating kinetics observed in the current and previous studies that the reduced sensitivity to drug block arises because the 1a/1b heteromer has fewer 1a N termini available to interact with the S4-S5 linker and stabilize inactivation 13. An intimate link exists between inactivation and drug block; in most cases, mutations that disable the inactivation mechanism dramatically reduce inhibition by hERG blockers 49-53. One study provides evidence for the importance of inactivation by demonstrating that the non-inactivating hERG relative, bovine-Eag, possesses the homologous drug-binding residues in its vestibule but is inhibited by hERG blockers only when inactivation is introduced by mutations at a separate site in the P-region 49. Our studies support the model that inactivation promotes hERG channel block, demonstrating that allosteric changes caused by structural domains far from the drug-binding site, such as the N terminus, may play an important role.
The differences in hERG 1a and 1a/1b gating kinetics allow us to speculate about the corresponding conformational changes. The similar fold-increases in activation and deactivation rates for hERG 1a/1b compared to 1a currents are consistent with the absence of a shift in the V1/2 of the activation curve and suggest that heteromers encounter a lower energy barrier in the transition between the closed and open states. Because the subunits are otherwise identical, this lower energy barrier is attributable to a mechanism involving the N termini of the hERG 1a and 1b channels or a simple reduction in the number of the longer hERG 1a N termini in the heteromer. The N terminus of hERG 1a interacts with or near the S4-S5 linker 13, 15, which couples movement of the voltage sensor with the opening and closing of the activation gate 54, 55. Perhaps only 1a N termini interact with the S4-S5 linker, creating a load or stiffness against opening and closing. Although the stoichiometry of hERG 1a/1b heteromers is not known, the heteromer provides fewer 1a N termini and thus the channel may open and close with less resistance. This model is consistent with the structure of the Kv1.2 S4-S5 linkers 56, which collectively encircle the residues forming the activation gate within the S6 tetrad 57, 58.
These are the first studies of hERG 1a/1b channels in mammalian cells recorded at near-physiological temperatures. Previous studies have established differences in temperature sensitivities of hERG 1a gating parameters such that the overall current profile obtained at lower temperatures differs quite markedly from that recorded at higher temperatures 10, 59. Such temperature differences are consistent with the much greater reductions in rectification we observe here compared with our previous observations of hERG 1a/1b currents in Xenopus oocytes 17 or those by Bauer and colleagues in clonal pituitary cells 60, both measured at room temperature. In a recent study of hERG 1a and 1b expressed in CHO cells, gating kinetics measured at room temperature show up to six-fold increases in deactivation time constants for hERG 1a vs. 1a/1b currents 61. Other gating parameters exhibited relatively minor differences, such as a roughly 50% faster apparent activation rate in hERG 1a/1b but only minor differences in inactivation or recovery from inactivation. Thus, it is critical to consider temperature when extrapolating results of channel function in heterologous expression systems to physiological systems.
How the A8V mutation reduces hERG 1b protein levels is not known. Whether the 1bA8V protein levels are reduced because they oligomerize inefficiently or are inherently unstable, or because the nucleotide mutation leads to transcriptional perturbation or mRNA instability, will require further study to understand. Despite the low levels of A8V protein on western blots, the mutant subunits do associate with hERG 1a subunits and alter their expression. Because the current properties of hERG 1a/A8V-1b channels were highly variable, we were unable within the scope of this study to precisely characterize the biophysical mutant phenotype. We are currently testing the hypothesis that the mutation reduces overall current levels by associating with hERG 1a subunits and rendering the complex unstable.
There are several unanswered questions regarding the relative disposition of hERG 1a and 1b in native tissues. Although in our previous western blot analysis we observed robust and roughly equal expression of hERG 1a and 1b subunits in human left ventricle, our more extensive experience in other species shows there is significant variability, the determinants of which we do not currently understand20. The stoichiometry is unknown in tissue, although a recent meeting report indicates that assembly in heterologous systems is random,62 raising the intriguing possibility that stoichiometry may be flexible and established by independent gene expression of the hERG 1a and 1b alternate transcripts. Such regulation may lead to differences in regional, transmural, sex-specific or developmental expression of the hERG 1b subunit and thus its importance may be context-specific.
Our findings have implications for both acquired and inherited long QT syndrome. Until now, the primary platform for safety testing drugs in development to prevent acquired LQTS has been HEK-293 cell lines stably expressing solely hERG1a homomeric channels 10, 63. Given that different drugs exhibit different pore-binding mechanisms 64, it will be important to screen a variety of drugs with known torsadogenic potential in cells expressing hERG 1a/1b heteromeric channels that may more closely approximate the composition and functional properties of those underlying native IKr. Our findings also suggest that, in addition to searching for disease mutations in the hERG 1b-specific exon, evaluating the cellular phenotypes of established LQT2-susceptibility mutations and KCNH2 polymorphisms at physiological temperatures in the context of a hERG 1a/1b heteromeric channel rather than the hERG 1a homomeric channel may reveal novel molecular mechanisms for both congenital and drug-induced LQTS.
Supplementary Material
Acknowledgments
The authors thank Dr. Srinivasan Venkatachalan, Dr. Mathew Jones, Dr. Eugenia Jones, Sarah Wynia and other members of the Robertson lab for helpful discussion and Rebecca Uelmen and Robert Zinkel for technical assistance.
This work was supported by NIH R01 HL081780 (GAR), the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program (MJA), and NIH/NHLBI RO1-HL49054 and Merit Award R37-HL33343 and The Fred Saigh Distinguished Professorship at Washington University (YR).
Contributor Information
Harinath Sale, Department of Physiology, University of Wisconsin-Madison, Madison, WI.
Jinling Wang, Department of Physiology, University of Wisconsin-Madison, Madison, WI.
Thomas J. O'Hara, Department of Biomedical Engineering, Washington University, St. Louis, MO.
David J. Tester, Departments of Medicine, Pediatrics, and Pharmacology, Mayo Clinic, Rochester, MN
Pallavi Phartiyal, Department of Physiology, University of Wisconsin-Madison, Madison, WI.
Jia-Qiang He, Department of Physiology, University of Wisconsin-Madison, Madison, WI.
Yoram Rudy, Department of Biomedical Engineering, Washington University, St. Louis, MO.
Michael J. Ackerman, Departments of Medicine, Pediatrics, and Pharmacology, Mayo Clinic, Rochester, MN
Gail A. Robertson, Department of Physiology, University of Wisconsin-Madison, Madison, WI
References
- 1.Sanguinetti MC, Jurkiewicz NK. Delayed rectifier outward K+ current is composed of two currents in guinea pig atrial cells. Am J Physiol. 1991;260:H393–399. doi: 10.1152/ajpheart.1991.260.2.H393. [DOI] [PubMed] [Google Scholar]
- 2.Shibasaki T. Conductance and kinetics of delayed rectifier potassium channels in nodal cells of the rabbit heart. J Physiol (Lond) 1987;387:227–250. doi: 10.1113/jphysiol.1987.sp016571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 4.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- 5.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 6.Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 7.Roden DM. Torsade de pointes. Clin Cardiol. 1993;16:683–686. doi: 10.1002/clc.4960160910. [DOI] [PubMed] [Google Scholar]
- 8.Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- 9.Wang S, Liu S, Morales MJ, Strauss HC, Rasmusson RL. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J Physiol (Lond) 1997;502:45–60. doi: 10.1111/j.1469-7793.1997.045bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J. 1998;74:230–241. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. J Gen Physiol. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schonherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol (Lond) 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Trudeau MC, Zappia AM, Robertson GA. Regulation of deactivation by an amino terminal domain in human ether-a- go-go-related gene potassium channels. J Gen Physiol. 1998;112:637–647. doi: 10.1085/jgp.112.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Viloria CG, Barros F, Giraldez T, Gomez-Varela D, de la Pena P. Differential Effects of Amino-Terminal Distal and Proximal Domains in the Regulation of Human erg K+ Channel Gating. Biophys J. 2000;79:231–246. doi: 10.1016/S0006-3495(00)76286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Myers CD, Robertson GA. Dynamic control of deactivation gating by a soluble amino-terminal domain in HERG K(+) channels. J Gen Physiol. 2000;115:749–758. doi: 10.1085/jgp.115.6.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lees-Miller JP, Kondo C, Wang L, Duff HJ. Electrophysiological characterization of an alternatively processed ERG K+ channel in mouse and human hearts. Circ Res. 1997;81:719–726. doi: 10.1161/01.res.81.5.719. [DOI] [PubMed] [Google Scholar]
- 17.London B, Trudeau MC, Newton KP, Beyer AK, Copeland NG, Gilbert DJ, Jenkins NA, Satler CA, Robertson GA. Two isoforms of the mouse ether-a-go-go-related gene coassemble to form channels with properties similar to the rapidly activating component of the cardiac delayed rectifier K+ current. Circ Res. 1997;81:870–878. doi: 10.1161/01.res.81.5.870. [DOI] [PubMed] [Google Scholar]
- 18.Phartiyal P, Jones EM, Robertson GA. Heteromeric assembly of human ether-a-go-go-related gene (hERG) 1a/1b channels occurs cotranslationally via N-terminal interactions. J Biol Chem. 2007;282:9874–9882. doi: 10.1074/jbc.M610875200. [DOI] [PubMed] [Google Scholar]
- 19.Phartiyal P, Sale H, Jones EM, Robertson GA. ER retention and rescue by heteromeric assembly regulate hERG 1a/1b surface channel composition. J Biol Chem. 2007;283:3702–3707. doi: 10.1074/jbc.M708999200. [DOI] [PubMed] [Google Scholar]
- 20.Jones EM, Roti Roti EC, Wang J, Delfosse SA, Robertson GA. Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. J Biol Chem. 2004;279:44690–44694. doi: 10.1074/jbc.M408344200. [DOI] [PubMed] [Google Scholar]
- 21.Lees-Miller JP, Guo J, Somers JR, Roach DE, Sheldon RS, Rancourt DE, Duff HJ. Selective knockout of mouse ERG1 B potassium channel eliminates I(Kr) in adult ventricular myocytes and elicits episodes of abrupt sinus bradycardia. Mol Cell Biol. 2003;23:1856–1862. doi: 10.1128/MCB.23.6.1856-1862.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McPate MJ, Duncan RS, Milnes JT, Witchell HJ, Hancox JC. The N588K-HERG K+ channel mutation in the ‘short QT syndrome’: mechanism of gain-in-function determined at 37 degrees C. Biochem Biophys Res Commun. 2005;334:441–449. doi: 10.1016/j.bbrc.2005.06.112. [DOI] [PubMed] [Google Scholar]
- 23.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 24.Tester DJ, Will ML, Ackerman MJ. Mutation detection in congenital long QT syndrome: cardiac channel gene screen using PCR, dHPLC, and direct DNA sequencing. Methods Mol Med. 2006;128:181–207. doi: 10.1385/1-59745-159-2:181. [DOI] [PubMed] [Google Scholar]
- 25.Splawski I, Shen J, Timothy KW, Vincent GM, Lehman MH, Keating MT. Genomic structure of three long QT syndrome genes: KVLQT1, HERG, and KCNE1. Genomics. 1998;51:86–97. doi: 10.1006/geno.1998.5361. [DOI] [PubMed] [Google Scholar]
- 26.Roti Roti EC, Myers CD, Ayers RA, Boatman DJ, Delfosse SA, Chan EK, Ackerman MJ, January CT, Robertson GA. Interaction with GM130 during HERG ion channel trafficking. Disruption by type 2 congenital long QT syndrome mutations. J Biol Chem. 2002;277:47779–47785. doi: 10.1074/jbc.M206638200. [DOI] [PubMed] [Google Scholar]
- 27.Silva J, Rudy Y. Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation. 2005;112:1384–1391. doi: 10.1161/CIRCULATIONAHA.105.543306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carmeliet E. Voltage- and time-dependent block of the delayed K+ current in cardiac myocytes by dofetilide. J Pharmacol Exp Ther. 1992;262:809–817. [PubMed] [Google Scholar]
- 29.Mitcheson JS, Chen J, Sanguinetti MC. Trapping of a methanesulfonanilide by closure of the HERG potassium channel activation gate. J Gen Physiol. 2000;115:229–240. doi: 10.1085/jgp.115.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stork D, Timin EN, Berjukow S, Huber C, Hohaus A, Auer M, Hering S. State dependent dissociation of HERG channel inhibitors. Br J Pharmacol. 2007;151:1368–1376. doi: 10.1038/sj.bjp.0707356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spector PS, Curran ME, Keating MT, Sanguinetti MC. Class III antiarrhythmic drugs block HERG, a human cardiac delayed rectifier K+ channel. Open-channel block by methanesulfonanilides. Circ Res. 1996;78:499–503. doi: 10.1161/01.res.78.3.499. [DOI] [PubMed] [Google Scholar]
- 32.Snyders DJ, Chaudhary A. High affinity open channel block by dofetilide of HERG expressed in a human cell line. Mol Pharmacol. 1996;49:949–955. [PubMed] [Google Scholar]
- 33.Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of HERG channel block. Mol Pharmacol. 2006;69:1709–1716. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT. Electrophysiological and pharmacological properties of HERG channels in a stably transfected human cell line. Biophysical Journal. 1997;72:WAMC7–WAMC7. [Google Scholar]
- 35.Fink M, Noble D, Virag L, Varro A, Giles WR. Contributions of HERG K+ current to repolarization of the human ventricular action potential. Prog Biophys Mol Biol. 2008;96:357–376. doi: 10.1016/j.pbiomolbio.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 36.ten Tusscher KH, Panfilov AV. Alternans and spiral breakup in a human ventricular tissue model. Am J Physiol Heart Circ Physiol. 2006;291:H1088–1100. doi: 10.1152/ajpheart.00109.2006. [DOI] [PubMed] [Google Scholar]
- 37.Priebe L, Beuckelmann DJ. Simulation study of cellular electric properties in heart failure. Circ Res. 1998;82:1206–1223. doi: 10.1161/01.res.82.11.1206. [DOI] [PubMed] [Google Scholar]
- 38.Faber GM, Rudy Y. Action potential and contractility changes in [Na(+)](i) overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78:2392–2404. doi: 10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitcheson JS, Hancox JC. An investigation of the role played by the E-4031-sensitive (rapid delayed rectifier) potassium current in isolated rabbit atrioventricular nodal and ventricular myocytes. Pflugers Arch. 999(438):843–850. doi: 10.1007/s004249900118. [DOI] [PubMed] [Google Scholar]
- 40.Weerapura M, Nattel S, Chartier D, Caballero R, Hebert TE. A comparison of currents carried by HERG, with and without coexpression of MiRP1, and the native rapid delayed rectifier current. Is MiRP1 the missing link? J Physiol (Lond) 2002;54:15–27. doi: 10.1113/jphysiol.2001.013296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang S. Isolation and characterization of I(Kr) in cardiac myocytes by Cs+ permeation. Am J Physiol Heart Circ Physiol. 2006;290:H1038–1049. doi: 10.1152/ajpheart.00679.2005. [DOI] [PubMed] [Google Scholar]
- 42.Wang L, Feng ZP, Kondo CS, et al. Developmental changes in the delayed rectifier K+ channels in mouse heart. Circ Res. 1996;79:79–85. doi: 10.1161/01.res.79.1.79. [DOI] [PubMed] [Google Scholar]
- 43.London B, Beyer AK, Newton KP, Trudeau MC, Robertson GA. Cloning a cardiac specific isoform of HERG from the mouse. Biophysical Journal. 1997;72:WAMC4–WAMC4. [Google Scholar]
- 44.Tester DJ, Ackerman MJ. Sudden infant death syndrome: how significant are the cardiac channelopathies? Cardiovasc Res. 2005;67:388–396. doi: 10.1016/j.cardiores.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 45.Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Wang DW, Rhodes TE, George AL, Swartz PJ. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361–367. doi: 10.1161/CIRCULATIONAHA.106.658021. [DOI] [PubMed] [Google Scholar]
- 46.Rhodes TE, Abraham RL, Welch RC, Vanoye CG, Crotti L, Arnestad M, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Rognum T, Roden DM, Schwartz PJ, George AL. Cardiac potassium channel dysfunction in sudden infant death syndrome. J Mol Cell Cardiol. 2008;44:571–581. doi: 10.1016/j.yjmcc.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lees-Miller JP, Duan Y, Teng GQ, Duff HJ. Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol Pharmacol. 2000;57:367–374. [PubMed] [Google Scholar]
- 48.Mitcheson J, Perry M, Stansfeld P, Sanguinetti MC, Witchell H, Hancox J. Structural determinants for high-affinity block of hERG potassium channels. Novartis Found Symp. 2005;266:136–150. discussion 150-138. [PubMed] [Google Scholar]
- 49.Ficker E, Jarolimek W, Brown AM. Molecular determinants of inactivation and dofetilide block in ether a-go-go (EAG) channels and EAG-related K(+) channels. Mol Pharmacol. 2001 Dec;60:1343–1348. doi: 10.1124/mol.60.6.1343. [DOI] [PubMed] [Google Scholar]
- 50.Ficker E, Jarolimek W, Kiehn J, Baumann A, Brown AM. Molecular determinants of dofetilide block of HERG K+ channels. Circ Res. 1998;82:386–395. doi: 10.1161/01.res.82.3.386. [DOI] [PubMed] [Google Scholar]
- 51.Herzberg IM, Trudeau MC, Robertson GA. Transfer of rapid inactivation and E-4031 sensitivity from HERG to M-EAG Channels. Journal of Physiology. 1998;511(1):3–14. doi: 10.1111/j.1469-7793.1998.003bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen J, Seebohm G, Sanguinetti MC. Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc Natl Acad Sci U S A. 2002;99:12461–12466. doi: 10.1073/pnas.192367299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. Modulation of HERG affinity for E-4031 by [K+]o and C-type inactivation. FEBS Lett. 1997;417:43–47. doi: 10.1016/s0014-5793(97)01245-3. [DOI] [PubMed] [Google Scholar]
- 54.Tristani-Firouzi M, Chen J, Sanguinetti MC. Interactions between S4-S5 linker and S6 transmembrane domain modulate gating of HERG K+ channels. J Biol Chem. 2002;277:18994–19000. doi: 10.1074/jbc.M200410200. [DOI] [PubMed] [Google Scholar]
- 55.Lu Z, Klem AM, Ramu Y. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J Gen Physiol. 2002;120:663–676. doi: 10.1085/jgp.20028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 57.del Camino D, Yellen G. Tight steric closure at the intracellular activation gate of a voltage-gated K(+) channel. Neuron. 2001;32:649–656. doi: 10.1016/s0896-6273(01)00487-1. [DOI] [PubMed] [Google Scholar]
- 58.Hackos DH, Chang TH, Swartz KJ. Scanning the intracellular S6 activation gate in the shaker K+ channel. J Gen Physiol. 2002;119:521–532. doi: 10.1085/jgp.20028569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vandenberg JI, Varghese A, Lu Y, Bursill JA, Mahaut-Smith MP, Huang CL. Temperature dependence of human Ether-a-go-go Related Gene (hERG) K+ currents. Am J Physiol Cell Physiol. 2006;291:C165–75. doi: 10.1152/ajpcell.00596.2005. [DOI] [PubMed] [Google Scholar]
- 60.Kirchberger NM, Wulfsen I, Schwarz JR, Bauer CK. Effects of TRH on heteromeric rat erg1a/1b K+ channels are dominated by the rerg1b subunit. J Physiol. 2006;571:27–42. doi: 10.1113/jphysiol.2005.101667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Larsen AP, Olesen SP, Grunnet M, Jespersen T. Characterization of hERG1a and hERG1b potassium channels - a possible role for hERG1b in the IKr current. Pflugers Arch. 2008 doi: 10.1007/s00424-008-0476-7. [DOI] [PubMed] [Google Scholar]
- 62.Pendon ZaT M. Biochemical and Spectroscopic Studies of Heteromeric Interactions of HERG1a and HERG1b Potassium Channel Subunits. Biophys J Supplement, 569-Pos. 2008 [Google Scholar]
- 63.Walker BD, Singleton CB, Tie H, Bursill JA, Wyse KR, Valenzuela SM, Breit SN, Campbell TJ. Comparative effects of azimilide and ambasilide on the human ether-a-go-go-related gene (HERG) potassium channel. Cardiovasc Res. 2000;48:44–58. doi: 10.1016/s0008-6363(00)00155-3. [DOI] [PubMed] [Google Scholar]
- 64.Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.