

Abstract
Parathyroid hormone-related protein (PTHrP) is expressed by human colon cancer tissue and cell lines; expression correlates with colon carcinoma severity. PTHrP is synthesized as a prepro isoform and contains two targeting sequences – a signal sequence and a nuclear localization signal (NLS). The signal peptide (SP) directs PTHrP to the secretory pathway, where it exerts autocrine/paracrine effects. The NLS directs PTHrP to the nucleus/nucleolus, where it exerts intracrine effects. In this study, we used the human colon cancer cell line LoVo as a model system to study the effects of autocrine/paracrine and intracrine PTHrP action on cell growth and survival, hallmarks of malignant tumor cells. We report that PTHrP increases cell growth and survival, protects cells from serum-starvation-induced apoptosis, and promotes anchorage-independent cell growth via an intracrine pathway. Conversely, autocrine/paracrine PTHrP action decreases cell growth and survival. We also show an inverse relationship between secreted and nuclear PTHrP levels, in that cells overexpressing NLS-deleted PTHrP secrete higher PTHrP levels than those overexpressing the wild-type isoform. Conversely, SP deletion results in higher nuclear PTHrP levels. These observations provide evidence of a link between intracrine PTHrP action and cell growth and survival. Targeting PTHrP production in colon cancer may thus prove therapeutically beneficial.
Keywords: Parathyroid hormone-related protein, LoVo (colon cancer cells), Apoptosis, Anchorage independence, Signal peptide, Nuclear localization signal
Introduction
Colorectal carcinoma accounts for over 90% of the malignant tumors of the large bowel, and is the second most common cause of death from malignant disease in the United States [1,2]. As many as 50% of patients with cancers of the colon and rectum eventually die from this disease [3]. The molecular processes of tumor progression are mediated via both inherent tumor cell characteristics and growth factors, matrix molecules and cytokines in the tumor environment [4–6]. One of these factors is parathyroid hormone-related protein (PTHrP). PTHrP expression correlates with the severity of colon carcinoma [7], and human specimens from Stage II colorectal carcinomas have increased PTHrP expression compared to specimens from tubulovillous adenoma (Stage 0) [8]. PTHrP significantly increases xenograft growth in a nude mouse model; these effects are accompanied by increased expression of the pro-invasive integrin α6β4 and activation of the phosphatidylinositol 3-kinase (PI3-K)/Akt pathway [8]. Since the gastrointestinal epithelium is prone to cancer development, particularly in the colon, understanding the mechanisms via which PTHrP exerts its effects in this system may provide important information for the treatment of colon cancer.
The mechanisms via which PTHrP exerts its effects in colon cancer are not fully understood. The PTHrP molecule is synthesized as a “prepro” isoform. The N-terminal region of prepro PTHrP includes a 36 amino acid sequence which functions as a signal peptide (SP), directing PTHrP to the secretory pathway. Mature secretory forms of PTHrP include N-terminal, mid-region, and C-terminal isoforms; these isoforms function via an autocrine/paracrine pathway. N-terminal PTHrP exerts its effects via interaction with the parathyroid hormone (PTH)/PTHrP 1 receptor (PTH1R) [9]. One of the consequences of this mode of PTHrP action is the syndrome of humoral hypercalcemia of malignancy (HHM). While colon cancer is not typically associated with hypercalcemia, colon cancer cells and tissue do express a functional PTH1R [10,11]. PTHrP also functions via an intracrine pathway after translocation to the nucleus or nucleolus. Translocation is mediated via a bipartite nuclear localization signal (NLS) located at amino acids 88–91 and 102–106 [12,13].
Oncogenic cell transformation is a multistage process in which multiple genetic lesions result in alterations in cellular physiology [14]. Both dysregulated cellular proliferation and decreased apoptosis play major roles in carcinogenesis [14]. Cancer cell migration from the primary site and invasion into surrounding tissues requires that the cells gain anchorage independence to escape apoptosis [14]. In fact, resistance to apoptosis increases in colon cancer cells with increasing metastatic potential [15,16]. The term “anoikis” has been coined to describe apoptosis induced by loss of anchorage during dissemination in lymph or blood [17]. This term was originally defined by Frisch to describe apoptotic cell death as a consequence of insufficient cell-matrix interactions [17], and has since been recognized as a significant player in tumor metastasis and angiogenesis [17–19].
In this study, we sought to distinguish between the autocrine/paracrine and intracrine pathways of PTHrP action in the regulation of cell growth and apoptosis. Given the correlation between anchorage-independent growth in vitro and cellular tumorigenicity in nude mice, we also investigated the pathway via which PTHrP increases anchorage independent cell growth, thereby decreasing anoikis. Understanding the role of PTHrP is crucial for the development of therapeutic strategies aimed at the control of colon cancer.
Materials and Methods
Materials
Fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Norcross, GA). Tissue culture supplies were purchased from Life Technologies, Inc. (Gaithersburg, MD). Antibodies for Western blot analysis and for cell treatment were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and from Cell Signaling Technology (Danvers, MA). The small interfering RNAs (siRNAs) targeting the PTH1R, as well as the non-targeting control (NTC) siRNA, were purchased from Dharmacon (Lafayette, CO).
Plasmid constructs
A cDNA encoding human PTHrP (obtained from Genentech, Inc., South San Francisco, CA) was digested with EcoRI and HindIII and subcloned in the sense orientation into the expression vector pcDNA3.1(+) (Invitrogen, San Diego, CA). This construct was used to prepare the PTHrP constructs deleted over the NLS (amino acids 88–91 and 102–106) and over the SP (amino acids −36 to −1). To allow efficient translation of the ΔSP PTHrP, an ATG coding for methionine was introduced in the +1 position.
Cell culture and transfection
LoVo cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were grown at 37°C in a humidified 95% air-5% CO2 atmosphere in F12 medium supplemented with 10% FBS and L-glutamine.
The cells were stably transfected using FuGENE™ 6 Transfection Reagent (Roche Molecular Biochemicals, Indianapolis, IN). pcDNA3.1 (+) was used as the empty vector control. Two days after transfection, 600 µg/ml G418 (Geneticin; Life Technologies Inc.) was added, and resistant clones were selected. Single clones of stably transfected cells, isolated by limiting dilution in 96-well plates, were transferred to individual flasks and cultured in medium containing 150 µg/ml G418. Individual clones overexpressing wild-type (WT) or mutated PTHrP were tested for PTHrP mRNA levels by reverse transcription/real-time PCR [20] and for PTHrP secretion using an immunoradiometric (IRMA) assay (Diagnostics Systems Laboratories, Webster, TX) [21].
Cell growth and apoptosis
Cells were plated in 96-well dishes (1 × 104 cells/well) in medium containing 10% FBS. Cell growth was measured after 96 h using the Quick Cell Proliferation Assay kit (Biovision; Mountain View, CA). In some experiments, the cells were transferred to 0% FBS 24 h after plating. Cell growth was measured after a further 72 h.
To measure apoptosis, cells were plated in 96-well dishes (1 × 104 cells/well) in medium containing 10% FBS. The level of apoptosis was measured after 72 h using the Cell Death Detection ELISA PLUS kit (Roche Applied Science, Indianapolis, IN). The cells were lysed for 30 min at room temperature by incubation with 200 µl of lysis buffer. After centrifugation (10 min at 200 × g), 20 µl of the supernatant was transferred onto a streptavidin-coated microplate for quantitation at 405 nm per the manufacturer’s protocol. In some experiments, the cells were transferred to 0% FBS after 24 h. Apoptosis was then measured after a further 48 h.
Specific gene silencing using siRNA
Cells were plated in 96-well dishes at 1 × 104 cells/well in medium containing 10% FBS. After 48 h, the cells were transfected with ON-Target plus siRNAs directed against the PTH1R (100 nM). These ON-target siRNAs are modified to decrease any off-target effects caused by both strands. To further eliminate the potential for off-target effects, two independent siRNA sequences were used. As a control, the cells were transfected with ON-Target plus NTC siRNAs. Transfections were performed using the DharmaFECT 3 transfection reagent (Dharmacon) following the manufacturer’s protocol. To measure mRNA levels, the cells were harvested 48 h after transfection. Cell apoptosis and growth were measured 48 h and 72 h after transfection, respectively.
Treatment with anti-PTHrP antibody
Cells were plated in 96-well dishes (1 × 104 cells/well) in medium containing 10% FBS. After 48 h, the cells were treated with a monoclonal antibody raised against PTHrP amino acids (1–34) (Santa Cruz Biotechnology). IgG was used as a control. In some experiments, the cells were incubated in the absence of FBS for 24 h before addition of the anti-PTHrP antibody. Cell apoptosis and growth were measured after 48 h and 72 h, respectively.
Real-time polymerase chain reaction analysis of PTHrP and PTH1R gene expression
Total RNA from LoVo cells was extracted using the RNAqueous® isolation kit (Ambion Inc., Austin, TX), per the manufacturer’s protocol. RNA concentrations were determined by spectrophotometry.
Separate tube (singleplex) real-time PCR was performed using 0.5 µg of RNA as template to detect PTHrP and PTH1R mRNA transcripts and 18S rRNA (endogenous control for normalization purposes). The following TaqMan inventoried products were used: PTHrP, Hs00174969_m1; PTH1R, Hs00174895_m1; and the pre-developed 18S rRNA primers (VIC™-dye labeled probe, TaqMan® assay reagent, P/N 4319413E). These products were obtained from Applied Biosystems, as was the universal PCR master mix reagent kit (P/N 4304437). The cycling parameters for real-time PCR were: UNG (uracil-DNA-glycosylase) activation at 50 °C for 2 min, AmpliTaq activation at 95 °C for 10 min, then denaturation at 95 °C for 15 sec and annealing/extension at 60 °C for 1 min (repeated 40 times) on an ABI7000 real-time PCR machine. Duplicate CT values were analyzed in Microsoft Excel using the comparative CT (ΔΔCT) method as described by the manufacturer (Applied Biosystems). The amount of target (2−ΔΔCT) was obtained by normalization to the endogenous reference (18S). Real-time PCR was performed using the facilities of the Sealy Center for Cancer Cell Biology’s Real-Time PCR core facility at the University of Texas Medical Branch; http://www.utmb.edu/scccb/pcr/index.htm. As negative controls, PCR reactions were performed without RNA.
Western blot analysis
Nuclear and cytoplasmic fractions were prepared using the NE-PER extraction kit (Pierce, Rockford, IL). In all experiments, fractionation was confirmed by probing the blots with anti-lamin B1 and anti-β-actin antibodies, specific for nuclear and cytoplasmic markers, respectively. Western blot analysis was performed as previously described [20]. Densitometric analysis was performed using the Alpha Innotech Image Analysis system (Alpha Innotech Corporation, San Leandro, CA). Values were normalized to the loading controls.
Anchorage-independent cell growth
Assays to determine colony formation in soft agar were performed in 60 mm dishes containing a bottom layer consisting of 1.5 ml culture medium plus 0.4% (w/v) agar (BMA, Rockland, ME). Cells (1 × 104) were plated in a top layer of culture medium plus 0.3% agar. After the top agar had solidified, 1 ml of culture medium was added. Two days after plating, 5 fields/well were counted to ensure that the plating efficiencies of the different clones were similar. The medium was replaced every 3 days. Photographs were taken after 2 weeks at 10 × magnification to measure clone frequency. Five fields per plate were photographed and all clones in focus > 50 µm in size were counted. The imaging software ImageJ (NIH) was used. Two independent experiments for each of three independent clones were performed in triplicate.
Statistics
Numerical data are presented as the mean ± SEM. The data were analyzed by ANOVA, followed by a Bonferroni post-test to determine the statistical significance of differences. All statistical analyses were performed using Instat Software (GraphPad Software, Inc., San Diego, CA). P < 0.05 was considered significant.
Results
Characterization of cell lines overexpressing wild-type or mutated PTHrP
To study the mechanisms via which PTHrP alters cell growth and apoptosis, we established LoVo cells overexpressing WT PTHrP or PTHrP deleted over the NLS or the SP. LoVo cells transfected with the empty vector served as controls. These cells were characterized in terms of PTHrP mRNA levels, as well as secreted and intracellular protein levels. Transfection with the constructs expressing WT, ΔNLS or ΔSP PTHrP resulted in significant (~30-fold) increases in PTHrP mRNA levels (Fig. 1A). Secreted PTHrP levels were significantly higher in the cells overexpressing WT or NLS-deleted PTHrP, compared to empty vector transfectants and cells overexpressing ΔSP PTHrP (Fig. 1B). PTHrP secretion was higher in cells overexpressing ΔNLS PTHrP than in the WT PTHrP-overexpressing cells (Fig. 1B). PTHrP secretion was comparable in cells overexpressing ΔSP PTHrP and in control cells (Fig. 1B).
Figure 1. Characterization of LoVo cells overexpressing wild-type (WT) PTHrP or PTHrP deleted over the nuclear localization signal (ΔNLS) or the signal peptide (ΔSP).
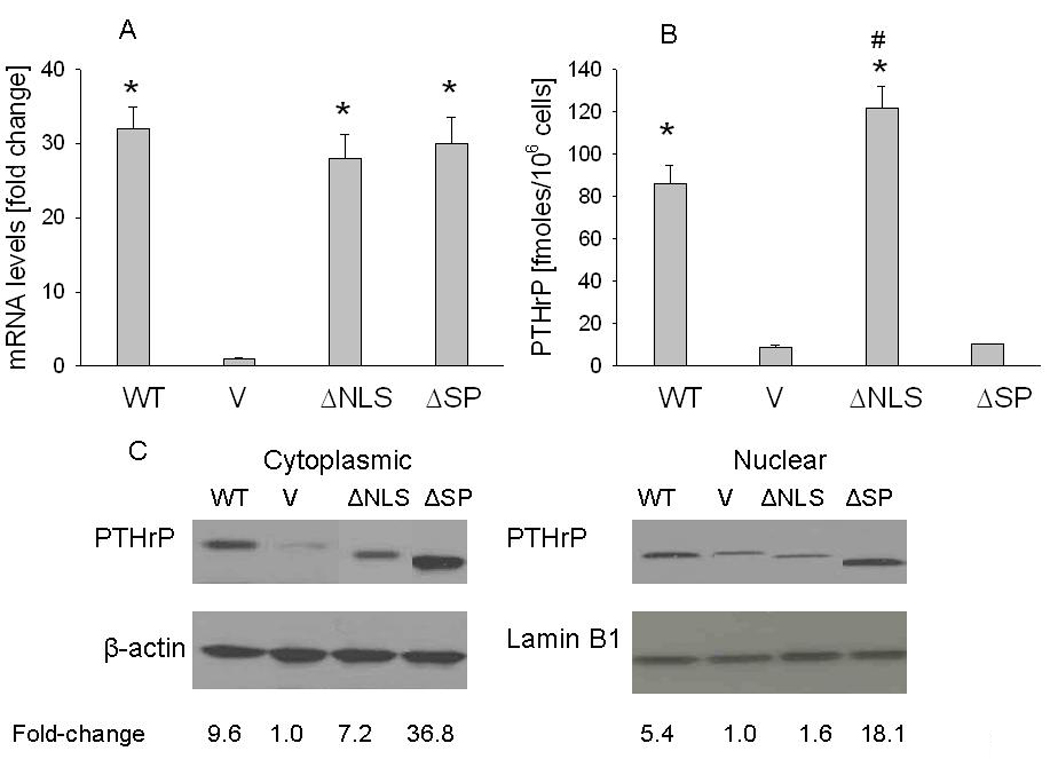
V = empty vector controls. (A) mRNA levels were measured by reverse transcription/real-time PCR. Values are expressed relative to the corresponding V value, set arbitrarily at 1.0. (B) PTHrP secretion was measured using an immunoradiometric assay. Values are expressed as PTHrP secreted in fmoles/106 cells. In (A) and (B), each bar is the mean ± SEM of three independent experiments for each of three independent clones. * = Significantly different from the control (V) value (P < 0.001); # = significantly different from the WT value (P < 0.001). (C) Western blot analysis of cytoplasmic and nuclear PTHrP fractions. WT PTHrP was detected as an ~ 18 kDa protein. Equal loading was confirmed by re-probing for cytoplasmic (β-actin) or nuclear (lamin B1) markers. The densitometric scanning data represents the mean fold-change obtained from two experiments for each of three independent clones. Values are expressed relative to the corresponding V value, set arbitrarily at 1.0.
Intracellular PTHrP levels were monitored by Western blot analysis of nuclear and cytoplasmic fractions. Overexpressing the WT or mutated PTHrP isoforms significantly increased cytoplasmic PTHrP levels, compared to vector controls (Fig. 1C). The level of cytoplasmic PTHrP was ~ 4-fold higher in cells overexpressing ΔSP PTHrP than in WT PTHrP-overexpressing cells (Fig. 1C). Nuclear PTHrP levels were ~ 3-fold higher in WT PTHrP-overexpressing cells than in NLS-mutated PTHrP-overexpressing cells (Fig. 1C). ΔSP PTHrP-overexpressing cells had ~ 3-fold higher nuclear PTHrP levels than WT PTHrP-overexpressing cells (Fig. 1C). Re-probing the blots with antibodies for lamin B1 and β-actin, specific for nuclear and cytoplasmic markers, respectively, confirmed that there was no cross-contamination of the nuclear or cytoplasmic fractions (data not shown). There was no difference in PTHrP mRNA and secreted protein levels in empty vector transfectants vs. parental cells (data not shown).
The growth-promoting and anti-apoptotic effects of PTHrP are mediated via an intracrine pathway
We compared the growth of cells overexpressing WT PTHrP to that of cells overexpressing ΔNLS or ΔSP PTHrP. The growth rate of cells overexpressing WT PTHrP was significantly higher than that of the control cells under both serum replete and serum-depleted conditions (Fig. 2A). Deletion of the NLS negated these effects of PTHrP (Fig. 2A). The growth of ΔSP PTHrP-overexpressing cells was higher than that of cells overexpressing the WT isoform under both serum replete and serum-depleted conditions, although this difference only reached statistical significance under serum depleted conditions. In addition, the presence of FBS did not significantly affect the growth of cells overexpressing ΔSP PTHrP (Fig. 2A).
Figure 2. Cell growth (A) and apoptosis (B) of LoVo cells overexpressing wild-type (WT) PTHrP or PTHrP deleted over the nuclear localization signal (ΔNLS) or signal peptide (ΔSP).
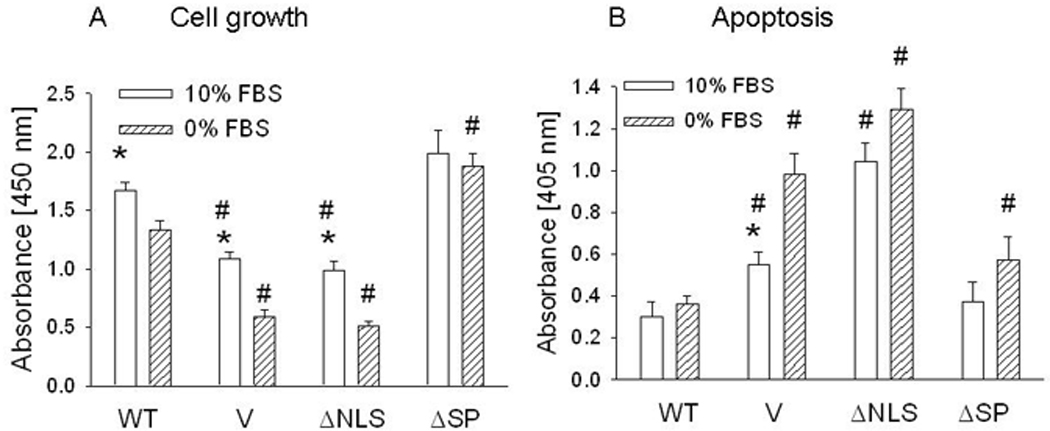
V = empty vector control. Cells were plated in 10% FBS. Cell growth and apoptosis under serum replete conditions were measured 96 h and 72 h after plating, respectively. For experiments under serum-depleted conditions, cells were transferred to 0% FBS 24 h after plating. Cell growth and apoptosis were measured after a further 72 h and 48 h, respectively. Each bar is the mean ± SEM of three independent experiments for each of three independent clones. * = Significantly different from the corresponding 0% FBS value (P < 0.001); # = significantly different from the corresponding WT value (P < 0.001).
We also measured apoptosis in cells overexpressing WT or mutated PTHrP. Under serum-replete conditions, the basal level of apoptosis was lower in WT PTHrP-overexpressing cells than in the control cells (Fig. 2B). Deleting the SP did not decrease the protective effects of PTHrP on apoptosis under these serum-replete conditions (Fig. 2B). In contrast, deleting the NLS abolished the protective effects of PTHrP (Fig. 2B). Serum starvation induced apoptosis in the control LoVo cells (Fig. 2B). WT and ΔSP-deleted PTHrP protected the cells from apoptosis, and there was no significant difference in apoptosis under serum-replete and serum-starved conditions in these cells. ΔNLS PTHrP did not protect cells from apoptosis; the level of apoptosis in these cells was also significantly elevated under serum replete conditions (Fig. 2B).
Effect of suppressing PTH1R signaling on cell growth and apoptosis
The data in Fig. 2 indicate that secreted PTHrP acting via an autocrine/paracrine pathway does not play a major role in the growth-regulatory and anti-apoptotic effects of PTHrP. Since N-terminal PTHrP activity is mediated via the PTH1R, we tested the effect of suppressing PTH1R expression on cell growth and apoptosis of parental LoVo cells. PTH1R expression was suppressed by transfection with an siRNA targeting the PTH1R. To decrease the potential for off-target effects, two independent PTH1R-specific siRNAs were used. Transfection with either of these siRNAs caused an ~75% decrease in PTH1R mRNA levels, compared to the NTC control (measured by reverse transcription/real-time PCR) (Fig. 3A).
Figure 3. Effect of suppressing PTH1R expression or neutralizing secreted PTHrP on LoVo cell apoptosis.
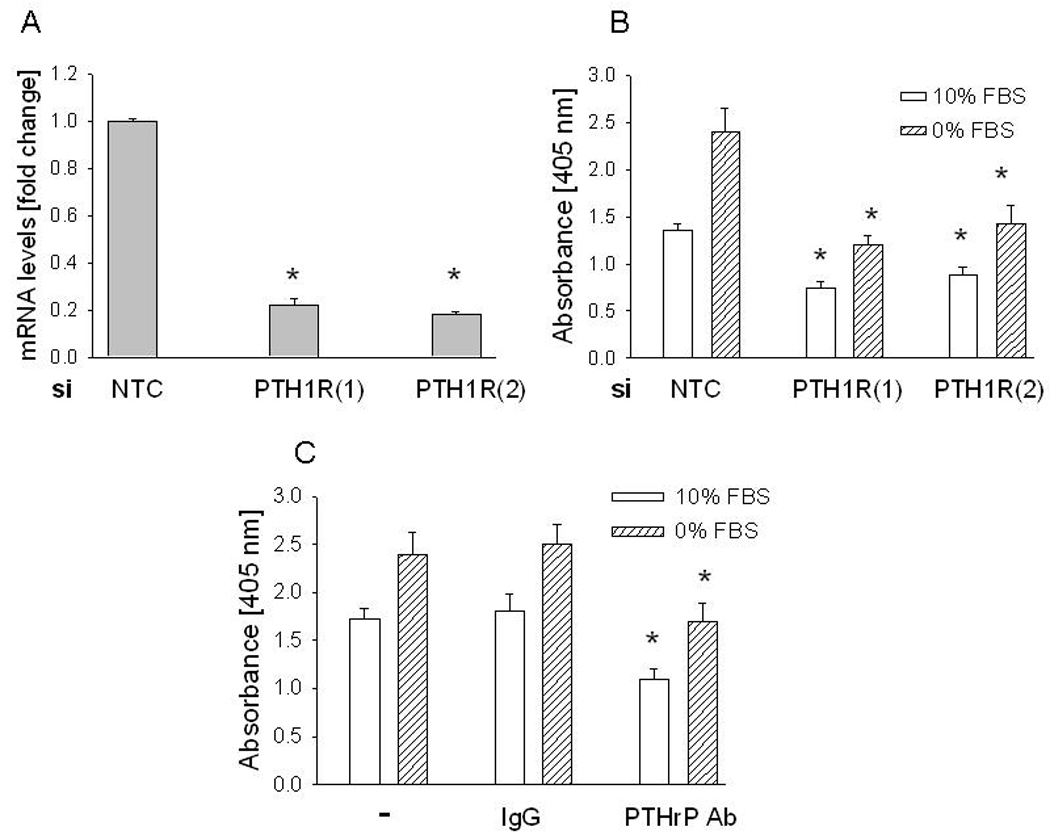
(A) PTH1R expression was suppressed by transfection with an siRNA targeting PTH1R. Two independent siRNAs (1 and 2) were used. As control, cells were transfected with a non-targeting control (NTC) siRNA. mRNA levels were measured 48 h after transfection. Values are expressed relative to the NTC value, set arbitrarily at 1.0. Each bar is the mean ± S.E.M. for three independent experiments. * = Significantly different from the NTC value (P < 0.001). (B) Apoptosis after transfection with siRNAs targeting PTH1R. Apoptosis was measured 48 h after transfection. Each bar is the mean ± SEM of three independent experiments for each of two independent siRNAs. * = Significantly different from the respective NTC control value (P < 0.001). (C) Apoptosis after neutralizing secreted PTHrP with an anti-PTHrP antibody. Cells were treated with an anti-PTHrP antibody (final concentration 2 µg/ml). Apoptosis was measured after 48 h. − = no treatment; IgG = IgG control. Each bar is the mean ± SEM of three independent experiments. * = Significantly different from the respective IgG control value (P < 0.001).
Suppression of PTH1R expression decreased cell apoptosis under both serum-replete and serum-depleted conditions, measured 48 h after transfection with the PTH1R siRNA (Fig. 3B). Downregulation of secreted PTHrP levels by incubation with an anti-PTHrP (1–34) antibody (2 µg/ml for 48 h) produced the same effects as suppression of PTH1R expression (Fig. 3C).
Autocrine/paracrine PTHrP action also decreased LoVo cell growth, measured under serum-depleted conditions. Thus, suppression of PTH1R expression caused an ~ 25% increase in LoVo cell growth, measured 72 h after transfection with the PTH1R siRNA (Fig. 4A). A similar effect on cell growth was obtained when cells were incubated with an anti-PTHrP (1–34) antibody (Fig. 4B). Under serum-replete conditions, the effect of suppression of PTH1R expression was less pronounced (~ 10% increase; data not shown).
Figure 4. Effect of suppressing PTH1R expression or neutralizing secreted PTHrP on LoVo cell growth.
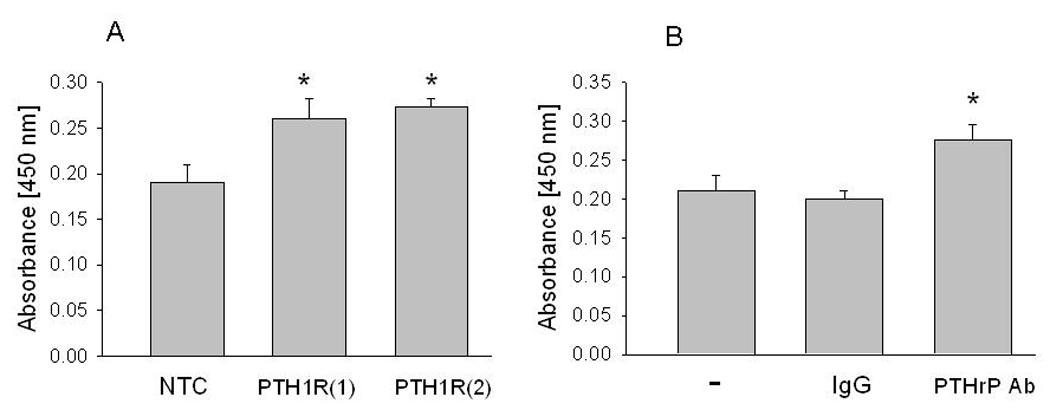
(A) Cell growth after transfection with an siRNA targeting PTH1R. Two independent siRNAs (1 and 2) were used. As control, cells were transfected with a non-targeting control (NTC) siRNA. Cell growth was measured 72 h after transfection. (B) Cell growth after neutralizing secreted PTHrP with an anti-PTHrP anti-body. At 48 h after plating, cells were treated with an anti-PTHrP antibody. Cell growth was measured after 72 h. − = no treatment; IgG = IgG control. Each bar is the mean ± SEM of three independent experiments for each of two independent siRNAs (A) or three independent experiments (B). (A) * = Significantly different from the NTC control value (P < 0.001). (B) * = Significantly different from the IgG control value (P < 0.001).
Intracrine PTHrP increases anchorage-independent cell growth
To investigate whether the anti-apoptotic effects of PTHrP in the cell monolayer system translate into increased cell survival under anchorage-independent conditions, we measured the ability of cells expressing the different PTHrP isoforms to grow as colonies in soft agar. Cells overexpressing WT or mutated PTHrP were grown in soft agar for 15 days. WT PTHrP facilitated soft agar clone formation; deletion of the SP had no effect on this process. In contrast, deletion of the NLS negated the PTHrP effect on anchorage-independent cell growth, in that there was no significant difference in the number and size of the colonies formed from cells overexpressing ΔNLS PTHrP and the empty vector controls (Fig. 5). Increasing the incubation time from 15 to 21 days did not increase the number of colonies formed by any of the clones (data not shown).
Figure 5. Effects of wild-type PTHrP and PTHrP deleted over the nuclear localization signal or signal peptide on anchorage-independent cell growth.
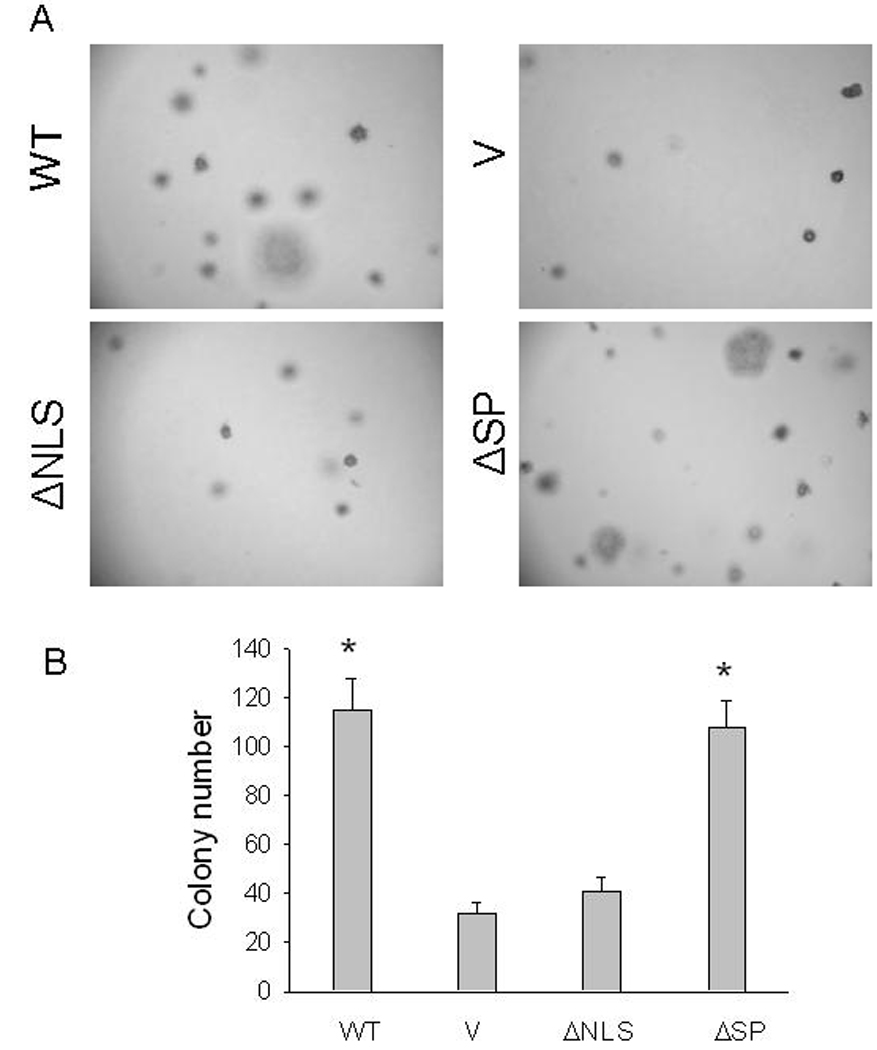
(A) Representative colonies after 2 weeks in culture. Magnification, ×10. (B) Each bar is the mean ± SEM of 20 colonies for each of three independent clones overexpressing WT PTHrP (WT), NLS-deleted PTHrP (ΔNLS), or SP-deleted PTHrP (ΔSP), or transfected with empty vector (V). * = Significantly different from the control (V) value (P < 0.001).
Discussion
PTHrP was originally identified as the factor responsible for humoral hypercalcemia of malignancy, a frequent syndrome in patients with cancers of the breast and lung, among others [reviewed in 22]. These effects of PTHrP are attributed to its role as a calciotropic hormone. Thus, both PTHrP and the PTH1R are expressed in bone cells and chondrocytes. PTHrP plays a major role in bone development, and targeted disruption of the PTHrP gene in mice results in skeletal dysplasia [reviewed in 23]. However, PTHrP exerts a much broader spectrum of effects in both normal physiology and disease states. The protein regulates cell growth, cell survival, smooth muscle relaxation and development [22,23]. PTHrP also plays a more general role in cancer cell progression independent of its calciotropic effects. Our main findings are that PTHrP increases LoVo cell growth and survival via an intracrine pathway. Intracrine PTHrP action also results in increased anchorage-independent cell growth. Given the correlation between anchorage-independent growth in vitro and cellular tumorigenicity in nude mice [24], these findings may explain the consequences of PTHrP expression in colon cancer.
The prepro form of the PTHrP molecule contains two targeting sequences – an N-terminal signal sequence for endoplasmic reticulum targeting and a nuclear localization signal in the mid-region of the molecule. The relationship between secreted and nuclear PTHrP levels is complex. An inverse relationship has been reported between SP cleavage and nuclear localization of PTHrP both in vitro and in a cell culture system [25]. The NLS has an inhibitory effect on signal sequence cleavage, and the efficiency of nuclear localization is inversely related to cleavage of the signal sequence [25]. This relationship between secreted and nuclear PTHrP levels was also evident in the LoVo cell model (Fig. 1). Thus, deletion of the SP or NLS may directly influence the autocrine/paracrine or intracrine effects of PTHrP, respectively, as well as exert effects secondary to changes in the levels of nuclear and secreted PTHrP levels resulting from indirect alterations in prepro-PTHrP processing and therefore secretion or subcellular localization of the protein.
Many growth factors, including PTHrP, regulate tumorigenesis by exerting positive effects on cell growth and survival. Both the autocrine/paracrine and intracrine pathway of PTHrP action have been implicated in the effects of the protein on these parameters [12,13,20,21,26]. Opposing effects of PTHrP on cell growth and apoptosis have been reported; the net result is dependent on the predominant pathway via which PTHrP exerts its effects in a cell type-specific manner [12,13]. Here we show (Fig. 2 and Fig. 4) that the growth-promoting effects of PTHrP are mediated predominantly via an intracrine pathway. Since serum deprivation has no significant effect on the growth of cells overexpressing ΔSP PTHrP, high nuclear PTHrP levels may overcome the effects of growth factor deprivation.
We show that PTHrP protects LoVo cells from apoptosis induced by serum deprivation via an intracrine pathway (Fig. 2). Similar effects of PTHrP have been observed in the prostate cancer cell lines C4-2 and PC-3, where PTHrP exerts a protective effect on apoptosis induced by the DNA-intercalating agent doxorubicin [27]. In view of the observation that autocrine/paracrine PTHrP action decreases cell survival, the higher level of apoptosis in NLS-deleted PTHrP-overexpressing cells vs. control cells cultured under serum replete conditions may be attributed to the higher secreted PTHrP levels after NLS deletion, as discussed above [25]. The effects of PTHrP on apoptosis under serum-depleted conditions are more complex. Both WT and SP-deleted PTHrP protect cells from apoptosis induced by serum deprivation. The higher level of apoptosis after NLS deletion, as compared to that in control cells, may again be attributed to higher secreted PTHrP levels in NLS-deleted PTHrP-overexpressing cells [25], which in turn results in increased apoptosis via the autocrine/paracrine pathway.
A requirement for tumor progression and metastasis is the ability of cells to migrate and invade through the basement membrane into surrounding tissues. This process requires anchorage independence [24], and can lead to apoptosis if not counterbalanced by increased survival [14]. Anoikis, which describes apoptosis induced by loss of anchorage, limits the spread of cells outside the tissue environment [17]. Here we report that WT and SP-deleted PTHrP increase LoVo cell growth as colonies in soft agar in the absence of adhesion to the extracellular matrix. The anti-apoptotic effects of WT and ΔSP PTHrP likely play a major role in the enhanced anchorage-independent growth of LoVo cells overexpressing these PTHrP isoforms. Since the level of anoikis correlates with in vivo tumorigenicity and metastatic capability [19], one mechanism via which PTHrP may enhance tumor progression and metastasis is through decreased anoikis.
This study underlies the critical role played by nuclear PTHrP action in colon cancer cell growth and survival in vitro. Recently, it has also been shown that knock-in mice expressing PTHrP deleted over the NLS/C-terminal domain exhibit retarded growth, early senescence and malnutrition, leading to a rapid demise postnatally [29]. These effects were accompanied by increased p21 levels and inhibition of cyclin E/Cdk2 and cyclin D1/Cdk4/Cdk6 activities, leading to cell cycle arrest in the G1 phase. Mice expressing PTHrP deleted over the NLS/C-terminal domain also had lower nuclear levels of Bim-1, a protein which promotes cell proliferation and suppresses genes that induce cellular senescence and cell death [30]. Taken together, these studies underline the importance of intracrine PTHrP action in vitro and in vivo.
In conclusion, the results presented here demonstrate that PTHrP increases cell growth, survival and anchorage-independent cell growth via an intracrine pathway. PTHrP also protects cells against cellular stress, such as that induced by serum starvation. These results establish intracrine PTHrP action as a major contributor to colon cancer progression. Strategies aimed at decreasing PTHrP production in colon cancer may thus provide therapeutic benefits.
Acknowledgements
This work was supported by NIH grant DK035608.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parker SL, Tong T, Bolden S, Wingo PA. Cancer statistics. CA Cancer J. Clin. 1996;46:5–27. doi: 10.3322/canjclin.46.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Martin R, Paty P, Fong Y, Grace A, Cohen A, DeMatteo R, Jarnagin W, Blumgart L. Simultaneous liver and colorectal resections are safe for synchronous colorectal liver metastasis. J. Am. Coll. Surg. 2003;197:233–241. doi: 10.1016/S1072-7515(03)00390-9. [DOI] [PubMed] [Google Scholar]
- 3.Declan Fleming RY. Colorectal cancer screening and follow-up. Surg. Oncol. 1998;7:125–137. doi: 10.1016/s0960-7404(99)00034-1. [DOI] [PubMed] [Google Scholar]
- 4.Kohn EC, Liotta LA. Molecular insights into cancer invasion: strategies for prevention and intervention. Cancer Res. 1995;55:1856–1862. [PubMed] [Google Scholar]
- 5.Goltzman D. Mechanisms of development of osteoblastic metastases. Cancer (Suppl.) 1997;80:1581–1587. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1581::aid-cncr8>3.3.co;2-8. [DOI] [PubMed] [Google Scholar]
- 6.Yoneda T, Sasaki A, Mundy GR. Osteolytic bone metastasis in breast cancer. Breast Cancer Res. Treat. 1994;32:73–84. doi: 10.1007/BF00666208. [DOI] [PubMed] [Google Scholar]
- 7.Nishihara M, Ito M, Tomioka T, Ohtsuru A, Tagashi T, Kanematsu T. Clinicopathological implications of parathyroid hormone-related protein in human colorectal tumours. J. Path. 1999;187:217–222. doi: 10.1002/(SICI)1096-9896(199901)187:2<217::AID-PATH210>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 8.Shen X, Rychahou PG, Evers BM, Falzon M. PTHrP increases xenograft growth and promotes integrin α6β4 expression and Akt activation in colon cancer. Cancer Lett. 2007;258:241–252. doi: 10.1016/j.canlet.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mannstadt M, Jüppner H, Gardella TJ. Receptors for PTH and PTHrP. Am J Physiol. 1999;277:F665–F675. doi: 10.1152/ajprenal.1999.277.5.F665. [DOI] [PubMed] [Google Scholar]
- 10.Shen X, Falzon M. PTH-related protein enhances LoVo colon cancer cell proliferation, adhesion, and integrin expression. Reg. Peptides. 2005;125:17–27. doi: 10.1016/j.regpep.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 11.Carron JA, Fraser WD, Gallagher JA. PTHrP and the PTH/PTHrP receptor are co-expressed in human breast and colon tumours. Br. J. Cancer. 1997;76:1095–1098. doi: 10.1038/bjc.1997.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henderson JE, Amikuza N, Warshawsky H, Biasotto D, Lanske BMK, Goltzman D, Karaplis AC. Nucleolar localization of parathyroid hormone-related peptide enhances survival of chondrocytes under conditions that promote apoptotic cell death. Mol. Cell. Biol. 1995;15:4064–4075. doi: 10.1128/mcb.15.8.4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massfelder T, Dann P, Wu TL, Vasavada R, Helwig J-J, Stewart AF. Opposing mitogenic and antimitogenic actions of parathyroid hormone-related protein in vascular smooth muscle cells: a critical role for nuclear targeting. Proc. Natl. Acad. Sci. USA. 1997;94:13630–13635. doi: 10.1073/pnas.94.25.13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 15.Coll ML, Rosen K, Ladeda V, Filmus J. Increased Bcl-XL expression mediates v-Src-induced resistance to anoikis in intestinal epithelial cells. Oncogene. 2002;21:2908–2913. doi: 10.1038/sj.onc.1205388. [DOI] [PubMed] [Google Scholar]
- 16.Windham TC, Parikh NU, Siwak DR, Summy JM, McConkey DJ, Kraker AJ, Gallick GE. Src activation regulates anoikis in human colon tumor cell lines. Oncogene. 2002;21:7797–7807. doi: 10.1038/sj.onc.1205989. [DOI] [PubMed] [Google Scholar]
- 17.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell. Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frisch SM, Screaton RA. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001;13:555–562. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 19.Berezovskaya O, Schimmer AD, Glinskii AB, Pinilla C, Hoffman RM, Reed JC, Glinsky GV. Increased expression of apoptosis inhibitor protein XIAP contributes to anoikis resistance of circulating human prostate cancer metastasis precursor cells. Cancer Res. 2005;65:2378–2386. doi: 10.1158/0008-5472.CAN-04-2649. [DOI] [PubMed] [Google Scholar]
- 20.Shen X, Mula RVR, Li J, Weigel NL, Falzon M. PTHrP contributes to the anti-proliferative and integrin α6β4-regulating effects of 1,25-dihydroxyvitamin D3. Steroids. 2007;72:930–938. doi: 10.1016/j.steroids.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Falzon M, Du P. Enhanced growth of MCF-7 breast cancer cells overexpressing parathyroid hormone-related protein. Endocrinology. 2000;141:1882–1892. doi: 10.1210/endo.141.5.7470. [DOI] [PubMed] [Google Scholar]
- 22.Strewler GI. Mechanisms of disease: the physiology of parathyroid hormone-related protein. N. Engl. J. Med. 2000;342:177–185. doi: 10.1056/NEJM200001203420306. [DOI] [PubMed] [Google Scholar]
- 23.Maioli E, Fortino V. The complexity of parathyroid hormone-related protein signaling. Cell. Mol. Life Sci. 2004;61:257–262. doi: 10.1007/s00018-003-3233-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell. 1974;3:355–359. doi: 10.1016/0092-8674(74)90050-6. [DOI] [PubMed] [Google Scholar]
- 25.Amaya Y, Nakai T, Komaru K, Tsuneki M, Miura S. Cleavage of the ER-targeting signal sequence of parathyroid hormone-related protein is cell-type-specific and regulated in cis by its nuclear localization signal. J. Biochem. 2008;143:569–579. doi: 10.1093/jb/mvn002. [DOI] [PubMed] [Google Scholar]
- 26.Shen X, Mula RV, Evers BM, Falzon M. Increased cell survival, migration, invasion and Akt expression in PTHrP-overexpressing LoVo colon cancer cell clones. Reg. Peptides. 2007;141:61–72. doi: 10.1016/j.regpep.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhatia V, Mula RV, Weigel NL, Falzon M. Parathyroid hormone-related protein regulates cell survival pathways via integrin α6β4-mediated activation of PI3-K/Akt signaling. Mol. Cancer Res. doi: 10.1158/1541-7786.MCR-08-0568. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosen K, Rak J, Leung T, Dean NM, Kerbel RS, Filmus J. Activated ras prevents downregulation of Bcl-XL triggered by detachment from the extracellular matrix: a mechanisms of ras-induced resistance to anoikis in intestinal epithelial cells. J. Cell. Biol. 2000;149:447–456. doi: 10.1083/jcb.149.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miao D, Su H, He B, Gao J, Xia Q, Zhu M, Gu Z, Goltzman D, Karaplis AC. Severe growth retardation and early lethality in mice lacking the nuclear localization sequence and C-terminus of PTH-related protein. Proc Natl Acad Sci USA. 2008;105:20309–20314. doi: 10.1073/pnas.0805690105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]