

Abstract
As part of our continuing investigation of azo-flavonoid derivatives as potential anticancer drug candidates, a series of 2-aryl-6,7-methylenedioxyquinolin-4-one analogs was designed and synthesized. The design combined structural features of 2-(2-fluorophenyl)-6,7-methylenedioxyquinolin-4-one (CHM-1), a previously discovered compound with potent in vivo antitumor activity, and 2-arylquinolin-4-ones identified by CoMFA models. The newly synthesized analogs were evaluated for cytotoxicity against seven human cancer cell lines, and structure-activity relationship (SAR) correlations were established. Analogs 1, 37, and 39 showed potent cytotoxicity against different cancer cell lines. Compound 1 demonstrated selective cytotoxicity against Hep 3B (hepatoma) cells. Compound 37 was cytotoxic against HL-60 (leukemia), HCT-116 (colon cancer), Hep 3B (hepatoma), and SK-MEL-5 (melanoma) cells. Compound 39 exhibited broad cytotoxicity against all seven cancer cell lines, with IC50 values between 0.07–0.19 µM. Results from mechanism of action studies revealed that these new quinolone derivatives function as antitubulin agents.
Keywords: 2-Arylquinolin-4-ones, CHM-1, Cytotoxicity, Tubulin inhibitor
Introduction
Microtubules of eukaryotic cells are known to play an important role in mitosis.1 Therefore, compounds that target microtubules have long been investigated as anticancer drugs. Typically, such compounds arrest cells, at least transiently, in the mitotic phase of the cell cycle. Currently, two groups of antimitotic agents are used clinically for cancer treatment. Vinca alkaloids and estramustine inhibit tubulin polymerization into microtubules, while taxoids and epothilones stabilize microtubules and consequently interfere with their normal dynamic instability.2, 3 Colchicine is another well-known antimitotic agent used for treating gout and inflammatory diseases. However, due to its high toxicity, colchicine is not used in cancer therapy. Combretastatin A-4, with a modified colchicine-like structure, exhibits a lower toxicity profile, while it still targets the same binding site as colchicine.4, 5 The phosphate prodrug of combretastatin A-4 (CA-4) is currently in phase III clinical trials as an anticancer agent.6, 7 Although several antimitotic agents are available, due to the structural complexity of vinca alkaloids and taxoids, there is still a need to identify novel anticancer drugs that target microtubules but have simplified structures, because such compounds can be readily prepared in a more cost-effective way.
In recent years, we have designed and synthesized three series of azo-flavonoids as new classes of antimitotic agents: 2-arylquinolone-4-ones,8–15 2-arylnaphthyridin-4-ones,16–18 and 2-arylquinazolin-4-ones 19, 20 (Figure 1). Some of these compounds, especially analogs belonging to the 2-arylnaphthyridin-4-one family, demonstrated significant antitumor and anti-tubulin activities. In general, a good correlation was found between the cytotoxicity of these analogs and their inhibitory effects on tubulin polymerization. Additional mechanism of action studies revealed that the most active of these compounds are also potent inhibitors of colchicine binding to tubulin. Further modification of the 2-phenylquinolin-4-one series was directed by computer modeling studies combined with SAR results from previously synthesized compounds. In the current study, we report the design and syntheses of novel 2-arylquinolone-4-one analogs, which showed potent and selective inhibitory activities towards different human cancer cell lines.
Figure 1.

Three series of structural skeletons of azo-flavonoids: 2-arylquinolone-4-ones, 2-arylnaphthyridin-4-ones, and 2-arylquinazolin-4-ones.
Design
Both Conventional Comparative Molecular Field Analysis (CoMFA) and g2 GRS CoMFA were used to identify structural requirements that may be essential for increasing the binding affinity of 2-phenylquinolin-4-one and 2-phenylnaphthyridin-4-one analogs for the colchicine site of tubulin. The training set of the models contained 51 compounds, and the cross-validated R2 (q2) values for conventional CoMFA and g2 GRS CoMFA were 0.637 and 0.692, respectively. These QSAR models predicted that a larger heterocyclic aromatic ring at the C-2 position should enhance activity. The predictive power of the models was validated by the prediction for a test set of 53 compounds with known anti-tubulin potencies, and the predictive R2 values were 0.546 and 0.426, respectively. Based on this study, new analogs were designed in both compound series (Figure 2).21 As a proof of concept, the newly designed arylnaphthyridine analog 2-(3-benzo[b]thienyl)naphthyridin-4-one (A), which has a benzothienyl ring at the C-2 position of the naphthyridine, exhibited potent cytotoxicity in the low micromolar to nanomolar concentration range. Further, a mechanism of action study revealed that the compound also inhibited tubulin polymerization with an IC50 value of 0.37 µM. These results verified the success of our modeling design.18 Thus, the same modifications were carried out at the C-2 position of the 2-arylquinolin-4-one series in the present study.
Figure 2.

Proposed models and newly designed compounds. Conventional Comparative Molecular Field Analysis (CoMFA) and g2 GRS CoMFA are two computational programs performed to identify the essential structure requirements for increasing affinity at the colchicine site of tubulin. Based on the results of the QSAR study, we proposed two concept models. As the proof of concept, analog A was synthesized and tested against cancer cell lines and for binding to tubulin.
In addition, when we re-analyzed results from our prior in vivo antitumor study of selected 2-phenylquinolin-4-one compounds, we found that analogs with a methylenedioxy functional group at the C-6 and -7 positions showed superior antitumor and safety profiles compared with C-6 mono-modified compounds. For instance, 2-(3-methoxyphenyl)-6-pyrrolinylquinolin-4-one (B, Figure 3) exhibited only moderate antitumor activity at the maximum tolerated dose (MTD, 25 mg/kg, ip, once weekly for 3 courses) against the OVCAR-3 xenograft model in nude mice. In contrast, 2-(2-fluorophenyl)-6,7-methylenedioxyquinolin-4-one (CHM-1, Figure 3) extended the life span of tumor-bearing mice by 124%, 133% and 79%, at dosages of 200, 134 and 79 mg/kg (ip, once weekly for 3 courses), respectively. Additionally, the maximum tolerated dose of CHM-1 was not reached at the highest tested dosage of 200 mg/kg.22 Currently, the phosphate prodrug of CHM-1 (CHM-1-P-Na, Figure 4) is under preclinical study.22, 23 Therefore, the methylenedioxy ring of CHM-1 was also incorporated into our newly designed 2-arylquinolone-4-one series, which led to the syntheses of 2-aryl-6,7-methylenedioxyquinolin-4-one analogs (1, 36–45). Compound 56, a ring isomer of analog 39, was also synthesized, because ring isomerization is a common approach in structure-activity relationship studies of a compound series.
Figure 3.

Structures of compound B and CHM-1.
Figure 4.

Metabolic pathway of CHM-1-P-Na.
Chemistry
The synthetic procedure for target compounds (1, 36–45) is illustrated in Scheme 1. The starting arylcarboxylic acids (3–13) were first treated with oxalyl chloride to form the corresponding acid chlorides (14–24), which were then reacted with 2-amino-4,5-methylenedioxyacetophenone (2) to give the corresponding amides (25–35). The intermediates (25, 27–34) were subjected to a cyclization reaction in t-BuOH, in the presence of t-BuOK, to afford the corresponding 2-arylquinolin-4-one analogs (1, 37–44). Cyclization was also carried out with 1,4-dioxane as solvent, in the presence of NaOH, which yielded the target products (1, 36, 39 and 45) in yields of 40%–74%. When the starting compounds (25–26, 28–32 and 34) were treated with t-BuOK in toluene at 90 °C for 72 h, the corresponding deacetylated products 46–53 were obtained unexpectedly.
Scheme 1.

Reagents and conditions: (a) toluene/DMF, 22–25 °C; (b) toluene/triethylamine, 22–25 °C; (c) t-BuOK/ t-BuOH or NaOH/1,4-dioxane, reflux; (d) t-BuOK/toluene, 90 °C, 72 h.
Scheme 2 depicts the synthesis of compound 56. The starting material 2 was first treated with 1-naphthylmethyl chloride (54) to give the corresponding amide (55). Compound 55 was then subjected to a cyclization reaction in 1,4-dioxane, in the presence of NaOH, which yielded 4-methyl-6,7-methylenedioxy-3-(1-naphthyl)quinolin-2-one (56) as the final product.
Scheme 2.

Reagents and conditions: (a) toluene/triethylamine, 22–25 °C; (b) NaOH/1,4-dioxane, reflux.
Results and Discussion
The newly synthesized 2-aryl-6,7-methylenedioxyquinolin-4-ones (1, 36–45) and the ring isomer 56 were assayed for in vitro cytotoxicity against seven human cancer cell lines, including HL-60 (leukemia), HCT-116 (colon cancer), A549 (non-small cell lung carcinoma), Hep 3B (hepatoma), KB (epidermoid carcinoma of the nasopharynx), KB-VIN (p-glycoprotein- expressing epidermoid carcinoma of the nasopharynx), and DU145 (prostate cancer). The cytotoxicity results are summarized in Table 1. Compounds 46–53 were tested against the HL-60, HCT-116, H226, A549, Hep G2 and A498 cancer cell lines; however, none of the compounds showed significant activity (IC50 values were >10 µM).
Table 1.
IC50 (µM) Values from In Vitro Cytotoxicity Testing of 1, 36-45 and 56.
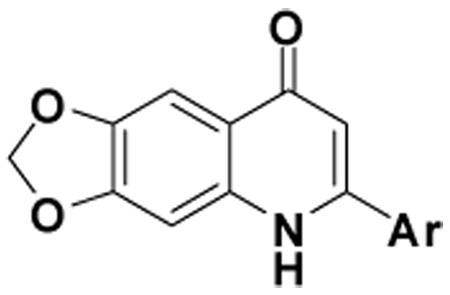 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Ar | HL-60 | HCT-116 | A549 | Hep 3B | KB | Kb-VIN | DU145 |
| 1 | 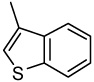 |
0.17 | 0.14 | >0.25 | 0.06 | >0.25 | >0.25 | >0.25 |
| 36 | 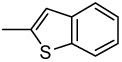 |
>1.00 | >1.00 | >1.00 | >1.00 | >1.00 | >1.00 | >1.00 |
| 37 | 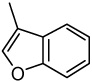 |
0.03 | 0.05 | 2.98 | 0.09 | 1.05 | 0.59 | 1.87 |
| 38 | 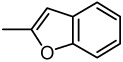 |
>1.00 | >1.00 | >1.00 | >1.00 | >1.00 | >1.00 | >1.00 |
| 39 | 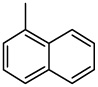 |
0.07 | 0.07 | 0.13 | 0.07 | 0.13 | 0.19 | 0.13 |
| 40 | 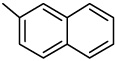 |
0.50 | >1.00 | >1.00 | >1.00 | >1.00 | >1.00 | >1.00 |
| 41 | 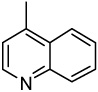 |
>5.00 | >5.00 | >5.00 | >5.00 | >5.00 | >5.00 | >5.00 |
| 42 | 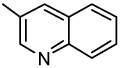 |
>0.10 | >0.10 | >0.10 | >0.10 | >0.10 | >0.10 | >0.10 |
| 43 | 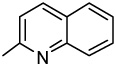 |
>5.00 | >5.00 | >5.00 | >5.00 | >5.00 | >5.00 | >5.00 |
| 44 | 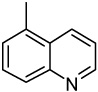 |
>10.00 | >10.00 | >10.00 | >10.00 | >10.00 | >10.00 | >10.00 |
| 45 | 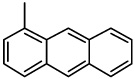 |
0.53 | 1.60 | NA* | 2.20 | NA | NA | NA |
| 56 | 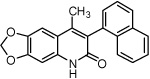 |
>10.00 | >10.00 | NA | 4.30 | NA | NA | NA |
Not assayed
Compound 1 exhibited significant cytotoxicity against Hep 3B cells, with an IC50 value of 0.06 µM, and moderate activity against HL-60 and HCT-116 cells, with IC50 values of 0.17 and 0.14 µM, respectively. This result, along with the prior cytotoxicity data obtained with compound A, confirmed the prediction generated by conventional and g2 GRS CoMFA that a larger heterocyclic aromatic ring should be preferred at the C-2 position. In comparison, replacement of the 3-benzo[b]thienyl group (1) with a 2-benzo[b]thienyl group (36) resulted in significantly reduced cytotoxicity, demonstrating the importance of the exact linkage of the aromatic ring moiety.
Bioisosteric replacement of 3-benzo[b]thienyl (1) with 3-benzo[b]furanyl (37) led to increased potency against two cancer cell lines. Specifically, compared with 1, the cytotoxic activity of 37 increased five-fold against HL-60 (IC50 0.03 µM) and three-fold against HCT-116 (IC50 0.05 µM) cells. The two compounds had similar potency against Hep 3B cells. Most importantly, 37 retained potency (IC50: 0.59 µM) against KB-VIN cells, a vincristine-resistant epidermoid carcinoma of the nasopharynx cell line, compared with KB cells (IC50 1.05 µM). As with 1 and 36, moving the attachment of the benzofuran moiety from 3′ in 37 to 2′ in 38 was detrimental to cytotoxic activity.
Compound 39, with a 1-naphthyl group rather than a 3-benzo[b]thienyl group (1) at the C-2 position, exhibited potent cytotoxicity towards all seven cancer cell lines, with IC50 values ranging from 0.07 to 0.19 µM. It should be noted that although 39 showed similar activity towards the HL-60, HCT-116, and Hep 3B cancer cell lines as compared with 1 and 37, it showed significantly increased cytotoxicity towards A549, KB, KB-VIN and DU145 cells. Compound 40, the ring positional isomer of 39, was much less cytotoxic, as was the case with compounds 36 and 38.
Surprisingly, cytotoxicity was reduced significantly when the C-2 aryl moiety was a quinoline group, no matter how it was attached (41–44). In addition, expanding the naphthalene ring of 39 to an anthracene (45) lowered the cytotoxicity remarkably. Thus, we speculate that the size of the C-2 substituted aromatic ring plays an important role in antitumor activity and that the binding pocket for this portion of the drug molecule is quite small.
Compound 56 was designed as a ring isomer of 39. The in vitro bioassay data showed that moving the aromatic ring to C-3 and the carbonyl to C-2 to give a cyclic amide completely abolished cytotoxicity of the compound. Non-cyclic amide analogs 46–53 also exhibited poor in vitro activity.
In addition to the in vitro cytotoxicity study, we also performed initial mechanism of action studies with the newly synthesized 2-aryl-6,7-methylenedioxyquinolin-4-one analogs (1, 37–45). As noted above, 2-arylquinolin-4-ones are azo-flavonoids, which were shown to inhibit tubulin assembly and the binding of colchicine to tubulin. Therefore, compounds 1 and 37–45 were tested for their in vitro activities in these assays in comparison with combretastatin A-4 (CA-4). The results are summarized in Table 2. The data showed that 1, 37, and 39, the three compounds with the greatest in vitro cytotoxicity, were potent inhibitors of tubulin assembly, with IC50 values of 0.76, 0.58, and 0.64 µM, respectively. Although all three compounds were better assembly inhibitors than CA-4 (IC50 1.2 µM), they were less effective than CA-4 in inhibiting colchicine binding to tubulin.
Table 2.
Anti-tubulin Data for CA-4, 1 and 37-45.
| Compound | Inhibition of Tubulin Assemblya IC50 (µM) ± SD |
Inhibition of Colchicine Bindingb % inhibition at tested concentration |
|
|---|---|---|---|
| 1 µM | 5 µM | ||
| CA-4 | 1.2 ± 0.2 | 91 | 99 |
| 1 | 0.76 ± 0.06 | 25 | 61 |
| 37 | 0.58 ± 0.02 | 39 | 51 |
| 38 | > 4 | - | - |
| 39 | 0.64 ± 0.07 | 33 | 67 |
| 40 | > 40 | - | - |
| 41 | > 40 | - | - |
| 42 | > 4 | - | - |
| 43 | > 40 | - | - |
| 44 | 27 ± 3 | - | - |
| 45 | > 40 | - | - |
Assembly assay contained 10 µM tubulin.
Colchicine binding assay contained 1 µM tubulin and 5 µM [3H]colchicine.
Furthermore, results from a pharmacokinetic study in a mice model revealed that the 6,7-methylenedioxy moiety of CHM-1-P-Na is metabolized to an ortho-quinone (D, Figure 4, unpublished data). It is known that the para-quinone moiety in mitomycin C can be subjected to one-electron reduction by NADPH-cytochrome C (P450) reductase to form the corresponding semiquinone radical anion.24 We postulate that similar reduction of the ortho-quinone moiety of D may take place to form the radical anion product E, which may be further metabolized or broken down into more cytotoxic metabolites in hypoxic cells. Because severe hypoxia is a common property of locally advanced solid tumors, this postulate may explain our finding that 6,7-methylenedioxyquinoline analogs (e.g. CHM-1) showed enhanced in vivo activity profiles compared with 6-monosubstituted analogs.
Among the active analogs, 37, which showed potent cytotoxicity and reasonable solubility, was chosen for submission to the National Cancer Institute (NCI, USA) for further screening against 60 human tumor cell lines. In preliminary screening, 37 showed potent selective cytotoxicity against many leukemia cell lines (Figure 5). In addition, it also significantly inhibited the growth of several colon cancer (HCC-2998 and KM-12), CNS cancer (SF-539 and SNB-75), melanoma (SK-MEL-5), and ovarian cancer (IGROV1 and OVCAR-3) cell lines. Based upon these results, 37 appears to be an attractive candidate for further development as a potential anticancer agent in the clinic.
Figure 5.

Dose response curves of 37 against different leukemia cell lines.
In conclusion, a series of 2-aryl-6,7-methylenedioxyquinolin-4-one analogs (1, 36–45) were designed, synthesized, and evaluated for in vitro cytotoxicity. This design combined two structural features: larger heterocyclic aromatic rings at the C-2 position as predicted to be favorable by CoMFA models and a 6,7-methylenedioxy moiety rather than C-6 mono-substituted analog based on the improved cytotoxicity of CHM-1 and the metabolic pathway of CHM-1-P-Na. In our studies, 1 showed selective cytotoxicity against Hep 3B cells, 37 was active against HL-60, HCT-116, Hep 3B and KB-VIN cells, and 39 had potent cytotoxicity against all seven cancer cell lines with IC50 values between 0.07–0.19 µM. A mechanism of action study demonstrated that 1, 37, and 39 also function as antitubulin agents. Compound 37 was further selected for evaluation against 60 human cancer cell lines and was active against several types of cancer cells. The significant in vitro cytotoxicity of 37 and 39 suggested that they can be further developed as anticancer drugs.
Experimental Section
Chemistry
General Experimental Procedures
Reagents and solvents were obtained commercially and used without further purification. Reactions were monitored by thin-layer chromatography, using Merck plates with fluorescent indicator (TLC Silica gel 60 F254). Flash column chromatography was performed on silica gel (Merck Slica gel 60, 40–63 µm) using a mixture of CH2Cl2 and EtOH as eluant. Melting points were determined on a Yanaco MP-500D melting point apparatus and were uncorrected. IR spectra were recorded on Shimadzu IRPrestige-21 spectrophotometers as KBr pellets. 1H NMR spectra were obtained on a Bruker NMR AV 400 spectrometer in DMSO. The following abbreviations are used: s, singlet; d, doublet; t, triplet; m, multiplet; dd, double doublet; td, triple doublet; ddd, double double doublet. EI-MS spectra were measured with an HP 5995 GC–MS instrument. ESI-MS spectra were measured with a Bruker HCT ultra PTM Discovery system (Proteineer fc, UltiMate 3000). Elemental analyses (C, H, and N) were performed on a Perkin-Elmer 2400 Series II CHNS/O analyzer, and the results were within ±0.4% of the calculated values.
Preparation of arylcarbonyl chlorides (14–24)
Arylcarboxylic acids (3–13) were suspended in dry toluene (150 mL) at 20 ± 2 °C. Oxalyl chloride (2.2 eq) was added dropwise. The reaction mixtures were stirred for 30 min at 20 ± 2 °C, then DMF (2 drops) was added. The mixtures were stirred for 6 h, and then evaporated to dryness. The residues were washed with petroleum ether and used directly in the next step.
Preparation of carboxamides (25–35, 55)
Into solutions of 14–24 (5.1 mmol) in 200 mL of dry toluene were added triethylamine (4 mL) and 2-amino-4,5-methylenedioxy acetophenone (2) (5 mmol). The mixtures were stirred at 20 ± 2 °C for 24 h, then evaporated. The residues were washed with acetone and EtOH, and then recrystallized from acetone or EtOH to form the pure carboxamides.
N-(6-Acetyl-1,3-benzodioxol-5-yl)-1-benzothiophene-3-carboxamide (25)
Obtained as a pale yellow solid from 2-amino-4,5-methylenedioxyacetophenone (2) and benzo[b]thiophene-3-carbonyl chloride (14). mp 213–214 °C; MS (ESI) 340 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.63 (3H, s), 6.18 (2H, s), 7.43–7.53 (2H, m), 7.66 (1H, s), 8.10 (1H, dd, J = 1.2, 7.2 Hz), 8.30 (1H, s), 8.47 (1H, dd, J = 1.2, 7.2 Hz), 8.53 (1H, s), 12.75 (1H, s). IR (KBr): 1638, 1668 (C=O) cm−1.
N-(6-Acetyl-1,3-benzodioxol-5-yl)-1-benzothiophene-2-carboxamide (26)
Obtained as a grayish white solid from 2 and benzo[b]thiophene-2-carbonyl chloride (15). mp 233–235 °C; MS (ESI) 340 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.69 (3H, s), 6.19 (2H, s), 7.46–7.57 (2H, m), 7.71 (1H, s), 8.09–8.13 (2H, m), 8.14 (1H, s), 8.25 (1H, s), 13.11 (1H, s). IR (KBr): 1640, 1655 (C=O) cm−1.
N-(6-Acetyl-1,3-benzodioxol-5-yl)-1-benzofuran-3-carboxamide (27)
Obtained as a pale yellow solid from 2 and benzo[b]furan-3-carbonyl chloride (16). mp 144–145 °C; MS (ESI) 324 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.63 (3H, s), 6.19 (2H, s), 7.41–7.50 (2H, m), 7.68 (1H, s), 7.75 (1H, dd, J = 1.6, 6.8 Hz), 8.15 (1H, dd, J = 2.0, 8.8 Hz), 8.27 (1H, s), 8.71 (1H, s), 12.63 (1H,s). IR (KBr): 1635, 1677 (C=O) cm−1.
N-(6-Acetyl-1,3-benzodioxol-5-yl)-1-benzofuran-2-carboxamide (28)
Obtained as a grayish white solid from 2 and benzo[b]thiophene-2-carbonyl chloride (17). mp 184–185 °C; MS (ESI) 324 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.69 (3H, s), 6.20 (2H, s), 7.40 (1H, t, J = 7.6 Hz), 7.55 (1H, td, J = 1.2, 7.8 Hz), 7.72–7.78 (3H, m), 7.84 (1H, d, J = 8.0 Hz), 8.36 (1H, s) 13.21 (1H, s). IR (KBr): 1667, 1682 (C=O) cm−1.
N-(6-Acetyl-1,3-benzodioxol-5-yl)naphthalene-1-carboxamide (29)
Obtained as a grayish white solid from 2 and naphthalene-1-carbonyl chloride (18). mp 143–144 °C; MS (ESI) 334 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.59 (3H, s), 6.20 (2H, s), 7.60–7.68 (4H, m), 7.87 (1H, d, J = 7.2 Hz), 8.05–8.07 (1H, m), 8.15 (1H, d, J = 8.0 Hz), 8.33–8.38 (2H, m), 12.52 (1H, s). IR (KBr): 1647, 1672 (C=O) cm−1.
N-(6-Acetyl-1,3-benzodioxol-5-yl)naphthalene-2-carboxamide (30)
Obtained as a pale yellow solid from 2 and naphthalene-2-carbonyl chloride (19). mp 172–173 °C; MS (ESI) 334 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.68 (3H, s), 6.19 (2H, s), 7.64–7.72 (3H, m), 7.99–8.06 (2H, m), 8.15 (2H, d, J = 8.8 Hz), 8.42 (1H, s), 8.58 (1H, s), 13.09 (1H, s). IR (KBr): 1636, 1670 (C=O) cm−1.
N-(6-Acetyl-1,3-benzodioxol-5-yl)quinoline-4-carboxamide (31)
Obtained as a grayish white solid from 2 and quinoline-4-carbonyl chloride (20). mp 166–167 °C; MS (ESI) 335 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.59 (3H, s), 6.21 (2H, s), 7.69 (1H, s), 7.72 (1H, ddd, J = 1.2, 7.2 Hz), 7.81 (1H, d, J = 4.4 Hz), 7.87 (1H, ddd, J = 1.2, 7.4 Hz), 8.15 (1H, d, J = 8.4 Hz), 8.24 (1H, s), 8.31 (1H, d, J = 8.4 Hz), 9.09 (1H, d, J = 4.2 Hz), 12.48 (1H, s). IR (KBr): 1645, 1680 (C=O) cm−1.
N-(6-Acetyl-1,3-benzodioxol-5-yl)quinoline-3-carboxamide (32)
Obtained as a pale yellow solid from 2 and quinoline-3-carbonyl chloride (21). mp 215–216 °C; MS (ESI) 335 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.66 (3H, s), 6.21 (2H, s), 7.73 (1H, s), 7.76 (1H, ddd, J = 1.2, 7.4 Hz), 7.94 (1H, ddd, J = 1.6, 6.8 Hz), 8.15 (1H, d, J = 8.4 Hz), 8.23 (1H, d, J = 7.2 Hz), 8.34 (1H, s), 8.92 (1H, s), 9.38 (1H, s), 12.05 (1H, s). IR (KBr): 1639, 1670 (C=O) cm−1.
N-(6-Acetyl-1,3-benzodioxol-5-yl)quinoline-2-carboxamide (33)
Obtained as a yellow solid from 2 and quinoline-2-carbonyl chloride (22). mp 210–211 °C; MS (ESI) 335 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.69 (3H, s), 6.21 (2H, s), 7.73 (1H, s), 7.79 (1H, ddd, J = 1.2, 7.2 Hz), 7.95 (1H, ddd, J = 1.6, 7.6 Hz), 8.15 (1H, d, J = 7.2 Hz), 8.22 (1H, d, J = 8.0 Hz), 8.28 (1H, d, J = 8.8 Hz), 8.55 (1H, s), 8.65–8.68 (1H, d, J = 8.8). IR (KBr): 1647, 1670 (C=O) cm−1.
N-(6-Acetyl-1,3-benzodioxol-5-yl)quinoline-5-carboxamide (34)
Obtained as a yellow solid from 2 and quinoline-5-carbonyl chloride (23). mp 210–211 °C; MS (ESI) 335 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.61 (3H, s), 6.21 (2H, s), 7.70 (1H, s), 7.82 (1H, dd, J = 4.4, 8.8 Hz), 8.03 (1H, t, J = 7.8 Hz), 8.11 (1H, d, J = 7.2 Hz), 8.30–8.35 (2H, m), 9.03 (1H, d, J = 8.4 Hz), 9.13 (1H, d, J = 4.4 Hz), 12.59 (1H, s). IR (KBr): 1636, 1672 (C=O) cm−1.
N-(6-Acetyl-1,3-benzodioxol-5-yl)anthracene-1-carboxamide (35)
Obtained as a pale yellow solid from 2 and anthracene-1-carbonyl chloride (24). mp 206–207 °C; MS (ESI) 384 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.59 (3H, s), 6.21 (2H, s), 7.55–7.61 (2H, m), 7.62–7.67 (1H, m), 7.70 (1H, s), 7.90 (1H, d, J = 6.8 Hz), 8.13–8.16 (2H, m), 8.32 (1H, d, J = 8.4 Hz), 8.46 (1H, s), 8.73 (1H, s), 9.03 (1H, s), 12.60 (1H, s). IR (KBr): 1643, 1674 (C=O) cm−1.
N-(6-Acetyl-1,3-benzodioxol-5-yl)-2-(naphthalen-1-yl)acetamide (55)
Obtained as a white solid from 2 and 1-naphthylmethyl chloride (54). mp 89–90 °C; MS (ESI) 384 m/z [M+H]+. 1H NMR (400 MHz, DMSO-d6, δ): 2.53 (3H, s), 4.21 (2H, s) 6.13 (2H, s), 7.50–7.61 (5H, m), 7.90 (1H, d, J = 8.0 Hz), 7.95 (1H, dd, J = 2.0, 7.2 Hz), 8.03 (1H, dd, J = 1.2, 8.0 Hz), 8.15 (1H, s), 11.89 (1H, s). IR (KBr): 1641, 1684 (C=O) cm−1.
Preparation of 2-aryl-6,7-methylenedioxyquinolin-4-ones (1, 37–44)
Method A
Into a suspension of 25, 27–34 (2.95 mmol) in t-butyl alcohol (100 mL) was added potassium t-butoxide (1.66 g, 14.7 mmol). The mixture was refluxed under argon for 12 h, cooled and poured into a 10% ammonium chloride solution (100 mL). The solid precipitate was collected and washed with EtOH. The crude product was purified by flash chromatography (silica gel, CH2Cl2:EtOH 16:1–10:1).
2-(3-Benzo[b]thienyl)-6,7-methylenedioxyquinolin-4-one (1)
35% yield from 25 as a white solid. mp >330 °C; MS (ESI) 322 m/z [M+H]+. 1H NMR (DMSO-d6, δ): 6.15 (2H, s), 6.19 (1H, s), 7.11 (1H, s), 7.43 (1H, s), 7.47–7.54 (2H, m), 7.93 (1H, d, J = 7.6 Hz), 8.14 (1H, d, J = 7.6 Hz), 8.24 (1H, s), 11.80 (1H, s). IR (KBr): 1616 (C=O) cm−1. Anal. Calcd for C18H11NO3S: C, 67.00; H, 3.48; N, 4.25. Found: C, 67.28; H, 3.45; N, 4.36.
2-(3-Benzo[b]furyl)-6,7-methylenedioxyquinolin-4-one (37)
17% yield from 27 as a pale yellow solid. mp >315 °C; MS (ESI) m/z 306 [M+H]+. 1H NMR (DMSO-d6, δ): 6.12 (2H, s), 6.49 (1H, s), 7.13 (1H, s), 7.36–7.45 (3H, m), 7.69 (1H, d, J = 8.0 Hz), 8.14 (1H, s), 8.52 (1H, s). IR (KBr): 1626 (C=O) cm−1. Anal. Calcd for C18H11NO4: C, 70.82; H, 3.63; N, 4.59. Found: C, 70.52; H, 3.95; N, 4.21.
2-(2-Benzo[b]furyl)-6,7-methylenedioxyquinolin-4-one (38)
28% yield from 28 as a grayish white solid. mp >320 °C; MS (EI, 70 eV) m/z 305 [M]+. 1H NMR (DMSO-d6, δ): 6.19 (2H, s), 6.75 (1H, s), 7.30 (1H, s), 7.35 (1H, t, J = 7.6 Hz), 7.40 (1H, s), 7.45 (1H, t, J = 7.6 Hz), 7.71–7.79 (3H, m). IR (KBr): 1630 (C=O) cm−1. Anal. Calcd for C18H11NO4: C, 70.82; H, 3.63; N, 4.59. Found: C, 70.62; H, 3.84; N, 4.24.
2-(1-Naphthalenyl)-6,7-methylenedioxyquinolin-4-one (39)
52% yield from 29 as a grayish white solid. mp >350 °C; MS (ESI) m/z 316 [M+H]+. 1H NMR (DMSO-d6, δ): 6.08 (1H, s), 6.15 (2H, s), 7.03 (1H, s), 7.46 (1H, s), 7.56–7.63 (2H, m), 7.63–7.70 (2H, m), 7.83 (1H, d, J = 7.6 Hz), 8.06 (1H, d, J = 7.6 Hz), 8.11 (1H, d, J = 7.6 Hz), 11.90 (1H, s). IR (KBr): 1653 (C=O) cm−1. Anal. Calcd for C20H13NO3: C, 76.18; H, 4.16; N, 4.44. Found: C, 75.60; H, 3.94; N, 4.29.
2-(2-Naphthalenyl)-6,7-methylenedioxyquinolin-4-one (40)
48% yield from 30 as a white solid. mp >330 °C; MS (ESI) m/z 316 [M+H]+. 1H NMR (DMSO-d6, δ): 6.16 (2H, s), 6.49 (1H, s), 7.25 (1H, s), 7.42 (1H, s), 7.59–7.64 (2H, m), 7.88–7.95 (1H, m), 8.00–8.03 (1H, m), 8.06–8.12 (2H, m), 8.42 (1H, s). IR (KBr): 1616 (C=O) cm−1. Anal. Calcd for C20H13NO3: C, 76.18; H, 4.16; N, 4.44. Found: C, 76.04; H, 4.28; N, 4.28.
2-(4-Quinolinyl)-6,7-methylenedioxyquinolin-4-one (41)
54% yield from 31 as a pale yellow solid. mp >320 °C; MS (ESI) m/z 317 [M+H]+. 1H NMR (DMSO-d6, δ): 6.12–6.29 (3H, m), 7.06 (1H, s), 7.47 (1H, s), 7.65–7.70 (2H, m), 7.86 (1H, t, J = 7.6 Hz), 7.93 (1H, d, J = 8.4 Hz), 8.15 (1H, d, J = 8.0 Hz), 9.03 (1H, s). IR (KBr): 1618 (C=O) cm−1. Anal. Calcd for C19H12N2O3: C, 72.15; H, 3.82; N, 8.86. Found: C, 72.35; H, 4.03; N, 8.60.
2-(3-Quinolinyl)-6,7-methylenedioxyquinolin-4-one (42)
65% yield from 32 as a grayish white solid, mp >320 °C; MS (ESI) m/z 317 [M+H]+. 1H NMR (DMSO-d6, δ): 6.19 (2H, s), 6.62 (1H, s), 7.23 (1H, s), 7.44 (1H, s), 7.73 (1H, t, J = 7.6 Hz), 7.87 (1H, t, J = 7.6 Hz), 8.11–8.17 (2H, m), 8.88 (1H, s), 9.38 (1H, s). IR (KBr): 1618 (C=O) cm−1. Anal. Calcd for C19H12N2O3: C, 72.15; H, 3.82; N, 8.86. Found: C, 71.86; H, 3.76; N, 8.63.
2-(2-Quinolinyl)-6,7-methylenedioxyquinolin-4-one (43)
48% yield from 33 as a pale yellow solid. mp 345–347 °C; MS (EI, 70 eV) m/z 316 [M]+. 1H NMR (DMSO-d6, δ): 6.18 (2H, s), 6.98 (1H, s), 7.43 (1H, s), 7.63 (1H, s), 7.73 (1H, t, J = 7.6 Hz), 7.91 (1H, t, J = 7.6 Hz), 8.11 (1H, d, J = 8.0 Hz), 8.27 (1H, d, J = 8.4 Hz), 8.32 (1H, d, J = 8.8 Hz), 8.60 (1H, d, J = 8.8 Hz), 11.85 (1H, s). IR (KBr): 1611 (C=O) cm−1. Anal. Calcd for C19H12N2O3: C, 72.15; H, 3.82; N, 8.86. Found: C, 71.76; H, 3.88; N, 8.46.
2-(5-Quinolinyl)-6,7-methylenedioxyquinolin-4-one (44)
47% yield from 34 as a pale yellow solid. mp >330 °C; MS (ESI) m/z 317 [M+H]+. 1H NMR (DMSO-d6, δ): 6.03–6.19 (3H, m), 7.03 (1H, s), 7.47 (1H, s), 7.60 (1H, dd, J = 4.0, 8.4 Hz), 7.81 (1H, d, J = 6.8 Hz), 7.91 (1H, t, J = 7.8 Hz), 8.20 (1H, d, J = 8.4 Hz), 8.30 (1H, d, J = 6.4 Hz), 8.99 (1H, s), 11.91 (1H, s). IR (KBr): 1614 (C=O) cm−1. Anal. Calcd for C19H12N2O3: C, 72.15; H, 3.82; N, 8.86. Found: C, 72.08; H, 3.94; N, 8.66.
Preparation of 2-aryl-6,7-methylenedioxyquinolin-4-ones (1, 36, 39, 45) and 4-methyl-6,7-methylenedioxy-3-(1-naphthyl)quinolin-2-one (56)
Method B
Following the same procedure described in method A, except for the use of 1,4-dioxane as solvent in place of t-butylalcohol, and the use of NaOH in place of potassium t-butoxide. Compounds 1 and 39 were confirmed by comparison of mp and TLC with those of a sample obtained from method A.
2-(3-Benzo[b]thienyl)-6,7-methylenedioxyquinolin-4-one (1)
40% yield from 25 as a pale yellow solid.
2-(2-Benzo[b]thienyl)-6,7-methylenedioxyquinolin-4-one (36)
74% yield from 26 as a white solid. mp >350 °C; MS (ESI) m/z 322 [M+H]+. 1H NMR (DMSO-d6, δ): 6.20 (2H, s), 7.21 (1H, s), 7.39–7.45 (3H, m), 7.96–8.16 (3H, m), 11.55 (1H, s). IR (KBr): 1616 (C=O) cm−1. Anal. Calcd for C18H11NO3S: C, 67.00; H, 3.48; N, 4.25. Found: C, 67.21; H, 3.47; N, 4.33.
2-(1-Naphthalenyl)-6,7-methylenedioxyquinolin-4-one (39)
58% yield from 29 as a grayish white solid.
2-(1-Anthracenyl)-6,7-methylenedioxyquinolin-4-one (45)
48% yield from 35 as a yellow solid. mp >320 °C; MS (ESI) m/z 366 [M+H]+. 1H NMR (DMSO-d6, δ): 6.15 (3H, s), 7.03 (1H, s), 7.43–7.70 (5H, m), 8.08 (2H, t, J = 7.6 Hz), 8.25 (1H, d, J = 8.4 Hz), 8.47 (1H, s), 8.70 (1H, s), 11.98 (1H, s). IR (KBr): 1632 (C=O) cm−1. Anal. Calcd for C24H15NO3: C, 78.89; H, 4.14; N, 3.83. Found: C, 78.24; H, 4.40; N, 3.37.
4-Methyl-6,7-methylenedioxy-3-(1-naphthyl)quinolin-2-one (56)
61% yield from 55 as a white solid. mp >260 °C; MS (EI, 70 eV) m/z 329 [M]+. 1H NMR (DMSO-d6, δ): 2.02 (3H, s), 6.13 (2H, s), 6.91 (1H, s), 7.29–7.32 (2H, m), 7.39–7.47 (2H, m), 7.52 (1H, ddd, J = 1.6, 6.4 Hz), 7.58 (1H, t, J = 8.0 Hz), 7.93–8.01 (2H, m), 11.84 (1H, s). IR (KBr): 1632 (C=O) cm−1. Anal. Calcd for C21H15NO3: C, 76.58; H, 4.59; N, 4.25. Found: C, 75.88; H, 5.07; N, 4.06.
Deacetylation of compounds 25, 26, 28–32 and 34
Into a suspension of compound (2.95 mol) in dry toluene (150 mL) was added potassium t-butoxide (1.66 g, 14.75 mol). The mixture was heated under argon at 90 °C for 72 h, and then concentrated and neutralized with 20% HOAc The resulting solid precipitate was collected and purified by flash chromatography (silica gel, CH2Cl2-EtOH) to afford 46–53.
N-(1,3-Benzodioxol-5-yl)-1-benzothiophene-3-carboxamide (46)
28% yield from 25 as a white solid. mp 188−189 °C; MS (EI, 70 eV) m/z 297 [M]+. 1H NMR (DMSO-d6, δ): 6.01 (2H, s), 6.90 (1H, d, J = 8.4 Hz), 7.17 (1H, dd, J = 2.0, 8.4 Hz), 7.40−7.51 (3H, m), 8.06 (1H, dd, J = 1.2, 7.2 Hz), 8.37 (1H, dd, J = 0.8, 8.0 Hz), 8.46 (1H, s), 10.29 (1H, s). IR (KBr): 1647 (C=O) cm−1. Anal. Calcd for C16H11NO3S: C, 64.63; H, 3.73; N, 4.71. Found: C, 64.32; H, 3.94; N, 4.50.
N-(1,3-Benzodioxol-5-yl)-1-benzothiophene-2-carboxamide (47)
36% yield from 26 as a grayish white solid. mp 171−173 °C; MS (EI, 70 eV) m/z 297 [M]+. 1H NMR (DMSO-d6, δ): 6.01 (2H, s), 6.91 (1H, d, J = 8.4 Hz), 7.15 (1H, dd, J = 2.0, 8.4 Hz), 7.38 (1H, d, J = 2.4 Hz), 7.41−7.54 (2H, m), 7.96–8.05 (2H, m), 8.28 (1H, s), 10.41 (1H, s). IR (KBr): 1634 (C=O) cm−1. Anal. Calcd for C16H11NO3S: C, 64.63; H, 3.73; N, 4.71. Found: C, 64.28; H, 3.98; N, 4.46.
N-(1,3-Benzodioxol-5-yl)-1-benzofuran-2-carboxamide (48)
28% yield from 28 as a yellow solid. mp 156-158 °C; MS (EI, 70 eV) m/z 281 [M]+. 1H NMR (DMSO-d6, δ): 6.03 (2H, s), 6.93 (1H, d, J = 8.4 Hz), 7.27 (1H, dd, J = 2.0, 8.4 Hz), 7.37 (1H, t, J = 7.6 Hz), 7.47 (1H, d, J = 2.0 Hz), 7.51 (1H, ddd, J = 2.0, 8.4 Hz), 7.71–7.75 (2H, m), 7.82 (1H, d, J = 8.0 Hz), 10.49 (1H, s). IR (KBr): 1668 (C=O) cm−1. Anal. Calcd for C16H11NO4: C, 68.32; H, 3.94; N, 4.98. Found: C, 68.22; H, 4.14; N, 4.22.
N-(1,3-Benzodioxol-5-yl)naphthalene-1-carboxamide (49)
32% yield from 29 as a white solid. mp 204–206 °C; MS (EI, 70 eV) m/z 291 [M]+. 1H NMR (DMSO-d6, δ): 6.02 (2H, s), 6.91 (1H, d, J = 8.4 Hz), 7.22 (1H, dd, J = 2.0, 8.4 Hz), 7.51 (1H, d, J = 2.0 Hz), 7.56–7.63 (3H, m), 7.72 (1H, d, J = 6.4 Hz), 7.99–8.03 (1H, m), 8.06 (1H, d, J = 8.4 Hz), 8.16–8.20 (1H, m), 10.46 (1H, s). IR (KBr): 1643 (C=O) cm−1. Anal. Calcd for C18H13NO3: C, 74.22; H, 4.50; N, 4.81. Found: C, 73.83; H, 4.82; N, 4.26.
N-(1,3-Benzodioxol-5-yl)naphthalene-2-carboxamide (50)
21% yield from 30 as a pale yellow solid. mp 184–185 °C; MS (EI, 70 eV) m/z 291 [M]+. 1H NMR (DMSO-d6, δ): 6.02 (2H, s), 6.94 (1H, d, J = 8.4 Hz), 7.24 (1H, dd, J = 2.0, 8.4 Hz), 7.50 (1H, d, J = 2.0 Hz), 7.60–7.68 (2H, m), 7.98–8.15 (4H, m), 8.56 (1H, s), 10.38 (1H, s). IR (KBr): 1636 (C=O) cm−1. Anal. Calcd for C18H13NO3: C, 74.22; H, 4.50; N, 4.81. Found: C, 74.19; H, 4.62; N, 4.56.
N-(1,3-Benzodioxol-5-yl)quinoline-4-carboxamide (51)
31% yield from 31 as a white solid. mp 167–168 °C; MS (EI, 70 eV) m/z 292 [M]+. 1H NMR (DMSO-d6, δ): 6.02 (2H, s), 6.93 (1H, d, J = 8.4 Hz), 7.16 (1H, dd, J = 1.6, 8.4 Hz), 7.43 (1H, d, J = 2.0 Hz), 7.66–7.72 (2H, m), 7.84 (1H, t, J = 8.0 Hz), 8.11 (2H, d, J = 8.8 Hz), 9.01 (1H, s), 10.65 (1H, s). IR (KBr): 1636 (C=O) cm−1. Anal. Calcd for C17H12N2O3: C, 69.86; H, 4.14; N, 9.58. Found: C, 69.65; H, 4.32; N, 9.34.
N-(1,3-Benzodioxol-5-yl)quinoline-3-carboxamide (52)
23% yield from 32 as a yellow solid. mp 254–256 °C; MS (EI, 70 eV) m/z 292 [M]+. 1H NMR (DMSO-d6, δ): 6.05 (2H, s), 6.95 (1H, d, J = 8.4 Hz), 7.23 (1H, dd, J = 2.0, 8.4 Hz), 7.49 (1H, d, J = 2.0 Hz), 7.74 (1H, ddd, J = 1.2, 8.0 Hz), 7.90 (1H, ddd, J = 1.6, 8.4 Hz), 8.10–8.18 (2H, m), 8.92 (1H, s), 9.33 (1H, s), 10.56 (1H, s). IR (KBr): 1659 (C=O) cm−1. Anal. Calcd for C17H12N2O3: C, 69.86; H, 4.14; N, 9.58. Found: C, 69.73; H, 4.35; N, 9.42.
N-(1,3-Benzodioxol-5-yl)quinoline-5-carboxamide (53)
20% yield from 34 as a white solid. mp 202–203 °C; MS (EI, 70 eV) m/z 292 [M]+. 1H NMR (DMSO-d6, δ): 6.02 (2H, s), 6.95 (1H, d, J = 8.4 Hz), 7.21 (1H, dd, J = 2.0, 8.0 Hz), 7.50 (1H, d, J = 2.0 Hz), 7.63 (1H, dd, J = 4.0, 8.4 Hz), 7.83–7.90 (2H, m), 8.16–8.20 (1H, m), 8.63 (1H, d, J = 8.0 Hz), 8.98 (1H, dd, J = 1.6, 4.0 Hz), 10.58 (1H, s). IR (KBr): 1643 (C=O) cm−1. Anal. Calcd for C17H12N2O3: C, 69.86; H, 4.14; N, 9.58. Found: C, 69.68; H, 4.33; N, 9.52.
Biological Evaluations
MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays 25,26
HL-60, HCT-116 and Hep 3B cells were treated with tested compounds for the indicated periods. After treatment, cells were washed once with PBS and incubated with MTT (Sigma, St. Louis, MO, USA) for 2 h. The formazan precipitate was dissolved in 150 µL of DMSO, and the absorbance was measured with an ELISA reader at 570 nm.
SRB assays 27
A549, KB, KB-VIN and DU145 cells were treated with tested compounds for the indicated periods. After additional incubation with DMSO or compounds, cells were fixed with 10% TCA, and SRB at 0.4% (w/v) in 1% acetic acid was added to stain cells. Unbound SRB was washed out with 1% acetic acid, and SRB bound to the cells was solubilized with 10 mM Trizma base. Absorbance was read at 515 nm.
Tubulin assays
Tubulin assembly was measured by turbidimetry as described in detail previously.28 Assay mixtures contained 1.0 mg/mL (10 µM) tubulin and varying drug concentrations and were preincubated 15 min at 30 °C in the absence of GTP. The samples were placed on ice, and 0.4 mM GTP was added. Reaction mixtures were transferred to cuvettes held at 0 °C, and turbidity development was followed for 20 min at 30 °C after a rapid temperature jump. Drug concentrations that inhibited increase in turbidity by 50% relative to a control sample were determined.
Inhibition of the binding of [3H]colchicine to tubulin was measured as described in detail previously.29 Incubation of 1.0 µM tubulin with 5.0 µM [3H]colchicine and either 1.0 or 5.0 µM inhibitor was for 10 min at 37 °C. At this time point, approximately 40–60% of maximum colchicine binding occurs.
Acknowledgments
We would like to acknowledge special appreciation and esteem for support received from Mr. Wen-Chih Chang, who passed away because of nasopharyngeal carcinoma before this paper was completed. The investigation was supported by research grants from the National Science Council of the Republic of China (NSC 96-2323-B-039-001 and NSC 97-2323-B-039-001) awarded to S. C. Kuo. Thanks are also due to support (in part) by grant CA 17625 from the National Cancer Institute, NIH (K. H. Lee).
References
- 1.Wordeman L, Mitchison TJ. Dynamics of microtubule assembly in vivo. In: Hyams J, Lloyd C, editors. In Microtubules. Wiley-Liss, Inc.; New York: 1994. [Google Scholar]
- 2.Rowinsky EK, Donehower RC. The clinical pharmacology and use of antimicrotubule agents in cancer chemotherapeutics. Pharmacol. Ther. 1991;52:35–84. doi: 10.1016/0163-7258(91)90086-2. [DOI] [PubMed] [Google Scholar]
- 3.Verweij J, Clavel M, Chevalier B. Paclitaxel (taxol) and docetaxel (taxotere): not simply two of a kind. Ann. Oncol. 1994;5:495–505. doi: 10.1093/oxfordjournals.annonc.a058903. [DOI] [PubMed] [Google Scholar]
- 4.Lin CM, Ho HH, Pettit GR. Antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the ,echanism of their inhibition of the binding of colchicine to tubulin. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 5.Pettit GR, Singh SB, Boyd MR, Hamel E, Pettit RK, Schmidt JM, Hogan F. Antineoplastic agents. 291. Isolation and synthesis of combretastatins A-4, A-5, and A-6(1a) J. Med. Chem. 1995;38:1666–1672. doi: 10.1021/jm00010a011. [DOI] [PubMed] [Google Scholar]
- 6.Pettit GR, Temple C, Jr, Narayanan VL, Varma R, Simpson MJ, Boyd MR, Rener GA, Bansal N. Antineoplastic agents 322. Synthesis of combretastatin A-4 prodrugs. Anticancer Drug Des. 1995;10:299–309. [PubMed] [Google Scholar]
- 7.Vincent L, Kermani P, Young LM, Cheng J, Zhang F, Shido K, Lam G, Bompais-Vincent H, Zhu Z, Hicklin DJ, Bohlen P, Chaplin DJ, May C, Rafii S. Combretastatin A4 phosphate induces rapid regression of tumor neovessels and growth through interference with vascular endothelial-cadherin signaling. J. Clin. Invest. 2005;115:2992–3006. doi: 10.1172/JCI24586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuo SC, Lee HZ, Juang JP, Lin YT, Wu TS, Chang JJ, Lednicer D, Paull KD, Lin CM, Hamel E, et al. Synthesis and cytotoxicity of 1,6,7,8-substituted 2-(4'-substituted phenyl)-4-quinolones and related compounds: identification as antimitotic agents interacting with tubulin. J. Med. Chem. 1993;36:1146–1156. doi: 10.1021/jm00061a005. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Wang HK, Kuo SC, Wu TS, Lednicer D, Lin CM, Hamel E, Lee KH. Antitumor agents. 150. 2',3',4',5',5,6,7-Substituted 2-phenyl-4-quinolones and related compounds: their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J. Med. Chem. 1994;37:1126–1135. doi: 10.1021/jm00034a010. [DOI] [PubMed] [Google Scholar]
- 10.Li L, Wang HK, Kuo SC, Wu TS, Mauger A, Lin CM, Hamel E, Lee KH. Antitumor agents. 155. Synthesis and biological evaluation of 3',6,7-substituted 2-phenyl-4-quinolones as antimicrotubule agents. J. Med. Chem. 1994;37:3400–3407. doi: 10.1021/jm00046a025. [DOI] [PubMed] [Google Scholar]
- 11.Xia Y, Yang ZY, Xia P, Bastow KF, Tachibana Y, Kuo SC, Hamel E, Hackl T, Lee KH. Antitumor agents. 181. Synthesis and biological evaluation of 6,7,2',3',4'-substituted-1,2,3,4-tetrahydro-2-phenyl-4-quinolones as a new class of antimitotic antitumor agents. J. Med. Chem. 1998;41:1155–1162. doi: 10.1021/jm9707479. [DOI] [PubMed] [Google Scholar]
- 12.Xia Y, Yang ZY, Xia P, Hackl T, Hamel E, Mauger A, Wu JH, Lee KH. Antitumor agents. 211. Fluorinated 2-phenyl-4-quinolone derivatives as antimitotic antitumor agents. J. Med. Chem. 2001;44:3932–3936. doi: 10.1021/jm0101085. [DOI] [PubMed] [Google Scholar]
- 13.Xia Y, Yang ZY, Xia P, Bastow KF, Nakanishi Y, Nampoothiri P, Hamel E, Brossi A, Lee KH. Antitumor agents. Part 226. Synthesis and cytotoxicity of 2-phenyl-4-quinolone acetic acids and their esters. Bioorg. Med. Chem. Lett. 2003;13:2891–2893. doi: 10.1016/s0960-894x(03)00624-3. [DOI] [PubMed] [Google Scholar]
- 14.Lai YY, Huang LJ, Lee KH, Xiao Z, Bastow KF, Yamori T, Kuo SC. Synthesis and biological relationships of 3',6-substituted 2-phenyl-4-quinolone-3-carboxylic acid derivatives as antimitotic agents. Bioorg. Med. Chem. 2005;13:265–275. doi: 10.1016/j.bmc.2004.09.041. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura S, Kozuka M, Bastow KF, Tokuda H, Nishino H, Suzuki M, Tatsuzaki J, Morris Natschke SL, Kuo SC, Lee KH. Cancer preventive agents, part 2: Synthesis and evaluation of 2-phenyl-4-quinolone and 9-oxo-9,10-dihydroacridine derivatives as novel antitumor promoters. Bioorg. Med. Chem. 2005;13:4396–4401. doi: 10.1016/j.bmc.2005.04.078. [DOI] [PubMed] [Google Scholar]
- 16.Chen K, Kuo SC, Hsieh MC, Mauger A, Lin CM, Hamel E, Lee KH. Antitumor agents. 174. 2',3',4',5,6,7-Substituted 2-phenyl-1,8-naphthyridin-4-ones: their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J. Med. Chem. 1997;40:2266–2275. doi: 10.1021/jm960858s. [DOI] [PubMed] [Google Scholar]
- 17.Chen K, Kuo SC, Hsieh MC, Mauger A, Lin CM, Hamel E, Lee KH. Antitumor agents. 178. Synthesis and biological evaluation of substituted 2-aryl-1,8-naphthyridin-4(1H)-ones as antitumor agents that inhibit tubulin polymerization. J. Med. Chem. 1997;40:3049–3056. doi: 10.1021/jm970146h. [DOI] [PubMed] [Google Scholar]
- 18.Zhang SX, Bastow KF, Tachibana Y, Kuo SC, Hamel E, Mauger A, Narayanan VL, Lee KH. Antitumor agents. 196. Substituted 2-thienyl-1,8-naphthyridin-4-ones: their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J. Med. Chem. 1999;42:4081–4087. doi: 10.1021/jm990208z. [DOI] [PubMed] [Google Scholar]
- 19.Hour MJ, Huang LJ, Kuo SC, Xia Y, Bastow K, Nakanishi Y, Hamel E, Lee KH. 6-Alkylamino- and 2,3-dihydro-3'-methoxy-2-phenyl-4-quinazolinones and related compounds: their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J. Med. Chem. 2000;43:4479–4487. doi: 10.1021/jm000151c. [DOI] [PubMed] [Google Scholar]
- 20.Xia Y, Yang ZY, Hour MJ, Kuo SC, Xia P, Bastow KF, Nakanishi Y, Nampoothiri P, Hackl T, Hamel E, Lee KH. Antitumor agents. Part 204: Synthesis and biological evaluation of substituted 2-arylquinazolinones. Bioorg. Med. Chem. Lett. 2001;11:1193–1196. doi: 10.1016/s0960-894x(01)00190-1. [DOI] [PubMed] [Google Scholar]
- 21.Zhang SX, Feng J, Kuo SC, Brossi A, Hamel E, Tropsha A, Lee KH. Antitumor agents. 199. Three-dimensional quantitative structure-activity relationship study of the colchicine binding site ligands using comparative molecular field analysis. J. Med. Chem. 2000;43:167–176. doi: 10.1021/jm990333a. [DOI] [PubMed] [Google Scholar]
- 22.Chou LC, Chen CT, Lee JC, Way TD, Huang SM, Teng CM, Yamori T, Wu TS, Sun CM, Chien DS, Huang LJ, Morris-Natschke SL, Qian K, Lee KH, Kuo SC. Synthesis of monosodium phosphate salt of 2-(2-fluorophenyl)-6,7-methylenedioxyquinolin-4-one as a potent anticancer clinical trials candidate. Mol. Pharmacol. 2009 doi: 10.1021/jm901292j. (Submitted) [DOI] [PubMed] [Google Scholar]
- 23.Lee JC, Chou LC, Chen WC, Huang CH, Kuo DH, Huang LJ, Lee KH, Teng CM, Kuo SC, Way TD. CHM-1, a novel microtubule-destabilizing agent exhibits antitumor activity via inducing the expression of SIRT2 in human breast cancer cells. Clin. Cancer Res. 2009 doi: 10.1016/j.cbi.2018.04.007. (Submitted) [DOI] [PubMed] [Google Scholar]
- 24.Belcourt MF, Hodnick WF, Rockwell S, Sartorelli AC. Differential toxicity of mitomycin C and porfiromycin to aerobic and hypoxic Chinese hamster ovary cells over expressing human NADPH: cytochrome C (P-450) reductase. Proc. Natl. Acad. Sci. U.S.A. 1996;93:456–460. doi: 10.1073/pnas.93.1.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen CJ, Hsu MH, Kuo SC, Lai YY, Chung JG, Huang LJ. (2E)-N,N-Dibutyl-3-(4-hydroxy-3-methoxyphenyl)acrylamide induces apoptosis and cell cycle arrest in HL-60 cells. Anticancer Res. 2007;27:343–349. [PubMed] [Google Scholar]
- 26.Hsu MH, Chen CJ, Kuo SC, Chung JG, Lai YY, Teng CM, Pan SL, Huang LJ. 2-(3-Fluorophenyl)-6-methoxyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (YJC-1) induces mitotic phase arrest in A549 cells. Eur. J. Pharmacol. 2007;559:14–20. doi: 10.1016/j.ejphar.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 27.Chen YC, Lu PH, Pan SL, Teng CM, Kuo SC, Lin TP, Ho YF, Huang YC, Guh JH. Quinolone analogue inhibits tubulin polymerization and induces apoptosis via Cdk1-involved signaling pathways. Biochem. Pharmacol. 2007;74:10–19. doi: 10.1016/j.bcp.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 28.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003;38:1–22. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 29.Verdier-Pinard P, Lai J-Y, Yoo H-H, Yu J, Márquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol. Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]