

Abstract
Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) has many beneficial physiological functions ranging from enhancing fatty acid catabolism, improving insulin sensitivity, inhibiting inflammation and increasing oxidative myofibers allowing for improved athletic performance. Thus, given the potential for targeting PPARβ/δ for the prevention and/or treatment of diseases including diabetes, dyslipidemias, metabolic syndrome and cancer, it is critical to clarify the functional role of PPARβ/δ in cell proliferation and associated disorders such as cancer. However, there is considerable controversy whether PPARβ/δ stimulates or inhibits cell proliferation. This review summarizes the literature describing the influence of PPARβ/δ on cell proliferation, with an emphasis toward dissecting the data that give rise to opposing hypotheses. Suggestions are offered to standardize measurements associated with these studies so that interlaboratory comparisons can be accurately assessed.
Keywords: peroxisome proliferator-activated receptor-β/δ, cell proliferation, apoptosis, differentiation, cancer
1. Introduction
Peroxisome proliferator-activated receptor-β/δ (also referred to as PPARβ and PPARδ) is one of three PPARs, including PPARα and PPARγ that are part of the nuclear receptor superfamily of transcription factors. PPARβ/δ can be activated by endogenous compounds, which are thought to act as natural ligands (Fig. 1). For example, PPARβ/δ is activated by μM concentrations of oleic acid and linoleic acid [1]. Additionally, PPARβ/δ may be activated by prostaglandin I2 (PGI2 or prostacyclin) [2] or stable analogues of prostacyclin such as carbaprostacyclin [1]. High affinity synthetic ligands for PPARβ/δ, produced by the pharmaceutical industry, include L165041, GW501516 and GW0742 (Fig. 1); all of which activate PPARβ/δ at concentrations in the low nM - μM range [3, 4]. Regulation of gene expression by PPARβ/δ is mediated by ligand activation of the receptor (reviewed in [5]). Briefly, ligand binding induces a conformational change in protein structure that causes dissociation of co-repressors, heterodimerization with RXRα, recruitment of co-activators and RNA polymerase, remodeling of histones near regions of DNA containing direct repeat 1 motifs (AGGTCANAGGTCA) and culminating with increased transcription of target genes (Fig. 2). In addition to this classic mechanism of gene regulation by transcription factors, PPARβ/δ can also interact directly with the p65 subunit of NFκB leading to inhibition of NFκB-dependent signaling and resultant anti-inflammatory activities (reviewed in [5]). Anti-inflammatory activities of PPARβ/δ may also be mediated through direct interaction with ERK5 or STAT3 [6, 7]. Regulation mediated by direct interactions of PPARβ/δ with other transcription factors is one mechanism by which PPARβ/δ can cause down-regulation of target gene expression. There is also evidence that PPARβ/δ may repress transcription of some target genes through binding to DNA response elements in association with co-repressors [8]. Combined, there are a number of different mechanisms whereby PPARβ/δ can modulate cellular homeostasis including direct up-regulation of target genes and down-regulation of gene expression mediated by other transcription factors (Fig. 2).
Fig. 1.
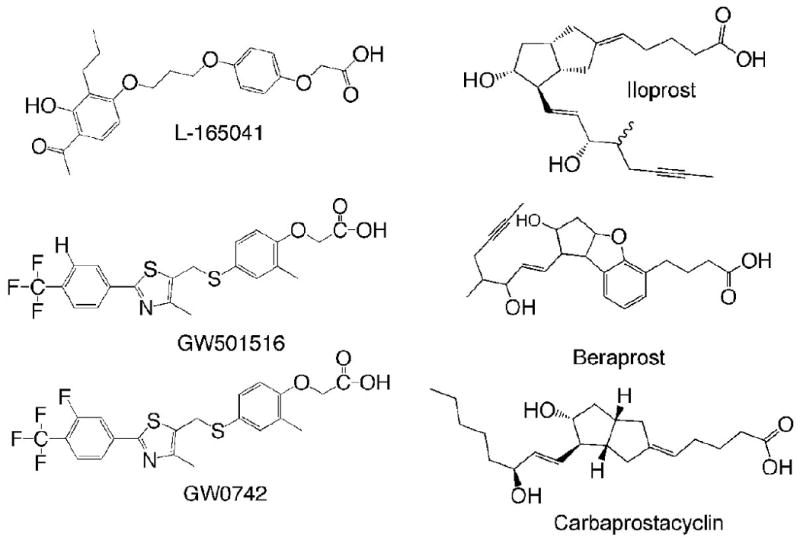
Chemical structure of ligands for PPARβ/δ.
Fig. 2.
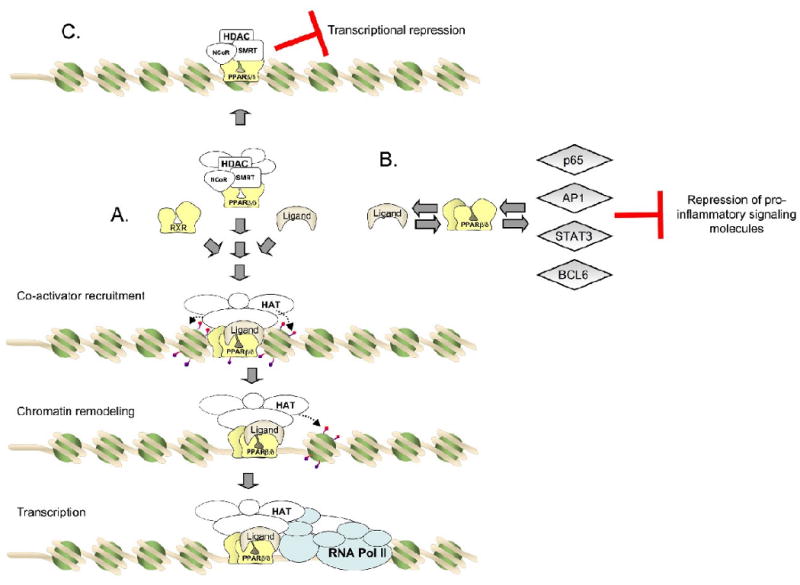
Modulation of gene expression by PPARβ/δ. (A) Transcriptional up-regulation of direct PPARβ/δ target genes. (B) Epigenetic regulation of gene expression by PPARβ/δ. PPARβ/δ can interfere with other transcription factors by physical interactions and inhibit expression of these gene products. This type of inhibition has been demonstrated for a number of inflammatory signaling cascades, including NF-κB, STAT3, BCL6 and AP1. (C) Transcriptional repression
2. Functional roles of PPARβ/δ
In addition to target gene specificity, tissue-specific expression patterns of the PPARs can distinguish their biological functions. In contrast to PPARα and PPARγ that are preferentially expressed in liver and kidney, and adipose tissues, respectively, PPARβ/δ is expressed in most tissues, with varying levels in certain cell types. For example, expression of PPARβ/δ is highest in small intestine, colon and liver where it is found predominantly in the nucleus and can be co-immunoprecipitated with its heterodimerization partner RXRα [9]. While expression of PPARβ/δ is not strikingly high in whole skin, it is found at very high level in keratinocytes, one of the main differentiated cell types found in skin [9]. Thus, there is the potential for PPARβ/δ to have functional role in tissues where it is expressed in high levels and even in tissues that may not exhibit high expression patterns by gross examination due to the low content of specific cell types.
Potential functional roles for PPARβ/δ have emerged in recent years. The first biological effect described for a PPARβ/δ agonist was an increase in serum high density lipoprotein cholesterol (HDLc) in diabetic mice following administration of L165041 [10]. An increase in serum HDLc was also found in obese non-human primates administered another potent PPARβ/δ ligand, GW501516 [11]. Further, ligand activation of PPARβ/δ also reduced serum low density lipoprotein cholesterol (LDLc), triglycerides and fasting insulin concentrations [11]. Studies in a mouse model of obesity determined that ligand activation of PPARβ/δ by GW501516 can increase expression of enzymes that catabolize fatty acids in skeletal muscle, and significantly decrease serum glucose concentrations [12]. Mice fed a high fat diet exhibit improvement in insulin sensitivity in response to ligand activation of PPARβ/δ, and this effect is not found in Pparβ/δ-null mice, thus demonstrating that PPARβ/δ is required to improve insulin sensitivity [13]. These observations demonstrate that PPARβ/δ ligands have great potential for the treatment of diseases associated with altered lipid and glucose homeostasis including diabetes. More recently, it was shown that combining ligand activation of PPARβ/δ with exercise increases the number of oxidative myofibers in muscle, resulting in enhanced exercise performance [14]. This observation suggests that PPARβ/δ ligands could be developed to improve athletic performance; this raises ethical questions regarding the potential for illicit use. PPARβ/δ and its ligand also have potent anti-inflammatory activities (reviewed in [5, 15]). The anti-inflammatory activities of PPARβ/δ and its PPARβ/δ ligands may be important for prevention of liver toxicity/cancer [6, 16-19] or neurotoxicity [20].
In contrast to the mechanisms elucidated for PPARβ/δ in metabolism and inflammation, the role of PPARβ/δ in cell growth remains controversial due to conflicting reports in the literature. Some studies indicate that activating PPARβ/δ causes increased cell proliferation and anti-apoptotic signaling, while others found that activating PPARβ/δ inhibits cell proliferation by inducing terminal differentiation and/or pro-apoptotic signaling. A critical analysis of these differences is the focus of this review. The remaining sections will focus on the role(s) reported for PPARβ/δ in specific cell types beginning with normal cell types and ending with cancer models.
3. Regulation of cell proliferation in skin and keratinocytes by PPARβ/δ
Regulation of cell proliferation in skin and keratinocytes has been extensively studied. There is evidence from three different null mouse models that PPARβ/δ inhibits cell proliferation in mouse keratinocytes [21-23]. A role for PPARβ/δ in cell proliferation in skin was revealed by the observations that phorbol ester-induced hyperplasia is exacerbated in PPARβ/δ-deficient mouse skin in vivo using two distinctly different Pparβ/δ-null mouse models [21, 22]. Epidermal thickness and proliferating cell nuclear antigen (PCNA)-positive cells is also increased in mice where PPARβ/δ expression was conditionally disrupted in keratinocytes [23]. These observations suggest that PPARβ/δ attenuates cell proliferation in keratinocytes. In agreement with these studies, inhibition of cell proliferation was found in normal human epidermal keratinocytes, HaCaT keratinocytes, N/TERT-1 and NCTC 2544 human keratinocyte cell lines in response to ligand activation of PPARβ/δ [24-27]. Similarly, increased cell proliferation was found in Pparβ/δ-null mouse keratinocytes in vitro [28] and ligand activation of PPARβ/δ inhibits cell proliferation of cultured mouse keratinocytes through a mechanism that requires a functional receptor [29]. Exacerbated hyperplasia was also noted in Pparβ/δ-null mouse skin following treatment with phorbol ester [30, 31] and ligand activation of PPARβ/δ inhibits phorbol ester-induced hyperplasia in mouse skin in vivo [30]. In one study, topical application of PPARβ/δ ligands had no effect on epidermal cell proliferation [32]. In contrast to these studies, one report suggested that ligand activation of PPARβ/δ increased cell proliferation of human keratinocytes. Treating cultured human keratinocytes from psoriasis patients with L165041 or GW501516 caused an increased in cell proliferation, and this increase was thought to be mediated by increased expression of heparin-binding EGF-like growth factor [33]. However, this study was limited to analysis of only one timepoint and a single concentration of either L165041 (1 μM) or GW501516 (5 μM); concentrations that are also known to cause activation of PPARα and PPARγ [3, 34]. Thus, to date there are thirteen published studies that have specifically examined cell proliferation in skin and keratinocytes with eleven showing inhibition of cell proliferation of human and mouse keratinocytes by PPARβ/δ, one report showing no effect on cell proliferation and one report showing enhanced cell proliferation of human keratinocytes from psoriasis patients (Table 1). A number of mechanisms have been postulated to explain these effects.
Table 1.
Regulation of cell proliferation in skin and keratinocytes by PPARβ/δ.
| Model | Effect | Reference |
|---|---|---|
| Homozygous Pparβ/δ-null mouse skin (disrupted by neomycin cassette in last exon of ligand binding domain) treated topically with phorbol ester | Enhanced hyperplasia in null mouse skin as compared to wild-type mouse skin | [22] |
| Heterozygous Pparβ/δ-null mouse skin (disrupted by neomycin cassette between exon 4 and 5 encoding the DNA binding domain) treated topically with phorbol ester | Enhanced hyperplasia in null mouse skin as compared to wild-type mouse skin | [21] |
| Homozygous Pparβ/δ-null mouse skin (disrupted by deleting exon 4 of the DNA binding domain) | Increased epidermal thickness and PCNA-positive keratinocytes in null mouse skin as compared to wild-type mouse skin | [23] |
| Mouse skin | Enhanced hyperplasia in null mouse skin as compared to wild-type mouse skin | [31] |
| Mouse skin | Enhanced hyperplasia in null mouse skin as compared to wild-type mouse skin, prevented by ligand activation of PPARβ/δ; an effect not found in null mouse skin | [30] |
| Normal human keratinocytes | 0.05 – 0.5 μM L165041 and 0.2 - 5.0 μM TTA inhibit cell proliferation | [26] |
| N/TERT-1 human keratinocyte cell line | 1.0 μM GW0742 inhibits cell proliferation | [25] |
| Human NCTC 2544 keratinocyte cell line | 0.05 – 1.0 mM DEHP inhibits cell proliferation | [27] |
| Human HaCaT keratinocyte cell line | 1.0 – 10 μM μM GW0742 inhibits cell proliferation and increases apoptosis; no effect on serum withdrawal-induced apoptosis | [24] |
| Mouse primary keratinocytes | 0.1 – 1.0 μM μM GW0742 inhibits cell proliferation | [24] |
| Mouse primary keratinocytes | Enhanced cell proliferation in null keratinocytes as compared to wild-type | [28] |
| Mouse primary keratinocytes | 0.01 - 0.1 μM μM GW0742 inhibits cell proliferation | [29] |
| Mouse skin | Topical application of 4 mM GW1514 has no effect on epidermal cell proliferation | [32] |
| Human primary keratinocytes from psoriasis patients | 1.0 μM L165041 and 5.0 μM GW501516 increases cell proliferation | [33] |
3. 1. PPARβ/δ-dependent regulation of differentiation in keratinocytes
The first suggestion that PPARβ/δ regulates terminal differentiation in skin was revealed by increased expression of PPARβ/δ that correlated with elevated levels of skin terminal differentiation markers transglutaminase-1 and small proline-rich proteins [35]. Subsequent work by four independent laboratories supported this finding by showing that ligand activation of PPARβ/δ increased terminal differentiation of mouse and human keratinocytes using both in vivo and in vitro models [24-26, 29, 30, 32, 36]. Expression of mRNAs and proteins required for terminal differentiation of human and mouse basal keratinocytes to cornified keratinocytes was increased by PPARβ/δ ligands (L165041, GW0742, tetradecylthioacetic acid (TTA)) [25, 26, 29, 30, 32] and these changes in gene expression were also reflected by an increase in cornified envelopes [29], the functional end product of keratinocyte terminal differentiation. That these effects require PPARβ/δ was demonstrated by studies showing the absence of these effects in null mouse models [25, 29, 30, 36]. These findings represent the most definitive response described to date for PPARβ/δ in cell proliferation and clearly establish that ligand activation of PPARβ/δ promotes terminal differentiation in mouse and human keratinocytes. The mechanisms that mediate this effect remain unclear as the PPARβ/δ target genes that promote terminal differentiation have not been identified to date. PPARβ/δ-dependent induction of keratin 10 (K10) in keratinocytes [30, 32, 36] may contribute to the mechanisms underlying PPARβ/δ-mediated inhibition of cell proliferation in keratinocytes since increased expression of K10 inhibits cell proliferation of human HaCaT keratinocytes through a retinoblastoma-dependent mechanism involving the nonhelical terminal domain of K10 [37]. Since the induction of terminal differentiation is known to be associated with withdrawal from the cell cycle, it is likely that PPARβ/δ-mediated induction of terminal differentiation in keratinocytes is a key mechanism that causes inhibition of cell proliferation observed in most skin/keratinocyte models studied to date.
3.2. PPARβ/δ-dependent regulation of apoptosis in keratinocytes
There is a relative unique balance between differentiation and apoptosis in keratinocytes that could also influence cell proliferation of keratinocytes. Keratinocytes undergo terminal differentiation to form a cornified cell and this process is associated with increased activity of caspases including caspase 3 and caspase 14 [38, 39]. Caspase 3 is known to be central in the process of apoptosis and increased caspase 3 activity occurs during keratinocyte terminal differentiation [39]. However, differentiating and fully differentiated keratinocytes (e.g cornified cells) do not exhibit shrinkage, DNA fragmentation and are not phagocytosed [40, 41]. Additionally, caspase 14 does not participate in apoptotic signaling but instead modulates terminal differentiation [38]. These observations demonstrate that apoptotic signaling observed in differentiating keratinocytes is relatively unique as compared to most cells because differentiated (cornified) keratinocytes have a more sustained lifespan whereas apoptosis usually leads to cell death and phagocytosis. Consistent with these observations, a PPARβ/δ-dependent increase in caspase 3 activity and TUNEL-positive cells was found in phorbol ester-treated mouse skin [31]. Increased annexin V-positive human HaCaT keratinocytes, indicative of apoptosis, was also reported following treatment with GW0742 [24]. However, increased caspase 3 activity was not found in N/TERT-1 human keratinocytes following treatment with GW0742 or in three mouse skin tumor cell lines [25, 30]. The effect of ligand activation of PPARβ/δ on the expression of caspase 14 in skin or keratinocytes has not been examined to date. Combined, there is compelling evidence for PPARβ/δ-dependent induction of apoptotic-like signaling in keratinocytes, that is likely related to induction of terminal differentiation and may also be involved in the inhibitory effects of PPARβ/δ on cell growth observed in mouse and human skin.
In contrast to strong evidence from multiple independent groups showing PPARβ/δ-mediated terminal differentiation of keratinocytes and the well-established relationship between terminal differentiation and apoptotic-like signaling, a paradoxical regulatory pathway has also been described suggesting that PPARβ/δ potentiates anti-apoptotic signaling in keratinocytes [42]. In response to ligand activation of PPARβ/δ by L165041 in cultured mouse keratinocytes or human HaCaT keratinocytes, the expression of phosphatase and tensin homolog deleted on chromosome Ten (PTEN) is decreased while expression of 3-phosphoinositide-dependent-protein kinase 1 (PDPK1) and integrin-linked kinase (ILK) is decreased [42]. The combined effect of this PPARβ/δ-dependent regulation led to increased phosphorylation of protein kinase B (AKT) causing inhibition of apoptosis and cell survival. Subsequent work by others using HaCaT human keratinocytes supports this putative anti-apoptotic signaling mediated by PPARβ/δ as shown by increased expression of PDPK1 and phosphorylation of AKT, but ILK or PTEN expression were not examined [43]. Inhibition of tumor necrosis factor-α (TNFα)-induced apoptosis has also been reported in response to L165041 in human HaCaT keratinocytes treated with and without epidermal growth factor (EGF) [44]. However, the high concentration of L165041 (10 μM) used for these studies could potentially activate PPARγ [3] and thus it is not possible to conclude that this effect was due solely to activation of PPARβ/δ. Similar changes in the PTEN/PDPK1/ILK/AKT pathway are not consistently observed in response to ligand activation of PPARβ/δ in mouse and human keratinocytes. For example, increased expression of the known PPARβ/δ target gene adipocyte differentiation-related protein (ADRP) is increased in mouse keratinocytes or human N/TERT-1 keratinocytes in response to GW0742 but no changes in the expression of PTEN, PDPK1 or ILK, or phosphorylation of AKT were observed [25]. Similarly, expression of two known PPARβ/δ target genes, angiopoietin-like protein 4 (ANGPTL4) and ADRP were increased by GW0742 in human HaCaT keratinocytes but no change in the expression of PDPK1 or phosphorylation of AKT were noted [24]. There are other examples where no changes in this putative PPARβ/δ-dependent PTEN/PDPK1/ILK/AKT pathway were found (described elsewhere in this review). One likely explanation for the lack of concordance between laboratories is/are the difference(s) in the model used. The studies suggesting a putative PPARβ/δ-dependent PTEN/PDPK1/ILK/AKT pathway in mouse keratinocytes used cells that lacked the typical cell morphology of primary keratinocyes and importantly, did not exhibit basal expression of keratin 6 (K6) [36, 42]. Since it is well documented that primary keratinocytes express K6 [45, 46], this suggests that the putative PPARβ/δ-dependent PTEN/PDPK1/ILK/AKT pathway may only function in cells that have undergone some molecular changes not associated with normal keratinocytes. While there is some evidence that this pathway may function during wound healing, the evidence to date is more compelling that it is not functional in normal mouse or human keratinocytes in response to ligand activation of PPARβ/δ. Whether this pathways functions in other cells types is also uncertain as some reports suggest it does, while others indicate that it does not.
3.3. PPARβ/δ-dependent regulation of cell signaling in keratinocytes
In addition to the induction of terminal differentiation, there is also evidence suggesting that PPARβ/δ attenuates cell proliferation in keratinocytes by modulating kinase signaling. In phorbol ester-stimulated skin and keratinocytes, enhanced cell proliferation is observed in the absence of PPARβ/δ expression [21, 22, 28, 30, 31]. In Pparβ/δ-null mouse skin treated with phorbol ester, increased expression of Ubiquitin C does not occur leading to a significant decrease in free ubiquitin (UB) and reduced UB-dependent turnover of protein kinase C α (PKCα) [28, 31]. Whether ligand activation of PPARβ/δ can increase UB-dependent turnover of PKCα or other kinases through this mechanism has not been examined. These findings suggest that this mechanism may contribute to PPARβ/δ-mediated inhibition of keratinocyte cell proliferation.
3.4. Effect of retinoic acid on cell proliferation in keratinocytes
Recent reports suggested that shuttling of retinoic acid between retinoic acid receptor (RAR) and PPARβ/δ occurs based on the relative expression of two RA-binding proteins, cellular retinoic acid binding protein II (CRABP-II) and fatty acid binding protein 5 (FABP5) [43, 47]. In this hypothetical model, retinoic acid is preferentially targeted to RAR in cells expressing more CRABPII leading to inhibition of cell growth, while retinoic acid is preferentially targeted to PPARβ/δ in cells expressing more FABP5 leading to enhanced cell growth [43]. This idea was supported by the reported increased PDPK1 expression in human HaCaT keratinocytes treated with either retinoic acid or the PPARβ/δ ligand GW501516. In contrast to these findings, recent work shows that retinoic acid does not bind to or activate PPARβ/δ as shown in cells with a high FABP5:CRABPII ratio as well as with ligand-mediated binding assays [48]. Similarly, retinoic acid inhibits cell growth of keratinocytes independent of PPARβ/δ expression, and ligand activation of PPARβ/δ does not increase expression of PDPK1 but rather inhibits proliferation of cells with a high FABP5:CRABPII ratio [24]. These observations are in line with inhibition of cell proliferation observed in HaCaT keratinocytes, other human keratinocyte cell lines, and various human cancers following treatment with retinoic acid [49-53]. Retinoic acid also inhibits proliferation of mouse primary keratinocytes, which is consistent with previous studies [54] and with inhibition of skin cancer by retinoids observed in several mouse models [55-61]. Combined, these observations are highly inconsistent with the proposed pathway suggesting that activating PPARβ/δ by retinoic acid will enhance cell growth of human keratinocytes [43, 47]. Given these inconsistencies, the aforementioned hypothesis that retinoic acid activates PPARβ/δ to promote cell survival should be rigorously re-evaluated.
4. Regulation of cell proliferation in other somatic cells by PPARβ/δ
Regulation of cell proliferation by PPARβ/δ in somatic cells other than skin and keratinocytes has also been studied, including endothelial cells, vascular smooth muscle cells, fibroblasts, hepatocytes and stellate cells. Blood vessels are composed of a number of cell types including endothelial cells (EC) that line the interior surface and vascular smooth muscle cells (VSMC) the major cell type in the cell walls. EC and VSMC can interact through the production of growth factors and signaling molecules that have paracrine and autocrine activities. Notably, EC produce prostacyclin, which may function as an endogenous PPARβ/δ ligand that could potentially act on both EC and VSMC. The effect of PPARβ/δ on cell proliferation in both of these cells types has been examined.
A stable prostacyclin analog (ciprostene) inhibits proliferation and associated 3H-thymidine incorporation in bovine EC at a concentration of 50-100 μM [62]. In the absence of PPARβ/δ expression, mouse EC proliferation is significantly increased as compared to EC that express PPARβ/δ [63], but the effect of ligand activation was not examined in this model. The inhibitory effect of PPARβ/δ in mouse EC is mediated by PPARβ/δ-dependent up-regulation of Cdkn1c, which encodes the cell cycle inhibitor p57Kip2 [63]. Activation of PPARβ/δ by 100 μM carbaprostacyclin or 50 μM L165041 does not change annexin V-positive cells or caspase 3 activity in human EC [64]. In contrast to these reports, activation of PPARβ/δ with 0.1 μM GW501516 in human EC and an EC line (EAHy926) is reported to cause a marked increase in cell proliferation, with almost a doubling of cell number [65]. Interestingly, this effect is found only after 72 hours of treatment with no changes in cell proliferation being noted at earlier timepoints [65]. In another study, 0.01-0.1 μM GW501516 was reported to increase cell proliferation of human EC after up to 14 days of culture, but the kinetics of cell proliferation are uncertain because data from only one timepoint was reported [66]. The authors of this study suggested that the increased EC proliferation may be mediated by elevated expression of vascular endothelial growth factor (VEGF) and its receptor FLT1 [66]. However, this hypothesis was not examined in detail and was based solely on mRNA data without further mechanistic evaluation. In endothelial progenitor cells (EPCs), an increase in cell proliferation and the percentage of cells undergoing the S phase of the cell cycle was found by 0.1-1.0 μM GW501516 treatment [67]. These changes found in cultured endothelial cells treated with GW501516 are reflected by angiogenic effects found in other models including increased endothelial cell tube formation, increased aortic ring outgrowths and increased tube formation in matrigel plug assays suggesting that activating PPARβ/δ with GW501516 stimulates angiogenesis by increasing expression of VEGF [65, 67]. Thus, there is evidence that PPARβ/δ ligands have inhibitory and proliferative effects in EC (Table 2).
Table 2.
Role of PPARβ/δ on cell proliferation and apoptosis in somatic cells
| Cell type | Effect | Reference |
|---|---|---|
| Bovine endothelial cells | 50 –100 μM ciprostene (prostacyclin analog) inhibits cell proliferation | [62] |
| Mouse primary endothelial cells | Increased cell proliferation in Pparβ/δ-null endothelial cells as compared to wild-type | [63] |
| Human primary endothelial cells | 100 μM carbaprostacyclin or 50 μM L165041 has no effect on annexin V-positive cells or caspase 3 activity | [64] |
| Human endothelial cells and a human endothelial cell line (EAHy926) | 0.1 μM GW501516 causes an increase in cell proliferation after 72 hours | [65] |
| Human primary endothelial cells | 0.01 – 0.1 μM GW501516 causes an increase in cell proliferation after 14 days | [66] |
| Human primary endothelial progenitor cells | 0.1 – 1.0 μM GW501516 causes an increase in cell proliferation after 8 days | [67] |
| Human primary endothelial cells | 1 – 100 μM carbaprostacyclin and 10-50 μM L165041 decrease H2O2-induced apoptosis | [64, 68] |
| Human primary endothelial progenitor cells | 0.1 – 1 μM GW501516 decrease hypoxia-induced apoptosis | [67] |
| Rat vascular smooth muscle cells | 1.0 μM iloprost (prostacyclin analog) inhibits cell proliferation | [71] |
| Rat vascular smooth muscle cells | Overexpression of prostacyclin synthase inhibits cell proliferation | [70] |
| Bovine vascular smooth muscle cells | 3 –100 μM ciprostene (prostacyclin analog) inhibits cell proliferation | [62] |
| Human vascular smooth muscle cells | 0.1 – 10 μM prostacyclin analogs (iloprost, cicaprost, UT-15) inhibits cell proliferation | [69] |
| Rat vascular smooth muscle cells | 1.0 μM beraprost (prostacyclin analog) inhibits cell proliferation; mediated by PPARβ/δ | [73] |
| Rat vascular smooth muscle cells | 5 – 10 μM L165041 inhibits PDGF-induced cell proliferation inhibits cell proliferation | [72] |
| Mouse primary hepatocytes | TNFα-induced incorporation of BrdU is enhanced in Pparβ/δ-null hepatocytes as compared to wild-type | [19] |
| Rat primary stellate cells | 10 μM L165041 increase cell proliferation | [74] |
| Mouse primary stellate cells | Increased cell proliferation in Pparβ/δ-null stellate cells as compared to wild-type | [19] |
| Human lung fibroblasts | 0.1 – 10 μM treprostinil (prostacyclin analog) and 0.1 – 100 μM GW0742 inhibit cell proliferation | [75] |
| Mouse lung fibroblasts | 100 μM treprostinil (prostacyclin analog) inhibits cell proliferation; effect not found in Pparβ/δ-null fibroblasts | [75] |
| Rat cardiac fibroblasts | 10 μM GW501516 inhibits cell proliferation | [76] |
| Rat cardiac fibroblasts | 10 μM GW501516 inhibits cell proliferation | [77] |
| Mouse embryonic fibroblasts | Overexpression of PPARβ/δ increases cell proliferation | [78] |
| Human proximal tubular epithelial cell line (HK-2) | 0.2 – 1.0 μM L165041 decrease H2O2-induced apoptosis | [79] |
| Human embryonic kidney epithelial cell line (HEK-293) | Increased intracellular prostacyclin increases apoptosis; mediated by PPARβ/δ | [81] |
| Mouse inner medullary collecting duct cell line (IMCD-K2) | 10 μM GW501516 inhibits cell proliferation; 0.01 – 1.0 μM GW501516 has no effect on anisomycin-induced apoptosis | [82] |
Anti-apoptotic activity resulting from activating PPARβ/δ was also reported in some EC models when apoptosis is induced. In human endothelial cells, 1-100 μM carbaprostacyclin and 10-50 μM L165041 decrease hydrogen peroxide-induced apoptosis as shown by decreased annexin V-positive cells, caspase 3 activity and PARP cleavage [64, 68]. Increased expression of 14-3-3ε mediated by activation of PPARβ/δ is thought to be the mechanisms underlying these effects [64, 68], which causes inhibition of BAD translocation and downstream events thereby preventing apoptotic signaling that is typically induced in EC by treatment with hydrogen peroxide. Ligand activation of PPARβ/δ was also reported to prevent hypoxia-induced apoptosis in human EPC by increasing phosphorylation of AKT, but this increase in phospho-AKT was not mediated by increased expression of ILK [67]. While expression of PDPK1 or PTEN was not assessed in this study, this is an example where the previously described PPARβ/δ-dependent modulation of PTEN/PDPK1/ILK/AKT does not appear to function as reported in a keratinocyte model [42].
There is contradictory evidence indicating that PPARβ/δ can either inhibit cell proliferation, potentiate cell proliferation or have anti-apoptotic activity under certain circumstances in EC. In contrast, reports examining the effect of PPARβ/δ in VSMC cell proliferation all indicate that activating this receptor will inhibit cell growth (Table 2). Prostacyclin is thought to be an endogenous PPARβ/δ ligand and synthetic prostacyclin analogs are routinely used for this purpose for the treatment of pulmonary hypertension. There is strong evidence from several laboratories that prostacyclin and prostacyclin analogs inhibit VSMC proliferation [62, 69-71]. In addition to membrane bound IP receptors that are known to mediate the effect of prostacyclin, recent evidence suggests that PPARβ/δ can mediate the inhibitory effect of prostacyclin analogs in VSMC [72, 73]. For example, cell proliferation of rat VSMC is inhibited by 1 μM beraprost and this effect is reduced by PPARβ/δ antagonism [73]. Additionally, platelet-derived growth factor (PDGF)-induced cell proliferation of rat VSMC was inhibited by 5-10 μM L165041 and this inhibition was associated with reduced expression of CYCLIN D1, CDK4 and reduced phosphorylation of ERK1/2 [72]. No change in PDGF-induced phosphorylation of AKT was noted in response to L165041 [72].
There are a limited number of reports examining the effect of ligand activation in other somatic cell types (Table 2). TNFα causes an increase in bromodeoxyuridine (BrdU) labeling in primary mouse hepatocytes, and this effect is increased in Pparβ/δ-null hepatocytes [19]. Primary stellate cells from rat exhibit a modest increase in cell proliferation following treatment with 10 μM L165041 [74]. In contrast, Pparβ/δ-null mouse primary stellate cells exhibit increased cell proliferation as compared to wild-type stellate cells [19]. The mechanisms underlying these effects have not been elucidated nor have the reason(s) for the differences observed between rat and mouse stellate cells. Cell proliferation of human lung fibroblasts is inhibited with the prostacyclin analog, Treprostinil, with concentrations greater than 0.1 μM and with GW0742 at concentrations greater than 1.0 μM [75]. PPARβ/δ is required for the inhibitory effect of Treprostinil in lung fibroblasts as this effect is mitigated in Pparβ/δ-null lung fibroblasts [75]. In cardiac fibroblasts, ligand activation of PPARβ/δ with 10 μM GW501516 caused an inhibition in cell proliferation that was associated with PPARβ/δ-dependent inhibition of collagen synthesis [76]. Activation of PPARβ/δ by GW501516 in rat cardiac fibroblasts caused inhibition of BrdU incorporation and increased expression of the cell cycle inhibitor gene G0/G1 switch gene 2 (G0S2) [77]. While the former three studies indicate that ligand activation of PPARβ/δ in fibroblasts inhibits cell proliferation, there is one recent report showing increased cell proliferation in embryonic fibroblasts over-expressing PPARβ/δ [78]; however the effect of ligand activation in these cells was not reported. In the same study, a modest increase in cell proliferation of normal human thyroid cells was also observed and it was suggested to be due to a CYCLIN E-dependent mechanism [78]. Anti-apoptotic activity was also reported to occur in a human kidney cell line following co-treatment with hydrogen peroxide and 0.2-1.0 μM L165041 [79] and in a rat cardiomyoblast cell line following co-treatment with hydrogen peroxide and 0.1-1.0 μM GW501516 [80]. In contrast, overexpressing prostacyclin synthase, which increases intracellular prostacyclin, increases apoptosis in a human embryonic kidney epithelial cell line through a mechanism that requires PPARβ/δ [81]. In a mouse inner medullary collecting duct cell line, 10 μM GW501516 inhibits cell proliferation whereas 0.01-1.0 μM GW501516 has no effect on anisomycin-induced apoptosis [82].
5. Regulation of cell proliferation in cancer models and cancer cell lines by PPARβ/δ
The effect of activating PPARβ/δ in cancer models and cancer cell lines has been examined, and there are considerable differences in the responses reported in the literature (Table 3). Colon cancer models are among the more extensively studied and this subject has been reviewed in greater detail elsewhere [5]. There are two opposing mechanisms by which PPARβ/δ has been postulated to regulate colon cancer. One view is that PPARβ/δ is up-regulated by the adenomatous polyposis coli (APC)/β-CATENIN/transcription factor 4 (TCF4) pathway, similar to what is observed with c-MYC and CYCLIN D1 [83]. It was hypothesized that PPARβ/δ could then be activated by cyclooxygenase-2 (COX2)-derived ligands such as prostacyclin [2, 83], leading to expression of yet-to-be identified target genes that cause increased cell proliferation and tumor promotion. Consistent with this hypothesis, ligand activation of PPARβ/δ with GW501516 was reported to cause no change in colon tumorigenesis but an increase in small intestinal tumors in APC min heterozygous mice [84], and cause a PPARβ/δ-dependent increase in intestinal and colon tumorigenesis in APC min heterozygous mice [85]. Serum withdrawal-induced apoptosis was also reportedly inhibited by GW501516 (0.1-5.0 μM) in LS174T and HCT116 colon cancer cell lines and mediated by PPARβ/δ-dependent up-regulation of VEGF and phosphorylation of AKT [84-86]. Interestingly, the changes observed in apoptotic signaling using serum withdrawal with in vitro models were in line with the observed reduction of TUNEL-positive cells in intestinal tumors from mice treated with GW501516 [84, 85]. This is of interest because increased signaling for apoptosis is not typically found in tumors cells. However, other studies found no correlation between the APC/β-CATENIN/TCF4 pathway and increased PPARβ/δ expression ([87], reviewed in [5]). Some studies revealed that ligand activation of PPARβ/δ had no effect on cell growth of HT29, SW480 and HCA7 cells [66]. Ligand activation of PPARβ/δ in LS174T, HCT116 or HT29 colon cancer cell had no effect on cell proliferation with 0.1 μM GW501516 or GW0742 but inhibited cell proliferation at concentrations from 1.0 - 10.0 μM of the same ligands [88]. Importantly, there was also no change in cell proliferation, expression of VEGF, PARP cleavage or phosphorylation of AKT in response to ligand activation of PPARβ/δ following serum-withdrawal [88]. Further, a PPARβ/δ-dependent increase in TUNEL-positive colonocytes was reported to occur in response to administration of GW0742 in mice [89]. Collectively, these findings and others (reviewed in [5]) are inconsistent with the idea that PPARβ/δ mediates anti-apoptotic signaling in colon cancer cell lines.
Table 3.
Role of PPARβ/δ on cell proliferation and apoptosis in cancer cell lines
| Cell type | Effect | Reference |
|---|---|---|
| HCT116 human colon cancer cell line | 0.1 – 5.0 μM GW501516 inhibits serum withdrawal-induced apoptosis | [84] |
| LS174T human colon cancer cell line | 0.1 – 1.0 GW501516 inhibits serum withdrawal-induced apoptosis | [86] |
| LS174T human colon cancer cell line | 1.0 GW501516 inhibits serum withdrawal-induced apoptosis | [85] |
| HT29, SW480 and HCA7 human colon cancer cell lines | 0.01 μM compound F has no effect on cell proliferation | [66] |
| LS174T, HCT116, HT29 human colon cancer cell lines | 0.1 μM GW501516 or GW0742 has no effect on cell proliferation. 1.0 – 10 μM GW501516 or GW0742 inhibits cell proliferation. 0.1 – 10 μM GW501516 or GW0742 has no effect on serum withdrawal-induced apoptosis. | [88] |
| HCT116 human colon cancer cell line | Silencing PPARβ/δ expression increases cell proliferation | [90] |
| MCF7, T47D human breast cancer cell lines (estrogen receptor-positive) | 0.01 μM compound F or up to 0.05 μM GW501516 increase cell proliferation | [66] |
| BT20, MDA-MB-231 human breast cancer cell lines (estrogen receptor-negative) | 0.01 μM compound F has no effect on cell proliferation | [66] |
| MCF7 human cancer cell line | No effect of cell proliferation with 0.1 – 1.0 μM GW501516 or GW0742, inhibition of cell proliferation with 10 μM GW501516 or GW0742 | [96] |
| A549 human lung cancer cell line | 10 – 40 μM L165041 inhibits cell proliferation | [97] |
| H157 human lung cancer cell line | 1.0 – 10.0 μM GW501516 increases cell proliferation | [101] |
| 1838 human lung cancer cell line | 1.0 μM GW501516 increases cell proliferation | [102] |
| A549, H23, H157 human lung cancer cell lines | 0.001 – 0.01 μM GW501516 increase cell proliferation; 0.01 – 0.1 μM GW501516 inhibits cisplatin-induced apoptosis | [103] |
| A549, H1838 human lung cancer cell lines | 0.1 – 10.0 μM GW501516 has no effect on cell proliferation | [104] |
| HepG2 human liver cancer cell line | 0.001 – 0.1 μM GW501516 increases cell proliferation | [105] |
| HepG2, Huh7, Hep3B human liver cancer cell lines | 0.005 – 0.05 μM GW501516 increases cell proliferation | [106] |
| CCLP1, HuCCT1, SG231 human cholangiocarcinoma cell lines | 0.001 μM – 0.05 μM GW501516 increases cell proliferation | [107] |
| HepG2, Huh7 human liver cancer cell lines | No effect of cell proliferation with 0.1 – 1.0 μM GW501516 or GW0742, inhibition of cell proliferation with 10 μM GW501516 or GW0742 | [88] |
| BT4Cn rat glioma cell line | 1.0 μM L165041 increases cell proliferation | [108] |
| BT4Cn rat glioma cell line | 100 μM TTA inhibits cell proliferation | [108] |
| UACC903 human melanoma cell line | 1.0 – 10.0 μM GW501516 or GW0742 inhibit cell proliferation | [96] |
| U266 human myeloma cell line and primary human myeloma cells | 10 – 50 μM carbaprostacyclin inhibits cell proliferation | [109] |
| SH-SYSY human neuroblastoma cell line | 1.0 μM GW0742 inhibits cell proliferation | [110] |
| 786-O human renal carcinoma cell line | 20 – 30 μM GW501516 inhibits cell proliferation | [111] |
| PNT1A, LnCaP human prostate cancer cell lines | 0.01 μM compound F increases cell proliferation | [66] |
| DU145, PC3 human prostate cancer cell lines | 0.01 μM compound F has no effect on cell proliferation | [66] |
An alternative hypothesis has also been postulated to explain how modulation of PPARβ/δ activity influences colon cancer (reviewed in [5]). In this model, ligand activation of PPARβ/δ mediates terminal differentiation of colonocytes and inhibition of cell proliferation thereby preventing tumor promotion. In support of this view, silencing PPARβ/δ expression in HCT116 cells caused increased cell proliferation [90]. In vivo disruption of PPARβ/δ in mice caused an increase in colon tumorigenesis in both a genetic and chemically-induced model [91], and ligand activation of PPARβ/δ with GW0742 inhibited chemically-induced colon cancer [89, 92]. Bezafibrate administration was also reported to inhibit colon cancer in mouse models and human colon cancer patients [93, 94]. This is of interest because while bezafibrate can activate PPARα, PPARβ/δ and PPARγ, the affinity of bezafibrate for human PPARβ/δ is greater as compared to human PPARα and PPARγ [34]. Well-characterized PPARβ/δ target genes associated with terminal differentiation of colonocytes are also increased by ligand activation of PPARβ/δ, including fatty acid binding protein (Fabp) and Adrp [89]. Since the induction of terminal differentiation is known to be associated with inhibition of cell proliferation, these findings are consistent with the observed inhibition of cell proliferation found in human colon cancer cell lines by 1.0-10.0 μM GW501516 or GW0742 [88]. While induction of terminal differentiation that may be mediated in part by increased expression of the PPARβ/δ target genes FABP and ADRP could contribute to the mechanisms leading to inhibition of tumor promotion in the colon, the involvement of other target genes remains uncertain.
Three studies have examined the effect of ligand activation of PPARβ/δ in breast cancer models. In estrogen receptor-positive human breast cancer cell lines MCF7 and T47D, ligand activation of PPARβ/δ by 0.01 μM compound F or up to 0.05 μM GW501516 caused an increase in cell proliferation after 12 days of culture, but this effect was not found in the estrogen receptor-negative human breast cancer cell lines BT20 and MDA-MB-231 [66]. The increase in cell proliferation was associated with an increase in cells undergoing S phase of the cell cycle, and increased expression of CDK2, VEGF and FLT1 mRNAs in cells treated with 0.025 μM GW501516 in medium with low (0.1%) serum [66]. In vivo, dietary administration of GW501516 at a concentration of 0.005% caused an increased in DMBA-induced mammary squamous cell carcinomas [95]. In tumors from these mice, PPARβ/δ co-immunoprecipitated with PDPK1 following GW501516 administration but this was only evaluated in two samples [95]. Critical examination of the functional significance of this putative interaction is uncertain and has not been examined in other cell types to date. In contrast to these reports, ligand activation of PPARβ/δ in MCF7 cells with either GW0742 or GW501516 does not increase cell proliferation, but rather, an inhibition of cell proliferation was observed with a concentration of 10 μM for either ligand [96]. These effects were independent of either PPARβ/δ ligand and were not influenced by the presence or absence of culture medium serum. Since previous studies used low serum culture conditions [66], these recent findings [96] suggest that the differences observed between laboratories is not due to the differences in ligand used or the presence/absence of culture medium serum.
The first report examining the effect of PPARβ/δ ligands in a lung cancer model demonstrated inhibition of proliferation with an A549 human lung cancer cell line by 10-40 μM L165041 that was associated with down-regulation of CYCLIN D1 and PCNA, and a block in the G1 phase of the cell cycle [97]. Combining ligand activation of PPARβ/δ with administration of indomethacin caused enhanced inhibition of cell proliferation in A549 cells that was associated with a block in the G1 phase of the cell cycle and increased apoptotic signaling [97]. These observations are consistent with results from in vivo models showing exacerbated Raf-dependent lung tumorigenesis in the absence of PPARβ/δ expression [98], and inhibition of lung tumorigenesis by increasing the production of the PPARβ/δ ligand prostacyclin [99, 100]. In contrast to these studies, others revealed that activating PPARβ/δ with GW501516 caused increased cell proliferation of human lung cancer cell lines. For example, ligand activation of PPARβ/δ with 0.001-10 μM GW501516 was reported to cause increased proliferation of A549, H23, H157, H1838 and H2106 human lung cancer cell lines [101-103]. Several mechanisms were postulated to explain this effect including PI3 kinase/AKT-mediated up-regulation of the EP4 receptor for PGE2, PPARβ/δ-dependent down-regulation of PTEN and increased phosphorylation of AKT [101-103]. However, analysis by others is inconsistent with these mechanisms as no changes in proliferation of A549 or H1838 cells are observed in response to 0.1-10.0 μM GW0742 or GW501516, and no changes in expression of PTEN, PDPK1 or phosphorylation of AKT were found following ligand activation of PPARβ/δ in these cells [104].
Four studies have examined the effect of ligand activation in liver cancer models. Culturing the human hepatoma cell line HepG2 with 0.001-0.1 μM GW501516 caused an increase in cell proliferation that was associated with elevated expression of COX2 [105]. A subsequent study reported similar findings with 0.005-0.05 μM GW501516 causing an increase in proliferation of HepG2, Huh7 and Hep3B cells [106]. These authors provided evidence suggesting that ligand activation of PPARβ/δ caused an increase in COX2 expression thereby increasing PGE2 and cytosolic phosholipase A2 (cPLA2) activity thus providing a potential feedback loop to drive tumor cell growth [106]. The same group proposed a similar mechanism for cholanigiocarcinomas [107]. However, this putative mechanism is based on the idea that ligand activation of PPARβ/δ increases expression of COX2 and when examined critically in the same liver cancer cell lines (HepG2 and Huh7), no change in COX2 expression was observed, despite confirming specific ligand activation of PPARβ/δ [88]. Further, only inhibition of cell proliferation by ligand activation of PPARβ/δ with 0.1-10.0 μM GW0742 or GW501516 was observed in a separate study, while no increased cell proliferation was noted [88].
In addition to colon, breast, lung and liver cancer models, there are also limited reports describing the effect of ligand activation of PPARβ/δ in other cancer models. In a rat glioma cell line (BT4Cn), modestly increased cell proliferation is found in response to 1.0 μM L165041 [108]. However, inhibition of BT4Cn cell proliferation was observed following culture in TTA [108], a compound that can activate PPARβ/δ. However, this study did not specifically determine whether this effect was due to PPARβ/δ but rather focused on PPARγ and found that PPARγ-independent mechanisms likely explained the growth inhibition by TTA. In a human melanoma cell line, UACC903, ligand activation of PPARβ/δ inhibits cell proliferation through an unknown mechanism [96]. Carbaprostacyclin inhibits cell proliferation of primary human myeloma cells and a human myeloma cell line and PPARβ/δ mediates inhibition of balcalein-induced inhibition of human myeloma cell proliferation, which may be due in part to inhibition of NF-κB activity [109]. Culturing the human neuroblastoma cell line SH-SYSY with 1.0 μM GW0742 causes inhibition of cell proliferation that is associated with down-regulation of CYCLIN D1, PCNA, BCL2, increased expression of p16 and induction of terminal differentiation [110]. Inhibition of cell proliferation is reported in 786-O renal carcinoma cells following culture in 20-30 μM GW501516 through unknown mechanisms [111]. Increased cell proliferation is found in some (PNT1A, LnCaP) but not other (DU145, PC3) human prostate cancer cell lines following culture in 0.01 μM compound F [66].
6. Regulation of PPARβ/δ expression as a modulator of cell proliferation
Some reports examining the effect of PPARβ/δ have focused on the expression of the receptor rather than on ligand activation. Many of these studies have inherent limitations. For example, there are reports suggesting that increased expression of PPARβ/δ is negatively associated with cancer progression [2, 112-116]. Some of these studies base the negative association between PPARβ/δ and cancer on mRNA expression or non-quantitative examination of PPARβ/δ protein. Importantly, none of these studies demonstrated that the relative presence of PPARβ/δ caused increased transcriptional activity of target genes. In the absence of these data, it is possible that the reported increase in PPARβ/δ mRNA is not reflected at the protein and/or functional level. Some studies have also suggested similar negative associations between PPARβ/δ expression and cancer using immunohistochemistry [114, 115]. Unfortunately, even highly-specific PPARβ/δ antibodies exhibit significant non-specific immunoreactivity [9]. For this reason, immunohistochemical analysis of PPARβ/δ must be viewed with caution. Indeed, using samples obtained from the Ouyang study, recent collaborative findings demonstrate with quantitative western blotting, that expression of PPARβ/δ is not higher in APC min heterozygous mouse colon [87], as previously suggested based solely on immunohistochemistry [115]. This also reinforces the notion that direct evidence of alterations in well characterized PPARβ/δ target genes should be included in analysis examining the effect of PPARβ/δ on cell growth. Lastly, some reports suggest that non-steroidal anti-inflammatory drugs (NSAIDs) can inhibit cancer cell growth by down-regulating expression of PPARβ/δ [117, 118]. However, these studies base their conclusions on extremely limited datasets and are inconsistent with more recent quantitative analysis showing either no change or increased expression of PPARβ/δ by NSAIDs in the same cancer cell lines [87]. Importantly, the latter studies also demonstrated consistent changes in expression of known PPARβ/δ target genes that correlated well with expression of PPARβ/δ [87], while the former studies provide no such comparison [117, 118]. Combined, studies examining the role of PPARβ/δ in cell proliferation that base their conclusions in part on expression of PPARβ/δ without direct evidence of distinct modulation of PPARβ/δ in their model system should be viewed with caution.
7. Gaps in Knowledge
It is clear from review of the literature that our understanding of the role of PPARβ/δ in cell proliferation and the mechanisms involved remain limited. There are a number of important questions that must be answered before this issue can be clarified. Why does ligand activation of PPARβ/δ cause inhibition of cell proliferation in normal mouse and human keratinocytes but increased cell proliferation in human keratinocytes from psoriasis patients? What are the direct target genes that mediate PPARβ/δ-dependent terminal differentiation in keratinocytes? How can PPARβ/δ inhibit cell proliferation in some models but increase cell proliferation in others? This is of particular interest for EC and VSMC since some studies indicate that ligand activation of PPARβ/δ will promote cell proliferation in EC but inhibit cell proliferation in VSMC. As PGI2 is produced in EC and can function as a PPARβ/δ ligand, why are anti-apoptotic activity and/or increased cell growth observed in EC but inhibition of cell proliferation is found in VSMC that are juxtaposed to EC? Is there a molecular difference in PPARβ/δ between these cell types that explain this difference? Are there species differences in the response to ligand activation of PPARβ/δ? This is a possibility that might explain why the literature suggests that ligand activation of PPARβ/δ in mouse and bovine EC may cause inhibition of cell proliferation while they increased proliferation in human EC. Severe pulmonary hypertension is associated with proliferation of ECs, and prostacyclin analogs are routinely used to treat this disease [119]. Since prostacyclin analogs can activate PPARβ/δ, why is EC proliferation not exacerbated in patients with severe pulmonary hypertension that are treated with prostacyclin analogs? Serum withdrawal-induced apoptosis in colon cancer cell lines can be inhibited by ligand activation of PPARβ/δ in some studies and this also reflected in vivo. Since the cellular milieu contains serum and growth factors, and apoptotic signaling in cancer cell lines is not typically affected by ligand activation of PPARβ/δ in the absence of apoptosis inducing agents, how can these differences be reconciled? Do anti-apoptotic effects observed in in vitro models when apoptosis is induced reflect physiological mechanisms? PPARβ/δ is expressed at high levels in liver, intestine and keratinocytes with evidence of constitutive gene activation activity, possibly due to the presence of endogenous ligands. There are a number of essential physiological roles for PPARβ/δ including regulating skeletal muscle fatty acid catabolism and glucose homeostasis. How can a nuclear receptor maintain essential physiological roles and also function to promote tumorigenesis? Is PPARβ/δ up-regulated by the APC/β-CATENIN/TCF4 pathway? Given the redundancy in DNA response elements for all three PPARs, how could activation of PPARγ inhibit tumorigenesis while activation of PPARβ/δ promotes tumorigenesis? Answering these questions with sound experimentation will provide significant insight for these disparities in the literature regarding the role of PPARβ/δ in cell proliferation.
8. Concluding remarks
Given the variation in responses reported for the same cell types and/or models, it is essential that critical examination of all endpoints be performed using highly stringent criteria, in order to clarify how activating PPARβ/δ influences cell proliferation. For example, specific demonstration of PPARβ/δ activity cannot rely solely on examination of mRNA expression of PPARβ/δ and should include examination of known PPARβ/δ target genes (e.g. ADRP, FABP, ANGPTL4, etc.). The use of knockout/knockdown approaches to confirm specificity is also highly suitable. The use of positive controls, in particular for examining PPARβ/δ expression since anti-PPARβ/δ antibodies exhibit significant non-specific immunoreactivity, should always be included.
There are a number of mechanisms described suggesting regulatory roles for PPARβ/δ in cell proliferation that have not been repeated and confirmed by independent laboratories. For example, the putative PPARβ/δ-dependent pathway described suggesting that PPARβ/δ causes down-regulation of PTEN expression, increased expression of ILK/PDPK1, increased phosphorylation of AKT and anti-apoptotic activity in keratinocytes [42] is based on data from cells that do not exhibit well characterized expression markers (e.g. K6). Careful re-examination of this pathway in mouse keratinocytes that express K6 demonstrated that this pathway does not function in normal mouse keratinocytes [25]. Changes in the PTEN/ILK/PDPK1/AKT pathway are also not found in human keratinocyte cell lines in response to ligand activation of PPARβ/δ [24, 25]. However, there are many reports in the literature that incorrectly cite this report [42] as having conclusively demonstrated PPARβ/δ-dependent anti-apoptotic activity without critically examining the literature. Similar examples exist for other molecular pathways that have not been replicated by independent laboratories including the idea that PPARβ/δ is up-regulated by the APC/β-CATENIN/TCF4 pathway ([83], reviewed in [5]), that PPARβ/δ up-regulates VEGF expression leading to increased phosphorylation of AKT and anti-apoptotic signaling [85, 88], that NSAIDs inhibit PPARβ/δ expression leading to anti-apoptotic activity [87, 117], that retinoic acid activates PPARβ/δ and potentiates cell proliferation [24, 43, 48], or other examples related to PPARβ/δ-dependent regulation of PDPK1 and AKT [102-104]. These reported differences emphasize the need to question all reported findings until they have been repeated and confirmed by independent laboratories. A collaborative effort between laboratories reporting disparate results is one approach that can clarify some of these misunderstandings. For example, the recent correction in the literature showing that PPARβ/δ expression is not up-regulated in APC min mouse colon or down-regulated by an NSAID [87] as once suggested [115] is a significant advance. Similar collaborations would also move this field forward.
Given the large differences reported in the literature, there is a need to critically examine basic, functional roles for PPARβ/δ. Collaborative efforts focusing on the effect of ligand activation of PPARβ/δ in a large variety of cells and cell lines using complementary quantitative analysis including dose-dependent examination of cell proliferation over time by multiple methods (Coulter counting, BrdU labeling, 3H-thymidine uptake, and assorted cell based systems), examination of dose-dependent changes in cell cycle kinetics (flow cytometry) and comparative analysis of PPARβ/δ-dependent changes in known PPARβ/δ target gene expression is clearly needed. Until such analyses is undertaken and confirmed by multiple independent laboratories, it is likely that the literature will remain confusing to those who are not experts in the field.
Acknowledgments
Work in our laboratories is supported by the National Institutes of Health
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc Natl Acad Sci U S A. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta RA, Tan J, Krause WF, Geraci MW, Willson TM, Dey SK, DuBois RN. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor δ in colorectal cancer. Proc Natl Acad Sci U S A. 2000;97:13275–13280. doi: 10.1073/pnas.97.24.13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger J, Leibowitz MD, Doebber TW, Elbrecht A, Zhang B, Zhou G, Biswas C, Cullinan CA, Hayes NS, Li Y, Tanen M, Ventre J, Wu MS, Berger GD, Mosley R, Marquis R, Santini C, Sahoo SP, Tolman RL, Smith RG, Moller DE. Novel peroxisome proliferator-activated receptor (PPAR) γ and PPARδ ligands produce distinct biological effects. J Biol Chem. 1999;274:6718–6725. doi: 10.1074/jbc.274.10.6718. [DOI] [PubMed] [Google Scholar]
- 4.Sznaidman ML, Haffner CD, Maloney PR, Fivush A, Chao E, Goreham D, Sierra ML, LeGrumelec C, Xu HE, Montana VG, Lambert MH, Willson TM, Oliver WR, Sternbach DD. Novel selective small molecule agonists for peroxisome proliferator-activated receptor δ (PPARδ)-synthesis and biological activity. Bioorg Med Chem Lett. 2003;13:1517–1521. doi: 10.1016/s0960-894x(03)00207-5. [DOI] [PubMed] [Google Scholar]
- 5.Peters JM, Hollingshead HE, Gonzalez FJ. Role of peroxisome-proliferator-activated receptor β/δ (PPARβ/δ) in gastrointestinal tract function and disease. Clin Sci (Lond) 2008;115:107–127. doi: 10.1042/CS20080022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kino T, Rice KC, Chrousos GP. The PPARδ agonist GW501516 suppresses interleukin-6-mediated hepatocyte acute phase reaction via STAT3 inhibition. Eur J Clin Invest. 2007;37:425–433. doi: 10.1111/j.1365-2362.2007.01796.x. [DOI] [PubMed] [Google Scholar]
- 7.Woo CH, Massett MP, Shishido T, Itoh S, Ding B, McClain C, Che W, Vulapalli SR, Yan C, Abe JI. ERK5 activation inhibits inflammatory responses via peroxisome proliferator-activated receptor δ (PPARδ) stimulation. J Biol Chem. 2006;281:32164–32174. doi: 10.1074/jbc.M602369200. [DOI] [PubMed] [Google Scholar]
- 8.Shi Y, Hon M, Evans RM. The peroxisome proliferator-activated receptor δ, an integrator of transcriptional repression and nuclear receptor signaling. Proc Natl Acad Sci U S A. 2002;99:2613–2618. doi: 10.1073/pnas.052707099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Girroir EE, Hollingshead HE, He P, Zhu B, Perdew GH, Peters JM. Quantitative expression patterns of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) protein in mice. Biochem Biophys Res Commun. 2008;371:456–461. doi: 10.1016/j.bbrc.2008.04.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leibowitz MD, Fievet C, Hennuyer N, Peinado-Onsurbe J, Duez H, Bergera J, Cullinan CA, Sparrow CP, Baffic J, Berger GD, Santini C, Marquis RW, Tolman RL, Smith RG, Moller DE, Auwerx J. Activation of PPARδ alters lipid metabolism in db/db mice. FEBS Lett. 2000;473:333–336. doi: 10.1016/s0014-5793(00)01554-4. [DOI] [PubMed] [Google Scholar]
- 11.Oliver WR, Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, Xu HE, Sternbach DD, Kliewer SA, Hansen BC, Willson TM. A selective peroxisome proliferator-activated receptor δ agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, Watanabe M, Magoori K, Ioka RX, Tachibana K, Watanabe Y, Uchiyama Y, Sumi K, Iguchi H, Ito S, Doi T, Hamakubo T, Naito M, Auwerx J, Yanagisawa M, Kodama T, Sakai J. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci U S A. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee CH, Olson P, Hevener A, Mehl I, Chong LW, Olefsky JM, Gonzalez FJ, Ham J, Kang H, Peters JM, Evans RM. PPARδ regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci U S A. 2006;103:3444–3449. doi: 10.1073/pnas.0511253103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, Kang H, Shaw RJ, Evans RM. AMPK and PPARδ agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kilgore KS, Billin AN. PPARβ/δ ligands as modulators of the inflammatory response. Curr Opin Investig Drugs. 2008;9:463–469. [PubMed] [Google Scholar]
- 16.Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, Yamazaki Y, Kuroda J, Shibata N. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARδ agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 17.Qin X, Xie X, Fan Y, Tian J, Guan Y, Wang X, Zhu Y, Wang N. Peroxisome proliferator-activated receptor-δ induces insulin-induced gene-1 and suppresses hepatic lipogenesis in obese diabetic mice. Hepatology. 2008;48:432–441. doi: 10.1002/hep.22334. [DOI] [PubMed] [Google Scholar]
- 18.Shan W, Nicol CJ, Ito S, Bility MT, Kennett MJ, Ward JM, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-β/δ protects against chemically induced liver toxicity in mice. Hepatology. 2008;47:225–235. doi: 10.1002/hep.21925. [DOI] [PubMed] [Google Scholar]
- 19.Shan W, Palkar PS, Murray IA, McDevitt EI, Kennett MJ, Kang BH, Isom HC, Perdew GH, Gonzalez FJ, Peters JM. Ligand activation of peroxisome proliferator-activated receptor β/δ (PPARβ/δ) attenuates carbon tetrachloride hepatotoxicity by downregulating proinflammatory gene expression. Toxicol Sci. 2008;105:418–428. doi: 10.1093/toxsci/kfn142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polak PE, Kalinin S, Dello Russo C, Gavrilyuk V, Sharp A, Peters JM, Richardson J, Willson TM, Weinberg G, Feinstein DL. Protective effects of a peroxisome proliferator-activated receptor-β/δ agonist in experimental autoimmune encephalomyelitis. Journal of neuroimmunology. 2005;168:65–75. doi: 10.1016/j.jneuroim.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 21.Michalik L, Desvergne B, Tan NS, Basu-Modak S, Escher P, Rieusset J, Peters JM, Kaya G, Gonzalez FJ, Zakany J, Metzger D, Chambon P, Duboule D, Wahli W. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)α and PPARβ mutant mice. J Cell Biol. 2001;154:799–814. doi: 10.1083/jcb.200011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters JM, Lee SST, Li W, Ward JM, Gavrilova O, Everett C, Reitman ML, Hudson LD, Gonzalez FJ. Growth, adipose, brain and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor b(d) Molecular and Cellular Biology. 2000;20:5119–5128. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Man MQ, Barish GD, Schmuth M, Crumrine D, Barak Y, Chang S, Jiang Y, Evans RM, Elias PM, Feingold KR. Deficiency of PPARβ/δ in the epidermis results in defective cutaneous permeability barrier homeostasis and increased inflammation. J Invest Dermatol. 2007;128:370–377. doi: 10.1038/sj.jid.5701026. [DOI] [PubMed] [Google Scholar]
- 24.Borland MG, Foreman JE, Girroir EE, Zolfaghari R, Sharma AK, Amin SM, Gonzalez FJ, Ross AC, Peters JM. Ligand Activation of Peroxisome Proliferator-Activated Receptor-β/δ (PPARβ/δ) Inhibits Cell Proliferation in Human HaCaT Keratinocytes. Mol Pharmacol. 2008;74:1429–1442. doi: 10.1124/mol.108.050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burdick AD, Bility MT, Girroir EE, Billin AN, Willson TM, Gonzalez FJ, Peters JM. Ligand activation of peroxisome proliferator-activated receptor-β/δ(PPARβ/δ) inhibits cell growth of human N/TERT-1 keratinocytes. Cell Signal. 2007;19:1163–1171. doi: 10.1016/j.cellsig.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westergaard M, Henningsen J, Svendsen ML, Johansen C, Jensen UB, Schroder HD, Kratchmarova I, Berge RK, Iversen L, Bolund L, Kragballe K, Kristiansen K. Modulation of keratinocyte gene expression and differentiation by PPAR-selective ligands and tetradecylthioacetic acid. J Invest Dermatol. 2001;116:702–712. doi: 10.1046/j.1523-1747.2001.01329.x. [DOI] [PubMed] [Google Scholar]
- 27.Martinasso G, Maggiora M, Trombetta A, Canuto RA, Muzio G. Effects of di(2-ethylhexyl) phthalate, a widely used peroxisome proliferator and plasticizer, on cell growth in the human keratinocyte cell line NCTC 2544. J Toxicol Environ Health A. 2006;69:353–365. doi: 10.1080/15287390500227522. [DOI] [PubMed] [Google Scholar]
- 28.Kim DJ, Murray IA, Burns AM, Gonzalez FJ, Perdew GH, Peters JM. Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) inhibits epidermal cell proliferation by down-regulation of kinase activity. J Biol Chem. 2005;280:9519–9527. doi: 10.1074/jbc.M413808200. [DOI] [PubMed] [Google Scholar]
- 29.Kim DJ, Bility MT, Billin AN, Willson TM, Gonzalez FJ, Peters JM. PPARβ/δ selectively induces differentiation and inhibits cell proliferation. Cell Death Differ. 2006;13:53–60. doi: 10.1038/sj.cdd.4401713. [DOI] [PubMed] [Google Scholar]
- 30.Bility MT, Devlin-Durante MK, Blazanin N, Glick AB, Ward JM, Kang BH, Kennett MJ, Gonzalez FJ, Peters JM. Ligand activation of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) inhibits chemically-induced skin tumorigenesis. Carcinogenesis. 2008;29:2406–2414. doi: 10.1093/carcin/bgn219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim DJ, Akiyama TE, Harman FS, Burns AM, Shan W, Ward JM, Kennett MJ, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor β (δ)-dependent regulation of ubiquitin C expression contributes to attenuation of skin carcinogenesis. J Biol Chem. 2004;279:23719–23727. doi: 10.1074/jbc.M312063200. [DOI] [PubMed] [Google Scholar]
- 32.Schmuth M, Haqq CM, Cairns WJ, Holder JC, Dorsam S, Chang S, Lau P, Fowler AJ, Chuang G, Moser AH, Brown BE, Mao-Qiang M, Uchida Y, Schoonjans K, Auwerx J, Chambon P, Willson TM, Elias PM, Feingold KR. Peroxisome proliferator-activated receptor (PPAR)-β/δ stimulates differentiation and lipid accumulation in keratinocytes. J Invest Dermatol. 2004;122:971–983. doi: 10.1111/j.0022-202X.2004.22412.x. [DOI] [PubMed] [Google Scholar]
- 33.Romanowska M, al Yacoub N, Seidel H, Donandt S, Gerken H, Phillip S, Haritonova N, Artuc M, Schweiger S, Sterry W, Foerster J. PPARδ enhances keratinocyte proliferation in psoriasis and induces heparin-binding EGF-like growth factor. J Invest Dermatol. 2008;128:110–124. doi: 10.1038/sj.jid.5700943. [DOI] [PubMed] [Google Scholar]
- 34.Shearer BG, Hoekstra WJ. Recent advances in peroxisome proliferator-activated receptor science. Curr Med Chem. 2003;10:267–280. doi: 10.2174/0929867033368295. [DOI] [PubMed] [Google Scholar]
- 35.Matsuura H, Adachi H, Smart RC, Xu X, Arata J, Jetten AM. Correlation between expression of peroxisome proliferator-activated receptor β and squamous differentiation in epidermal and tracheobronchial epithelial cells. Mol Cell Endocrinol. 1999;147:85–92. doi: 10.1016/s0303-7207(98)00214-7. [DOI] [PubMed] [Google Scholar]
- 36.Tan NS, Michalik L, Noy N, Yasmin R, Pacot C, Heim M, Fluhmann B, Desvergne B, Wahli W. Critical roles of PPARβ/δ in keratinocyte response to inflammation. Genes Dev. 2001;15:3263–3277. doi: 10.1101/gad.207501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paramio JM, Casanova ML, Segrelles C, Mittnacht S, Lane EB, Jorcano JL. Modulation of cell proliferation by cytokeratins K10 and K16. Mol Cell Biol. 1999;19:3086–3094. doi: 10.1128/mcb.19.4.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lippens S, Kockx M, Knaapen M, Mortier L, Polakowska R, Verheyen A, Garmyn M, Zwijsen A, Formstecher P, Huylebroeck D, Vandenabeele P, Declercq W. Epidermal differentiation does not involve the pro-apoptotic executioner caspases, but is associated with caspase-14 induction and processing. Cell Death Differ. 2000;7:1218–1224. doi: 10.1038/sj.cdd.4400785. [DOI] [PubMed] [Google Scholar]
- 39.Weil M, Raff MC, Braga VM. Caspase activation in the terminal differentiation of human epidermal keratinocytes. Curr Biol. 1999;9:361–364. doi: 10.1016/s0960-9822(99)80162-6. [DOI] [PubMed] [Google Scholar]
- 40.Gandarillas A, Goldsmith LA, Gschmeissner S, Leigh IM, Watt FM. Evidence that apoptosis and terminal differentiation of epidermal keratinocytes are distinct processes. Exp Dermatol. 1999;8:71–79. doi: 10.1111/j.1600-0625.1999.tb00350.x. [DOI] [PubMed] [Google Scholar]
- 41.Polakowska RR, Piacentini M, Bartlett R, Goldsmith LA, Haake AR. Apoptosis in human skin development: morphogenesis, periderm, and stem cells. Dev Dyn. 1994;199:176–188. doi: 10.1002/aja.1001990303. [DOI] [PubMed] [Google Scholar]
- 42.Di-Poi N, Tan NS, Michalik L, Wahli W, Desvergne B. Antiapoptotic role of PPARb in keratinocytes via transcriptional control of the Akt1 signaling pathway. Molecular Cell. 2002;10:721–733. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- 43.Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007;129:723–733. doi: 10.1016/j.cell.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang P, Jiang B, Huang X, Xiao W, Zhang P, Yang X, Long J, Xiao X. Anti-apoptotic role of EGF in HaCaT keratinocytes via a PPARβ-dependent mechanism. Wound Repair Regen. 2008;16:691–698. doi: 10.1111/j.1524-475X.2008.00419.x. [DOI] [PubMed] [Google Scholar]
- 45.Roop DR, Huitfeldt H, Kilkenny A, Yuspa SH. Regulated expression of differentiation-associated keratins in cultured epidermal cells detected by monospecific antibodies to unique peptides of mouse epidermal keratins. Differentiation. 1987;35:143–150. doi: 10.1111/j.1432-0436.1987.tb00162.x. [DOI] [PubMed] [Google Scholar]
- 46.Tyner AL, Fuchs E. Evidence for posttranscriptional regulation of the keratins expressed during hyperproliferation and malignant transformation in human epidermis. J Cell Biol. 1986;103:1945–1955. doi: 10.1083/jcb.103.5.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schug TT, Berry DC, Toshkov IA, Cheng L, Nikitin AY, Noy N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARβ/δ to RAR. Proc Natl Acad Sci U S A. 2008;105:7546–7551. doi: 10.1073/pnas.0709981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rieck M, Meissner W, Ries S, Muller-Brusselbach S, Muller R. Ligand-mediated regulation of peroxisome proliferator-activated receptor (PPAR) β/δ: a comparative analysis of PPAR-selective agonists and all-trans retinoic acid. Mol Pharmacol. 2008;74:1269–1277. doi: 10.1124/mol.108.050625. [DOI] [PubMed] [Google Scholar]
- 49.Chen WC, Sass JO, Seltmann H, Nau H, Orfanos CE, Zouboulis CC. Biological effects and metabolism of 9-cis-retinoic acid and its metabolite 9,13-di-cis-retinoic acid in HaCaT keratinocytes in vitro: comparison with all-trans-retinoic acid. Arch Dermatol Res. 2000;292:612–620. doi: 10.1007/s004030000189. [DOI] [PubMed] [Google Scholar]
- 50.Hansen LA, Sigman CC, Andreola F, Ross SA, Kelloff GJ, De Luca LM. Retinoids in chemoprevention and differentiation therapy. Carcinogenesis. 2000;21:1271–1279. [PubMed] [Google Scholar]
- 51.Kanekura T, Higashi Y, Kanzaki T. Inhibitory effects of 9-cis-retinoic acid and pyrrolidinedithiocarbamate on cyclooxygenase (COX)-2 expression and cell growth in human skin squamous carcinoma cells. Cancer Lett. 2000;161:177–183. doi: 10.1016/s0304-3835(00)00604-2. [DOI] [PubMed] [Google Scholar]
- 52.Klaassen I, Brakenhoff RH, Smeets SJ, Snow GB, Braakhuis BJ. Metabolism and growth inhibition of four retinoids in head and neck squamous normal and malignant cells. Br J Cancer. 2001;85:630–635. doi: 10.1054/bjoc.2001.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Memezawa A, Takada I, Takeyama K, Igarashi M, Ito S, Aiba S, Kato S, Kouzmenko AP. Id2 gene-targeted crosstalk between Wnt and retinoid signaling regulates proliferation in human keratinocytes. Oncogene. 2007;26:5038–5045. doi: 10.1038/sj.onc.1210320. [DOI] [PubMed] [Google Scholar]
- 54.Tong PS, Mayes DM, Wheeler LA. Differential effects of retinoids on DNA synthesis in calcium-regulated murine epidermal keratinocyte cultures. J Invest Dermatol. 1988;90:861–868. doi: 10.1111/1523-1747.ep12462107. [DOI] [PubMed] [Google Scholar]
- 55.Chen LC, Kirchhoff S, De Luca LM. Effect of excess dietary retinoic acid on skin papilloma and carcinoma formation induced by a complete carcinogenesis protocol in female Sencar mice. Cancer Lett. 1994;78:63–67. doi: 10.1016/0304-3835(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 56.Chen LC, Sly L, De Luca LM. High dietary retinoic acid prevents malignant conversion of skin papillomas induced by a two-stage carcinogenesis protocol in female SENCAR mice. Carcinogenesis. 1994;15:2383–2386. doi: 10.1093/carcin/15.10.2383. [DOI] [PubMed] [Google Scholar]
- 57.Tennenbaum T, Lowry D, Darwiche N, Morgan DL, Gartsbein M, Hansen L, De Luca LM, Hennings H, Yuspa SH. Topical retinoic acid reduces skin papilloma formation but resistant papillomas are at high risk for malignant conversion. Cancer Res. 1998;58:1435–1443. [PubMed] [Google Scholar]
- 58.Verma AK. Inhibition of both stage I and stage II mouse skin tumour promotion by retinoic acid and the dependence of inhibition of tumor promotion on the duration of retinoic acid treatment. Cancer Res. 1987;47:5097–5101. [PubMed] [Google Scholar]
- 59.Verma AK. Inhibition of tumor promoter 12-O-tetradecanoylphorbol-13-acetate-induced synthesis of epidermal ornithine decarboxylase messenger RNA and diacylglycerol-promoted mouse skin tumor formation by retinoic acid. Cancer Res. 1988;48:2168–2173. [PubMed] [Google Scholar]
- 60.Verma AK, Slaga TJ, Wertz PW, Mueller GC, Boutwell RK. Inhibition of skin tumor promotion by retinoic acid and its metabolite 5,6-epoxyretinoic acid. Cancer Res. 1980;40:2367–2371. [PubMed] [Google Scholar]
- 61.Xu H, Cheepala S, McCauley E, Coombes K, Xiao L, Fischer SM, Clifford JL. Chemoprevention of skin carcinogenesis by phenylretinamides: retinoid receptor-independent tumor suppression. Clin Cancer Res. 2006;12:969–979. doi: 10.1158/1078-0432.CCR-05-1648. [DOI] [PubMed] [Google Scholar]
- 62.Shirotani M, Yui Y, Hattori R, Kawai C. U-61,431F, a stable prostacyclin analogue, inhibits the proliferation of bovine vascular smooth muscle cells with little antiproliferative effect on endothelial cells. Prostaglandins. 1991;41:97–110. doi: 10.1016/0090-6980(91)90023-9. [DOI] [PubMed] [Google Scholar]
- 63.Müller-Brüsselbach S, Kömhoff M, Rieck M, Meissner W, Kaddatz K, Adamkiewicz J, Keil B, Klose KJ, Moll R, Burdick AD, Peters JM, Müller R. Deregulation of tumor angiogenesis and blockade of tumor growth in PPARβ-deficient mice. Embo J. 2007;26:3686–3698. doi: 10.1038/sj.emboj.7601803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liou JY, Lee S, Ghelani D, Matijevic-Aleksic N, Wu KK. Protection of endothelial survival by peroxisome proliferator-activated receptor-δ mediated 14-3-3 upregulation. Arterioscler Thromb Vasc Biol. 2006;26:1481–1487. doi: 10.1161/01.ATV.0000223875.14120.93. [DOI] [PubMed] [Google Scholar]
- 65.Piqueras L, Reynolds AR, Hodivala-Dilke KM, Alfranca A, Redondo JM, Hatae T, Tanabe T, Warner TD, Bishop-Bailey D. Activation of PPARβ/δ induces endothelial cell proliferation and angiogenesis. Arterioscler Thromb Vasc Biol. 2007;27:63–69. doi: 10.1161/01.ATV.0000250972.83623.61. [DOI] [PubMed] [Google Scholar]
- 66.Stephen RL, Gustafsson MC, Jarvis M, Tatoud R, Marshall BR, Knight D, Ehrenborg E, Harris AL, Wolf CR, Palmer CN. Activation of peroxisome proliferator-activated receptor δ stimulates the proliferation of human breast and prostate cancer cell lines. Cancer Res. 2004;64:3162–3170. doi: 10.1158/0008-5472.can-03-2760. [DOI] [PubMed] [Google Scholar]
- 67.Han JK, Lee HS, Yang HM, Hur J, Jun SI, Kim JY, Cho CH, Koh GY, Peters JM, Park KW, Cho HJ, Lee HY, Kang HJ, Oh BH, Park YB, Kim HS. Peroxisome proliferator-activated receptor-δ agonist enhances vasculogenesis by regulating endothelial progenitor cells through genomic and nongenomic activations of the phosphatidylinositol 3-kinase/Akt pathway. Circulation. 2008;118:1021–1033. doi: 10.1161/CIRCULATIONAHA.108.777169. [DOI] [PubMed] [Google Scholar]
- 68.Jiang B, Liang P, Zhang B, Huang X, Xiao X. Enhancement of PPAR-β activity by repetitive low-grade H2O2 stress protects human umbilical vein endothelial cells from subsequent oxidative stress-induced apoptosis. Free Radic Biol Med. 2009;46:555–563. doi: 10.1016/j.freeradbiomed.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 69.Clapp LH, Finney P, Turcato S, Tran S, Rubin LJ, Tinker A. Differential effects of stable prostacyclin analogs on smooth muscle proliferation and cyclic AMP generation in human pulmonary artery. Am J Respir Cell Mol Biol. 2002;26:194–201. doi: 10.1165/ajrcmb.26.2.4695. [DOI] [PubMed] [Google Scholar]
- 70.Hara S, Morishita R, Tone Y, Yokoyama C, Inoue H, Kaneda Y, Ogihara T, Tanabe T. Overexpression of prostacyclin synthase inhibits growth of vascular smooth muscle cells. Biochem Biophys Res Commun. 1995;216:862–867. doi: 10.1006/bbrc.1995.2701. [DOI] [PubMed] [Google Scholar]
- 71.Li RC, Cindrova-Davies T, Skepper JN, Sellers LA. Prostacyclin induces apoptosis of vascular smooth muscle cells by a cAMP-mediated inhibition of extracellular signal-regulated kinase activity and can counteract the mitogenic activity of endothelin-1 or basic fibroblast growth factor. Circ Res. 2004;94:759–767. doi: 10.1161/01.RES.0000121568.40692.97. [DOI] [PubMed] [Google Scholar]
- 72.Lim HJ, Lee S, Park JH, Lee KS, Choi HE, Chung KS, Lee HH, Park HY. PPARδ agonist L-165041 inhibits rat vascular smooth muscle cell proliferation and migration via inhibition of cell cycle. Atherosclerosis. 2009;202:446–454. doi: 10.1016/j.atherosclerosis.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 73.Lin H, Lee JL, Hou HH, Chung CP, Hsu SP, Juan SH. Molecular mechanisms of the antiproliferative effect of beraprost, a prostacyclin agonist, in murine vascular smooth muscle cells. Journal of cellular physiology. 2008;214:434–441. doi: 10.1002/jcp.21214. [DOI] [PubMed] [Google Scholar]
- 74.Hellemans K, Michalik L, Dittie A, Knorr A, Rombouts K, De Jong J, Heirman C, Quartier E, Schuit F, Wahli W, Geerts A. Peroxisome proliferator-activated receptor-β signaling contributes to enhanced proliferation of hepatic stellate cells. Gastroenterology. 2003;124:184–201. doi: 10.1053/gast.2003.50015. [DOI] [PubMed] [Google Scholar]
- 75.Ali FY, Egan K, Fitzgerald GA, Desvergne B, Wahli W, Bishop-Bailey D, Warner TD, Mitchell JA. Role of Prostacyclin Receptor Versus PPARβ with Treprostinil Sodium on Lung Fibroblast Proliferation. Am J Respir Cell Mol Biol. 2005;34:242–246. doi: 10.1165/rcmb.2005-0289OC. [DOI] [PubMed] [Google Scholar]
- 76.Zhang H, Pi R, Li R, Wang P, Tang F, Zhou S, Gao J, Jiang J, Chen S, Liu P. PPARβ/δ activation inhibits angiotensin II-induced collagen type I expression in rat cardiac fibroblasts. Arch Biochem Biophys. 2007;460:25–32. doi: 10.1016/j.abb.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 77.Teunissen BE, Smeets PJ, Willemsen PH, De Windt LJ, Van der Vusse GJ, Van Bilsen M. Activation of PPARδ inhibits cardiac fibroblast proliferation and the transdifferentiation into myofibroblasts. Cardiovasc Res. 2007;75:519–529. doi: 10.1016/j.cardiores.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 78.Zeng L, Geng Y, Tretiakova M, Yu X, Sicinski P, Kroll TG. Peroxisome proliferator-activated receptor-δ induces cell proliferation by a cyclin E1-dependent mechanism and is up-regulated in thyroid tumors. Cancer Res. 2008;68:6578–6586. doi: 10.1158/0008-5472.CAN-08-0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Letavernier E, Perez J, Joye E, Bellocq A, Fouqueray B, Haymann JP, Heudes D, Wahli W, Desvergne B, Baud L. Peroxisome proliferator-activated receptor β/δ exerts a strong protection from ischemic acute renal failure. J Am Soc Nephrol. 2005;16:2395–2402. doi: 10.1681/ASN.2004090802. [DOI] [PubMed] [Google Scholar]
- 80.Pesant M, Sueur S, Dutartre P, Tallandier M, Grimaldi PA, Rochette L, Connat JL. Peroxisome proliferator-activated receptor δ (PPARδ) activation protects H9c2 cardiomyoblasts from oxidative stress-induced apoptosis. Cardiovasc Res. 2006;69:440–449. doi: 10.1016/j.cardiores.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 81.Hatae T, Wada M, Yokoyama C, Shimonishi M, Tanabe T. Prostacyclin-dependent Apoptosis Mediated by PPARδ. J Biol Chem. 2001;276:46260–46267. doi: 10.1074/jbc.M107180200. [DOI] [PubMed] [Google Scholar]
- 82.Clark J, Nasrallah R, Hebert RL. The PPARδ Ligand GW501516 Reduces Growth but Not Apoptosis in Mouse Inner Medullary Collecting Duct Cells. PPAR research. 2009;2009:706283. doi: 10.1155/2009/706283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.He TC, Chan TA, Vogelstein B, Kinzler KW. PPARd is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gupta RA, Wang D, Katkuri S, Wang H, Dey SK, DuBois RN. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-δ accelerates intestinal adenoma growth. Nat Med. 2004;10:245–247. doi: 10.1038/nm993. [DOI] [PubMed] [Google Scholar]
- 85.Wang D, Wang H, Guo Y, Ning W, Katkuri S, Wahli W, Desvergne B, Dey SK, Dubois RN. Crosstalk between peroxisome proliferator-activated receptor δ and VEGF stimulates cancer progression. Proc Natl Acad Sci U S A. 2006;103:19069–19074. doi: 10.1073/pnas.0607948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang D, Wang H, Shi Q, Katkuri S, Walhi W, Desvergne B, Das SK, Dey SK, DuBois RN. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor δ. Cancer Cell. 2004;6:285–295. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 87.Foreman JE, Sorg JM, McGinnis KS, Rigas B, Williams JL, Clapper ML, Gonzales FJ, Peters JM. Regulation of peroxisome proliferator-activated receptor-β/δ by the APC/β-CATENIN pathway and nonsteroidal anti-inflammatory drugs. Molecular Carcinogenesis. 2009 doi: 10.1002/mc.20546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hollingshead HE, Killins RL, Borland MG, Girroir EE, Billin AN, Willson TM, Sharma AK, Amin S, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) ligands do not potentiate growth of human cancer cell lines. Carcinogenesis. 2007;28:2641–2649. doi: 10.1093/carcin/bgm183. [DOI] [PubMed] [Google Scholar]
- 89.Marin HE, Peraza MA, Billin AN, Willson TM, Ward JM, Kennett MJ, Gonzalez FJ, Peters JM. Ligand activation of peroxisome proliferator-activated receptor β/δ (PPARβ/δ) inhibits colon carcinogenesis. Cancer Res. 2006;66:4394–4401. doi: 10.1158/0008-5472.CAN-05-4277. [DOI] [PubMed] [Google Scholar]
- 90.Yang L, Zhou ZG, Zheng XL, Wang L, Yu YY, Zhou B, Gu J, Li Y. RNA Interference Against Peroxisome Proliferator-Activated Receptor δ Gene Promotes Proliferation of Human Colorectal Cancer Cells. Dis Colon Rectum. 2008;51:318–328. doi: 10.1007/s10350-007-9145-8. [DOI] [PubMed] [Google Scholar]
- 91.Harman FS, Nicol CJ, Marin HE, Ward JM, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-δ attenuates colon carcinogenesis. Nat Med. 2004;10:481–483. doi: 10.1038/nm1026. [DOI] [PubMed] [Google Scholar]
- 92.Hollingshead HE, Borland MG, Billin AN, Willson TM, Gonzalez FJ, Peters JM. Ligand activation of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) and inhibition of cyclooxygenase 2 (COX2) attenuate colon carcinogenesis through independent signaling mechanisms. Carcinogenesis. 2008;29:169–176. doi: 10.1093/carcin/bgm209. [DOI] [PubMed] [Google Scholar]
- 93.Kohno H, Suzuki R, Sugie S, Tanaka T. Suppression of colitis-related mouse colon carcinogenesis by a COX-2 inhibitor and PPAR ligands. BMC Cancer. 2005;5:46. doi: 10.1186/1471-2407-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tenenbaum A, Boyko V, Fisman EZ, Goldenberg I, Adler Y, Feinberg MS, Motro M, Tanne D, Shemesh J, Schwammenthal E, Behar S. Does the lipid-lowering peroxisome proliferator-activated receptors ligand bezafibrate prevent colon cancer in patients with coronary artery disease? Cardiovascular diabetology. 2008;7:18. doi: 10.1186/1475-2840-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yin Y, Russell RG, Dettin LE, Bai R, Wei ZL, Kozikowski AP, Kopleovich L, Glazer RI. Peroxisome proliferator-activated receptor δ and γ agonists differentially alter tumor differentiation and progression during mammary carcinogenesis. Cancer Res. 2005;65:3950–3957. doi: 10.1158/0008-5472.CAN-04-3990. [DOI] [PubMed] [Google Scholar]
- 96.Girroir EE, Hollingshead HE, Billin AN, Willson TM, Robertson GP, Sharma AK, Amin S, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) ligands inhibit growth of UACC903 and MCF7 human cancer cell lines. Toxicology. 2008;243:236–243. doi: 10.1016/j.tox.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fukumoto K, Yano Y, Virgona N, Hagiwara H, Sato H, Senba H, Suzuki K, Asano R, Yamada K, Yano T. Peroxisome proliferator-activated receptor δ as a molecular target to regulate lung cancer cell growth. FEBS Lett. 2005;579:3829–3836. doi: 10.1016/j.febslet.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 98.Muller-Brusselbach S, Ebrahimsade S, Jakel J, Eckhardt J, Rapp UR, Peters JM, Moll R, Muller R. Growth of transgenic RAF-induced lung adenomas is increased in mice with a disrupted PPARβ/δ gene. Int J Oncol. 2007;31:607–611. [PubMed] [Google Scholar]
- 99.Keith RL, Miller YE, Hoshikawa Y, Moore MD, Gesell TL, Gao B, Malkinson AM, Golpon HA, Nemenoff RA, Geraci MW. Manipulation of pulmonary prostacyclin synthase expression prevents murine lung cancer. Cancer Res. 2002;62:734–740. [PubMed] [Google Scholar]
- 100.Keith RL, Miller YE, Hudish TM, Girod CE, Sotto-Santiago S, Franklin WA, Nemenoff RA, March TH, Nana-Sinkam SP, Geraci MW. Pulmonary prostacyclin synthase overexpression chemoprevents tobacco smoke lung carcinogenesis in mice. Cancer Res. 2004;64:5897–5904. doi: 10.1158/0008-5472.CAN-04-1070. [DOI] [PubMed] [Google Scholar]
- 101.Han S, Ritzenthaler JD, Wingerd B, Roman J. Activation of peroxisome proliferator-activated receptor β/δ (PPARβ/δ) increases the expression of prostaglandin E2 receptor subtype EP4. The roles of phosphatidylinositol 3-kinase and CCAAT/enhancer-binding protein β. J Biol Chem. 2005;280:33240–33249. doi: 10.1074/jbc.M507617200. [DOI] [PubMed] [Google Scholar]
- 102.Han S, Ritzenthaler JD, Zheng Y, Roman J. PPARβ/δ agonist stimulates human lung carcinoma cell growth through inhibition of PTEN expression: the involvement of PI3K and NF-kappaB signals. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1238–1249. doi: 10.1152/ajplung.00017.2008. [DOI] [PubMed] [Google Scholar]
- 103.Pedchenko TV, Gonzalez AL, Wang D, DuBois RN, Massion PP. Peroxisome proliferator-activated receptor β/δ expression and activation in lung cancer. Am J Respir Cell Mol Biol. 2008;39:689–696. doi: 10.1165/rcmb.2007-0426OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.He P, Borland MG, Zhu B, Sharma AK, Amin S, El-Bayoumy K, Gonzalez FJ, Peters JM. Effect of ligand activation of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) in human lung cancer cell lines. Toxicology. 2008;254:112–117. doi: 10.1016/j.tox.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Glinghammar B, Skogsberg J, Hamsten A, Ehrenborg E. PPARδ activation induces COX-2 gene expression and cell proliferation in human hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2003;308:361–368. doi: 10.1016/s0006-291x(03)01384-6. [DOI] [PubMed] [Google Scholar]
- 106.Xu L, Han C, Lim K, Wu T. Cross-talk between Peroxisome Proliferator-Activated Receptor δ and Cytosolic Phospholipase A2α/Cyclooxygenase-2/Prostaglandin E2 Signaling Pathways in Human Hepatocellular Carcinoma Cells. Cancer Res. 2006;66:11859–11868. doi: 10.1158/0008-5472.CAN-06-1445. [DOI] [PubMed] [Google Scholar]
- 107.Xu L, Han C, Wu T. A novel positive feedback loop between PPARδ and PGE2 signaling pathways for human cholangiocarcinoma cell growth. J Biol Chem. 2006;281:33982–33996. doi: 10.1074/jbc.M600135200. [DOI] [PubMed] [Google Scholar]
- 108.Berge K, Tronstad KJ, Flindt EN, Rasmussen TH, Madsen L, Kristiansen K, Berge RK. Tetradecylthioacetic acid inhibits growth of rat glioma cells ex vivo and in vivo via PPAR-dependent and PPAR-independent pathways. Carcinogenesis. 2001;22:1747–1755. doi: 10.1093/carcin/22.11.1747. [DOI] [PubMed] [Google Scholar]
- 109.Otsuyama KI, Ma Z, Abroun S, Amin J, Shamsasenjan K, Asaoku H, Kawano MM. PPARβ-mediated growth suppression of baicalein and dexamethasone in human myeloma cells. Leukemia. 2007;21:187–190. doi: 10.1038/sj.leu.2404462. [DOI] [PubMed] [Google Scholar]
- 110.Di Loreto S, D'Angelo B, D'Amico MA, Benedetti E, Cristiano L, Cinque B, Cifone MG, Ceru MP, Festuccia C, Cimini A. PPARβ agonists trigger neuronal differentiation in the human neuroblastoma cell line SH-SY5Y. Journal of cellular physiology. 2007;211:837–847. doi: 10.1002/jcp.20996. [DOI] [PubMed] [Google Scholar]
- 111.Ou YC, Yang CR, Cheng CL, Raung SL, Hung YY, Chen CJ. Indomethacin induces apoptosis in 786-O renal cell carcinoma cells by activating mitogen-activated protein kinases and AKT. Eur J Pharmacol. 2007;563:49–60. doi: 10.1016/j.ejphar.2007.01.071. [DOI] [PubMed] [Google Scholar]
- 112.Delage B, Rullier A, Capdepont M, Rullier E, Cassand P. The effect of body weight on altered expression of nuclear receptors and cyclooxygenase-2 in human colorectal cancers. Nutr J. 2007;6:20. doi: 10.1186/1475-2891-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Feilchenfeldt J, Brundler MA, Soravia C, Totsch M, Meier CA. Peroxisome proliferator-activated receptors (PPARs) and associated transcription factors in colon cancer: reduced expression of PPARγ-coactivator 1 (PGC-1) Cancer Lett. 2004;203:25–33. doi: 10.1016/j.canlet.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 114.Knutsen HK, Olstorn HB, Paulsen JE, Husoy T, Goverud IL, Loberg EM, Kristiansen K, Alexander J. Increased levels of PPARβ/δ and cyclin D1 in flat dysplastic ACF and adenomas in Apc(Min/+) mice. Anticancer Res. 2005;25:3781–3789. [PubMed] [Google Scholar]
- 115.Ouyang N, Williams JL, Rigas B. NO-donating aspirin isomers downregulate peroxisome proliferator-activated receptor (PPAR)δ expression in APC(min/+) mice proportionally to their tumor inhibitory effect: Implications for the role of PPARδ in carcinogenesis. Carcinogenesis. 2006;27:232–239. doi: 10.1093/carcin/bgi221. [DOI] [PubMed] [Google Scholar]
- 116.Takayama O, Yamamoto H, Damdinsuren B, Sugita Y, Ngan CY, Xu X, Tsujino T, Takemasa I, Ikeda M, Sekimoto M, Matsuura N, Monden M. Expression of PPARδ in multistage carcinogenesis of the colorectum: implications of malignant cancer morphology. Br J Cancer. 2006;95:889–895. doi: 10.1038/sj.bjc.6603343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liou JY, Ghelani D, Yeh S, Wu KK. Nonsteroidal anti-inflammatory drugs induce colorectal cancer cell apoptosis by suppressing 14-3-3epsilon. Cancer Res. 2007;67:3185–3191. doi: 10.1158/0008-5472.CAN-06-3431. [DOI] [PubMed] [Google Scholar]
- 118.Shureiqi I, Jiang W, Zuo X, Wu Y, Stimmel JB, Leesnitzer LM, Morris JS, Fan HZ, Fischer SM, Lippman SM. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-δ to induce apoptosis in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003;100:9968–9973. doi: 10.1073/pnas.1631086100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tuder RM, Zaiman AL. Prostacyclin analogs as the brakes for pulmonary artery smooth muscle cell proliferation: is it sufficient to treat severe pulmonary hypertension? Am J Respir Cell Mol Biol. 2002;26:171–174. doi: 10.1165/ajrcmb.26.2.f230. [DOI] [PubMed] [Google Scholar]