

Summary
Fusion of HIV-1 and target cells is mediated by the envelope protein gp41 that undergoes a series of conformational changes during the process of infection. Knowledge of the structural biology of gp41 allows the design of potent peptide inhibitors that prevent the virus from entering lymphocytes and macrophages. The design of such inhibitors is the subject of this review.
Introduction
At the end of 2007, almost three decades after the discovery of HIV-1/AIDS, 33 million people were estimated to have this infection and 25 million individuals have died from the disease. In the design of new therapeutic strategies for HIV-1 infections an epicenter has been the synergy between peptide chemistry and structural biology. A milestone in AIDS therapy was the approval of the first peptide fusion inhibitor and this article will review the structural basis for the design of such molecules.
HIV-1 Biology
The rational design of anti-HIV-1 therapeutics must be based on a detailed knowledge of the biology of the virus (Figure 1). HIV-1 interacts with target cells using envelope glycoproteins (gp120 and gp41). These proteins are recognized by CD4 receptors and CCR5 (macrophage) or CXCR4 (T cell) co-receptors leading to membrane fusion followed by virus entry and subsequent integration of the viral and host genomes. Initial therapeutic strategies controlled the disease by preventing protein maturation and RNA replication to DNA. These events occur after cellular infection. This review focuses on molecular interactions involving gp41 and the structural biology of this critical membrane fusion determinant. Discussion will be limited to peptide-based fusion inhibitors. The literature pertinent to gp41-directed vaccines is not addressed. A comprehensive review of HIV entry inhibitors was published in 2007 [1].
Figure 1.
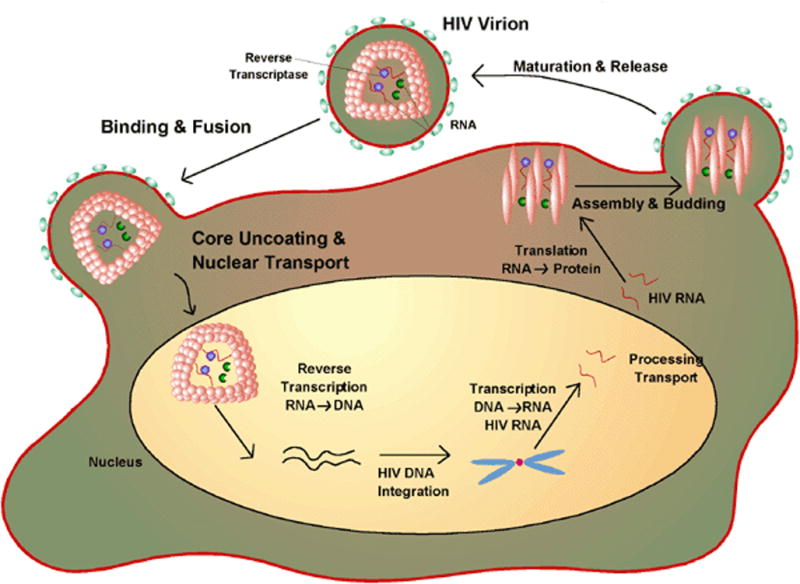
The life-cycle of HIV-1. The virus attaches to its target cell and subsequent to membrane fusion the RNA of the virus is reverse transcribed to DNA which is integrated into the cell’s genome. During it’s virulent phase the viral DNA is transcribed back to RNA which codes for various proteins required for virion assembly. Drugs can inhibit various events in the life cycle. The focus of the review is peptides that inhibit fusion to the target cell. Figure from http://www.wiley.com/legacy/college/boyer/0470003790/cutting_edge/aids_therapies/hiv_lifecycle.gif
Structures of gp41 and gp120
gp120 and gp41 are formed by proteolysis of gp160 and remain non-covalently associated in a spike on the outside of the viral membrane. gp120 is a surface subunit that interacts with CD4 inducing a conformational transformation that exposes gp120 sites that bind to either CCR5 or CXCR4. gp41 is a complex polypeptide with a fusion peptide (FP) region, two helical heptad repeat (HR) regions, an immunodominant loop region, a membrane proximal region (MPER), a transmembrane domain and a carboxyl terminal region (Figure 2). This envelope protein undergoes major conformational changes from a pre-fusion complex with gp120, to an extended structure, and finally to a hairpin fusion-active structure that juxtaposes the viral and host membranes (Figure 3A). Inhibition of this latter step prevents membrane fusion and thereby infection. Thus, the advantage of entry inhibitors is that as prophylactics they might prevent primary infection and the integration of the viral genome into the host cell genome.
Figure 2.
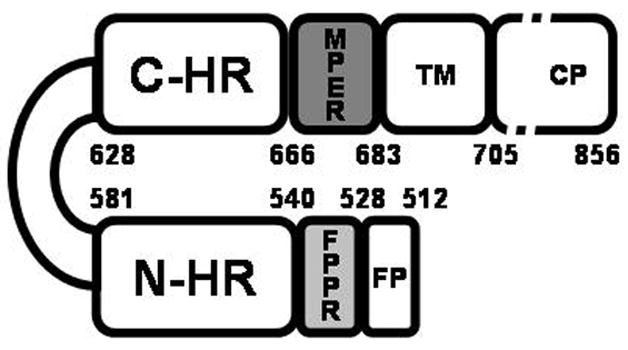
Schematic representation of HIV-1 gp41. The abbreviations represent the fusion peptide (FP); the fusion peptide proximal domain (FPPR); the N-terminal helical region (N-HR), the immunodominant loop region, the C-terminal helical region (C-HR), the membrane proximal region (MPER) and the transmembrane region (TM). The protein is numbered according to the HXB2 HIV-1 strain. The figure was adapted from [23].
Figure 3.

Mechanism of action of HIV-1 entry inhibitors. Panel A illustrates the involvement of gp41 in the fusion of the cell and viral membranes and the interference of peptide fusion inhibitors with this event. The inhibitors are believed to prevent formation of the 6-helix hairpin bundle necessary for juxtaposing the cell and viral membranes. C-HR peptides like T-20 bind to the N-HR trimeric core and N-peptides form a trimer which binds to the C-HR helix forming heterocomplexes that are defective in fusion. Panel B Schematic helical wheel diagram used in the design of C-peptides with increased helicity and water solubility. The positions that pack against the N-trimeric core (indicated in yellow) are less favored for replacement than those facing away (indicated in blue). Panel C. 5- Helix inhibitor. The hetero bundle lacks one C-peptide and thus provides a binding pocket for the C-HR peptide of gp41. Adapted from [13,38].
gp41 as a Drug Target- The Core Structure of gp41
A key finding in post-“cocktail” HIV-1 therapy was the molecular structure of the core of gp41 [2–4]. X-ray analysis of crystals formed from biosynthetic or synthetic peptide fragments of gp41 revealed the formation of a six-helix bundle (6-HB) composed of the N- and C- HR regions of gp41 (Figure 4). These domains oriented anti-parallel with N-HR which forms a core against which the C-HR peptides were tightly packed. The fact that N and C peptides containing 36 and 34 residues, respectively, formed stable crystals supported efforts to find peptide inhibitors of 6-HB bundle formation [5].
Figure 4.

X-ray structure of the gp41 6-helix bundle. The bundle was formed from synthetic N36 and C34 peptides (see Table 1). Panel A shows an end view of the 6-HB with the C-peptides in grey and the trimeric core in green. Panel B is a side view illustrating the trimeric N-core interacting with the C-peptides. The pocket binding domain (PDB; WMEWDREI) residues Trp, Trp and Ile of C34 are shown explicitly. These interact with the hydrophobic pocket of the N-trimeric core. The model was built using coordinates from the PDB 1AIK.
Finding Peptide Fusion Inhibitors
The original approach to the development of HIV-1 entry inhibitors used peptide synthesis to mimic various regions of gp41 [6–8]. Peptides homologous to the putative C-HR helical region of gp41 inhibited viral reproduction at ng/mL concentrations. One of these, originally designated DP-178, later T-20 and now FUZEON or Enfuviritide became the first approved fusion inhibitor for HIV-1 therapy. Although this drug is remarkably effective its method of administration, subcutaneous injection, is detrimental to treatment even in Western countries and it is limited to salvage treatment for cocktail resistant-patients
The Mechanism of Action of T-20 and C34
Early structure activity relationship studies, conducted in the absence of the high resolution structure of gp41, concluded that T-20 acted at the entry level, was likely a virus fusion inhibitor, and that this peptide interacted with the N-HR region of the Env protein. The availability of the high resolution structures of the gp41 core led to a putative mechanism of action wherein T-20 formed a heterocomplex that blocked the formation of the 6-HB hairpin required for membrane fusion [5]. This mechanism suggested that both C-HR and N-HR peptides could be effective inhibitors acting in a dominant negative manner to prevent viral entry. However, the structures of the gp41 core lacked some of the residues in T-20 and it was possible that this inhibitor acted differently from the C34 peptide that was used in crystal formation.
The X-ray structure of the gp41 core indicated that the trimeric N-HR peptides formed deep hydrophobic pockets that interacted with a complementary pocket binding domain (PBD) on the C-HR peptides [9]. The observation that the C-HR helices packed against the triple helical N-HR core suggested that a critical aspect of the inhibition of fusion was the propensity of the N-core to form a stable triple helix and the ability of peptides such as T-20 to assume a stable helical structure. However, analysis of T-20 and the closely related C-34 (Table 1) led to the conclusion that under physiological conditions neither peptide was highly helical [10,11]. On the basis of the structural observations and the relatively rapid mutation to resistance to T-20, attempts were made to improve the inhibitory efficacy by 1) increasing the inherent helicity of C-peptides, 2) inserting a PBD into T-20 to increase its affinity for the N-core and 3) designing N-peptides that would form a soluble, stable trimeric core.
Table 1.
Peptides Used in Studies on HIV-1 Fusion Inhibitors
  
|
Using helical wheels to guide the design (Figure 3B), analogs of C-34 were engineered with helix- promoting alanine residues and helix stabilizing salt bridges at selected (i,i+4) positions [12]. A relatively good correlation was found between the inhibitor’s inherent helicity and the Tm of the 6-HB it formed with a N-HR peptide, and its anti-HIV-1 propensity. Excitingly the T2635 analog (Table 1) was highly effective against HIV-1 strains that showed nearly complete resistance to T-20. Since T2635 has substitutions for nearly 50% of the wild-type residues, one can conclude that stable 6-HBs can form in the absence of primary sequence homology. The potency of one inhibitor T2544 (Table 1) was nearly retained for over 7 months after in vitro passaging of a clinical isolate through cultures in the presence of increasing concentrations of the drug, and despite multiple mutations in gp41. These findings suggest that resistance against these high-affinity, conformation-directed inhibitors will require so many mutations that the infectivity of the virus is diminished.
A cautionary note is that most of the above structural conclusions were based on low resolution analyses using CD, and the actual molecular contacts that are formed in various complexes are unknown. However, this caveat is tempered by a very similar study where C34 analogs with increased helicity were designed by formation of salt bridges on the solvent exposed face of the peptide [13]. The most promising inhibitor (SC34EK, Table 1) was again effective against a battery of resistant HIV-1 strains, and despite replacement of 13 of the C-34 residues it formed a crystal structure with N36 that was virtually identical to that reported for the native gp41 peptides [13]. A truncated analog of SC34EK, SC29EK and a C34 analog called Sifuviritide, both designed to have increased inherent helicity, maintained excellent anti-HIV-1 activity against primary isolates and T-20 resistant strains [14,15]. Thus, electrostatic stabilization of the C-HR helix is becoming a common strategy in entry inhibitor design.
Reevaluating the T-20 Inhibition mechanism
It has been suggested that the interaction of T-20 with HIV-1 appears to be complex and may involve multiple sites on the viral envelope proteins [1,16]. There is evidence that T-20 can bind to gp120 and that this binding is CD4 dependent [17,18] suggesting that conformational changes must precede drug-envelope protein interactions. The interaction involves the V3 loop and C4 sequences, is inhibited by C4-V3 and C4 peptides and has an electrostatic component. Although these observations are relevant to HIV-1 biology and point to a sequence of unfolding of the gp120/gp41 complex during the infectivity pathway, it is not clear whether they are germane to the therapeutic activity of T-20. If direct interactions of T-20 and gp120 at the C4/V3 domains were critical for anti-viral activity one would expect mutation to resistance at these sites. However, the major loci for T-20 resistance have been restricted to N-HR sequences [19].
C-34 contains a PBD sequence at its N-terminus (Table 1) wherein W628, W631, I635 are critical for interacting with the deep hydrophobic pocket in the N-HR trimeric core [9,20]. T-20 lacks these residues. There is evidence that a C34 like inhibitor (T649) interacts with both the LLSGIV sequence and hydrophobic pocket of N-HR and thus has a bi-modal docking mechanism with the N-HR region of gp41 resulting in increased potency [21]. Although, T-20 and C34 share 24 identical residues (Table 1) they differ significantly at their amine and carboxyl termini and C34 is often effective against T-20 resistant HIV-1 strains. Using a series of peptides that scanned through the C34 and T-20 sequences, Jiang and coworkers concluded that unlike C34, T-20 does not inhibit fusion by preventing formation of the 6-HB [16]. Rather, T-20 but not C-34 binds to lipids an interaction that is abolished by replacing its lipid binding domain sequence WNWF (LBD; blue Table 1) with ANAA [16]. Jiang has suggested that interaction of T-20 with HIV-1 may perturb formation of the membrane fusion pore.
The absolute requirement for the PBD in C34 and the LBD in T-20 suggested that more potent inhibitors might result by incorporating both of these domains in one molecule. Indeed such an analog T1249 (Table 1) was more potent than the parent compounds, had a longer half life and was active against resistant strains [1]. A systematic study showed that incorporation of both the PBD and LBD sequences into a C-HR peptide increased anti HIV-1 activity >30 fold compared to the HR peptide itself and from 6 to 15-fold compared to analogs with only the PBD or LBD, respectively [22]. Despite the fact that clinical development of T1249 has been discontinued the new knowledge from peptides containing the PBD and LBD in addition to HR sequences should help rational structure based design of even more effective inhibitors.
Extending the Boundaries
The design of many peptide entry inhibitors depended on the crystal structure of peptides spanning C34 (gp41628–661) and N36 (gp41546–581) and it was widely accepted that this represented the 6-HB core. Recent studies may be challenging this belief. When N-HR and C-HR peptides were designed to have nearly complete overlap, a T-20 like peptide (C43; in Table 1) formed a stable 6-HB with a complementary N-HR peptide (N42; Table 1) [23]. The sequences of these peptides included the MPER and FPPR regions of gp41 which were required to obtain a high Tm for the 6-HB. Evidence also exists that gp41 MPER and fusion peptide regions form a stable complex [24] and that MPER/FPPR act synergistically during virus fusion [25]. Interestingly, in the elegant studies of the Jiang group, T-20 did form a 6-HB with an N-HR peptide (N36F10; Table 1) that contained a portion of the FPPR [26], and T-20 formed an uncharacterized complex with a 54-residue peptide that contained the FPPR [27]. Our analysis suggests one must be very cautious in interpreting results with peptides that are surrogates for regions of gp41 and that careful matching of these peptides should be considered to avoid exposure of unpaired hydrophobic regions that may lead to non-specific aggregation and perhaps precipitation.
Another exciting finding was reported with N and C peptides extended into the immunodominant loop region. By including residues upstream of the PBD (CP621–652, Table 1) and engineering mutations to stabilize helicity and increase solubility (CP32M), C-HR peptides with pM potencies against a broad spectrum of both T-20 resistant and sensitive HIV-1 strains were obtained [28,29]. The finding that 621QIWNNMT627 stabilizes the 6-HB is consistent with the NMR structure of the SIV gp41 ectodomain, however, this structure does not provide information on most of the MPER or FPPR regions of gp41 [30]. Using a redesigned 5-Helix (see below) T-20 was found to bind to form a 6-HB with a 30 nM Kd [31]. However, the mode of binding differed significantly from that of C34.
An NMR study using a 15N/13C labeled extended form of T-20 showed this peptide had significant helical tendencies in water and that residues 657–669 were highly helical [32]. Although this conclusion conflicts with the extended structure of a similar gp41 peptide bound to the anti g41 antibody 2F5 [33], a recent analysis indicates that gp41663–683 forms a helix kinked at F673,N674 when bound to micelles and bicelles and that binding to the anti gp41 antibody 4E10 causes this C-HR region to undergo a conformational change [34]. Notably, X-ray and NMR analysis of gp41 from HIV and SIV indicate that some residues from the MPER domain of T-20 assume helices in the postfusion ectodomain structure [4,30]. It is clear to us that T-20 has strong helical tendencies, is conformationally flexible and can form a 6-HB with a proper partner. The design of gp41 fusion inhibitors should also consider the immunodominant loop, MPER and FPPR regions of the envelope protein. Many insights have been gained from CD, electrophoretic analyses and sedimentation studies. However, more high resolution work on the N-HR/C-HR 6-HB is needed to fully understand the atomic interactions contributed by the MPER and FPPR regions to the gp41 structure.
Peptide-Fatty Acid and Cholesterol Conjugates as gp41 Fusion Inhibitors
The exact role of the LBD domain in T-20 activity is not unequivocally defined. It is indisputable that T-20 binds to liposomes, suggesting that the LBD may act in part by binding to the viral membrane. Based on this observation a novel set of fusion inhibitors where the LBD was mimicked with fatty acids, to provide a potential membrane binding locus, were developed [35]. The parent peptide, termed DP (Table 1), contained 26 residues. DP alone was 5000-fold less effective than T-20 but modification with a C-16 carboxylic acid restored nearly complete activity. The activity correlated with the fatty acid chain length and the point of modification, with C terminal fatty acids more effective than N-terminal fatty acids. This investigation provides a potential lead compound for drug development. However, some of the conclusions of the paper require caution because no high resolution structures of these conjugates and gp41 or N-HR peptides are available. For example, the authors speculate that the reason that (C-16)-DP has a fairly high anti-HIV-1 activity may be due to an increase in the local inhibitor concentration near the viral membrane. However, when it is attached to the C-HR N-terminus the fatty acyl chain in the bound drug is positioned close to the site of the PBD of C-34 and may interact with the hydrophobic pocket of the N-HR core. Only high resolution structures will decide between these possibilities. In a very recent and elegant extension of the above, cholesterol was attached to C34 resulting in pM IC50 values and improved pharmokinetics [36]. The incorporation of cholesterol was hypothesized to target C34 to lipid rafts thereby increasing the drug concentration at the site of membrane fusion [36]. The C34-cholesterol conjugate had dramatically increased serum lifetime in mice. The concept of targeting peptides to membranes to increase receptor interactions has been long known to peptide chemists [37]. The increased local concentration could be especially beneficial if kinetics are a limiting factor in drug activity.
Fusion Inhibitors Based on the Trimeric Coiled-Coil Core
N-peptides mimicking the N-HR of gp41 should also be effective fusion inhibitors by targeting the C-HR region of gp41. The successful design of 5-Helix (Figure 3C), which contained a 6-HB lacking one C-peptide and inhibited HIV-1 fusion at nM concentrations, demonstrated that C-HR of gp41 was a viable target for entry inhibition [38]. However, IC50 values of isolated N-HR peptides were 1000-fold poorer than those of T-20. To overcome potential aggregation problems and to increase the formation of the trimeric coiled-coil, researchers fused soluble leucine zipper sequences to N-HR peptides. This led to a 1000-fold increase in potency and a clear indication that the anti-viral activity is related to the stability of the triple-helical core [39]. In parallel with these investigations an exposed trimeric coiled-coil stabilized by disulfide bonds at the C-terminus and attachment to a 6-HB was found to have an IC50 of 16 nM in a HIV-1 cell fusion assay [40]. Later the most potent inhibitor of Eckert and Kim (IZN17, Table 1) was further stabilized by covalent modification resulting in pM potency in an anti-viral assay [41]. Significantly the disulfide stabilized coiled-coils were 10-fold more potent than the non-covalently linked homologs indicating that the dissociation of the trimers at low concentrations likely impacted their inhibitory capacity. The covalently linked inhibitor had a broad HIV-1 inhibitory profile and acted synergistically with T-20.
Recently, single N-HR peptides were engineered to form soluble, stable trimeric coiled-coils, and their crystal structures were similar but not identical to the trimeric core found in the 6-HB [42]. The study demonstrated that N-peptides inhibit entry by binding C-HR peptides, that the predisposition of N-peptides to form trimeric coiled-coils may be more important for anti-viral activity than the Tm of the oligomer, and that binding of C-HR to N-HR may result in conformational changes. Since the kinetics of transition from the pre-hairpin state to the membrane fusion state of gp41 impacts the activity of entry inhibitors and neutralizing antibodies, knowledge of molecular interactions that affect this process is highly relevant. Used simultaneously, N-HR and C-HR based fusion inhibitors work synergistically [43] and might force the virus to make multiple mutations to maintain infectivity. However, the trimeric N-peptides are higher molecular weight, would have potentially higher immunogenicity and must be delivered by injection. Thus, they would not overcome some of the basic problems of the C-peptide fusion inhibitors.
Fusion Inhibitors based on D-Amino Acids
T-20 has poor serum stability and marginal solubility requiring twice daily subcutaneous injection of ~100 mg of peptide. Using mirror image phage display and panning with a synthetic all D-trimeric coiled coil (D-IQN-17) that was designed based on the crystal structure of the 6-HB core, an all D-12-residue peptide with a μM inhibition constant was identified [44]. The D-inhibitor had virtually identical binding to the trimeric core with a Cα rmsd of 0.65 Ǻ compared to the wild-type. More recently, highly potent pocket-specific D-peptide inhibitors based on an 8-residue sequence were found and trimeric versions of these exhibited high avidity and pM potencies. The authors found that antiviral potencies did not correlate with the strong binding observed and suggested that drug association kinetics were the limiting factor [45]. The extremely low KD’s observed provide a “resistance capacitor” that protects the drug from mutations that moderately decrease binding because these mutations would not affect the anti-viral efficacy of the drug [45]. These studies provide excellent leads for future optimization. Since D-peptides are highly resistant to protease degradation and may be absorbed by paracellular passage through the intestine, they may overcome the disadvantages of T-20. An exciting application would be as topical microbicides. Peptides containing both L and D-residues have also been used. A C34 analog (C34M3, Table 1) designed on the basis of a model docking C34 and the N36 trimer (Figure 5) had three strategically inserted D-residues, formed a 6-HB with N-36 and was nearly as potent as C34 [46]. Moreover it had a 6-fold increase in solubility and twice the serum stability of the C-34 parent peptide. Thus the use of D-residues and structure based design concepts is providing more stable and lower molecular weight leads that should result in less expensive and more easily administered drugs.
Figure 5.
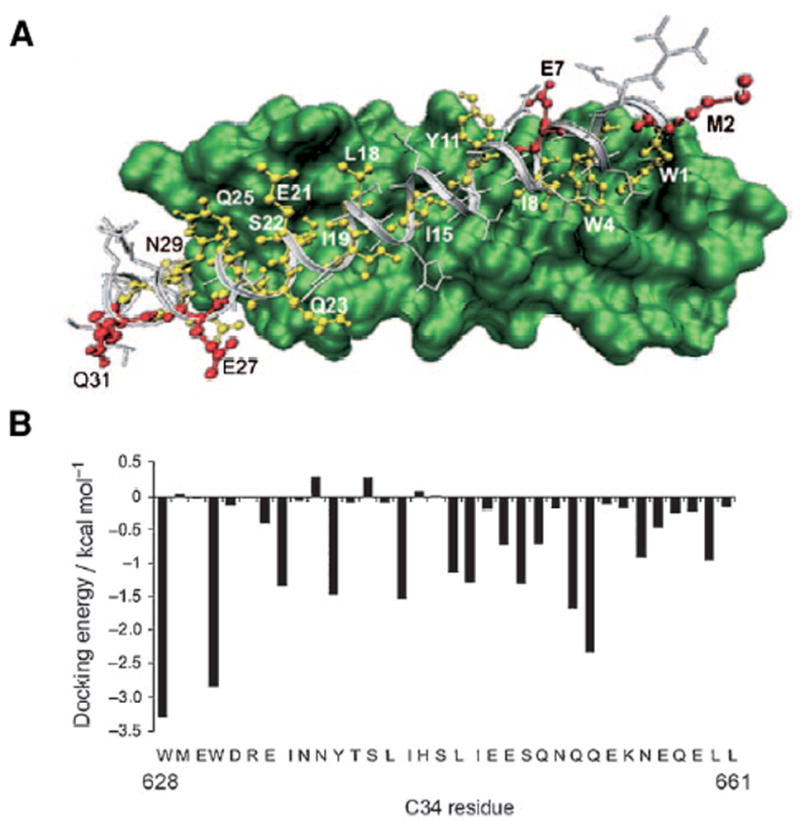
Docking of C34 into the N-trimeric core was used to choose residues for replacement by D-residues. The side chains indicated in red face away from the core and can be replaced with amino acids of opposite chirality. The docking energies provide additional guidance for positions that do not contribute to the binding affinity. Based on this structure C34M3 had D-residues inserted at positions 2, 27 and 31 and had potency comparable to C34 but was more soluble and more stable to protease digestion. Figure from [46].
Musings and Perspectives
It is a conundrum that high levels of T-20 are required for effective treatment of HIV-1 infections. IC50 values are in the low nM range in vitro, whereas serum concentrations of the drug have been estimated to be 4.6 μg/mL (~1 μM). Simply, the T-20 might be rapidly degraded, cleared by the kidneys, and/or bind to membranes or hydrophobic serum proteins lowering availability. However, perhaps the need for the high concentrations that are dosed also results from the short time frame (minutes) that the drug has to inhibit formation of the fusion-active hairpin. Measurements of binding indicate that C-peptides bind to a 5-helix with femptomolar dissociation constants [31]. Thus once the hairpin is formed it will not dissociate and infection is assured. The inhibitory efficacy of 5-helix was related to kinetics and has been shown to depend on the association rate constant rather than the IC50 [48]. A similar kinetic limit was posited for the potency of a cyclic D-peptide trimer [45]. This suggests that the fusion inhibitors must quickly trap the short-lived pre-hairpin intermediate before the fusion active hairpin forms. Since the rate of drug-gp41 interaction is likely pseudo first order with respect to inhibitor, high local concentrations of the drug are crucial. If the on-rate is in the diffusion controlled limit [108 M−1, sec−1], nM drug concentrations would result in rapid quenching of the prehairpin intermediate. However, if steric barriers cause significant decreases in the on-rate the inhibitor will be less effective. A steric defense of the N-trimer was recently revealed [47] although conjugates of albumin and C34 were nearly as effective as the free peptide in a human peripheral mononuclear cell anti-HIV1 assay [49]. The design of the next generation of fusion inhibitors with improved pharmokinetic profiles should consider all of the above, and despite impressive successes of computational modeling in predicting binding of C-HR peptides to the hydrophobic pocket [50], the clinically useful entry inhibitors must do more than bind strongly to its target.
The ideal HIV-1 entry inhibitor will be an inexpensive, stable, low molecular weight compound that is orally active and has a long serum half-life. Peptides that are currently administered do not meet any of these criteria. Nevertheless, the development of T-20, and analogs currently in clinical trials, represents a significant triumph for structural biology. Virtually every new drug is based on the knowledge of the gp41 6-HB and on fundamental information concerning those factors which stabilize helical structures and result in stable trimeric coiled coils. The finding that L-peptides containing 26 to 29 residues, and that even smaller D-peptides can result in activity equal or better than T-20 will result in more efficient syntheses and a reduction in cost. However, the recognition between C34 or T-20 and the N-HR requires that elements spanning tens of angstroms are involved in multi-atomic interactions. This suggests that it will be very challenging to find small molecules that can be effective. (see [1,51] for update on small molecule inhibitors). Small molecules have been resistant to co-crystallization with the gp41 trimetic core impeding structural analysis. An NMR method for screening the structures of bound ligands holds promise for improving structure based-design of such inhibitors [52]. Although combinations of small drugs designed to attack different sites on gp41 might work, entropic factors will impede such combinations and tethering to increase avidity, as was done with D-peptide inhibitors, may be required. Effective fatty acylated and cholesterol modified C-peptides are an exciting discovery. Ultimately, additional high resolution structures on complexes of the new lead compounds described herein together with an improved understanding of the fusion pathway will provide the knowledge necessary to fight this elusive and deadly pathogen.
Acknowledgments
We are grateful to Zohar Biron and Eran Noah for their suggestions, careful reading of the review and contributions to our understanding of the structure of gp41.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu S, Wu S, Jiang S. HIV entry inhibitors targeting gp41: from polypeptides to small-molecule compounds. Curr Pharm Des. 2007;13:143–162. doi: 10.2174/138161207779313722. Comprehensive review of the design of HIV-1 inhibitors targeting gp41 (133 references) [DOI] [PubMed] [Google Scholar]
- 2.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. Determination of the crystal structure of the gp41 core formed using synthetic peptides. [DOI] [PubMed] [Google Scholar]
- 3.Tan K, Liu J, Wang J, Shen S, Lu M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A. 1997;94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. Description of the crystal structure of the gp41 core using crystals formed from biosynthetic and synthetic peptides. [DOI] [PubMed] [Google Scholar]
- 5.Chan DC, Kim PS. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 6.Jiang S, Lin K, Strick N, Neurath AR. HIV-1 inhibition by a peptide. Nature. 1993;365:113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- 7.Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc Natl Acad Sci U S A. 1992;89:10537–10541. doi: 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci U S A. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan DC, Chutkowski CT, Kim PS. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci U S A. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. Identification of hydrophobic pocket in the six-helix bundle formed by gp41 N-HR and C-HR. This pocket is critical in the design of inhibitory peptides which target it. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Otaka A, Nakamura M, Nameki D, Kodama E, Uchiyama S, Nakamura S, Nakano H, Tamamura H, Kobayashi Y, Matsuoka M, et al. Remodeling of gp41-C34 peptide leads to highly effective inhibitors of the fusion of HIV-1 with target cells. Angew Chem Int Ed Engl. 2002;41:2937–2940. doi: 10.1002/1521-3773(20020816)41:16<2937::AID-ANIE2937>3.0.CO;2-J. Design of water soluble C34 analogs based on helical conformation and consideration of exposed and buried faces of the C-peptide. Designed peptides have increased solubility and are helical by CD. [DOI] [PubMed] [Google Scholar]
- 11.Lu M, Kim PS. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J Biomol Struct Dyn. 1997;15:465–471. doi: 10.1080/07391102.1997.10508958. [DOI] [PubMed] [Google Scholar]
- 12.Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc Natl Acad Sci U S A. 2007;104:12772–12777. doi: 10.1073/pnas.0701478104. Design of improved fusion peptides using conformation-based analogs that have increased helicity. The analogs were effective against T-20 resistant HIV-1 strains. Paper demonstrates that the topology of the interaction can be mimicked by peptides with major differences in sequence compared to the native C-HR peptide. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishikawa H, Nakamura S, Kodama E, Ito S, Kajiwara K, Izumi K, Sakagami Y, Oishi S, Ohkubo T, Kobayashi Y, et al. Electrostatically constrained alpha-helical peptide inhibits replication of HIV-1 resistant to enfuvirtide. Int J Biochem Cell Biol. 2009;41:891–899. doi: 10.1016/j.biocel.2008.08.039. Conformation directed design of Fuzeon-like peptide analogs based on electrostatic interactions. Paper has crystal structure showing that these highly modified gp41 surrogates form a crystal highly similar to that formed by native gp41 peptides. [DOI] [PubMed] [Google Scholar]
- 14.He Y, Xiao Y, Song H, Liang Q, Ju D, Chen X, Lu H, Jing W, Jiang S, Zhang L. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J Biol Chem. 2008;283:11126–11134. doi: 10.1074/jbc.M800200200. [DOI] [PubMed] [Google Scholar]
- 15.Naito T, Izumi K, Kodama E, Sakagami Y, Kajiwara K, Nishikawa H, Watanabe K, Sarafianos SG, Oishi S, Fujii N, et al. SC29EK, a peptide fusion inhibitor with enhanced alpha-helicity, inhibits replication of human immunodeficiency virus type 1 mutants resistant to enfuvirtide. Antimicrob Agents Chemother. 2009;53:1013–1018. doi: 10.1128/AAC.01211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu S, Jing W, Cheung B, Lu H, Sun J, Yan X, Niu J, Farmar J, Wu S, Jiang S. HIV gp41 C-terminal heptad repeat contains multifunctional domains. Relation to mechanisms of action of anti-HIV peptides. J Biol Chem. 2007;282:9612–9620. doi: 10.1074/jbc.M609148200. Provides convincing evidence that the C-34 and T-20 inhibitors inhibit fusion via different molecular interactions. [DOI] [PubMed] [Google Scholar]
- 17.Yuan W, Craig S, Si Z, Farzan M, Sodroski J. CD4-induced T-20 binding to human immunodeficiency virus type 1 gp120 blocks interaction with the CXCR4 coreceptor. J Virol. 2004;78:5448–5457. doi: 10.1128/JVI.78.10.5448-5457.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alam SM, Paleos CA, Liao HX, Scearce R, Robinson J, Haynes BF. An inducible HIV type 1 gp41 HR-2 peptide-binding site on HIV type 1 envelope gp120. AIDS Res Hum Retroviruses. 2004;20:836–845. doi: 10.1089/0889222041725181. [DOI] [PubMed] [Google Scholar]
- 19.Greenberg ML, Cammack N. Resistance to enfuvirtide, the first HIV fusion inhibitor. J Antimicrob Chemother. 2004;54:333–340. doi: 10.1093/jac/dkh330. [DOI] [PubMed] [Google Scholar]
- 20.Mo H, Konstantinidis AK, Stewart KD, Dekhtyar T, Ng T, Swift K, Matayoshi ED, Kati W, Kohlbrenner W, Molla A. Conserved residues in the coiled-coil pocket of human immunodeficiency virus type 1 gp41 are essential for viral replication and interhelical interaction. Virology. 2004;329:319–327. doi: 10.1016/j.virol.2004.08.025. Systematic molecular biology and modeling analysis of the interactions that occur in the deep hydrophobic pocket of the gp41 coiled-coil core. [DOI] [PubMed] [Google Scholar]
- 21.Chang DK, Hsu CS. Biophysical evidence of two docking sites of the carboxyl heptad repeat region within the amino heptad repeat region of gp41 of human immunodeficiency virus type 1. Antiviral Res. 2007;74:51–58. doi: 10.1016/j.antiviral.2006.12.006. Evidence for bi-modal binding of a C34 like peptide to the N-HR region of gp41 based on scanning analysis . [DOI] [PubMed] [Google Scholar]
- 22.Qi Z, Shi W, Xue N, Pan C, Jing W, Liu K, Jiang S. Rationally designed anti-HIV peptides containing multifunctional domains as molecule probes for studying the mechanisms of action of the first and second generation HIV fusion inhibitors. J Biol Chem. 2008;283:30376–30384. doi: 10.1074/jbc.M804672200. Systematic analysis of the role of PBD and LBD domains on fusion peptide inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noah E, Biron Z, Naider F, Arshava B, Anglister J. The membrane proximal external region of the HIV-1 envelope glycoprotein gp41 contributes to the stabilization of the six-helix bundle formed with a matching N′ peptide. Biochemistry. 2008;47:6782–6792. doi: 10.1021/bi7023139. Demonstration that T-20 forms a stable 6-HB with a properly matched N-HR peptide of gp41. Study has implications on the mechanism of action of T-20. It also indicates that conclusions pertaining to 6-HB formation should consider the matching of the C-HR and N-HR regions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorizate M, de la Arada I, Huarte N, Sanchez-Martinez S, de la Torre BG, Andreu D, Arrondo JL, Nieva JL. Structural analysis and assembly of the HIV-1 Gp41 amino-terminal fusion peptide and the pretransmembrane amphipathic-at-interface sequence. Biochemistry. 2006;45:14337–14346. doi: 10.1021/bi0612521. [DOI] [PubMed] [Google Scholar]
- 25.Bellamy-McIntyre AK, Lay CS, Baar S, Maerz AL, Talbo GH, Drummer HE, Poumbourios P. Functional links between the fusion peptide-proximal polar segment and membrane-proximal region of human immunodeficiency virus gp41 in distinct phases of membrane fusion. J Biol Chem. 2007;282:23104–23116. doi: 10.1074/jbc.M703485200. [DOI] [PubMed] [Google Scholar]
- 26.Liu S, Lu H, Niu J, Xu Y, Wu S, Jiang S. Different from the HIV fusion inhibitor C34, the anti-HIV drug Fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J Biol Chem. 2005;280:11259–11273. doi: 10.1074/jbc.M411141200. [DOI] [PubMed] [Google Scholar]
- 27.Wexler-Cohen Y, Johnson BT, Puri A, Blumenthal R, Shai Y. Structurally altered peptides reveal an important role for N-terminal heptad repeat binding and stability in the inhibitory action of HIV-1 peptide DP178. J Biol Chem. 2006;281:9005–9010. doi: 10.1074/jbc.M512475200. Insertion of D residues into T-20 alter the biological activity and interaction profile with an N-HR peptide. [DOI] [PubMed] [Google Scholar]
- 28.He Y, Cheng J, Lu H, Li J, Hu J, Qi Z, Liu Z, Jiang S, Dai Q. Potent HIV fusion inhibitors against Enfuvirtide-resistant HIV-1 strains. Proc Natl Acad Sci U S A. 2008;105:16332–16337. doi: 10.1073/pnas.0807335105. Incorporation of residues upstream of the pocket binding domain of C-HR peptides increases 6-HB stability and results in pM inhibition constants. Designed peptide has a broad range of anti-HIV-1 potency and is effective against peptides resistant to T-20 and C34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He Y, Cheng J, Li J, Qi Z, Lu H, Dong M, Jiang S, Dai Q. Identification of a critical motif for the human immunodeficiency virus type 1 (HIV-1) gp41 core structure: implications for designing novel anti-HIV fusion inhibitors. J Virol. 2008;82:6349–6358. doi: 10.1128/JVI.00319-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caffrey M, Cai M, Kaufman J, Stahl SJ, Wingfield PT, Covell DG, Gronenborn AM, Clore GM. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. Embo J. 1998;17:4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Champagne K, Shishido A, Root MJ. Interactions of HIV-1 inhibitory peptide T20 with the gp41 N-HR coiled coil. J Biol Chem. 2009;284:3619–3627. doi: 10.1074/jbc.M809269200. Use of a redesigned 5-helix to investigate the mode of binding of T-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biron Z, Khare S, Quadt SR, Hayek Y, Naider F, Anglister J. The 2F5 epitope is helical in the HIV-1 entry inhibitor T-20. Biochemistry. 2005;44:13602–13611. doi: 10.1021/bi0509245. Use of heteronuclear labeling to determine the NMR structure of an extended T-20 peptide in water. Study concludes that significant portions of T-20 assume a helical structure in water. This differs from the conclusions of CD analysis on this peptide. [DOI] [PubMed] [Google Scholar]
- 33.Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, Wyatt R, Kwong PD. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J Virol. 2004;78:10724–10737. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun ZY, Oh KJ, Kim M, Yu J, Brusic V, Song L, Qiao Z, Wang JH, Wagner G, Reinherz EL. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity. 2008;28:52–63. doi: 10.1016/j.immuni.2007.11.018. NMR and EPR investigation of gp41 peptide bound to micelles and 4E10 antibody. Provides insights into the membrane bound structure of gp41 peptides and to the mechanism of binding of a broadly neutralizing Ab to gp41. [DOI] [PubMed] [Google Scholar]
- 35.Wexler-Cohen Y, Shai Y. Demonstrating the C-terminal boundary of the HIV 1 fusion conformation in a dynamic ongoing fusion process and implication for fusion inhibition. Faseb J. 2007;21:3677–3684. doi: 10.1096/fj.07-8582com. Development of C-HR – fatty acid conjugates that have high anti HIV-1 potency. First demonstration that the peptide-peptide interaction in the formation of the 6-HB can be mimicked by a fatty acid. This has important implications for design of a peptidomimetic entry inhibitor. [DOI] [PubMed] [Google Scholar]
- 36.Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci U S A. 2009;106:5801–5806. doi: 10.1073/pnas.0901007106. Synthesis and characterization of C34-cholesterol conjugates that target the drug to the lipid raft domains that are involved in membrane fusion. The best conjugates had pM IC50 values and were active against a panel of primary isolates and had dramatically increased serum lifetimes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwyzer R. Peptide-membrane interactions and a new principle in quantitative structure-activity relationships. Biopolymers. 1991;31:785–792. doi: 10.1002/bip.360310624. [DOI] [PubMed] [Google Scholar]
- 38.Root MJ, Kay MS, Kim PS. Protein design of an HIV-1 entry inhibitor. Science. 2001;291:884–888. doi: 10.1126/science.1057453. Design of 5-Helix a C-HR directed HIV-1 fusion inhibitor composed of a 5-helix bundle. [DOI] [PubMed] [Google Scholar]
- 39.Eckert DM, Kim PS. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc Natl Acad Sci U S A. 2001;98:11187–11192. doi: 10.1073/pnas.201392898. The use of N-HR peptides as fusion inhibitors. Paper discusses increasing the stability of the trimeric core by attachment of soluble leucine zipper sequences. Activity of the peptides increases 1000-fold. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Louis JM, Bewley CA, Clore GM. Design and properties of N(CCG)-gp41, a chimeric gp41 molecule with nanomolar HIV fusion inhibitory activity. J Biol Chem. 2001;276:29485–29489. doi: 10.1074/jbc.C100317200. [DOI] [PubMed] [Google Scholar]
- 41.Bianchi E, Finotto M, Ingallinella P, Hrin R, Carella AV, Hou XS, Schleif WA, Miller MD, Geleziunas R, Pessi A. Covalent stabilization of coiled coils of the HIV gp41 N region yields extremely potent and broad inhibitors of viral infection. Proc Natl Acad Sci U S A. 2005;102:12903–12908. doi: 10.1073/pnas.0502449102. Disulfide linked coiled coils based on a fusion of the gp41 N-HR region and a soluble coiled-coil forming sequence exhibited broad antiviral activity and pM inhibition constants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dwyer JJ, Wilson KL, Martin K, Seedorff JE, Hasan A, Medinas RJ, Davison DK, Feese MD, Richter HT, Kim H, et al. Design of an engineered N-terminal HIV-1 gp41 trimer with enhanced stability and potency. Protein Sci. 2008;17:633–643. doi: 10.1110/ps.073307608. Engineering of isolated N-peptides that form a soluble trimeric coiled-coil. Study shows viral inhibition is related to formation of trimer and not necessarily to stability of the trimeric coiled coil. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hrin R, Montgomery DL, Wang F, Condra JH, An Z, Strohl WR, Bianchi E, Pessi A, Joyce JG, Wang YJ. Short communication: In vitro synergy between peptides or neutralizing antibodies targeting the N- and C-terminal heptad repeats of HIV Type 1 gp41. AIDS Res Hum Retroviruses. 2008;24:1537–1544. doi: 10.1089/aid.2008.0129. Demonstration of synergy between N-HR and C-HR based fusion inhibitors. [DOI] [PubMed] [Google Scholar]
- 44.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. Use of phage display and a structure based hook to identify D-peptide fusion inhibitors. [DOI] [PubMed] [Google Scholar]
- 45.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D-peptide inhibitors of HIV-1 entry. Proc Natl Acad Sci U S A. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. Optimization of small all D-fusion inhibitors with increased avidity and anti-HIV-1 potency. Study shows avidity is an important aspect of obtaining inhibitors with extremely high affinities. However, must take into account kinetic factors as well when designing antiviral entry inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaston F, Granados GC, Madurga S, Rabanal F, Lakhdar-Ghazal F, Giralt E, Bahraoui E. Development and Characterization of Peptidic Fusion Inhibitors Derived from HIV-1 gp41 with Partial D-Amino Acid Substitutions. ChemMedChem. 2009 doi: 10.1002/cmdc.200800390. [DOI] [PubMed] [Google Scholar]
- 47.Eckert DM, Shi Y, Kim S, Welch BD, Kang E, Poff ES, Kay MS. Characterization of the steric defense of the HIV-1 gp41 N-trimer region. Protein Sci. 2008;17:2091–2100. doi: 10.1110/ps.038273.108. The gp41 prehairpin trimer is sterically blocked from antibodies and large C-peptide inhibitors and the nature of this steric defense is asymmetric with respect to the viral and target cell membranes . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steger HK, Root MJ. Kinetic dependence to HIV-1 entry inhibition. J Biol Chem. 2006;281:25813–25821. doi: 10.1074/jbc.M601457200. Detailed analysis shows that there is a better correlation between inhibitory potency and association constants of inhibitors than there is with their binding affinities to 5-helix. This paper is important for the design of inhibitors as it shows that steric factors which influence binding kinetics may play a role in the efficacy of the inhibitor . [DOI] [PubMed] [Google Scholar]
- 49.Stoddart CA, Nault G, Galkina SA, Thibaudeau K, Bakis P, Bousquet-Gagnon N, Robitaille M, Bellomo M, Paradis V, Liscourt P, et al. Albumin-conjugated C34 peptide HIV-1 fusion inhibitor: equipotent to C34 and T-20 in vitro with sustained activity in SCID-hu Thy/Liv mice. J Biol Chem. 2008;283:34045–34052. doi: 10.1074/jbc.M805536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Strockbine B, Rizzo RC. Binding of antifusion peptides with HIVgp41 from molecular dynamics simulations: quantitative correlation with experiment. Proteins. 2007;67:630–642. doi: 10.1002/prot.21301. MD calculations of affinities of C-HR peptides to a conserved hydrophobic binding pocket correlated well with experimental binding constants. [DOI] [PubMed] [Google Scholar]
- 51.Liu K, Lu H, Hou L, Qi Z, Teixeira C, Barbault F, Fan BT, Liu S, Jiang S, Xie L. Design, synthesis, and biological evaluation of N-carboxyphenylpyrrole derivatives as potent HIV fusion inhibitors targeting gp41. J Med Chem. 2008;51:7843–7854. doi: 10.1021/jm800869t. Recent update on small molecule fusion inhibitors. Most lead compounds have micromolar inhibition constants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balogh E, Wu D, Zhou G, Gochin M. NMR Second Site Screening for Structure Determination of Ligands Bound in the Hydrophobic Pocket of HIV-1 gp41. J Am Chem Soc. 2009 doi: 10.1021/ja8094558. A novel NMR method based on paramagnetic relaxation effects to determine structure of small molecule inhibitors bound to the hydrophobic pocket og gp41. [DOI] [PMC free article] [PubMed] [Google Scholar]