

Abstract
Retroviral Gag polyprotein precursors are both necessary and sufficient for the assembly and release of virus-like particles (VLPs) from infected cells. It is well established that small Gag-encoded motifs, known as late domains, promote particle release by interacting with components of the cellular endosomal sorting and ubiquitination machinery. The Gag proteins of a number of different retroviruses are ubiquitinated; however, the role of Gag ubiquitination in particle egress remains undefined. In this study, we investigated this question by using a panel of equine infectious anemia virus (EIAV) Gag derivatives bearing the wild-type EIAV late domain, heterologous retroviral late domains or no late domain. Ubiquitin was fused in cis to the C-termini of these Gag polyproteins, and the effects on VLP budding were measured. Remarkably, fusion of ubiquitin to EIAV Gag lacking a late domain (EIAV/ΔYPDL-Ub) largely rescued VLP release. We also determined the effects of ubiquitin fusion on the sensitivity of particle release to budding inhibitors and to depletion of key endosomal sorting factors. Ubiquitin fusion rendered EIAV/ΔYPDL-Ub sensitive to depletion of cellular endosomal sorting factors Tsg101 and Alix and to overexpression of dominant-negative fragments of Tsg101 and Alix. These findings demonstrate that ubiquitin can functionally compensate for the absence of a retroviral late domain and provide insights into the host-cell machinery engaged by ubiquitin during particle egress.
Keywords: Alix, assembly and release, EIAV, Gag, retroviruses, Tsg101, ubiquitin
The essential and common retroviral component that drives particle assembly and release is the Gag precursor polyprotein. Within Gag lie short recognition motifs, known as late domains, that promote particle budding and release by interacting directly with endosomal sorting complexes required for transport (ESCRT) machinery and associated factors. Three distinct retroviral late domain sequences have been characterized thus far (1-3). The Pro-Thr/Ser-Ala-Pro (PT/SAP)-type late domain, found in the p6 region of HIV-1 Gag, interacts with the N-terminal ubiquitin E2 variant (UEV) domain of the ESCRT-I component Tsg101. Pro-Pro-Pro-Tyr (PPPY) motifs, which serve dominant functions in the release of a number of retroviruses, bind WW domains present in the Nedd4 family of E3 ubiquitin ligases. Finally, Tyr-Pro-Xn-Leu (YPXnL) motifs (where Xn represents 1-3 variable amino acids), found in the HIV-1 p6 and the p9 domain of equine infectious anemia virus (EIAV) Gag, bind the ESCRT-associated protein ALG2-interacting protein X (Alix or AIP1) (4-6).
Sorting of cargo proteins into the ESCRT pathway often requires the modification of the cargo proteins with ubiquitin. Posttranslational modification of proteins by attachment of a single ubiquitin frequently serves as a signal for protein internalization and endosomal sorting, whereas modification by attachment of multiple ubiquitin moieties typically targets proteins for proteasomal degradation. The recognition of monoubiquitin by the endosomal sorting machinery involves an interaction between ubiquitin-binding motifs, found in a wide variety of protein sorting factors, and a hydrophobic patch of residues in ubiquitin (7).
Although it is clear that ubiquitination plays an important role in the delivery of cargo proteins into multivesicular bodies (MVBs), the function of Gag ubiquitination in retroviral egress remains ill defined (2,6,8). Several lines of evidence support a role for Gag ubiquitination in retrovirus release: (i) high levels of free ubiquitin can be found in retroviral particles (9,10); (ii) retroviral Gag proteins are ubiquitinated (11-13), and the presence of different late domains can alter the levels of Gag ubiquitination (14,15); (iii) depletion of cellular pools of free ubiquitin by treatment with proteasome inhibitors impairs retrovirus release (16-18); (iv) PPPY-type late domains interact with Nedd4 family ubiquitin ligases (19); (v) overexpression of ubiquitin variants containing mutations in residues that regulate ubiquitin interactions inhibits HIV-1 release (15) and (vi) mutation of multiple lysine residues in HIV-1 and Rous sarcoma virus (RSV) Gag inhibits virus budding (20,21). Other observations suggest that Gag ubiquitination is not required for particle egress: (i) mutation or deletion of ubiquitin acceptor Lys residues in HIV-1 p6 (9,22,23) or in murine leukemia virus (MLV) Gag (9) does not significantly inhibit virus budding and (ii) non-ubiquitinated Gag mutants of primate foamy viruses are released efficiently relative to their ubiquitinated counterparts (24). Derivatives of these non-ubiquitinated mutants bearing a PPPY late domain retain a requirement for ubiquitin ligase expression (24), and a non-ubiquitinated mutant of HTLV-1 remains ubiquitin ligase dependent (25), suggesting that Gag ubiquitination is not sufficient to explain the stimulation of virus release mediated by the interaction between the PPPY late domain and the Nedd4 family ubiquitin ligases.
To clarify the role of Gag ubiquitination in retrovirus budding and release, we fused ubiquitin in cis to the C-termini of a panel of EIAV Gag mutants that differ only in the nature of their late domain (26,27). The strategy of ubiquitin fusion has been widely used to evaluate the role of ubiquitination in the trafficking and function of a variety of cellular proteins (28-33). Analysis of the ubiquitin-fused EIAV Gag chimeras indicated that ubiquitin fusion largely rescued the budding defect imposed by deletion of the YPXnL late domain. Ubiquitin fusion also sensitized late-domain-defective EIAV release to specific inhibitors of the endosomal sorting pathway, indicating that ubiquitin attached to Gag can serve as a signal for recognition by ESCRT machinery.
Results
Fusion of ubiquitin to the C-terminus of EIAV Gag lacking a late domain rescues virus release
To investigate the role of Gag ubiquitination in retroviral particle assembly and release, we made use of a series of constructs that express EIAV Gag either lacking a late domain (EIAV/ΔYPDL) or containing one of the three recognized retroviral late domains: YPDL (EIAV/WT), PTAP (EIAV/PTAP) or PPPY (EIAV/PPPY) (Figure 1) (26,27). We generated derivatives of these constructs in which the ubiquitin-coding region was fused in-frame to the C-terminus of Gag and measured the effect of ubiquitin fusion on virus-like particle (VLP) production. As reported previously (27), EIAV/WT, EIAV/PPPY and EIAV/PTAP show similar VLP release efficiencies, whereas EIAV/ΔYPDL exhibits a major (∼10-fold) defect in particle production (Figure 2). Interestingly, fusion of ubiquitin to the C-terminus of EIAV Gag lacking a late domain (EIAV/ΔYPDL-Ub) led to rescue of VLP release to 60-70% of the levels measured for wild-type (WT) EIAV Gag. Fusion of ubiquitin to the C-terminus of WT EIAV Gag consistently led to a 30-50% reduction in the efficiency of particle production. We also analyzed EIAV/PPPY-Ub and EIAV/PTAP-Ub and found that fusion of ubiquitin had no effect on their release (Figure 2). These data demonstrate that ubiquitin fusion is able to rescue the defect imposed by late domain deletion in the context of EIAV Gag.
Figure 1. Schematic representation of EIAV Gag constructs used in this study.

The EIAV Gag matrix (MA), capsid (CA), nucleocapsid (NC) and p9 domains are indicated. The p9 region of EIAV Gag bears the YPDL late domain. EIAV Gag constructs with native YPDL late domain (WT), no late domain (ΔYPDL) or heterologous (PTAP and PPPY) late domains are depicted with or without fusion of a single ubiquitin (Ub) moiety to the Gag C-terminus.
Figure 2. Ubiquitin fusion to the C-terminus of EIAV Gag lacking a late domain rescues virus particle production.
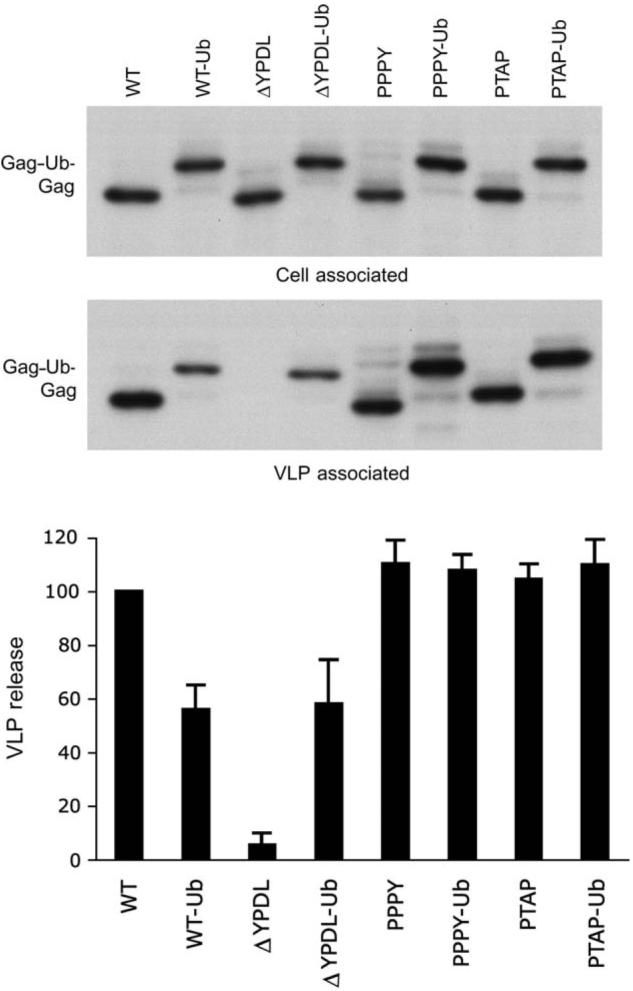
293T cells were transfected with plasmids expressing the EIAV Gag proteins depicted in Figure 1 and were labeled with [35S]Met/Cys for 5 h. Cell and VLP lysates were immunoprecipitated with horse anti-EIAV serum followed by resolution on SDS-PAGE gels. Levels of cell- and virion-associated Gag proteins were quantified by phosphorImager analysis. VLP release efficiency was calculated as (VLP Gag)/[total (cell + VLP) Gag] as a percentage of EIAV/WT release. Data represent mean ± SD, n = 6.
To determine the concentration of ubiquitinated Gag required to rescue the release of a late-domain-deficient Gag, cells were cotransfected with a constant amount of EIAV/ΔYPDL plasmid along with increasing amounts of EIAV/ΔYPDL-Ub DNA. We observed that coexpression of EIAV/ΔYPDL-Ub enhanced the release of EIAV/ΔYPDL Gag, with rescue being most effective at an approximately 1:1 DNA ratio (Figure 3).
Figure 3. An optimal amount of EIAV/ΔYPDL-Ub Gag is required for efficient rescue of ΔYPDL-Ub Gag.
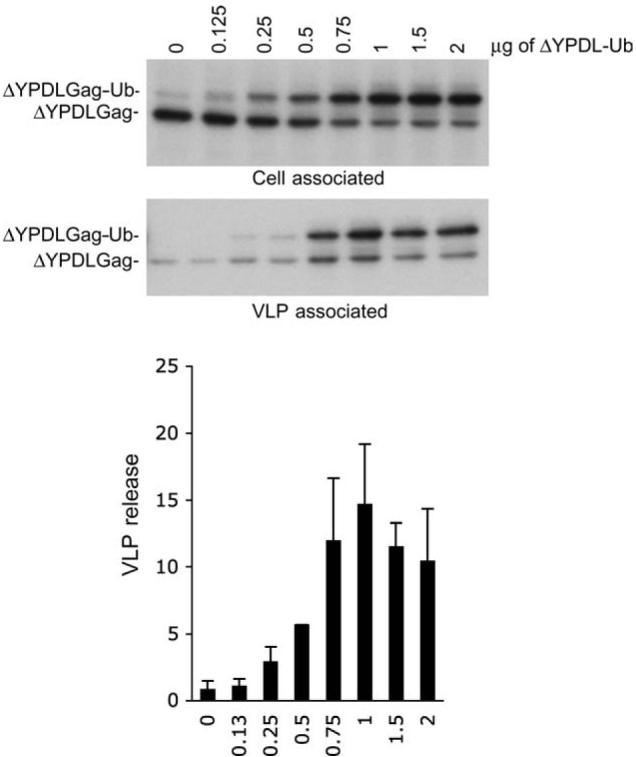
293T cells were transfected with a constant amount (0.75 µg) of plasmid expressing EIAV/ΔYPDL Gag along with increasing amounts of EIAV/ΔYPDL-Ub expression vector. VLP release efficiency was measured as described in Figure 2. Data represent mean ± SD, n = 3.
Ubiquitin rescue is conferred by enhanced VLP release
Several explanations could account for the apparent rescue of EIAV/ΔYPDL particle production by ubiquitin fusion. It is possible that ubiquitin fusion alters recognition of cell-associated Gag by anti-EIAV antiserum. We were able to exclude this possibility by using a previously described assay (34) based on comparing levels of Gag detection by immunoprecipitation before and after sample denaturation (data not shown). We also observed that ubiquitin fusion did not affect levels of EIAV/ΔYPDL Gag bound to membrane (Figure S1) nor did it discernibly alter Gag localization (Figure S2).
To determine whether ubiquitin fusion stimulates VLP budding and release, electron microscopy (EM) analysis was performed. As shown in Figure 4, a characteristic defective late-domain phenotype was evident in cells expressing EIAV/ΔYPDL Gag, with a large number of virions tethered to the plasma membrane. In contrast, this pinching-off defect was not observed in cells expressing EIAV/ΔYPDL-Ub Gag (Figure 4). Although ubiquitin fusion to EIAV/ΔYPDL Gag is able to stimulate particle budding, we note that the VLPs produced by ubiquitin-fused Gags were larger in size than the unfused Gag VLPs and displayed what appeared to be some discontinuities in the Gag shell. Together, these findings indicate that fusion of ubiquitin to a late domain-deficient EIAV Gag rescues VLP release.
Figure 4. Ubiquitin fusion to the C-terminus of EIAV Gag lacking a late domain rescues VLP release.

293T cells were transfected with the indicated EIAV Gag expression vectors and were fixed and examined by transmission EM. Scale bar = 100 nm.
Rescue of release by ubiquitin fusion requires an intact ubiquitin sorting motif
The data presented above suggest that rescue of VLP release by fusion of ubiquitin to EIAV/ΔYPDL Gag could occur through the recruitment of host factors containing ubiquitin-interacting motifs to the site of assembly. Mutation of residues in ubiquitin that are critical for binding to ubiquitin-interacting motifs should thus abrogate the rescue. To test this model, we mutated residues Leu-8 and Ile-44 in the fused ubiquitin moiety to Ala. These amino acids form part of a surface-exposed hydrophobic patch that acts as a docking platform for cellular sorting factors containing ubiquitin-binding motifs (32). We observed that introduction of these mutations into the ubiquitin moiety in EIAV/ΔYPDL-Ub Gag abolished rescue mediated by ubiquitin fusion (Figure 5). These data demonstrate that an intact hydrophobic surface patch in ubiquitin is required for the ubiquitin-mediated rescue of EIAV/ΔYPDL release. The approximately twofold reduction in particle release imposed by fusion of ubiquitin to EIAV/WT Gag is eliminated by the L8A/I44A ubiquitin mutations (Figure 5), suggesting that in the context of the YPDL late domain, recognition of the fused ubiquitin by host protein ubiquitin-binding domains is modestly inhibitory to particle production.
Figure 5. An intact sorting motif is required for rescue of VLP release by ubiquitin fusion.
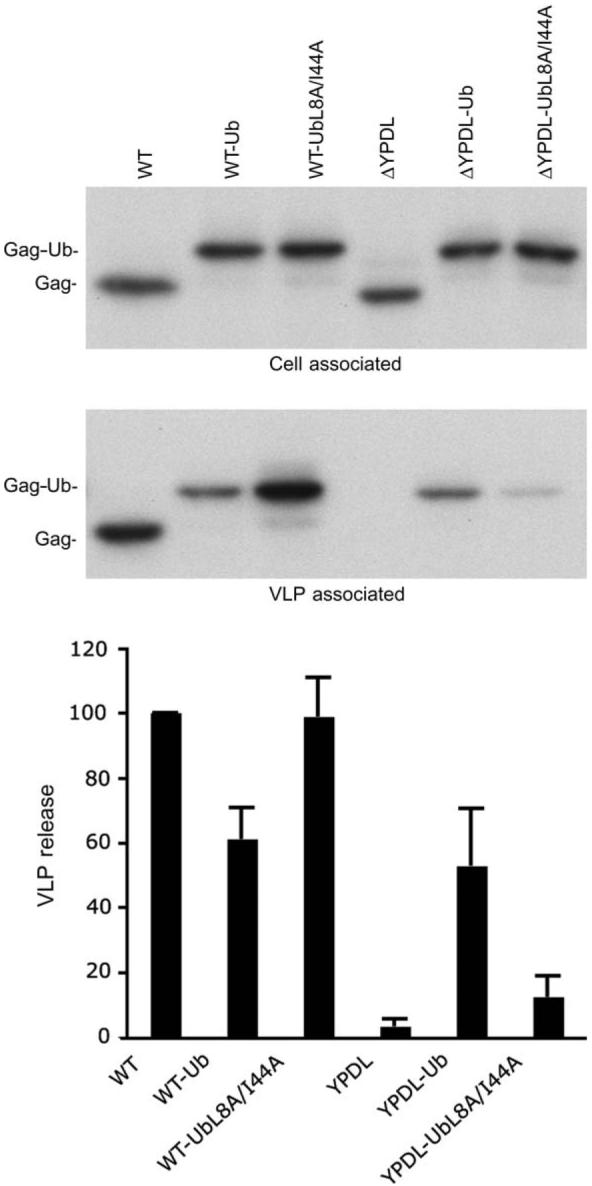
293T cells were transfected with the indicated EIAV Gag expression vectors. VLP release efficiency was determined as described in Figure 2. Data represent mean ± SD, n = 3.
Ubiquitin fusion selectively alters sensitivity to Tsg101-based budding inhibitors and proteasome inhibitors
To explore further the role of ubiquitin in retrovirus release, we examined the ability of several previously reported Tsg101-based virus budding inhibitors to disrupt the release of VLPs produced by the ubiquitin-fused Gags. These inhibitors include TSG-5′, an N-terminal Tsg101 fragment that inhibits release by binding to the PTAP motif (27,35-37); TSG-3′, a C-terminal Tsg101 fragment that blocks the release of PTAP- and PPPY-containing Gag but not YPDL-containing Gag and induces the formation of aggresome-like structures that sequester ubiquitin and ubiquitinated cargo (27,36,38); and full-length Tsg101 (TSG-F), whose overexpression disrupts retrovirus budding by inducing the formation of aberrant, swollen endosomes (27,36). Consistent with our previous report (27), WT EIAV release was insensitive to inhibition by TSG-F, TSG-5′ or TSG-3′, whereas EIAV/PTAP release was disrupted by all three of these inhibitors (Figure 6). EIAV/PPPY release was unaffected by TSG-5′, modestly reduced by TSG-F and strongly inhibited by TSG-3′ (Figure 6), again consistent with our previous report (27). Ubiquitin fusion had no significant effect on inhibition by TSG-F or TSG-5′ in the context of either WT EIAV or EIAV/PTAP but significantly reduced the sensitivity of EIAV/PPPY to TSG-3′. In contrast, fusion of ubiquitin to WT or EIAV/ΔYPDL Gag rendered them sensitive to inhibition by TSG-3′. Release of all Gag VLPs was potently inhibited by dominant-negative Vps4 (Vps4EQ) (Figure 6), which globally disrupts the cellular class E Vps machinery (39).
Figure 6. Ubiquitin fusion alters the sensitivity of EIAV Gag release to VLP budding inhibitors.

293T cells were cotransfected with the indicated EIAV Gag expression constructs and control plasmid or a 1:1 DNA ratio of vectors expressing TSG-F, TSG-5′, TSG-3′ or Vps4EQ. VLP release efficiency was calculated after radiolabeling and immunoprecipitation as described in Figure 2. For proteasome inhibitor treatment, transfected cells were pretreated for 30 min with media containing dimethyl sulfoxide (DMSO) alone or 10 µM MG-132 in DMSO and were metabolically labeled for 4 h in media containing or lacking MG-132. Data represent mean ± SD, n = 3 (TSG-F, TSG-5′, TSG-3′ and MG-132) or n = 2 (Vps4EQ).
We also examined the sensitivity of ubiquitin-fused Gags to proteasome inhibitors. We and others previously reported that WT EIAV release is insensitive to proteasome inhibitors (13,27,40); in contrast, we observed that release of EIAV/PPPY and EIAV/PTAP VLPs is significantly disrupted by these compounds (27). To evaluate the effect of ubiquitin fusion on sensitivity to proteasome inhibitors, we measured the release efficiency of unfused and ubiquitin-fused Gag VLPs in the presence or absence of MG-132 (Figure 6). Consistent with our published results (27), MG-132 did not inhibit the release of EIAV/WT but significantly reduced particle production for EIAV/PTAP and EIAV/PPPY. Fusion of ubiquitin to WT EIAV Gag had no effect on MG-132 sensitivity, whereas ubiquitin fusion to EIAV/PPPY and EIAV/PTAP resulted in a loss of sensitivity to the proteasome inhibitor. Interestingly, MG-132 treatment led to a significant reduction in the release of EIAV/ΔYPDL-Ub (Figure 6). These data demonstrate that in the context of PPPY- or PTAP-mediated virus release, ubiquitin fusion confers insensitivity to MG-132, whereas in the absence of a functional late domain ubiquitin fusion sensitizes VLP release to proteasome inhibition.
Overexpression of the Alix V domain potently inhibits release of EIAV/ΔYPDL-Ub VLPs
We previously reported that overexpression of the Gagbinding region from Alix, the so-called ‘V’ domain (Figure 7A), potently inhibits the release of HIV-1 and EIAV (41,42). Inhibition by Alix V domain overexpression requires a direct binding between the late domain sequence in Gag (YPXnL) and the V domain as the inhibition can be blocked by mutations in either p6 [L41A or L41R (42)] or the V domain itself [F676D (41)] that prevent Gag-Alix binding. To further define the host-cell machinery with which ubiquitin-fused Gags interact, we examined the sensitivity of the EIAV Gag constructs to inhibition by Alix V domain overexpression. In agreement with previous reports (42,43), we observed that the Alix V domain potently (by ∼10-fold) inhibits release of WT EIAV; this inhibition is maintained for the EIAV/WT-Ub Gag (Figure 7B). EIAV/ΔYPDL release was unaffected by Alix V domain overexpression (Figure 7B and Figure 8A), consistent with its lack of a YPXnL motif. EIAV/PTAP and EIAV/PPPY, in either ubiquitin-fused or unfused forms, were also not significantly inhibited by V-domain overexpression (Figure 7B). Surprisingly, however, release of EIAV/ΔYPDL-Ub VLPs was strongly blocked by the V domain, despite the absence of an YPXnL late domain in this Gag (Figure 7B and Figure 8A). EM analysis demonstrated that Alix V domain overexpression induced the accumulation of EIAV/ΔYPDL-Ub VLPs at the plasma membrane (data not shown). These findings indicate that fusion of ubiquitin to an EIAV Gag lacking a late domain renders EIAV release sensitive to inhibition mediated by the Alix V domain.
Figure 7. Alix V domain overexpression inhibits release of EIAV/ΔYPDL-Ub.
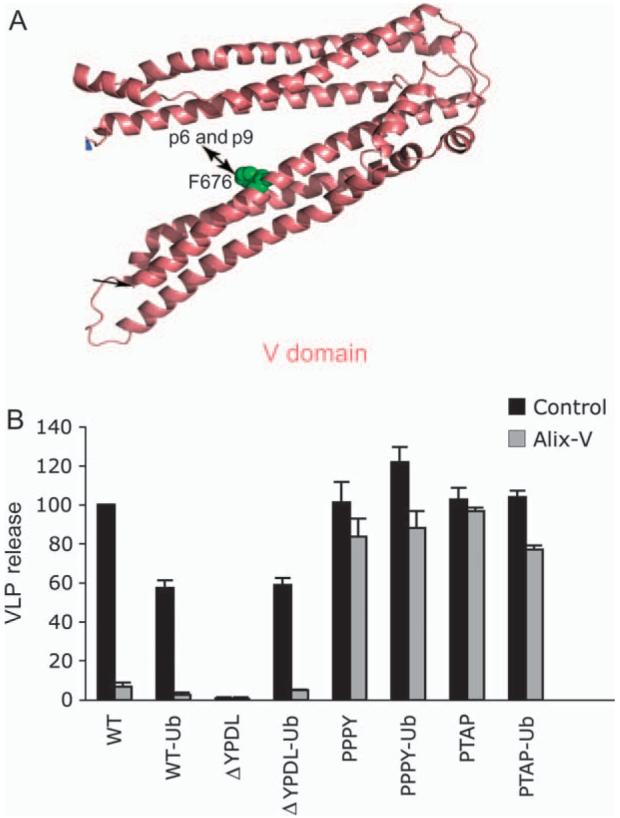
A) Structure of the Alix V domain, with residue F676 critical for binding to Gag YPXnL motifs in HIV-1 p6 and EIAV p9 indicated. B) 293T cells were cotransfected with the indicated EIAV Gag expression vectors and control vector or Alix V domain expression vector at a 1:1 DNA ratio. VLP release efficiency was measured as described in Figure 2. Data represent mean ± SD, n = 3. Panel A modified from Fujii et al. (4), with permission.
Figure 8. Alix V domain overexpression or Alix and Tsg101 depletion inhibits the release of EIAV/ΔYPDL-Ub.

A) 293T cells were transfected with EIAV/ΔYPDL or ΔYPDL-Ub Gag expression vectors along with control vector or vector expressing Alix V domain at a 1:1 DNA ratio. VLP release was determined as described in Figure 2. Release efficiencies for EIAV/ΔYPDL and EIAV/ΔYPDL-Ub in the absence of V-domain expression are each assigned a relative value of 100. B) Alix V domain expression inhibits the release of WT and EIAV/ΔYPDL-Ub VLPs by distinct mechanisms. Release of EIAV WT or ΔYPDL-Ub Gag was determined in the presence of an Alix V domain mutant (F676D) defective for binding to YPXnL motifs. C) Release of EIAV/ΔYPDL-Ub VLPs is sensitive to Alix and Tsg101 depletion. 293T cells were cotransfected with EIAV/ΔYPDL or EIAV/ΔYPDL-Ub Gag expression vectors along with control, Alix or Tsg101 siRNA. To allow accurate quantification of EIAV/ΔYPDL release, the transfections for panels A and C were scaled up 10-fold relative to the EIAV/ΔYPDL-Ub vector transfection. Data represent mean ± SD, n = 3 (A and C) or n = 2 (B). D) Tsg101 overexpression overcomes the inhibition of EIAV/ΔYPDL-Ub release mediated by Tsg101 depletion through ubiquitin binding. 293T cells were transfected with control or Tsg101 siRNA along with empty vector, or vectors expressing siRNA-resistant Tsg101 (denoted by*), or siRNA-resistant Tsg101 mutant defective in ubiquitin binding (ND45, 46AA). Twenty-four hour later, cells were transfected with the respective siRNAs along with EIAV/ΔYPDL-Ub DNA. Cells were labeled 24 h after the second transfection and virus release efficiency determined as described above. Data represent mean ± SD, n = 3.
Alix residue F676 plays a critical role in binding to YPXnL motifs in HIV-1 and EIAV Gag (41,44,45). To characterize the ability of the Alix V domain to inhibit the release of EIAV/ΔYPDL-Ub, we compared the effect of WT and F676D V-domain expression on the release of WT and ΔYPDL-Ub particles (Figure 8B). As anticipated, the release of WT EIAV was severely compromised by expression of the Alix V domain, and this inhibition was abolished by the F676D mutation. Interestingly, release of EIAV/ΔYPDL-Ub VLPs was comparably inhibited by both WT and F676D versions of the Alix V domain (Figure 8B). These data indicate that the Alix V domain inhibits the release of WT and EIAV/ΔYPDL-Ub VLPs by distinct mechanisms.
Inhibition of VLP release by Tsg101 and Alix depletion
We next determined the effect of Tsg101 and Alix depletion on the release of the EIAV Gag series. As anticipated, release of WT EIAV Gag was inhibited by Alix depletion but was unaffected by Tsg101 small interfering RNA (siRNA) (Figure S3); EIAV/PTAP was, as expected, disrupted by Tsg101 depletion (data not shown). Release of EIAV/PPPY was significantly inhibited by both Alix and Tsg101 depletion (Figure S3). This latter result is consistent with the observation that the release of some viruses that rely predominantly on a PPxY motif (e.g. Mason-Pfizer monkey virus and Marburg virus) is inhibited by Tsg101 depletion (46,47). Consistent with its lack of a functional late domain, EIAV/ΔYPDL release was unaffected by depletion of either Tsg101 or Alix. Interestingly, release of EIAV/ΔYPDL-Ub was significantly reduced by siRNAs specific for either Tsg101 or Alix (Figure 8C). Release of EIAV/ΔYPDL-UbL8A/I44A VLPs was also inhibited by Tsg101 and Alix depletion (data not shown). These results demonstrate that depletion of Tsg101 or Alix reverses the ability of ubiquitin fusion to rescue the release of a late domain-defective EIAV Gag mutant.
To define further the role of Tsg101 in ubiquitin-mediated enhancement of VLP release, siRNA depletion and Tsg101 add-back experiments were conducted either with WT Tsg101 or with a Tsg101 mutant defective in ubiquitin binding (46,48). As shown in Figure 8D, expression of siRNA-resistant Tsg101 rescued the defect in EIAV/ΔYPDL-Ub VLP release induced by Tsg101 depletion. In contrast, expression of a Tsg101 mutant (ND45, 46AA) defective in ubiquitin binding was unable to confer rescue (Figure 8D). These findings further support a role for Tsg101 in enhancement of EIAV/ΔYPDL-Ub VLP release, presumably through binding of Tsg101 to ubiquitin.
The Alix V domain specifically binds to ubiquitin-agarose
The observations presented above raised the possibility that the Alix V domain was capable of making non-YPXnL interactions with EIAV/ΔYPDL-Ub Gag, perhaps involving the fused ubiquitin. This hypothesis was tested by precipitating transfected cell lysates with ubiquitin-agarose beads. As demonstrated in Figure 9A, the Alix V domain, as well as full-length Alix, was efficiently precipitated by ubiquitin-agarose but not protein A-agarose beads. As specificity controls, we also tested the ability of ubiquitin-agarose to precipitate TSG-F or Tsg101 mutants containing substitutions (N45A and F88A) reported to reduce the interaction between the Tsg101 UEV domain and ubiquitin (49). We readily detected binding between TSG-F and ubiquitin-agarose but not protein A-agarose beads (Figure 9A). As expected (49), this binding was reduced but not eliminated by the N45A and F88A Tsg101 mutations (Figure 9A). These data suggest that the Alix V domain is capable of interacting with ubiquitin, either directly or through a cellular bridging protein. To determine whether binding of Alix V domain to ubiquitin required the surface-exposed hydrophobic residues (L8, I44) of ubiquitin, we performed coimmunoprecipitation analyses with Alix and EIAV/ΔYPDL-Ub Gag with L8A and I44A ubiquitin mutations. As indicated in Figure 9B, both the full-length Alix and the Alix V domain efficiently bound to EIAV/ΔYPDL-Ub Gag. This binding was blocked by the L8A/I44A ubiquitin mutations. As expected for the controls, Alix binding was seen for WT Gag but not for ΔYPDL Gag. These data demonstrate a role for the ubiquitin interaction motif in the binding between the EIAV/ΔYPDL-Ub Gag and the Alix V domain.
Figure 9. The Alix V domain binds ubiquitin-agarose beads.

A) Cells were transfected with the indicated HA-tagged expression plasmids. Twenty-four hour posttransfection, cell lysates were incubated with ubiquitin-agarose (Ub agarose) or protein A-agarose (PrA agarose) beads. Eluted complexes were resolved by SDS-PAGE and immunoblotted with anti-HA antibody. In the top ‘lysates’ panels, cell lysates were subjected directly to SDS-PAGE and immunoblotted with anti-HA antibody. Alix-F and TSG-F denote full-length Alix and Tsg101, respectively. Lanes C, mock-transfected controls. B) Binding of EIAV/ΔYPDL-Ub Gag to Alix requires an intact sorting motif. Cells were transfected with vectors expressing WT EIAV, EIAV/ΔYPDL, ΔYPDL-Ub or ΔYPDL-Ub L8A/I44A along with Alix full length (Alix-F) or Alix V (Alix-V) domain expression vector. Cell lysates were immunoprecipitated with anti-HA beads followed by western blot using anti-EIAV antibody (top). Western blot of cell lysates using anti-EIAV serum (middle) or anti-HA antibody (bottom).
Discussion
In this study, we examined the effect of Gag-ubiquitin fusion in the context of all three recognized retroviral late domains in an otherwise isogenic EIAV Gag background. We observed that fusion of ubiquitin to EIAV/ΔYPDL Gag rescues VLP release. Mechanistic studies suggested that the rescue of release occurred through recruitment of proteins bearing ubiquitin-binding motifs to the site of budding. Ubiquitin fusion also altered the sensitivity of individual Gag constructs to inhibitors of endosomal sorting and virus release. Our data demonstrate that in the presence of a functional late domain, fusion of ubiquitin does not significantly stimulate particle production, but in the absence of a late domain, ubiquitin fusion markedly enhances VLP release.
Studies by Patnaik et al. (16) demonstrated that fusion of ubiquitin to the C-terminus of RSV Gag overcame the inhibition mediated by proteasome inhibitors but did not rescue the release of a late domain-defective RSV mutant. In contrast, we observed that in the context of EIAV Gag, the late domain defect could be largely reversed by ubiquitin fusion. We further observed that ubiquitin fusion to EIAV/PPPY and EIAV/PTAP Gag conferred resistance to proteasome inhibition; however, in the absence of a late domain, ubiquitin fusion sensitized virus release to disruption of proteasome activity. The observation that release of EIAV/ΔYPDL-Ub VLPs is sensitive to the proteasome inhibitor MG-132 is consistent with the hypothesis that these compounds act at least in part by disrupting host-cell machinery involved in particle release rather than by preventing Gag ubiquitination. The findings that EIAV/ΔYPDL-Ub release is sensitive to depletion of Alix and Tsg101, whereas release of EIAV/ΔYPDL is not, and that ubiquitin fusion to EIAV/ΔYPDL and EIAV/PPPY promoted the incorporation of Alix into VLPs (unpublished results) suggest that ubiquitin mediates interactions, either direct or indirect, with host factors important for retrovirus budding. The observed sensitivity of EIAV/ΔYPDL-Ub release to Alix and Tsg101 disruption argues that ubiquitin fusion allows Gag to productively enter the budding pathway upstream of these factors. This hypothesis is further supported by the findings that inhibition of ΔYPDL-Ub Gag by Tsg101 depletion could be rescued by expression of siRNA-resistant WT Tsg101 but not by a mutant Tsg101 defective for ubiquitin binding.
When overexpressed, the Alix V domain is a potent inhibitor of HIV-1 and EIAV release (41-43). The inhibition observed in our prior studies requires a direct interaction between Gag and the V domain, as demonstrated by the finding that mutations in either HIV-1 p6 (42) or Alix (41) that block the interaction abrogate V domain-mediated disruption of particle release. We show in this study that release of WT EIAV is potently inhibited by V-domain overexpression, whereas EIAV/ΔYPDL, EIAV/PPPY and EIAV/PTAP, all of which lack a known Alix-binding site, are not inhibited by the V domain. Surprisingly, however, we observed that EIAV/ΔYPDL-Ub release is strongly inhibited by V-domain expression, suggesting a mechanism of inhibition that does not require a direct V-domain- YPXnL interaction. Indeed, a mutation (F676D) in the V domain that blocks late domain binding (41) has no effect on the ability of V-domain expression to block the release of EIAV/ΔYPDL-Ub VLPs. These results indicate that ubiquitin fusion in the absence of a functional late domain renders EIAV release sensitive to the Alix V domain by a mechanism that either does not require V domain-Gag binding or involves a novel binding interface. The experiments presented in Figure 9 suggest that the V domain interacts, either directly or indirectly, with ubiquitin. This interaction appears to involve residues that comprise the surface-exposed hydrophobic patch (which includes ubiquitin residue Leu-8 and Ile-44) involved in ubiquitin binding to ubiquitin-interacting motifs. Ubiquitin fusion is not sufficient to impose sensitivity to V-domain-mediated inhibition, as neither EIAV/PPPY-Ub nor EIAV/PTAP-Ub release is disrupted by the V domain. These results again indicate that the effect of ubiquitin attachment to Gag is determined in part by the presence or absence of a Gag-encoded late domain.
A common theme among retroviral late domains is the frequent presence of multiple late domains within an individual Gag protein. For example, HTLV-1 and M-PMV bear both PTAP and PPPY-type late domains (50,51), and MLV reportedly contains all three (PPPY-, PTAP- and YPXnL-type) (52) retroviral late domains. When more than one late domain coexist, one typically plays a dominant role in virus release. For example, HIV-1 contains a dominant Tsg101-binding PTAP-type late domain and an auxiliary Alix-binding YPXnL-type motif. The role of the YPXnL late domain in HIV-1 release is most evident when the dominant PTAP motif has been ablated and Alix is overexpressed (44,53). By analogy, our data suggest that ubiquitin serves a secondary late domain function that is revealed when the dominant late domain (YPXnL, in the case of EIAV) has been removed. The presence of multiple late domains likely arose to allow efficient virus budding to proceed when the primary late domain is not able to function to its full potential; for example, when its cellular binding partner is in short supply. It was reported recently that overexpression of the Nedd4-like ubiquitin ligase Nedd4L rescued the release of HIV-1 mutants lacking both PTAP and YPXnL late domains (46,54). This rescue required Nedd4L ubiquitin ligase activity and was blocked by Tsg101 depletion. It is conceivable that Nedd4L over-expression increases Gag ubiquitination, creating a situation analogous to that reported in this study in which Gag ubiquitination stimulates release of late-domain-deficient Gag in a Tsg101-dependent manner.
Retroviral late domains are able to function in trans (55), as we show in this study for fused ubiquitin, because not every molecule of Gag in an assembled particle must engage the cellular budding machinery. Indeed, we observe that while ubiquitin fusion is able to rescue the release of a late domain mutant, the resulting VLPs are morphologically aberrant, presumably because of the additional mass of ubiquitin attached to Gag. One would therefore expect that a limited amount of Gag ubiquitination would be sufficient to exert a positive effect on VLP release and that ubiquitin would be removed from Gag during the assembly/release process. Consistent with these expectations, under physiological conditions, only a small subset of Gag molecules are ubiquitinated (12) and abundant levels of free ubiquitin are detected in virus particles (10,12). The hypothesis that ubiquitin attached to Gag is able to recruit host sorting factors to the site of budding is supported by our observation that mutations in the hydrophobic patch of ubiquitin (comprised in part of Leu-8 and Ile-44) that interacts with ubiquitin-binding motifs not only abrogates rescue of release but also blocks EIAV/ΔYPDL-Ub interaction with Alix. This concept is also supported by our results indicating that ubiquitin fusion alters the sensitivity of VLP release to budding inhibitors and to depletion of cellular budding machinery (e.g. Tsg101 and Alix). Overall, our findings demonstrate that in the absence of an intact late domain, ubiquitin attached to Gag can promote particle release. Ubiquitin likely functions in this regard by recruiting to the site of virus assembly and release cellular factors that bear ubiquitin-binding domains.
Materials and Methods
Cell culture, plasmids, immunoprecipitations and western blotting analysis
293T cells were maintained in DMEM supplemented with 10% FBS. Transfections were performed in six-well dishes by using the Lipofectamine 2000 reagent (Invitrogen). Metabolic labeling was performed 24 h posttransfection essentially as described (56). Briefly, cells were washed once in Met-/Cys- medium and cultured for 5 h in the same medium supplemented with 10% FBS and 500 mCi/well [35S]Met/Cys protein labeling mix (PerkinElmer). Virus pellets were obtained after centrifugation of filtered culture supernatants for 45 min at 100 000 × g. Cell and virus lysates were immunoprecipitated with horse anti-EIAV serum (kindly provided by R. Montelaro, University of Pittsburgh); immunoprecipitated complexes were resolved by SDS-PAGE and detected by fluorography followed by phosphorImager analysis. Vectors expressing full-length (TSGF) (36) or truncated Tsg101 (TSG-5′ and TSG-3′) (35,36,57), WT or mutant Alix V domain (41,42) or a green fluorescent protein (GFP)-tagged, catalytically inactive (adenosine triphosphatase-deficient) mutant of Vps4 (Vps4EQ) (39) have been described elsewhere. Plasmids expressing siRNA-resistant WT Tsg101 and the Tsg101 mutant defective in ubiquitin binding were a kind gift of Dr Wesley Sundquist (University of Utah) (46,48). Western blotting for detection of haemagglutin (HA)-tagged proteins (TSGF, TSG-3′, TSG-5′ and Alix V domain) was performed using the anti-HA7 monoclonal antibody (Sigma). GFP-tagged proteins were detected with the anti-GFP peptide antibody (Sigma). Tsg101 was detected using the mouse monoclonal antibody C2 (Santa Cruz Biotechnologies). For detection of the EIAV Gag polyprotein by western blot, samples were probed with the anti-EIAV serum followed by detection using the horseradish peroxidaseconjugated anti-horse immunoglobulin G (Sigma). Quantification of immunoblot data was performed using an AlphaInnotech digital imager. For ubiquitin-agarose binding assays, cells were lysed 24 h posttransfection with the M-PER mammalian protein extraction reagent (Pierce). Cell lysates were then incubated with ubiquitin-agarose (Sigma) or protein A-agarose beads overnight at 4°C followed by three washes with Tris-buffered saline (TBS) containing 0.05% Tween-20. Complexes were then eluted in sample buffer [125 mM Tris-HCl (pH 6.8) containing 6% sodium dodecyl sulfate, 10% 2-mercaptoethanol and 20% glycerol] and boiled for 5 min. Eluates were resolved by SDS-PAGE followed by anti-HA immunoblot.
Generation of EIAV Gag-ubiquitin fusions
The generation of plasmids expressing WT EIAV Gag and mutant versions lacking a late domain (EIAV/ΔYPDL) or bearing heterologous late domains (EIAV/PPPY and EIAV/PTAP) has been described previously (26,27). To make these constructs, the QNLYPDLS sequence of EIAV Gag was changed to RPEPTAPP in EIAV/PTAP and QNLYPDLSEI was changed to ASAPPPPYVG in EIAV/PPPY (the underlines highlight the sequences that were changed). EIAV/ΔYPDL was constructed by deleting residues YPDL and replacing them with SRSA. Vectors expressing C-terminal Gag-ubiquitin fusions were constructed as follows: a polymerase chain reaction (PCR) product (reaction A) was generated from the EIAV Gag series using forward primer 5′-ACA ACA AAT CTG TTG TAC AAG AG-3′ and reverse primer 5′-AAA ATC TGC ATC TCC CAC AAA CTG TCC AGG TTG-3′'. The forward primer bears at its N-terminus a BsrGI site, and the reverse primer at its C-terminus bears the sequence encoding the N-terminus of ubiquitin. Simultaneously, a PCR product (reaction B) was derived from ubiquitin using forward primer 5′-GAC AGT TTG TGG GAG ATG CAG ATC TTC GTG AAG-3′ and reverse primer 5′ CCG AGC AAG CGG CCG CCA CCT TAA CCT CTG AGA CGG AGT ACC AG-3′. The ubiquitin forward primer at its N-terminus includes nucleotides from the C-terminus of EIAV Gag, and the reverse primer at its C-terminus contains a NotI restriction site. The 150 bp PCR product from reaction A was mixed with the 275 bp product from reaction B, filled in with Klenow and PCR amplified (reaction C) using primers Gag-F and Ubiquitin-R. This PCR product bears at its N-terminus the C-terminus of the EIAV-Gag-coding region and at its C-terminus a NotI restriction site. The EIAV Gag product was then digested in two separate reactions with enzymes XbaI/NotI and BsrGI/NotI. The PCR product from reaction C was digested with BsrGI/NotI. The above-digested fragments were then ligated, transformed and sequenced.
Immunofluorescence and EM analysis
Immunofluorescence analysis was performed essentially as described (58). Briefly, cells were seeded in chamber slides and transfected with the indicated plasmids. Twenty-four hour posttransfection, cells were fixed with 3.7% formaldehyde in sodium phosphate buffer, permeabilized with 0.1% Triton-X-100 in PBS and stained with EIAV anti-p26 antibody (kindly provided by R. Montelaro, University of Pittsburgh). Cells were mounted in Aqua Poly Mount (Polysciences, Inc) and visualized with the DeltaVision RT deconvolution microscope. For EM analysis, transfected cells were fixed in a 2% glutaraldehyde/100 mM sodium cacodylate solution, thin sectioned and analyzed with the transmission electron microscope.
Gag membrane binding and multimerization assays
Assays for Gag membrane binding and multimerization were conducted as described (34,59). Briefly, transfected cells were starved for 30 min in Cys-/Met- medium and then pulse-labeled for 5 min in medium containing [35S]Met/Cys followed by a chase of 15 min in complete, unlabeled medium. Cells were then washed with cold PBS, scrapped using a cell lifter and sonicated to achieve disruption. Postnuclear supernatants were placed at the bottom of 85.5%/65%/10% (w/v) sucrose gradients and subjected to equilibrium density centrifugation. Membrane (top) and non-membrane (bottom) fractions were isolated and lysed in 2× RIPA buffer. Fractions were then immunoprecipitated with anti-EIAV serum with or without prior denaturation by boiling in the presence of 2× sample buffer. Immunoprecipitated complexes were then resolved by SDS-PAGE followed by fluorography.
Proteasome inhibitor treatment
The peptide-based proteasome inhibitor N-carbobenzoxy-L-leucyl-L-leucyl-L-leucinal (zLLL or MG-132) (Calbiochem) was used in this study. Briefly, 24 h posttransfection, cells were treated for 30 min either with solvent (dimethyl sulfoxide) alone or with 10 µM MG-132 in DMEM containing 10% FBS. Thereafter, cells were treated with the same concentration of MG-132 in labeling medium for 4 h.
siRNA analysis
siRNAs against Tsg101 and Alix (AIP1), which were obtained from Qiagen as custom-synthesized oligonucleotides, have been described previously (43,48,60). The knockdown efficiency of the siRNAs was >80% for Tsg101 and >70% for Alix, as determined by quantitative western blot analysis.
Acknowledgments
We thank A. Ono and members of the Freed laboratory for helpful discussions and critical review of the manuscript and F. Soheilian for expert assistance with EM. We thank W. Sundquist for siRNA-resistant Tsg101 expression constructs, P. Woodman for the Vps4 expression vector, Z. Sun for TSG-5′ and TSG-3′ expression vectors and R. Montelaro for anti-EIAV Gag antiserum. This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (NIH), and by the Intramural AIDS Targeted Antiviral Program and was funded in part with federal funds from the National Cancer Institute, NIH, under contract N01-CO-12400.
Supplementary Material
References
- 1.Bieniasz PD. Late budding domains and host proteins in enveloped virus release. Virology. 2006;344:55–63. doi: 10.1016/j.virol.2005.09.044. [DOI] [PubMed] [Google Scholar]
- 2.Demirov DG, Freed EO. Retrovirus budding. Virus Res. 2004;106:87–102. doi: 10.1016/j.virusres.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Morita E, Sundquist WI. Retrovirus budding. Annu Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 4.Fujii K, Hurley JH, Freed EO. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat Rev Microbiol. 2007;5:912–916. doi: 10.1038/nrmicro1790. [DOI] [PubMed] [Google Scholar]
- 5.Gottlinger HG. How HIV-1 hijacks ALIX. Nat Struct Mol Biol. 2007;14:254–256. doi: 10.1038/nsmb0407-254. [DOI] [PubMed] [Google Scholar]
- 6.Martin-Serrano J. The role of ubiquitin in retroviral egress. Traffic. 2007;8:1297–1303. doi: 10.1111/j.1600-0854.2007.00609.x. [DOI] [PubMed] [Google Scholar]
- 7.Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochem J. 2006;399:361–372. doi: 10.1042/BJ20061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogt VM. Ubiquitin in retrovirus assembly: actor or bystander? Proc Natl Acad Sci U S A. 2000;97:12945–12947. doi: 10.1073/pnas.97.24.12945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ott DE, Coren LV, Chertova EN, Gagliardi TD, Schubert U. Ubiquitination of HIV-1 and MuLV Gag. Virology. 2000;278:111–121. doi: 10.1006/viro.2000.0648. [DOI] [PubMed] [Google Scholar]
- 10.Putterman D, Pepinsky RB, Vogt VM. Ubiquitin in avian leukosis virus particles. Virology. 1990;176:633–637. doi: 10.1016/0042-6822(90)90035-p. [DOI] [PubMed] [Google Scholar]
- 11.Heidecker G, Lloyd PA, Fox K, Nagashima K, Derse D. Late assembly motifs of human T-cell leukemia virus type 1 and their relative roles in particle release. J Virol. 2004;78:6636–6648. doi: 10.1128/JVI.78.12.6636-6648.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ott DE, Coren LV, Copeland TD, Kane BP, Johnson DG, Sowder RC, II, Yoshinaka Y, Oroszlan S, Arthur LO, Henderson LE. Ubiquitin is covalently attached to the p6Gag proteins of human immunodeficiency virus type 1 and simian immunodeficiency virus and to the p12Gag protein of Moloney murine leukemia virus. J Virol. 1998;72:2962–2968. doi: 10.1128/jvi.72.4.2962-2968.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ott DE, Coren LV, Sowder RC, II, Adams J, Nagashima K, Schubert U. Equine infectious anemia virus and the ubiquitin-proteasome system. J Virol. 2002;76:3038–3044. doi: 10.1128/JVI.76.6.3038-3044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin-Serrano J, Perez-Caballero D, Bieniasz PD. Context-dependent effects of L domains and ubiquitination on viral budding. J Virol. 2004;78:5554–5563. doi: 10.1128/JVI.78.11.5554-5563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strack B, Calistri A, Gottlinger HG. Late assembly domain function can exhibit context dependence and involves ubiquitin residues implicated in endocytosis. J Virol. 2002;76:5472–5479. doi: 10.1128/JVI.76.11.5472-5479.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patnaik A, Chau V, Wills JW. Ubiquitin is part of the retrovirus budding machinery. Proc Natl Acad Sci U S A. 2000;97:13069–13074. doi: 10.1073/pnas.97.24.13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schubert U, Ott DE, Chertova EN, Welker R, Tessmer U, Princiotta MF, Bennink JR, Krausslich HG, Yewdell JW. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc Natl Acad Sci U S A. 2000;97:13057–13062. doi: 10.1073/pnas.97.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strack B, Calistri A, Accola MA, Palu G, Gottlinger HG. A role for ubiquitin ligase recruitment in retrovirus release. Proc Natl Acad Sci U S A. 2000;97:13063–13068. doi: 10.1073/pnas.97.24.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin-Serrano J, Eastman SW, Chung W, Bieniasz PD. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J Cell Biol. 2005;168:89–101. doi: 10.1083/jcb.200408155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gottwein E, Jager S, Habermann A, Krausslich HG. Cumulative mutations of ubiquitin acceptor sites in human immunodeficiency virus type 1 gag cause a late budding defect. J Virol. 2006;80:6267–6275. doi: 10.1128/JVI.02177-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spidel JL, Craven RC, Wilson CB, Patnaik A, Wang H, Mansky LM, Wills JW. Lysines close to the Rous sarcoma virus late domain critical for budding. J Virol. 2004;78:10606–10616. doi: 10.1128/JVI.78.19.10606-10616.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demirov DG, Orenstein JM, Freed EO. The late domain of human immunodeficiency virus type 1 p6 promotes virus release in a cell type-dependent manner. J Virol. 2002;76:105–117. doi: 10.1128/JVI.76.1.105-117.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang M, Orenstein JM, Martin MA, Freed EO. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J Virol. 1995;69:6810–6818. doi: 10.1128/jvi.69.11.6810-6818.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhadina M, McClure MO, Johnson MC, Bieniasz PD. Ubiquitin-dependent virus particle budding without viral protein ubiquitination. Proc Natl Acad Sci U S A. 2007;104:20031–20036. doi: 10.1073/pnas.0708002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heidecker G, Lloyd PA, Soheilian F, Nagashima K, Derse D. The role of WWP1-Gag interaction and Gag ubiquitination in assembly and release of human T-cell leukemia virus type 1. J Virol. 2007;81:9769–9777. doi: 10.1128/JVI.00642-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li F, Chen C, Puffer BA, Montelaro RC. Functional replacement and positional dependence of homologous and heterologous L domains in equine infectious anemia virus replication. J Virol. 2002;76:1569–1577. doi: 10.1128/JVI.76.4.1569-1577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shehu-Xhilaga M, Ablan S, Demirov DG, Chen C, Montelaro RC, Freed EO. Late domain-dependent inhibition of equine infectious anemia virus budding. J Virol. 2004;78:724–732. doi: 10.1128/JVI.78.2.724-732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donaldson KM, Yin H, Gekakis N, Supek F, Joazeiro CA. Ubiquitin signals protein trafficking via interaction with a novel ubiquitin binding domain in the membrane fusion regulator, Vps9p. Curr Biol. 2003;13:258–262. doi: 10.1016/s0960-9822(03)00043-5. [DOI] [PubMed] [Google Scholar]
- 29.Mosesson Y, Shtiegman K, Katz M, Zwang Y, Vereb G, Szollosi J, Yarden Y. Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. J Biol Chem. 2003;278:21323–21326. doi: 10.1074/jbc.C300096200. [DOI] [PubMed] [Google Scholar]
- 30.Nakatsu F, Sakuma M, Matsuo Y, Arase H, Yamasaki S, Nakamura N, Saito T, Ohno H. A Di-leucine signal in the ubiquitin moiety. Possible involvement in ubiquitination-mediated endocytosis. J Biol Chem. 2000;275:26213–26219. doi: 10.1074/jbc.M907720199. [DOI] [PubMed] [Google Scholar]
- 31.Reggiori F, Pelham HR. Sorting of proteins into multivesicular bodies: ubiquitin-dependent and -independent targeting. EMBO J. 2001;20:5176–5186. doi: 10.1093/emboj/20.18.5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shih SC, Sloper-Mould KE, Hicke L. Monoubiquitin carries a novel internalization signal that is appended to activated receptors. EMBO J. 2000;19:187–198. doi: 10.1093/emboj/19.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Urbanowski JL, Piper RC. Ubiquitin sorts proteins into the intralumenal degradative compartment of the late-endosome/vacuole. Traffic. 2001;2:622–630. doi: 10.1034/j.1600-0854.2001.20905.x. [DOI] [PubMed] [Google Scholar]
- 34.Ono A, Waheed AA, Joshi A, Freed EO. Association of human immunodeficiency virus type 1 gag with membrane does not require highly basic sequences in the nucleocapsid: use of a novel Gag multimerization assay. J Virol. 2005;79:14131–14140. doi: 10.1128/JVI.79.22.14131-14140.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demirov DG, Ono A, Orenstein JM, Freed EO. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc Natl Acad Sci U S A. 2002;99:955–960. doi: 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goila-Gaur R, Demirov DG, Orenstein JM, Ono A, Freed EO. Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J Virol. 2003;77:6507–6519. doi: 10.1128/JVI.77.11.6507-6519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luttge BG, Shehu-Xhilaga M, Demirov DG, Adamson CS, Soheilian F, Nagashima K, Stephen AG, Fisher RJ, Freed EO. Molecular characterization of feline immunodeficiency virus budding. J Virol. 2008;82:2106–2119. doi: 10.1128/JVI.02337-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson MC, Spidel JL, Ako-Adjei D, Wills JW, Vogt VM. The C-terminal half of TSG101 blocks Rous sarcoma virus budding and sequesters Gag into unique nonendosomal structures. J Virol. 2005;79:3775–3786. doi: 10.1128/JVI.79.6.3775-3786.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bishop N, Woodman P. ATPase-defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol Biol Cell. 2000;11:227–239. doi: 10.1091/mbc.11.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patnaik A, Chau V, Li F, Montelaro RC, Wills JW. Budding of equine infectious anemia virus is insensitive to proteasome inhibitors. J Virol. 2002;76:2641–2647. doi: 10.1128/JVI.76.6.2641-2647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee S, Joshi A, Nagashima K, Freed EO, Hurley JH. Structural basis for viral late-domain binding to Alix. Nat Struct Mol Biol. 2007;14:194–199. doi: 10.1038/nsmb1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Munshi UM, Kim J, Nagashima K, Hurley JH, Freed EO. An Alix fragment potently inhibits HIV-1 budding: characterization of binding to retroviral YPXL late domains. J Biol Chem. 2007;282:3847–3855. doi: 10.1074/jbc.M607489200. [DOI] [PubMed] [Google Scholar]
- 43.Chen C, Vincent O, Jin J, Weisz OA, Montelaro RC. Functions of early (AP-2) and late (AIP1/ALIX) endocytic proteins in equine infectious anemia virus budding. J Biol Chem. 2005;280:40474–40480. doi: 10.1074/jbc.M509317200. [DOI] [PubMed] [Google Scholar]
- 44.Fisher RD, Chung HY, Zhai Q, Robinson H, Sundquist WI, Hill CP. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell. 2007;128:841–852. doi: 10.1016/j.cell.2007.01.035. [DOI] [PubMed] [Google Scholar]
- 45.Zhai Q, Fisher RD, Chung HY, Myszka DG, Sundquist WI, Hill CP. Structural and functional studies of ALIX interactions with YPX(n)L late domains of HIV-1 and EIAV. Nat Struct Mol Biol. 2008;15:43–49. doi: 10.1038/nsmb1319. [DOI] [PubMed] [Google Scholar]
- 46.Chung HY, Morita E, von Schwedler U, Muller B, Krausslich HG, Sundquist WI. NEDD4L overexpression rescues release and infectivity of HIV-1 constructs lacking PTAP and YPXL late domains. J Virol. 2008;82:4884–4897. doi: 10.1128/JVI.02667-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Urata S, Noda T, Kawaoka Y, Morikawa S, Yokosawa H, Yasuda J. Interaction of Tsg101 with Marburg virus VP40 depends on the PPPY motif, but not the PT/SAP motif as in the case of Ebola virus, and Tsg101 plays a critical role in the budding of Marburg virus-like particles induced by VP40, NP, and GP. J Virol. 2007;81:4895–4899. doi: 10.1128/JVI.02829-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 49.Pornillos O, Alam SL, Rich RL, Myszka DG, Davis DR, Sundquist WI. Structure and functional interactions of the Tsg101 UEV domain. EMBO J. 2002;21:2397–2406. doi: 10.1093/emboj/21.10.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gottwein E, Bodem J, Muller B, Schmechel A, Zentgraf H, Krausslich HG. The Mason-Pfizer monkey virus PPPY and PSAP motifs both contribute to virus release. J Virol. 2003;77:9474–9485. doi: 10.1128/JVI.77.17.9474-9485.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang H, Machesky NJ, Mansky LM. Both the PPPY and PTAP motifs are involved in human T-cell leukemia virus type 1 particle release. J Virol. 2004;78:1503–1512. doi: 10.1128/JVI.78.3.1503-1512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Segura-Morales C, Pescia C, Chatellard-Causse C, Sadoul R, Bertrand E, Basyuk E. Tsg101 and Alix interact with murine leukemia virus Gag and cooperate with Nedd4 ubiquitin ligases during budding. J Biol Chem. 2005;280:27004–27012. doi: 10.1074/jbc.M413735200. [DOI] [PubMed] [Google Scholar]
- 53.Usami Y, Popov S, Gottlinger HG. Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J Virol. 2007;81:6614–6622. doi: 10.1128/JVI.00314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Usami Y, Popov S, Popova E, Gottlinger HG. Efficient and specific rescue of human immunodeficiency virus type 1 budding defects by a Nedd4-like ubiquitin ligase. J Virol. 2008;82:4898–4907. doi: 10.1128/JVI.02675-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wills JW, Cameron CE, Wilson CB, Xiang Y, Bennett RP, Leis J. An assembly domain of the Rous sarcoma virus Gag protein required late in budding. J Virol. 1994;68:6605–6618. doi: 10.1128/jvi.68.10.6605-6618.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freed EO, Martin MA. Evidence for a functional interaction between the V1/V2 and C4 domains of human immunodeficiency virus type 1 envelope glycoprotein gp120. J Virol. 1994;68:2503–2512. doi: 10.1128/jvi.68.4.2503-2512.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun Z, Pan J, Hope WX, Cohen SN, Balk SP. Tumor susceptibility gene 101 protein represses androgen receptor transactivation and interacts with p300. Cancer. 1999;86:689–696. doi: 10.1002/(sici)1097-0142(19990815)86:4<689::aid-cncr19>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 58.Joshi A, Nagashima K, Freed EO. Mutation of dileucine-like motifs in the human immunodeficiency virus type 1 capsid disrupts virus assembly, gag-gag interactions, gag-membrane binding, and virion maturation. J Virol. 2006;80:7939–7951. doi: 10.1128/JVI.00355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ono A, Freed EO. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J Virol. 1999;73:4136–4144. doi: 10.1128/jvi.73.5.4136-4144.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hewitt EW, Duncan L, Mufti D, Baker J, Stevenson PG, Lehner PJ. Ubiquitylation of MHC class I by the K3 viral protein signals internalization and TSG101-dependent degradation. EMBO J. 2002;21:2418–2429. doi: 10.1093/emboj/21.10.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.