

Abstract
To examine base excision repair (BER) capacity in the context of living cells, we developed and applied a plasmid-based reporter assay. Non-replicating plasmids containing unique DNA base lesions were designed to express luciferase only after lesion repair had occurred, and luciferase expression in transfected cells was measured continuously during a repair period of 14 h. Two types of DNA lesions were examined: uracil opposite T reflecting repair primarily by the single-nucleotide BER sub-pathway, and the abasic site analogue tetrahydrofuran (THF) opposite C reflecting repair by long-patch BER. We found that the repair capacity for uracil-DNA in wild type mouse fibroblasts was very strong, whereas the repair capacity for THF-DNA, although strong, was slightly weaker. Repair capacity in DNA polymerase β (Pol β) null cells for uracil-DNA and THF-DNA was reduced by approximately 15% and 20%, respectively, compared to that in wild type cells. In both cases, the repair deficiency was fully complemented in Pol β null cells expressing recombinant Pol β. The effect of inhibition of poly(ADP-ribose) polymerase (PARP) activity on repair capacity was examined by treatment of cells with the inhibitor 4-amino-1,8-naphthalimide (4-AN). PARP inhibition decreased the repair capacity for both lesions in wild type cells, and this reduction was to the same level as that seen in Pol β null cells. In contrast, 4-AN had no effect on repair in Pol β null cells. The results highlight that Pol β and PARP function in the same repair pathway, but also suggest that there is repair independent of both Pol β and PARP activities. Thus, before the BER capacity of a cell can be predicted or modulated, a better understanding of Pol β and PARP activity-independent BER pathways is required.
Keywords: Base excision repair, DNA polymerase β, Poly(ADP-ribose) polymerase, Luciferase, Plasmid-based assay
1. Introduction
Mammalian cells have a variety of DNA repair systems that are essential for maintenance of genomic integrity [1]. Of these systems, base excision repair (BER) is critical for removal of modified or abnormal bases that arise spontaneously and after exposure to genotoxicants [2,3]. BER is considered to operate by two broad sub-pathways, single-nucleotide (SN-BER) and multi-nucleotide or long-patch (LP-BER), depending on the excision repair patch size as well as the enzymes and co-factors involved [4–6] In the current working model for the simplest type of SN-BER, repair is initiated by a damage-specific monofunctional DNA glycosylase, producing an apurinic/apyrimidinic (AP) or abasic site that is cleaved by AP endonuclease 1 (APE1). The resulting single-nucleotide gap is filled by DNA polymerase β (Pol β), the incised sugar phosphate is removed, and repair is completed by a DNA ligase [7–10]
There are many reports describing biochemical studies that suggest roles for Pol β in SN-BER and LP-BER. For example, purified Pol β has BER-related enzymatic activities, and BER activity is decreased in extracts from Pol β null cells or when Pol β is inhibited in wild type cell extracts [11–15]. However, experiments to understand the role of Pol β in BER in the context of a living cell are limited, and the measurements available thus far have been indirect. For example, after treatment of cells with a base damaging agent, assays for cell viability, chromosomal aberrations, mutagenesis, and persistence of strand breaks have revealed a protective role for Pol β consistent with its known BER-related enzymatic activities [12,14,16–19]. In addition, Pol β is recruited to sites of base damage in living cells [20]
In addition to concerns regarding the indirect nature of previous approaches for Pol β involvement in BER, most genotoxic agents used in the laboratory (e.g., methyl methanesulfonate [MMS]), produce several distinct forms of DNA damage. Therefore, interpretation of experiments regarding repair of specific types of lesions is complex. In the present study, we designed a plasmid-based assay to measure SN-BER and LP-BER capacity in living cells following transfection with plasmids bearing a single unique DNA lesion, either uracil-DNA and tetrahydofuran (THF)-DNA. These lesions were selected to reflect repair by SN-BER and LP-BER, respectively. The role of Pol β in both BER sub-pathways was evaluated along with the effect of inhibition of PARP activity on the observed repair.
The various sequential steps in the SN-BER and LP-BER sub-pathways appear to depend on coordination between the DNA intermediates of BER and the repair enzymes and co-factors, such that the intermediates are passed from one step to the next without triggering cell cycle arrest or cell death. The mechanism for this step-to-step coordination is not well understood, but it appears to be facilitated by co-factors including the abundantly expressed enzyme PARP-1. This enzyme has high affinity for DNA strand breaks, including those generated during BER [21]. Following PARP-1 binding at a BER intermediate strand break, PARP-1 is activated leading to autoribosylation, and activation of PARP-1 enables recruitment of other BER proteins, such as X-ray cross-complementing protein-1 (XRCC1) and Pol β, to the strand break-containing intermediate. PARP-1 can be released from the damaged site following its activation [22–27]. Surprisingly, PARP-1 deficient mouse fibroblasts cells are only moderately hypersensitive to DNA alkylating agents that produce base damage [28]. Yet, in sharp contrast, inhibitors of PARP-1 activity greatly increase hypersensitivity in response to DNA alkylating agents, e.g., wild type mouse fibroblasts are sensitized to MMS by approximately 100-fold [28]. This extreme sensitization indicates that PARP-1 activity is required in wild type mouse fibroblasts for efficient handling of MMS-induced DNA damage.
Uracil is a natural base lesion occurring in DNA by virtue of spontaneous deamination of cytosine or by dUMP insertion during replication [29,30]. It is known that uracil is removed by one of several uracil-DNA glycosylases depending on the base pairing status, among other factors [31–34]. After uracil removal, the resultant AP-site is incised by APE1, and it appears from cell extract-based experiments that both SN-BER and LP-BER sub-pathways operate in parallel to repair the BER intermediate [6,35–37].
In biochemical studies of the LP-BER sub-pathway, THF-DNA is frequently used as a model DNA substrate in place of DNA with the natural deoxyribose. Since Pol β cannot remove the 5'-THF phosphate group using its deoxyribose phosphate (dRP) lyase activity, repair is shunted into the LP-BER sub-pathway, and the 5'-THF-containing flap is generated and removed by flap endonuclease 1 (FEN1). Coordination between FEN1 and gap-filling polymerase(s) results in replacement of 2 to ~10 nucleotides via LP-BER [38,39].
Here, we describe the design and use of plasmids containing two types of unique base lesions. These plasmids can be introduced into cells for evaluation of DNA repair capacity by virtue of synthesis of a reporter gene product that depends strictly on repair of the DNA lesion. The system allows continuous measurement of the reporter gene product (luciferase) in cell culture, so as to provide quantification of cellular repair capacity over time. This is an improvement over methodology that requires preparation of a cell extract for analysis of repair at each time point. We report features of the assay system and demonstrate that repair capacities for uracil-DNA and THF-DNA vary as a function of Pol β expression. Further, our results using the PARP inhibitor 4-AN suggest that Pol β and PARP-1 activities may contribute to the same repair pathway in vivo.
2. Materials and methods
2.1. Cell lines and culture
The mouse embryonic fibroblast cell lines used in these experiments were wild type (MB16.3tsA), Pol β null (MB19.4tsA) and Pol β null cells complemented by expression of a FLAG epitope-tagged Pol β (Pol β-comp); these cell lines have been described previously [25]. Wild type and Pol β null cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) (HyClone, Logan, UT) containing 10% fetal bovine serum (FBS) (HyClone), 4 mM GlutaMAX-I (Invitrogen, Carlsbad, CA) and 80 μg/ml hygromycin (Invitrogen) in a 10% CO2 incubator at 34°C. For the maintenance of Pol β null cells expressing recombinant Pol β, the medium was additionally supplemented with 600 μg/ml G418 (Invitrogen). All cells were routinely tested and found to be free of mycoplasma contamination.
2.2. Oligonucleotides
Lesion-free oligonucleotides were from Operon Biotechnologies, Inc. (Huntsville, AL). Oligonucleotides containing uracil or THF were synthesized by Integrated DNA Technologies (Coralville, IA). The oligonucleotides were annealed to complementary strands and used as insert fragments for plasmid construction.
2.3. Preparation of plasmids
The plasmid AM1 was constructed by replacing the Renilla luciferase gene of pGL4.75 (Promega, Madison, WI) with the Chroma-Luc™ gene of pCBG68-control plasmid (Promega) at HindIII and XbaI sites. The BsaXI site was introduced in the Chroma-Luc™ gene by site-directed mutagenesis, and the resultant plasmid was digested by BsaXI (New England Biolabs, Ipswich, MA) and purified with a gel extraction kit (Qiagen, Valencia, CA) after 1% agarose gel electrophoresis. In the descriptions to follow, the underlined residues illustrate the altered codon base pairs in the luciferase gene. The 5'-phosphorylated oligonucleotide BsaXI fragment, the 30-mer uracil containing strand, 5'- GTGCTCGGAACUACATCTCACCAGCGGTGA annealed with the complementary strand, 5'-CCGCTGGTGAGATGTTGTTCCGAGCACTGC, was inserted into the BsaXI-digested AM1 plasmid vector (4897 bp) to prepare AM1-U. For a positive control, AM1-P, 5'-GTGCTCGGAACAACATCTCACCAGCGGTGA was annealed with the complementary strand, 5'-CCGCTGGTGAGATGTTGTTCCGAGCACTGC. For a negative control, AM1-N, 5'-GTGCTCGGAACTACATCTCACCAGCGGTGA was annealed with the complementary strand, 5'-CCGCTGGTGAGATGTAGTTCCGAGCACTGC.
Another plasmid series for THF, termed AM2-F, AM2-P and AM2-N, was prepared as described below. The CMV promoter gene was introduced into pCBG68-basic vector (Promega) in KpnI and HindIII sites. This plasmid, pCBG68-CMV, was digested with PsrI (SibEnzyme, West Roxbury, MA) and purified with a gel extraction kit after 1% agarose gel electrophoresis (5477 bp). The 5'-phosphorylated 32-mer oligonucleotide containing THF for the AM2-F plasmid, 5'-CTGGGCCFTAGATCACGTTCTTTTCGCGTTTC, and the oligonucleotide for the AM2-P plasmid, 5'-CTGGGCCGTAGATCACGTTCTTTTCGCGTTTC were annealed with 5'-GCGAAAAGAACGTGATCTACGGCCCAGAACCA. For AM2-N, the oligonucleotide, 5'-CTGGGCCCTAGATCACGTTCTTTTCGCGTTTC annealed with 5'-GCGAAAAGAACGTGATCTAGGGCCCAGAACCA was used as an insert fragment for the ligation reaction.
All ligation reactions were conducted at a molar ratio of insert to vector of 1:1 and were carried out at 16°C for ~12 h with T4 DNA ligase (New England Biolabs). After incubation, the ligation product was electrophoresed in a 1% agarose gel and visualized using a BlueView Transilluminator. Recircularized DNA plasmids were isolated and extracted using a gel extraction kit and suspended in TE buffer. Plasmids were characterized by restriction enzyme digestion. Reaction mixtures containing 100 ng plasmid DNA and 2 U BsaXI were incubated for 1 h at 37°C.
2.4. Immunoblotting
For immunoblotting, whole cell extracts were prepared as described [40]. Briefly, cells were grown to near-confluency, harvested by scraping, and collected by centrifugation, and the cell pellet was suspended in Buffer I [10 mM Tris-HCl, pH 7.8, 200 mM KCl, and protease inhibitor cocktail (Roche Molecular Diagnostics, Pleasanton, CA)]. An equal volume of Buffer II [10 mM Tris-HCl, pH 7.8, 200 mM KCl, 2 mM EDTA, 40% glycerol, 0.2% Nonidet P-40, 2 mM dithiothreitol, and protease inhibitor cocktail] was added. The cell suspension was mixed on a rocking platform for 1 h at 4°C, centrifuged at 14,000 rpm for 15 min at 4°C, and the supernatant fraction was collected. The protein concentration in the supernatant fraction was determined by Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA) with bovine serum albumin as standard. Immunoblotting analysis was conducted as described [41]. The cell extract (40 μg) was loaded onto NuPAGE 4–12% Bis-Tris gel (Invitrogen), separated by electrophoresis and transferred onto a nitrocellulose membrane. The membrane was incubated with 5% nonfat dry milk in Tris-buffered saline containing 0.1% (v/v) Tween 20 (TBST) to reduce non-specific binding. The membrane was probed with monoclonal antibody to Pol β (18S) [9], PARP-1 (BD Pharmingen, San Diego, CA), FEN1 (GeneTex, Inc.), Proliferating Cell Nuclear Antigen (PCNA) (EMD Chemicals Inc., Gibbstown, NJ) or G3PDH (Alpha Diagnostic, San Antonio, TX) at an appropriate dilution in 1% nonfat dry milk in TBST for 2 h at room temperature. After washing with TBST, the membrane was incubated with secondary antibody, either goat anti-mouse IgG (H+L)-horseradish peroxidase (HRP) conjugate (Bio-Rad Laboratories) or goat anti-rabbit IgG (H+L)-horseradish peroxidase (HRP) conjugate (Bio-Rad Laboratories) for 1 h at room temperature. The HRP activity was detected by enhanced chemiluminescence using SuperSignal West Pico Chemiluminescent substrate (Pierce Biotechnology Inc., Rockford, IL). The antibodies on the membrane were stripped by incubation with Restore Western Blot Stripping Buffer (Pierce Biotechnology Inc.) for 30 min at room temperature. After washing with TBST, the membrane was subjected to immunodetection with another antibody.
2.5. Use of luciferase expression in cultured cells
Wild type, Pol β null, and Pol β-comp cells were seeded in complete culture medium (10,000 cells/well) in a 96-well flat clear bottom polystyrene tissue culture-treated plate (Corning, Lowell, MA). After 20 h, the cells (still in logarithmic phase) were washed, and the medium was replaced with Opti-MEM I Reduced Serum Medium without phenol red (Invitrogen). Each well of cells was co-transfected with a plasmid mixture containing 200 ng internal control plasmid (pGL4.75) and 200 ng plasmid construct using PLUS Reagent (Invitrogen) and Lipofectamine LTX Reagent (Invitrogen) in the Opti-MEM I Reduced Serum Medium without phenol red. Transfections were performed on ice for 3 h with or without the PARP inhibitor 4-AN (10 μM), followed by medium change to DMEM without phenol red containing 10% FBS, 4 mM GlutaMAX-I, 25 mM HEPES and 0.2 mM D-luciferin potassium salt (MP Biomedicals, Solon, OH) plus 4-AN where indicated. After continuous measurement of luciferase activity for a repair period of 14 h at 34°C using a Tropix TR717 microplate luminometer (Applied Biosystems Inc., Foster City, CA), each well was washed with phosphate buffered saline. In some cases, as indicated, cell lysates were prepared and activities were assayed using the Dual-Glo Luciferase assay system (Promega).
3. Results
3.1. Construction and use of plasmids containing unique, site-specific DNA lesions
To investigate the contribution of Pol β to BER capacity in vivo for a specific type of base damage, we prepared non-replicating plasmids with a single unique base lesion in the luciferase reporter gene, either uracil opposite T for study of SN-BER/LP-BER or THF opposite C for study of LP-BER. Both of these lesion-containing plasmids were designed to result in a translational stop codon in the absence of repair. This situation was created using mismatches (U/T or THF/C), such that luciferase would be expressed only if substitution at the U or THF sites, respectively, had occurred during repair (Fig. 1 and Fig. 6). For example, uracil in codon 27 would be changed to A during repair and this codon would then encode for the original leucine residue; THF in codon 10 would be changed to G and would encode for the original tyrosine after repair. The U/T mismatch was chosen, in part, because it was expected to be a poor substrate for mismatch repair. Positive and negative control plasmids also were prepared in each case, such that they contained the original leucine or tyrosine codon (positive control) or the termination codon (negative control). The final plasmid preparations were routinely confirmed by restriction enzyme digestion. AM1-P and AM1-U do not have the BsaXI site that is present in AM1. As expected, AM1 was linearized by BsaXI treatment, but AM1-P and AM1-U were not, indicating that any AM1 contamination was below the detection limit (Fig. 1C).
Fig. 1.

(A) Transcriptional base substitution in the luciferase gene for the study of SN-BER. The codon 27 of the luciferase gene was modified to a stop codon by introduction of a uracil residue, as indicated. The uracil will be converted to a leucine codon after DNA repair, resulting in luciferase protein expression. (B) The construction of plasmid DNA containing uracil is described under “Materials and methods.” The Renilla luciferase gene of pGL4.75 was replaced by the Chroma-Luc™ gene, and the oligonucleotide fragment containing uracil was ligated at the BsaXI site introduced by site-directed mutagenesis in the Chroma-Luc™ gene. (C) Confirmation of plasmid preparations. Plasmids AM1, AM1-P (positive) and AM1-U (uracil) were treated with BsaXI at 37°C for 1 h, and then the mixtures were analyzed by 1% agarose gel electrophoresis. AM1 was used as reference for closed circular (lane 1) and linear DNA (lane 2).
Fig. 6.

Transcriptional base substitution in the luciferase gene for the study of LP-BER. THF was placed in codon 10 of the luciferase gene, resulting in a stop codon. The repaired codon will encode tyrosine, allowing expression of the luciferase gene product (protein).
During transfection using these plasmids, the cells were held on ice for 3 h and then were incubated for repair and luciferase expression in normal culture medium at 34°C. Chroma-Luciferase was used for continuous measurement of luciferase expression in each culture well (~20,000 cells), and the luciferase signal was measured as a function of time in culture. The incubation period and plasmid DNA amount routinely used were determined from experiments similar to those shown in Fig. 2. After the transfection of wild type cells with a positive control plasmid, the luciferase signal was followed for 42 h (Fig. 2A). Luciferase expression was first observed after ~4 h of culture and then increased in a linear fashion from ~5 to 15 h approaching a plateau, then after a plateau phase, the expression level decreased. This phenomenon of different phases including a linear increase, plateau and decrease was observed in all cell lines. Luciferase expression within the linear phase also was measured using different amounts of positive control plasmid or lesion-containing plasmid in wild type cells, and the results were plotted at the 10 h time point (Fig. 2B). This time was chosen because of the uniform slope linearity at this time and also because of the sufficient difference in luciferase expression between positive control plasmids and lesion-containing plasmids. There were no differences between the two plasmids with less than or equal to 200 ng transfected DNA, and luciferase expression increased linearly, depending on plasmid amount (Fig. 2B). This was not the case with 400 and 800 ng DNA where expression from both plasmids was no longer linear with DNA amount (Fig. 2B). Therefore, 200 ng plasmid DNA was selected for the transfections, and repair activity was compared at the 10 h point.
Fig. 2.

(A) Luciferase expression with the positive control plasmid. Expression from AM1-P was measured repeatedly in wild type cells for 42 h at 34°C. (B) Effect of amount of transfection plasmid DNA on luciferase expression. A range of plasmid quantities (10, 50, 100, 200, 400 and 800 ng) of the positive control (AM1-P, diamonds) or uracil-DNA (AM1-U, circles) plasmids were used for transfection, and the luciferase expression at the 10 h time point was plotted.
Typical results for the time courses of luciferase expression in the set of three cell cultures and representing transfection with the positive and negative control plasmids corresponding to each type of lesion are shown in Fig. 3. In this experiment, after transfection with positive and negative control plasmids, the cells were cultured at 34°C for 14 h. The results revealed that luciferase expression was never observed in cells containing the negative control plasmids. With the positive control plasmids, the rate of increase in luciferase activity and extent of the increase varied slightly from experiment to experiment and also as a function of the cell line used. These features are illustrated in Fig. 3A and B for wild type, Pol β null, and Pol β-comp cells. Both the extent and rate of increase in luciferase activity reflected the amount of luciferase gene product in the cells. For the experiments to be described below with lesion-containing plasmids, the expression (i.e., luciferase activity) of the respective positive control plasmid was taken to be equivalent to 100% repair. This simple method of assessment of repair gave results that were essentially equivalent to values obtained from complex models based on the linear rate of increase in luciferase or the level at the plateau phase.
Fig. 3.


Triplicate experiments illustrating variations in expression of luciferase observed with the positive and negative control plasmids. (A) Expression observed with the uracil-DNA positive control (diamonds) and negative control (triangles) plasmids. The three cell lines used are indicated at the top. (B) Expression observed with the THF-DNA positive control (diamonds) and negative control (triangles) plasmids. The three cell lines are indicated.
3.2. Characterization of cell lines for protein expression
In order to use a plasmid-based reporter assay system for measurement of cellular BER capacity, it is important to characterize expression of key BER enzymes in the cell lines used. Immunoblotting analysis was conducted to examine expression of Pol β, PARP-1, FEN1 and PCNA (Fig. 4). As expected, expression of Pol β was absent in the Pol β null cell line; and, expression was abundant in the Pol β-comp cell line (Fig. 4, 1st panel). Expression levels of PARP-1 (2nd panel), FEN1 (3rd panel) and PCNA (4th panel) were not affected by the Pol β expression status.
Fig. 4.
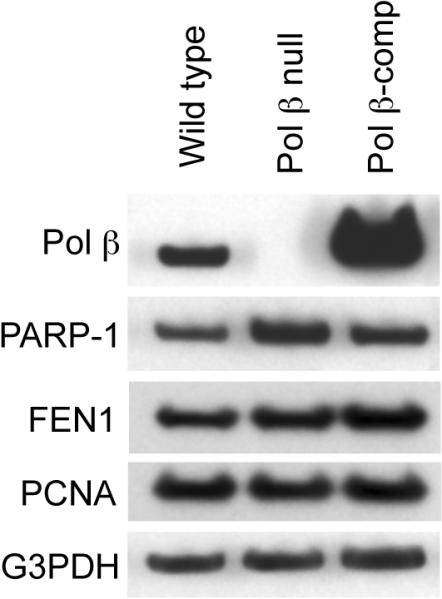
Immunoblotting for characterization of Pol β, PARP-1, FEN1 and PCNA expression. Whole cell extract was prepared from wild type, Pol β null, and Pol β-comp cells, and immunoblotting was with antibodies for each protein as indicated. The blots also were probed for anti-G3PDH as a loading control. Typical results are shown.
3.3. Repair efficiency for uracil-DNA in vivo
To quantify in vivo repair of uracil-DNA, AM1-U was co-transfected along with pGL4.75 into wild type cells, Pol β null cells or Pol β-comp cells. Experiments were conducted in parallel with AM1-P, the positive control plasmid containing the A/T base pair (used as reference for a completely repaired plasmid), with AM1-N, the negative control plasmid containing the T/A base pair (used as reference for an un-repaired plasmid) and with AM1-U, the lesion-containing plasmid. The level and time course of luciferase expression from AM1-U in wild type and in Pol β-comp cells was similar, and the expression in each case was identical to that of the positive control plasmid (Fig. 5A and C). These results indicate that repair capacity for uracil-DNA in both of these cell lines was strong. In Pol β null cells, there was a difference between results obtained with the uracil-DNA plasmid and the positive control plasmid (Fig. 5B). The relative repair capacity for uracil was evaluated by calculating the ratio of luciferase activity with the uracil-DNA plasmid versus that with the positive control plasmid. Complete results are summarized in Table 1, and a representative experiment is illustrated in Fig. 5B. These data represent mean ± SEM of seven independent experiments with three replicate cultures in each at the 10 h time point after the beginning of the repair incubation. The results show that the repair capacity for uracil-DNA in Pol β null cells was ~85% (P = 0.003), compared with the 100% uracil-DNA repair capacity of wild type and Pol β-comp cells. Finally, to confirm the results obtained from continuous luciferase measurements, cell extracts were prepared and assayed using the traditional Dual-Glo Luciferase assay system. This also allowed us to confirm transfection efficiency using the pGL4.75 plasmid. The results for repair capacity obtained by these extract-based measurements were consistent with the results obtained by continuous luciferase measurements in intact cells (data not shown).
Fig. 5.

Effect of uracil-DNA on luciferase expression in transfected cells. (A) Wild type (closed symbols), (B) Pol β null (open symbols) and (C) Pol β-comp (gray symbols) mouse fibroblast cells were transfected with a positive (+) control (diamonds), uracil-DNA (circles) or negative (−) control (triangles) plasmid on ice for 3 h, then luciferase activity was measured continuously for 14 h at 34°C. Typical results are shown, except the results with Pol β null cells illustrate an example of several experiments, but do not reflect the average from all experiments, as summarized in Table 1.
Table 1.
Relative repair activities for uracil-DNA and THF-DNA in Pol β null and Pol β-comp cells.
| Cell type | Relative repair activitya for uracil-DNA (%) | Relative repair activitya for THF-DNA (%) |
|---|---|---|
| Pol β null | 85 ± 3.1 (P = 0.003)b | 82 ± 4.7 (P = 0.011)b |
| Pol β-comp | 101 ± 5.0 (P = 0.82)b | 129 ± 5.8 (P = 0.005)b |
The relative repair activity was calculated by dividing the ratio (positive control vs. experimental) of luciferase activity for uracil-DNA or THF-DNA in Pol β null cells or Pol β-comp cells by the ratio in wild type cells. Values represent the mean ± SEM of 7 independent experiments, with three replicate cultures in each, at the 10 h time point after the beginning of the repair incubation.
P-value in student t-test was calculated by the comparison with wild type cells.
3.4. Repair efficiency of THF-DNA in vivo
The THF-containing plasmid, AM2-F, was used to assess LP-BER capacity in vivo (Fig. 6). In the same manner as the uracil-DNA plasmid experiments, AM2-F and the respective positive and negative control plasmids were co-transfected along with pGL4.75 into the three mouse fibroblast cell lines. Fig. 7 shows results for time-dependent luciferase expression of AM2-F, AM2-P and AM2-N. In contrast to the results obtained with the uracil-DNA plasmid, the repair efficiency of the THF-DNA plasmid in wild type cells did not allow luciferase expression to reach the level of the positive control plasmid (Fig. 7A). A difference between the THF plasmid and the positive control plasmid was even more significant in Pol β null cells, indicating a deficiency in repair relative to wild type cells. The luciferase expression difference between the THF plasmid and the positive control plasmid was the smallest in Pol β-comp cells (Fig. 7B and C). As reported in Table 1, the repair of THF-DNA in Pol β-comp cells was higher than in wild type cells (129 ± 5.8%) (P = 0.005) and the THF-DNA repair capacity of Pol β null cells was about 20% lower than in wild type cells (P = 0.011).
Fig. 7.

Effect of THF-DNA on luciferase expression in transfected cells. (A) Wild type, (B) Pol β null and (C) Pol β-comp cells. Symbol notations are as described in Fig. 5. Typical results are shown, except the results with Pol β null cells illustrate an example of several experiments, but do not reflect the average from all experiments, as summarized in Table 1.
3.5. Effect of PARP inhibition on repair capacity in vivo
We next examined the effect of PARP inhibition on in vivo repair in our assay system. The cells were treated continuously with the inhibitor 4-AN during the transfection period and subsequent culture while repair was analyzed (Fig. 8). First, we confirmed that 4-AN treatment had no effect on the transfection efficiency in experiments using the positive control plasmid (data not shown). In experiments with the uracil-DNA plasmid and wild type cells (Fig. 8A, closed bars), repair was decreased to ~20% relative to cells without 4-AN treatment (P = 0.044). This level of repair in 4-AN-treated wild type cells was similar to the level observed in Pol β null cells without 4-AN. In contrast, with the uracil-DNA plasmid in Pol β null cells, there was no difference in repair with and without 4-AN. In Pol β-comp cells, 4-AN treatment produced a slight reduction in uracil-DNA repair. Results with the THF-DNA plasmid showed a pattern similar to that observed with the uracil-DNA plasmid (Fig. 8B). Repair in wild type cells was decreased by PARP inhibition (P = 0.061), whereas repair in Pol β null cells was not; and repair of the THF-DNA plasmid in Pol β-comp cells was slightly reduced by 4-AN treatment (Fig. 8B).
Fig. 8.

Effects of PARP inhibition on BER activity. The PARP inhibitor 4-AN was present during the transfection and throughout the continuous luciferase expression measurement. The expression level was normalized for wild type cells without 4-AN treatment 10 h after the start of analysis. Experiments were conducted as described under “Materials and methods.” Shown are (A) repair of the uracil-DNA plasmid and (B) repair of the THF-DNA plasmid. The closed bars represent wild type cells, the open bars Pol β null cells, and the gray bars Pol β-comp cells. Typical results are shown. Asterisks show the P-values calculated by student t-test; uracil, P = 0.044; THF, P = 0.061.
4. Discussion
We examined BER capacity in the context of living cells using an assay system where Chroma-Luciferase expression representing synthesis of a reporter gene product is continuously measured. This approach has the benefit of providing a large number of data points on luciferase production with the economy of a single culture well. Use of this assay made it possible for us to observe subtle time-dependent changes in expression of luciferase associated with the repair of a specific single lesion-containing plasmid DNA in mouse fibroblast cells. We used two types of base lesion with the aim of examining both SN-BER and LP-BER sub-pathways. We viewed the uracil-DNA plasmid as a substrate for predominantly SN-BER and the THF-DNA plasmid as a substrate for LP-BER. As expected, the repair capacity for both uracil-DNA and THF-DNA was found to be lower in Pol β null cells compared with that in wild type cells (Table 1); yet, in all of our experiments, the measured reduction in repair in Pol β null cells was never more than ~50%. This reflected a high level of residual repair activity in the absence of Pol β, and the finding was surprising to us, based on earlier reports indicating that Pol β is the major DNA polymerase in SN-BER responsible for BER gap-filling synthesis and dRP lyase activity [9,10].
The residual repair in the absence of Pol β could be as a result of back-up DNA polymerases, back-up DNA repair pathways, or an intrinsic background in our assay system. First, regarding the latter possibility, there are reports on the effects of abasic site lesions on RNA polymerase II (RNAPII) transcription involving experiments with purified systems and cell extracts [42]. The abasic site lesion has been found to be a block for RNAPII [43], and the single strand break generated from the conversion of these damaged sites by DNA glycosylase and/or APE1 activities blocks RNAPII transcription elongation [44,45]. In our system, however, even if transcription occurs by lesion bypass before repair, the effects on the residual activity will be minimal because of the use of a stop codon and the rare C and U misinsertions opposite any bypass of the THF lesion (see Fig. 6). Hence, this possible explanation for the residual repair in the absence of Pol β seems unlikely.
Next, concerning the possibility of back-up DNA polymerases, there appears to be a number of options available to the cell. DNA polymerase λ (Pol λ) is an X family polymerase, like Pol β, and is known to have the DNA polymerase and dRP lyase activities required for SN-BER [46–48]. Although kinetic analyses demonstrated that the DNA synthesis turnover number of Pol λ is lower than that of Pol β, it seems clear that Pol λ can play a back-up role for Pol β, and furthermore, expression of Pol λ is strong in the mouse fibroblasts used here [49,50]. Pol λ interacts with PCNA [51], suggesting it may be involved in LP-BER as well as SN-BER, even when uracil-DNA is the substrate. In addition, DNA polymerases ι and θ have been suggested to function in BER [52–54], and DNA polymerase δ and/or ε have been suggested to participate specifically in LP-BER [55]. Accordingly, several DNA polymerases could compensate for a repair deficiency in the absence of Pol β, especially since the amount of damage used in the plasmid-based luciferase assay is expected to be lower than that following treatment of cells with most commonly used genotoxic agents. Nevertheless, despite the presence of these potential back-up polymerases, repair in Pol β null cells was not as strong as in wild type cells.
Finally, regarding the possibility that repair pathways other than BER participate in the Pol β-independent residual repair activity observed, mismatch repair does not seem to be a strong candidate for uracil/T, one reason that this mispair was chosen. The T/T mismatch is known to be a relatively poor substrate for mismatch repair [56–59]. Based on the structural similarity between uracil and T, we expect this also to be a poor mismatch repair substrate. Transcription-coupled repair (TCR), initially described as a sub-pathway of nucleotide excision repair, also has been suggested to act on non-bulky base lesions like oxidative base damage [42,60–62]. Hence, TCR may function to back-up repair in the Pol β-deficient mouse cells used here [63–66]. Nevertheless, a better understanding of the Pol β-independent BER activity observed in these experiments will require further study.
In the presence of a PARP inhibitor, the repair capacity of wild type cells was decreased, and this reduction was to the same level as that found in Pol β null cells without inhibitor (Fig. 8). Moreover, the PARP inhibitor did not affect repair capacity in Pol β null cells. This indicates that PARP and Pol β activities are involved in the same repair process. Although not unexpected, these results are the first observation that the repair capacity for defined BER substrates is decreased by PARP inhibition in vivo. In contrast, cell sensitivity experiments have indicated that PARP activity plays a strong protective role in the response of replicating cells to alkylating agent-induced genotoxic stress, but this effect on cell sensitivity is not epistatic with the protective role of Pol β (i.e., the effects were additive). Taken together, it appears that Pol β and PARP may be partners in BER, as shown here, but that PARP activity has additional, Pol β-independent, roles in cellular protection against alkylating agent-induced damage.
Our data also demonstrated no differences in luciferase expression between the uracil-DNA plasmid and the positive control plasmid in wild type cells. This indicated rapid repair of uracil-DNA in wild type cells (Fig. 5A). In contrast, the repair of THF-DNA in wild type cells was slower (Fig. 7A). In the case of THF-DNA, the repair capacity in Pol β null cells expressing recombinant Pol β was higher than that of wild type cells, reflecting a situation where over-expression of Pol β can facilitate LP-BER to a level greater than that seen in wild type cells (Fig. 7). Thus, the level in wild type cells may be limiting for the type of LP-BER measured, since the Pol β level in wild type cells was lower than in Pol β-comp cells (Fig. 4).
It is noteworthy that we did not find differential effects of PARP inhibition on SN-BER versus LP-BER in these experiments. Favaudon et al. [67] reported that the LP-BER sub-pathway was not affected by the absence of PARP-1 expression, whereas the SN-BER sub-pathway was affected by the deficiency. This interpretation was based on in vivo recruitment analysis at sites of single strand breaks and did not include direct measurements of DNA repair. The role of PARP-1 and PARP activity in BER is still not fully understood, however, the results of our experiments are consistent with a scheme where PARP-1 and/or other PARP isoforms first bind to protect the nicked AP-site BER intermediate, then becomes automodified and leaves DNA allowing subsequent Pol β-dependent DNA synthesis. The effects of inhibition of PARP activity are in sharp contrast to a deficiency in PARP-1 expression [24,67,68].
There are many versions of “unique lesion” plasmid-based assay systems for measurement of DNA repair (or lesion by-pass) in vivo. However, most of these assay systems involve preparation of large amounts of plasmid containing unique damage and the use of extracts from multiple culture wells to measure a reporter gene product. In these systems, the reporter signals from well to well are normalized by use of another plasmid, but cell conditions may not be identical from one culture well to the next. A notable difference between the system described here and previous systems is that continuous measurement allows use of a minimal amount of plasmid, and signal tracking within the same culture well allows more precision in measurements. For future advancement of this system, it may be possible to measure reported gene products from both a positive control plasmid and the lesion-containing plasmid within the same culture well by using signals that are measured at different wavelengths; this should provide even more accurate repair comparisons. Finally, in previous plasmid assays, repair efficiency generally was calculated with much longer culture periods after transfection (e.g., 24 or 48 h), whereas the present system involved use of a shorter culture period and comparison of repair in the log phase of cell growth.
In summary, we identified a reduction in cellular base lesion repair as a result of Pol β deficiency by making use of lesion-containing plasmids, along with a precise reporter gene expression assay. There was high residual repair activity in the absence of Pol β, probably due to back-up DNA polymerases and/or repair systems. Inhibition of cellular PARP activity resulted in a similar reduction in the repair capacity of wild type cells. The results highlight the existence of redundant BER pathways that require a better understanding before the BER capacity of a cell can be predicted or modulated. This assay system should facilitate future comparison of repair capacity in wild type and repair protein-deficient cells, and also testing of designed small molecule repair inhibitors that may alter repair capacity. In addition, it should be possible to evaluate repair of various other lesions mimicking those introduced by endogenous stress.
Acknowledgements
We thank Kenjiro Asagoshi, Michael Carrozza, Graciela Spivak and Philip Hanawalt for discussions and critical reading of the manuscript and Ms. Bonnie Mesmer for editorial assistance. This research was supported by Research Project Numbers Z01-ES050158 and Z01-ES050159 in the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement The authors declare that there are no conflicts of interest.
REFERENCES
- [1].Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, DC: 2005. [Google Scholar]
- [2].Hickson ID. Base Excision Repair of DNA Damage. Chapman & Hall; New York: 1997. [Google Scholar]
- [3].Moldave K, Mitra S, McCullough AK, Lloyd RS, Wilson SH. Base Excision Repair (Progress in Nucleic Acid Research and Molecular Biology) Academic Press; London, New York, San Diego: 2001. [Google Scholar]
- [4].Beard WA, Wilson SH. Structure and mechanism of DNA polymerase β. Chemical Reviews. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- [5].Dianov GL, Prasad R, Wilson SH, Bohr VA. Role of DNA polymerase β in the excision step of long patch mammalian base excision repair. J. Biol. Chem. 1999;274:13741–13743. doi: 10.1074/jbc.274.20.13741. [DOI] [PubMed] [Google Scholar]
- [6].Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- [7].Cappelli E, Taylor R, Cevasco M, Abbondandolo A, Caldecott K, Frosina G. Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J. Biol. Chem. 1997;272:23970–23975. doi: 10.1074/jbc.272.38.23970. [DOI] [PubMed] [Google Scholar]
- [8].Prasad R, Singhal RK, Srivastava DK, Molina JT, Tomkinson AE, Wilson SH. Specific interaction of DNA polymerase β and DNA ligase I in a multiprotein base excision repair complex from bovine testis. J. Biol. Chem. 1996;271:16000–16007. doi: 10.1074/jbc.271.27.16000. [DOI] [PubMed] [Google Scholar]
- [9].Singhal RK, Prasad R, Wilson SH. DNA polymerase β conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J. Biol. Chem. 1995;270:949–957. doi: 10.1074/jbc.270.2.949. [DOI] [PubMed] [Google Scholar]
- [10].Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-β in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- [11].Fortini P, Parlanti E, Sidorkina OM, Laval J, Dogliotti E. The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J. Biol. Chem. 1999;274:15230–15236. doi: 10.1074/jbc.274.21.15230. [DOI] [PubMed] [Google Scholar]
- [12].Horton JK, Joyce-Gray D, Pachkowski BF, Swenberg JA, Wilson SH. Hypersensitivity of DNA polymerase β null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair (Amst) 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- [13].Podlutsky AJ, Dianova I, Wilson SH, Bohr VA, Dianov GL. DNA synthesis and dRPase activities of polymerase β are both essential for single-nucleotide patch base excision repair in mammalian cell extracts. Biochemistry. 2001;40:809–813. doi: 10.1021/bi002064s. [DOI] [PubMed] [Google Scholar]
- [14].Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH. The lyase activity of the DNA repair protein β-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000;405:807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- [15].Tano K, Nakamura J, Asagoshi K, Arakawa H, Sonoda E, Braithwaite EK, Prasad R, Buerstedde JM, Takeda S, Watanabe M, Wilson SH. Interplay between DNA polymerases β and λ in repair of oxidation DNA damage in chicken DT40 cells. DNA Repair (Amst) 2007;6:869–875. doi: 10.1016/j.dnarep.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cabelof DC, Guo Z, Raffoul JJ, Sobol RW, Wilson SH, Richardson A, Heydari AR. Base excision repair deficiency caused by polymerase β haploinsufficiency: accelerated DNA damage and increased mutational response to carcinogens. Cancer Res. 2003;63:5799–5807. [PubMed] [Google Scholar]
- [17].Fortini P, Pascucci B, Belisario F, Dogliotti E. DNA polymerase β is required for efficient DNA strand break repair induced by methyl methanesulfonate but not by hydrogen peroxide. Nucleic Acids Res. 2000;28:3040–3046. doi: 10.1093/nar/28.16.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Frechet M, Canitrot Y, Bieth A, Dogliotti E, Cazaux C, Hoffmann JS. Deregulated DNA polymerase β strengthens ionizing radiation-induced nucleotidic and chromosomal instabilities. Oncogene. 2002;21:2320–2327. doi: 10.1038/sj.onc.1205295. [DOI] [PubMed] [Google Scholar]
- [19].Kunkel TA. The mutational specificity of DNA polymerase-β during in vitro DNA synthesis. Production of frameshift, base substitution, and deletion mutations. J. Biol. Chem. 1985;260:5787–5796. [PubMed] [Google Scholar]
- [20].Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc. Natl. Acad. Sci. USA. 2004;101:13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Woodhouse BC, Dianov GL. Poly ADP-ribose polymerase-1: an international molecule of mystery. DNA Repair (Amst) 2008;7:1077–1086. doi: 10.1016/j.dnarep.2008.03.009. [DOI] [PubMed] [Google Scholar]
- [22].Lindahl T, Satoh MS, Poirier GG, Klungland A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995;20:405–411. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- [23].Dantzer F, de La Rubia G, Menissier-De Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- [24].Horton JK, Watson M, Stefanick DF, Shaughnessy DT, Taylor JA, Wilson SH. XRCC1 and DNA polymerase β in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008;18:48–63. doi: 10.1038/cr.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J. Biol. Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- [26].Prasad R, Lavrik OI, Kim SJ, Kedar P, Yang XP, Vande Berg BJ, Wilson SH. DNA polymerase β-mediated long patch base excision repair. Poly(ADP-ribose)polymerase-1 stimulates strand displacement DNA synthesis. J. Biol. Chem. 2001;276:32411–32414. doi: 10.1074/jbc.C100292200. [DOI] [PubMed] [Google Scholar]
- [27].Sukhanova MV, Khodyreva SN, Lebedeva NA, Prasad R, Wilson SH, Lavrik OI. Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase β and poly(ADP-ribose) polymerase 1: interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res. 2005;33:1222–1229. doi: 10.1093/nar/gki266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Horton JK, Wilson SH. Hypersensitivity phenotypes associated with genetic and synthetic inhibitor-induced base excision repair deficiency. DNA Repair (Amst) 2007;6:530–543. doi: 10.1016/j.dnarep.2006.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Brynolf K, Eliasson R, Reichard P. Formation of Okazaki fragments in polyoma DNA synthesis caused by misincorporation of uracil. Cell. 1978;13:573–580. doi: 10.1016/0092-8674(78)90330-6. [DOI] [PubMed] [Google Scholar]
- [30].Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- [31].Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- [32].Masaoka A, Matsubara M, Hasegawa R, Tanaka T, Kurisu S, Terato H, Ohyama Y, Karino N, Matsuda A, Ide H. Mammalian 5-formyluracil-DNA glycosylase. 2. Role of SMUG1 uracil-DNA glycosylase in repair of 5-formyluracil and other oxidized and deaminated base lesions. Biochemistry. 2003;42:5003–5012. doi: 10.1021/bi0273213. [DOI] [PubMed] [Google Scholar]
- [33].Pearl LH. Structure and function in the uracil-DNA glycosylase superfamily. Mutat. Res. 2000;460:165–181. doi: 10.1016/s0921-8777(00)00025-2. [DOI] [PubMed] [Google Scholar]
- [34].Visnes T, Akbari M, Hagen L, Slupphaug G, Krokan HE. The rate of base excision repair of uracil is controlled by the initiating glycosylase. DNA Repair (Amst) 2008;7:1869–1881. doi: 10.1016/j.dnarep.2008.07.012. [DOI] [PubMed] [Google Scholar]
- [35].Fortini P, Pascucci B, Parlanti E, Sobol RW, Wilson SH, Dogliotti E. Different DNA polymerases are involved in the short- and long-patch base excision repair in mammalian cells. Biochemistry. 1998;37:3575–3580. doi: 10.1021/bi972999h. [DOI] [PubMed] [Google Scholar]
- [36].Horton JK, Prasad R, Hou E, Wilson SH. Protection against methylation-induced cytotoxicity by DNA polymerase β-dependent long patch base excision repair. J. Biol. Chem. 2000;275:2211–2218. doi: 10.1074/jbc.275.3.2211. [DOI] [PubMed] [Google Scholar]
- [37].Hou EW, Prasad R, Asagoshi K, Masaoka A, Wilson SH. Comparative assessment of plasmid and oligonucleotide DNA substrates in measurement of in vitro base excision repair activity. Nucleic Acids Res. 2007;35:e112. doi: 10.1093/nar/gkm639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Liu Y, Beard WA, Shock DD, Prasad R, Hou EW, Wilson SH. DNA polymerase β and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J. Biol. Chem. 2005;280:3665–3674. doi: 10.1074/jbc.M412922200. [DOI] [PubMed] [Google Scholar]
- [40].Biade S, Sobol RW, Wilson SH, Matsumoto Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J. Biol. Chem. 1998;273:898–902. doi: 10.1074/jbc.273.2.898. [DOI] [PubMed] [Google Scholar]
- [41].Kedar PS, Kim SJ, Robertson A, Hou E, Prasad R, Horton JK, Wilson SH. Direct interaction between mammalian DNA polymerase β and proliferating cell nuclear antigen. J. Biol. Chem. 2002;277:31115–31123. doi: 10.1074/jbc.M201497200. [DOI] [PubMed] [Google Scholar]
- [42].Tornaletti S. Transcription arrest at DNA damage sites. Mutat. Res. 2005;577:131–145. doi: 10.1016/j.mrfmmm.2005.03.014. [DOI] [PubMed] [Google Scholar]
- [43].Tornaletti S, Maeda LS, Hanawalt PC. Transcription arrest at an abasic site in the transcribed strand of template DNA. Chem. Res. Toxicol. 2006;19:1215–1220. doi: 10.1021/tx060103g. [DOI] [PubMed] [Google Scholar]
- [44].Kathe SD, Shen GP, Wallace SS. Single-stranded breaks in DNA but not oxidative DNA base damages block transcriptional elongation by RNA polymerase II in HeLa cell nuclear extracts. J. Biol. Chem. 2004;279:18511–18520. doi: 10.1074/jbc.M313598200. [DOI] [PubMed] [Google Scholar]
- [45].Kuraoka I, Endou M, Yamaguchi Y, Wada T, Handa H, Tanaka K. Effects of endogenous DNA base lesions on transcription elongation by mammalian RNA polymerase II. Implications for transcription-coupled DNA repair and transcriptional mutagenesis. J. Biol. Chem. 2003;278:7294–7299. doi: 10.1074/jbc.M208102200. [DOI] [PubMed] [Google Scholar]
- [46].Garcia-Diaz M, Bebenek K, Gao G, Pedersen LC, London RE, Kunkel TA. Structure-function studies of DNA polymerase λ. DNA Repair (Amst) 2005;4:1358–1367. doi: 10.1016/j.dnarep.2005.09.001. [DOI] [PubMed] [Google Scholar]
- [47].Moon AF, Garcia-Diaz M, Batra VK, Beard WA, Bebenek K, Kunkel TA, Wilson SH, Pedersen LC. The X family portrait: structural insights into biological functions of X family polymerases. DNA Repair (Amst) 2007;6:1709–1725. doi: 10.1016/j.dnarep.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Uchiyama Y, Takeuchi R, Kodera H, Sakaguchi K. Distribution and roles of X-family DNA polymerases in eukaryotes. Biochimie. 2008 doi: 10.1016/j.biochi.2008.07.005. [DOI] [PubMed] [Google Scholar]
- [49].Braithwaite EK, Kedar PS, Lan L, Polosina YY, Asagoshi K, Poltoratsky VP, Horton JK, Miller H, Teebor GW, Yasui A, Wilson SH. DNA polymerase λ protects mouse fibroblasts against oxidative DNA damage and is recruited to sites of DNA damage/repair. J. Biol. Chem. 2005;280:31641–31647. doi: 10.1074/jbc.C500256200. [DOI] [PubMed] [Google Scholar]
- [50].Braithwaite EK, Prasad R, Shock DD, Hou EW, Beard WA, Wilson SH. DNA polymerase λ mediates a back-up base excision repair activity in extracts of mouse embryonic fibroblasts. J. Biol. Chem. 2005;280:18469–18475. doi: 10.1074/jbc.M411864200. [DOI] [PubMed] [Google Scholar]
- [51].Maga G, Blanca G, Shevelev I, Frouin I, Ramadan K, Spadari S, Villani G, Hubscher U. The human DNA polymerase λ interacts with PCNA through a domain important for DNA primer binding and the interaction is inhibited by p21/WAF1/CIP1. FASEB J. 2004;18:1743–1745. doi: 10.1096/fj.04-2268fje. [DOI] [PubMed] [Google Scholar]
- [52].Bebenek K, Tissier A, Frank EG, McDonald JP, Prasad R, Wilson SH, Woodgate R, Kunkel TA. 5'-Deoxyribose phosphate lyase activity of human DNA polymerase ι in vitro. Science. 2001;291:2156–2159. doi: 10.1126/science.1058386. [DOI] [PubMed] [Google Scholar]
- [53].Petta TB, Nakajima S, Zlatanou A, Despras E, Couve-Privat S, Ishchenko A, Sarasin A, Yasui A, Kannouche P. Human DNA polymerase ι protects cells against oxidative stress. EMBO J. 2008;27:2883–2895. doi: 10.1038/emboj.2008.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yoshimura M, Kohzaki M, Nakamura J, Asagoshi K, Sonoda E, Hou E, Prasad R, Wilson SH, Tano K, Yasui A, Lan L, Seki M, Wood RD, Arakawa H, Buerstedde JM, Hochegger H, Okada T, Hiraoka M, Takeda S. Vertebrate PolQ and Polβ cooperate in base excision repair of oxidative DNA damage. Mol. Cell. 2006;24:115–125. doi: 10.1016/j.molcel.2006.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Stucki M, Pascucci B, Parlanti E, Fortini P, Wilson SH, Hubscher U, Dogliotti E. Mammalian base excision repair by DNA polymerases δ and ε. Oncogene. 1998;17:835–843. doi: 10.1038/sj.onc.1202001. [DOI] [PubMed] [Google Scholar]
- [56].Brown TC, Jiricny J. Different base/base mispairs are corrected with different efficiencies and specificities in monkey kidney cells. Cell. 1988;54:705–711. doi: 10.1016/s0092-8674(88)80015-1. [DOI] [PubMed] [Google Scholar]
- [57].David P, Efrati E, Tocco G, Krauss SW, Goodman MF. DNA replication and postreplication mismatch repair in cell-free extracts from cultured human neuroblastoma and fibroblast cells. J. Neurol. Sci. 1997;17:8711–8720. doi: 10.1523/JNEUROSCI.17-22-08711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kramer B, Kramer W, Fritz HJ. Different base/base mismatches are corrected with different efficiencies by the methyl-directed DNA mismatch-repair system of E. coli. Cell. 1984;38:879–887. doi: 10.1016/0092-8674(84)90283-6. [DOI] [PubMed] [Google Scholar]
- [59].Su SS, Lahue RS, Au KG, Modrich P. Mispair specificity of methyl-directed DNA mismatch correction in vitro. J. Biol. Chem. 1988;263:6829–6835. [PubMed] [Google Scholar]
- [60].Frosina G. The current evidence for defective repair of oxidatively damaged DNA in Cockayne syndrome. Free Radical Biol. Med. 2007;43:165–177. doi: 10.1016/j.freeradbiomed.2007.04.001. [DOI] [PubMed] [Google Scholar]
- [61].Le Page F, Klungland A, Barnes DE, Sarasin A, Boiteux S. Transcription coupled repair of 8-oxoguanine in murine cells: the ogg1 protein is required for repair in nontranscribed sequences but not in transcribed sequences. Proc. Natl. Acad. Sci. USA. 2000;97:8397–8402. doi: 10.1073/pnas.140137297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tornaletti S, Maeda LS, Lloyd DR, Reines D, Hanawalt PC. Effect of thymine glycol on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. J. Biol. Chem. 2001;276:45367–45371. doi: 10.1074/jbc.M105282200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Stevnsner T, Muftuoglu M, Aamann MD, Bohr VA. The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mech. Ageing Dev. 2008;129:441–448. doi: 10.1016/j.mad.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Thorslund T, von Kobbe C, Harrigan JA, Indig FE, Christiansen M, Stevnsner T, Bohr VA. Cooperation of the Cockayne syndrome group B protein and poly(ADP-ribose) polymerase 1 in the response to oxidative stress. Mol. Cell. 2005;25:7625–7636. doi: 10.1128/MCB.25.17.7625-7636.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wong HK, Muftuoglu M, Beck G, Imam SZ, Bohr VA, Wilson DM., 3rd Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007;35:4103–4113. doi: 10.1093/nar/gkm404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Spivak G, Hanawalt PC. Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syndrome but normal in UV-sensitive syndrome fibroblasts. DNA Repair (Amst) 2006;5:13–22. doi: 10.1016/j.dnarep.2005.06.017. [DOI] [PubMed] [Google Scholar]
- [67].Godon C, Cordelieres FP, Biard D, Giocanti N, Megnin-Chanet F, Hall J, Favaudon V. PARP inhibition versus PARP-1 silencing: different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res. 2008;36:4454–4464. doi: 10.1093/nar/gkn403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Horton JK, Stefanick DF, Naron JM, Kedar PS, Wilson SH. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest after DNA methylating agent exposure. J. Biol. Chem. 2005;280:15773–15785. doi: 10.1074/jbc.M413841200. [DOI] [PubMed] [Google Scholar]