

Abstract
Proliferation and migration of vascular smooth muscle cells (VSMCs) lead to intimal thickening and influence the long-term patency of venous graft post coronary arterial bypass graft. There is increasing evidence that connexins are involved in the development of intimal hyperplasia and restenosis. We assessed connexin 43 (Cx43) expression and its role in angiotensin II-induced proliferation and migration of smooth muscle cells and the signal pathways involved in human saphenous vein bypass conduits. Angiotensin-II significantly increased gap junctional intercellular communication and induced the expression of Cx43 in human saphenous vein SMCs in a dose- and time-dependent manner through angiotensin II type 1 receptor. The effect of angiotensin-II was blocked by siRNA of ERK 1/2, p38 MAPK and JNK, respectively. Overexpression of Cx43 markedly increased the proliferation of saphenous vein SMCs. However, siRNA for Cx43 inhibited angiotensin-II-induced proliferation, cyclin E expression and migration of human saphenous vein SMCs. In dual-luciferase reporter assay, angiotensin-II markedly activated AP-1 transcription factor, which was significantly attenuated by a dominant negative AP-1 (A-Fos) with subsequent inhibition of angiotensin-II-induced transcriptional expression of Cx43. These data demonstrate the role of Cx43 in the proliferation and migration of human saphenous vein SMCs and angiotensin-II induced Cx43 expression via mitogen-activated protein kinases (MAPK)-AP-1 signaling pathway.
Keywords: Angiotensin II, Atherosclerosis, Connexin, Gap junctions, Mitogen-activated protein kinase, Restenosis, Vascular Smooth Muscle Cells
1. Introduction
Intimal hyperplasia is a proliferative vasculopathy that causes stenosis in the venous bypass graft, a pathophysiologic feature leading to occlusion of the vessel lumen [1], resulting in failure rates of 20% and 50% at 5 years and 10 years, respectively [2]. Proliferation and migration of vascular smooth muscle cells (VSMCs) into the sub-intimal space play an important role in intimal thickening in atherosclerosis and restenosis and influences the long-term patency of the venous graft [3]. Initiation and progression of the atherosclerotic plaque and restenotic vessel involve complex patterns of interaction between the cells of the arterialwall, in which cytokines, chemokines, and growth factors are known to play a critical role [4, 5]. For example, angiotensin II (Ang II), a potent growth factor, stimulates proliferation and migration of VSMCs, and induces the accumulation and deposition of collagen through Ang II type 1 (AT1) receptor; all factors contributing to the pathogenesis of atherosclerosis [6].
Another form of cell-to-cell interaction involves direct exchange of small cytosolic components via gap junctions, which are composed of connexon hemichannels, each of which are made of 6 connexin (Cx) molecules [7]. Three subtypes of Cx, Cx37, Cx40, and Cx43, are found in the cells of the vasculature, with Cx43 being the predominant one [7, 8]. Changes in connexin expression and function have been found in association with phenotypic changes in VSMCs during the progression of atherosclerotic lesions and following injury in large arteries [9]. Also, Ang II can activate the expression of Cx43 in the myocardial cells of rats in vitro [10]. However, little is known about the relationship between Ang II and Cx43 expression in VSMCs of the human saphenous vein (hSV). In this study, we examined the Cx43 expression and its role in the proliferation and migration of VSMCs. Further, we delineated the signaling pathways involved in the proliferation of smooth muscle cells in hSV bypass conduits following stimulation with Ang II.
2. Methods
2.1 Specimen Collection, Processing and Culture of Smooth Muscle Cell
The experimental protocol for this study was approved by the Institutional Review Board of Creighton University. The hSV specimens (mean patient age 71.5 ± 3.7 yrs) left over from the bypass surgical procedure were used for the study. Specimens were collected fresh in ice-cold University of Wisconsin solution, which is used to maintain cellular and tissue integrity in the organs for transplantation [11]. The hSV conduits were immediately brought to the laboratory where all of the procedures were carried out under sterile conditions. SMCs from the hSV conduits were isolated by a method previously reported from our laboratory [2]. Briefly, after gentle removal of the endothelial cells and adventitia, the specimen was minced and digested with digestion media (containing elastase, collagenase, M199SF medium, 1 mg/L glucose, HEPES, and FBS). The isolated cells were removed by centrifugation at 900g for 10 min at 4°C, suspended in M199SF and incubated at 37°C in a humidified 5% CO2 atmosphere for 10–14 days and passaged. The subcultured VSMCs were used between passages 3–6. The confluent cells showed the characteristic hill-and-valley pattern associated with spindle-shaped VSMCs. The purity of isolated VSMCs was examined with positive immunostaining to smooth muscle α-actin and caldesmon. Prior to stimulation experiments, cells were serum-starved for 24 h in DMEM.
2.2 Scrape Loading/Dye Transfer
Levels of gap junctional intercellular communication (GJIC) in control and treated cells in culture were determined by SL/DT (Scrape Loading/Dye Transfer) technique using a fluorescent dye, Lucifer Yellow (LY) (Invitrogen, Eugene, OR) [12, 13]. hSV SMCs were washed thoroughly with PBS in which Ca2+ was omitted to prevent uncoupling of the cells. Scrape loading was performed applying three cuts on cell monolayer with a razor blade, and then 0.5% LY dye was added to the cells. The dye was rinsed away after 5 min. Cells were washed three times with PBS, fixed with 4% paraformaldehyde, and cells stained with LY dye were detected by fluorescence emission with an inverted fluorescent microscope equipped with a camera. Cells that received the LY dye from the scrape-loaded cells were considered communicating with each other. The numbers of communicating cells in the untreated and treated cells were counted under a light microscope (magnification 10×). GJIC was expressed as percentage of the control.
2.3 pcDNA™3.1 - Cx43 Expression Vector Construction and Transfection of Vascular Smooth Muscle Cells
Reverse transcription polymerase chain reaction (RT-PCR) was performed following standard protocol. PCR was performed in a Perkin–Elmer GeneAMP PCR System 2400 as a hot-start PCR. Primer sequences for Cx43 were 5′-GGATCCATGGGTGACTGGAGCGCC-3′ (Forward primer), 5′-GCGGCCGC CTAGATCTCCAGGTCATCAGGC-3′ (Reverse primer) (1149bp). The primers were purchased from IDT and the Genbank Accession Code for Cx43 was NM000165. After successful amplification of cDNA from human SV SMCs, the Cx43 product (1149bp) was cloned into pcDNA™3.1(+) (5.4kb) (Invitrogen, OR, USA) using BamHI and NotI restriction enzyme sites according to the manufacture’s instruction. Plasmids prepared using QIAprep Spin Miniprep Kit (Qiagene Science, Maryland, USA) and plasmid constructs were confirmed by direct sequence using an ABI PRISM™ 310 Genetic Analyzer (PE Biosystems Japan, Chiba, Japan).
Transfection of VSMCs was performed using the nucleofector device and solutions (Amaxa biosystems, Amaxa Inc, USA). Nucleofector device is an apparatus which delivers unique electrical stimuli to the target cells. The resulting transfection efficiency was more than 70% in VSMCs.
2.4 Small Interfering RNA
The small interfering RNAs (siRNA) with following sense and antisense sequences were for Cx43: sense, 5′-CCUAGUCCAUCAGAUCAUGtt-3′; and antisense, 5′-CAUGAUCUGAUGGACUAGGtc-3′; for control siRNA (non-silencing): sense, 5′-UUCUCCGAACGUGUCACGUdTdT-3′; and antisense, 5′-ACGUGACACGUUCGGAGAAdTdT-3′. siRNAs for Cx43 were purchased from Ambion (Austin, USA), siRNA of ERK ½ P38 and c-jun were purchased from Santa Cruz Biotechnology (California, USA).
2.5 Proliferation and Migration Assay
The BrdU incorporation assay was performed using a cell proliferation ELISA BrdU kit (Roche Applied Science, Germany). The cells were labeled with 10 μM of BrdU solution and incubated for 24 h at 37°C. The cells were dried and fixed, and the cellular DNA was denatured with FixDenat solution for 30 min at room temperature. A mouse anti-BrdU monoclonal antibody conjugated with peroxidase was added to each well and the plates were incubated again at room temperature for 2 h. Tetramethylbenzidine was added and the cells were incubated for 30 min at room temperature. Finally, the absorbance of the samples was measured by a microplate reader at 450 nm. Cell numbers were counted in a hemocytometer using light microscopy. Briefly, a suspension of hSV SMCs was prepared and treated with Ang II ((10−7 mol/L) with pcDNA™3.1-Cx43 plasmid or Cx43-siRNA using 3 wells per group. Cells were then incubated for 48 h after experimental stimulation, and final cell number in each well was determined.
VSMC migration assay was performed using Transwell (Costar, MA, USA) system, which allows cells to migrate through a 5 μm pores sized polycarbonate membrane [14]. Briefly, cells were collected by trypsinization and 1×105 cells were added to the upper chamber of transwells. Serum-free DMEM with chemoattractant (100nM Ang II) was added to the lower compartment. After 24 h in a standard CO2 incubator at 37 °C, the membrane was removed from the chamber and placed into methanol for 5 min. Cells were scraped from the upper side of the membrane and the membrane was subsequently placed into stain with 0.2% crystal violet for 30 seconds. The membrane was mounted on a slide and the cells present on the underside of the membrane were counted under a light microscope (10× magnification).
2.6 Western Blot Analysis
VSMCs were collected and lysed in lysis buffer (60 mmol/L Tris HCl pH 7.4, 1% NP-40, 0.15% sodium deoxycholate, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L phenylmethylsulfonyl fluoride, 5 mmol/L sodium orthovanadate, 5 mmol/L NaF, 20 μg/mL leupeptin, and 5 μg/mL aprotinin), and the protein concentration of the lysate was determined by BioRad protein assay. Proteins were resolved by SDS-PAGE and transferred to Immobilon P membrane. Non-specific proteins were blocked by incubation in blocking buffer (5% milk powder/Tris-buffered saline, pH 7.8/0.1% Tween 20) and the membranes were incubated overnight at 4°C with blocking buffer containing antibodies to Cx43, cyclin E, ERK1/2, p38 MAPK and c-jun. Membranes were washed and incubated with HRP-conjugated secondary antibody and detected using an ECL kit (Pierce, Rockford, IL). As a loading control, GAPDH was probed and visualized in all blots.
2.7 Activation of Transcription Factor AP-1
VSMCs were transfected with 5μg of pAP-1-Luc (Panomics, Reswood City, CA) designed to monitor the AP-1 signal transduction pathway with or without dominant-negative AP-1 (A-Fos) (kindly provided by Dr. Charles Vinson) and 0.2μg of Renilla luciferase reporter control vector (Promega, Madison, WI) to normalize transfection efficiencies. Twenty-four hours after transfection, the cells were stimulated with Ang II for 3 hours following which the cells were harvested and lysates prepared. Luciferase activity was measured using a luminometer and a Dual Luciferase Reporter Assay System (Promega, Madison, WI) as per the manufacturer’s protocol. All values were normalized to Renilla luciferase activity.
2.8 Statistical Analysis
Data are presented as mean ± SE, Statistical analysis was performed using Student’s t- test or ANOVA when appropriate to analyze statistically significant differences between groups. For this study P < 0.05 was considered significant.
3. Results
3.1 Ang II Induced Cx43 Expression Through AT1 Receptors in the SMCs of Human Venous Bypass Vessels
Cx43 protein expression in hSV SMCs was evaluated in a time- and dose-dependent manner. After treatment with Ang II (10−7 mol/L), the Cx43 expression was increased significantly at 0.5 h, peaked at 1 h and remained at an elevated level even after 3 h in the SV samples (Fig. 1A). Also, Ang II (10−7 and 10−6 mol/L) induced a significant maximal response in SV at 1 h in a dose-dependent manner (Fig. 1B).
Fig. 1.

Ang II induced Cx43 protein expression through AT1 receptor in VSMCs. (A) Ang II (10−7 mol/L) induced Cx43 protein expression in SV SMCs in a time-dependent manner. (B) Ang II induced Cx43 protein expression of SV SMCs at 1 h in a dose-dependent manner. The relative amount of Cx43 protein expression was measured by Western blot. (C) Ang II receptor antagonists, losartan (10−6 mol/L, AT1 receptor-specific) and PD-123319 (10−6 mol/L, AT2 receptor-specific), were added to the cultured medium for 30 minutes followed by stimulation of the cells with 10−7 mol/L Ang II for an additional 24 hours. The relative amount of Cx43 protein expression was measured by Western blot. Each bar represents the ratio of Cx43/GAPDH (mean ± SE) from three independent experiments. *P<0.05 compared with control group, §P<0.01 compared with control group, #P<0.01 compared with Ang II group.
To determine which AT receptor subtype is involved in Ang II-induced Cx43 protein expression in hSV SMCs, the cells were pretreated for 30 minutes with either AT1 receptor antagonist, losartan (10−6 mol/L) or the AT2 receptor antagonist, PD-123319 (10−6 mol/L) followed by stimulation with Ang II (10−7 mol/L). Ang II-induced expression of Cx43 was completely inhibited by pretreatment with losartan but not by PD-123319 (Fig. 1C), suggesting that Ang-induced Cx43 expression in VSMCs is mediated through AT1 receptors.
3.2 Ang II Increased Gap Junctional Intercellular Communication in SMCs of Saphenous Vein
To determine the extent to which Cx43 expression was related to gap junctional intercellular communication in VSMCs, we performed Scrape Loading/Dye Transfer assays using the gap junction permeable fluorescent dye lucifer yellow. As assessed by this technique, serum-starved cells were communication- competent and transferred lucifer yellow to numerous cells distant to the wound edge. We added Ang II (10−7 mol/L) resulting in a significant increased GJIC in hSV SMCs (Fig. 2).
Fig. 2.
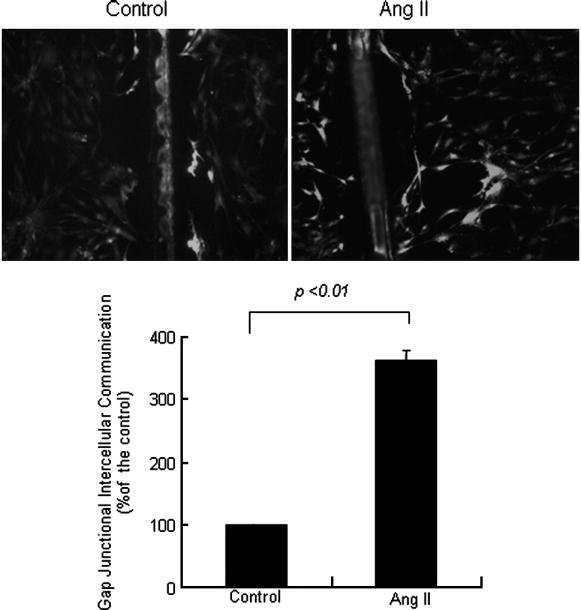
Ang II increased gap junctional intercellular communication in saphenous vein smooth muscle cells. SV SMCs were treated with Ang II (10−7 mol/L) for 1 hour, and gap junctional intercellular communication capacity was assayed by the Scrape Loading/Dye Transfer method. The number of communicating cells in the untreated and treated cultures was counted under a light microscope (magnification 10×). GJIC was expressed as percentage of the control. The bar represents percentage of the control (mean ± SE) for three independent experiments.
3.3 Overexpression of Cx43 Increased the Proliferation of Saphenous Vein SMCs
We performed transient transfection analysis using construct pcDNA™3.1 –Cx43 plasmid and investigated the Cx43 function in VSMCs. After transfection of the cells with pcDNA™3.1- Cx43 for 48 h, the total protein expression of Cx43 was significantly increased in VSMCs (Fig. 3). Cell proliferation ELISA was designed as a precise, fast and simple colorimetric alternative to quantify cell proliferation based on the measurement of BrdU incorporation during DNA synthesis in proliferating cell. In addition, cell numbers were also counted using light microscopy. The interesting finding was that the proliferation of VSMCs was induced significantly by overexpression of pcDNA™3.1 –Cx43 plasmid. However, there was no significant difference in the VSMC proliferation between empty and pcDNA™3.1-Cx43 plasmid transfected cells after Ang II stimulation (Fig. 4).
Fig. 3.
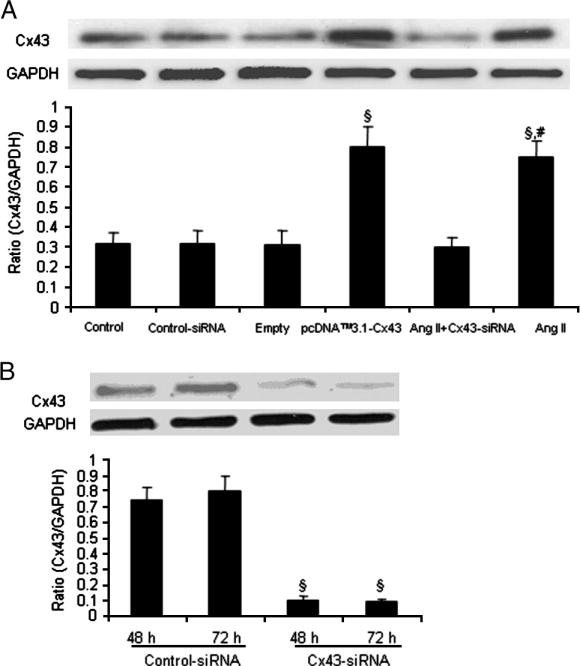
Effect of Cx43 siRNA and overexpression of pcDNA™3.1- Cx43 on the Cx43 expression in saphenous vein smooth muscle cells. (A) VSMCs were transfected with control-siRNA or empty plasmid or pcDNA™3.1- Cx43 or Cx43-siRNA for 48 h. (B) SV SMCs were transfected with siRNA against Cx43 and non-silencing siRNA and total protein expression of Cx43 was determined by Western blot analysis after 48h and 72h. The cells which were transfected with Cx43-siRNA were treated with Ang II (10−7 mol/L) for 24 h, lysed and protein expression of Cx43 was measured. Values are mean ± SE for three independent experiments. §P<0.01 compared with control group. # P<0.01 compared with Ang II+Cx-siRNA group.
Fig. 4.
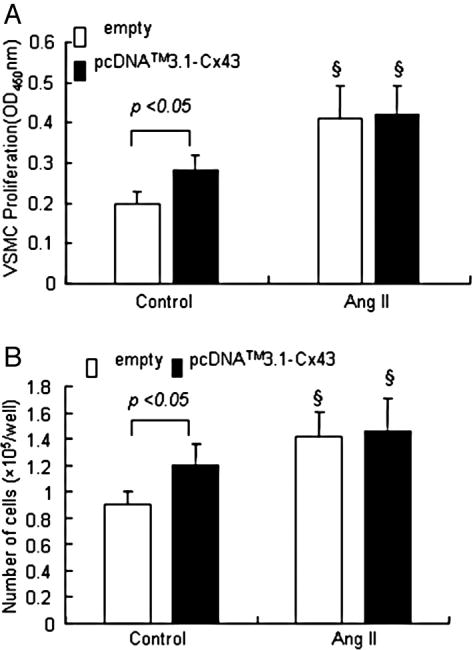
Effect of Cx43 overexpression on the proliferation of VSMCs. After transfection with pcDNA™3.1- Cx43 and empty vector, VSMCs were treated with Ang II (10−7 mol/L) for 24 h. (A) BrdU incorporation into VSMC was increased by Cx43 overexpression or Ang II stimulation. (B) Cell number of VSMC was significantly increased by Cx43 overexpression or Ang II stimulation. Values are mean± SE for three independent experiments. §P<0.01 compared with control group.
3.4 Cx43 Modulates Ang II-Induced Proliferation and Migration of Saphenous Vein SMCs
To obtain more insight into the role of Cx43 in Ang II-induced proliferation and migration of SMCs of saphenous vein, we knocked down Cx43 using siRNA. Initially, VSMCs were transfected with siRNA for Cx43 and non-silencing siRNA and the protein expression of Cx43 was determined by Western blot analysis. A marked reduction in Cx43 protein expression was observed at 48 h and 72 h following siRNA treatment (Fig. 3B). Cx43-siRNA also decreased the Ang II-induced expression of Cx43 protein (Fig. 3A). We then treated the VSMCs with siRNA for Cx43 and subsequently exposed them to Ang II (10−7 mol/L) for 24 h. Ang II significantly induced the proliferation of VSMCs and cyclin E expression in the control non-silencing siRNA transfected cells. Interestingly, the proliferation and cyclin E protein expression of VSMCs was significantly attenuated after transfection of the cells with Cx43-siRNA following Ang II stimulation (Fig. 5). These results provide further evidence as to the involvement of Cx43 in the proliferation of VSMCs.
Fig. 5.

Effect of Cx43–siRNA on Ang II-induced-proliferation of VSMCs and expression of cyclin E protein. After transfection with Cx43-siRNA and control-siRNA, VSMCs were treated with Ang II (10−7 mol/L) for 24 h. (A) BrdU incorporation into VSMC was inhibited by transfection with Cx43-siRNA. (B) Cell number of VSMC was significantly decreased by transfection with Cx43-siRNA. (C) Expression of cyclin E was inhibited by transfection of the cells with Cx43-siRNA. Values are mean± SE for three independent experiments.
Ang II also significantly induced the migration of VSMCs in the control non-silencing siRNA transfected cells, the migration of VSMCs could not be significantly induced after transfection of the cells with Cx43-siRNA following Ang II stimulation (Fig. 6). These results provide evidence as to the involvement of Cx43 in the migration of VSMCs.
Fig. 6.

Effect of Cx43–siRNA on the migration of VSMCs. After transfection with Cx43-siRNA and control-siRNA, VSMCs were treated with Ang II (10−7 mol/L) for 24 h and subsequently studied cells migration by using transwell apparatus. Photograph of migrated SMC of saphenous vein were shown under microscope (magnification 10×). Each bar represents mean ± SE of cell migrated per high-power microscope for three independent experiments.
3.5 Ang II Induced Activation of ERK 1/2, p38 and JNK Signaling Pathway–Role in Cx43 Expression in Saphenous Vein
We further investigated the potential signaling pathways in Ang II-induced Cx43 expression in hSV SMCs. Ang II markedly induced the phosphorylation of ERK ½, p38 and JNK (Fig. 7A). After transfection of the cells with siRNA for ERK, p38, c-jun, the protein expression of ERK, p38 and c-jun was significantly attenuated at 48 h and 72 h. There was no significant effect of control non-silencing siRNA (data not shown). However, Ang II-induced Cx43 protein expression was blocked by siRNA of ERK ½, p38 MAPK or JNK (Fig. 7B). These findings demonstrate that ERK 1/2, p38 MAPK and JNK mediated AngII-induced expression of Cx43 in hSV SMCs.
Fig. 7.

Effect of siRNA for ERK1/2, p38 and JNK on Ang II -induced expression of Cx43 in SMCs of saphenous vein. (A) hSV SMCs were stimulated with Ang II (10−7 mol/L) for 10 min (ERK and p38) and 30 min (JNK). The cells were lysed and protein expression of p-ERK, p-p38 and p-c-jun was examined. (B) After transfection of the cells with siRNA for ERK 1/2, p38, and c-jun and control-siRNA, VSMCs were treated with Ang II (10−7 mol/L) for 24 h. Proteins were quantified and each well was loaded with 20 μg of protein. Each bar represents the ratio of Cx43/GAPDH (mean ± SE) from three independent experiments. §P<0.01 compared with control group. # P<0.01 compared with Ang II group.
3.6 Involvement of the Activation of Transcription Factor AP-1 in the Ang II-induced Cx43 Expression
In order to confirm the involvement and activation of transcription factor AP-1 in Ang II-induced Cx43 expression, VSMCs were cotransfected with pAP-1-Luc and dominant-negative AP-1 (A-Fos). The dominant negative AP-1 (A-Fos) significantly blocked Ang II–induced activation of transcription factor AP-1 (Fig. 8A). Furthermore, dominant negative AP-1 (A-Fos) markedly inhibited Ang II-induced Cx43 protein (Fig. 8B). These results demonstrate that Ang II induces the Cx43 expression via activation of transcription factor AP-1.
Fig. 8.
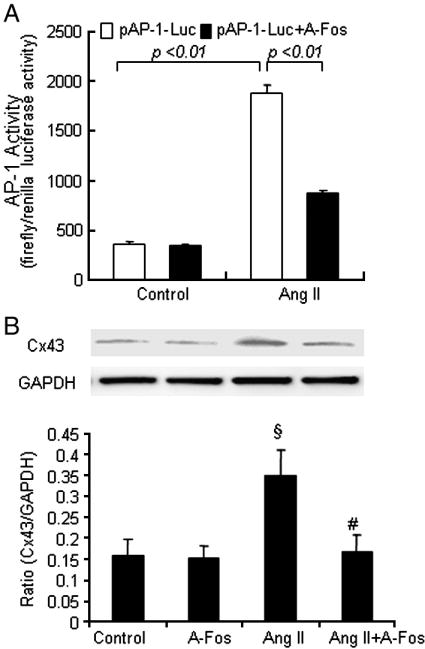
Involvement of activation of transcription factor AP-1 in Ang II induced connexin43 expression in saphenous vein SMCs. (A) VSMCs were co-transfected with pAP-1-Luc and 0.2 μg of Renilla luciferase reporter control vector in the absence or presence of dominant-negative AP-1 (A-Fos). Twenty-four hours after transfection, the cells were treated with Ang II (10−7 mol/L) for 3 hours, the cells were lysed and luciferase activities were measured. VSMCs were transfected with either control empty plasmid or dominant-negative AP-1 (A-Fos). After overnight recovery, cells were starved for 24 h and then either left untreated or treated with Ang II (10−7 mol/L) for 24 hours. (B) The cells were lysed and Cx43 protein was measured. Data are mean ± SE of five observations. §P<0.01 compared with control group. #P<0.01 compared with Ang II group.
4. Discussion
The hSV is the most commonly employed CABG conduit. Unfortunately, intimal hyperplasia leading to stenosis and occlusion of hSV commonly limit long-term results following coronary artery bypass graft (CABG) surgery [15]. The mechanisms involved in the intimal hyperplasia are proliferation and migration of medial smooth muscle cells towards the sub-intimal space [16]. There is increasing evidence that connexins are involved in the development of intimal hyperplasia and restenosis involving mouse and human atherosclerotic lesions [17]. Mensink and colleagues [18] demonstrated an increase in GJIC in VSMCs incubated with basic fibroblast growth factor and GJIC was a candidate for mediating the corresponding effect in vivo. We recently reported that increased Cx43 expression following stimulation of VSMCs with Ang II and insulin-like growth factor-1 (IGF-1) contributed to more proliferation in the SMCs of saphenous vein than in the internal mammary artery [2]. Gap junctions are the structural basis of intercellular communication between two adjacent cells and serves as channel for direct intercellular exchange of ions, small molecular metabolites, and as secondary messengers [19]. In most VSMCs, Cx43 expression and function seem dominant [20]. However, the regulation of Cx43 expression is not clear. In the present study, we investigated the effect of Ang II on the Cx43 expression and function and found that Cx43 was involved in the proliferation and migration of hSV SMCs. In addition, we provide new evidence that the Ang II-dependent increase in Cx43 expression is regulated by transcription factor, AP-1, via the ERK ½, p38 MAP kinases and JNK.
Over the past several years, a number of observations have suggested that Ang II plays an important role in various cardiovascular disease associated with VSMC proliferation such as hypertension, atherosclerosis, stenosis following balloon angioplasty and restenosis post-CABG [2, 21]. Recent studies revealed that the upregulation of Cx43 may relate to Ang II [22]. In our study, we demonstrate that Ang II induces increased protein expression of Cx43 in the hSV SMCs in a dose- and time-dependent manner via AT1 receptor. The increased expression of Cx43 may result in the increase in GJIC in VSMCs. Ang II is a multifunctional peptide that has numerous actions on VSMCs including proliferation [2, 23] and regulation of Cx43 expression [24]. In cardiac cells, Ang II induced activation of ERK1/2 and p38 pathways and is thought to play an important role in the upregulation of gap junctions under patho-physiological conditions [25]. In rat hepatic epithelial cells, Ang II upregulates the expression of Cx43, which subsequently results in the changes of cellular metabolic conditions and secondary messengers [26]. In atherosclerotic aorta of rabbits, statins reduce Cx40 and Cx43 expression by inhibition of angiotensin receptor [27]. We have recently reported that under ex vivo conditions connexin43 expression in both saphenous vein and internal mammary artery was influenced by angiotensin II and IGF-1 and may contribute to the pathogenesis of vein graft disease following coronary artery bypass grafting [2]. These results correlate with the findings in the literature that under in vitro conditions, angiotensin II can increase gap junctions and intercellular communication in cardiac cells [14] and connexin43 expression in WB rat liver epithelial cells [24]. Increased connexin43 expression in human saphenous veins in culture is also associated with intimal hyperplasia [1]. Thus, the regulation of Ang II-induced Cx43 expression in hSV SMCs may provide an explanation or elucidate the cause for the selective failure of hSV bypass grafts due to restenosis following CABG.
In our study, we found that the transfection of Cx43 alone was able to induce the proliferation of hSV SMCs. Conversely, silencing of Cx43 expression was able to inhibit the proliferation and migration induced by Ang II. These results are consistent with previous reports to show the participation of Cx43 in cell growth, differentiation and the development of intimal hyperplasia [1]. Also, Christos et al. also found that with a reduction in Cx43 expression of hypercholesterolemic mice, there is limited neointima formation after acute vascular injury accompanied by a decrease in the inflammatory response, and reduced VSMC migration and proliferation [28]. Thus, Cx43-mediated GJIC between VSMCs may play a role in the proliferative and migratory responses. The underlying mechanisms are unclear. The findings in our previous study have shown that cyclin E play an important role in the proliferation of VSMCs in human atherosclerotic plaque [3]. In this study, we found that cyclin E was involved in the Cx43-induced proliferation of VSMC after Ang II stimulation. Cx43 may also regulate cell cycle inhibitor proteins via p27 and p21, since the upregulation of Cx43 accompanies an increase in p27 and p21 expression levels in cancer cells [29]. These results provide insight into the direct correlation of Cx43 expression and cell cycle proteins. In our study we have not examined the relationship between Cx43 expression and p27 and p21 proteins in VSMCs. However, increased expression and activity of Cx43 in VSMCs increased proliferation, which is different from the studies in cancer cells [29]. None-the-less, Gap junction channels provide an enclosed conduit for direct exchanges of signaling molecules, including ions and small metabolites, between cells [1]. It is possible that this system of communication allows cells to review the functional state of their neighbors and regulate MAP kinases in a feedback manner to induce cell proliferation.
The mechanisms underlying migration and proliferation in VSMCs are different. The processes of migration include remodeling of the cytoskeleton, changing the adhesiveness of the cell to the matrix, and activating motor proteins. The key intracellular molecules that are involved in the migration process include phosphatidylinositol 3-kinase, calcium-dependent protein kinase, Rho-activated protein kinase, p21-activated protein kinases, LIM kinase, and mitogen-activated protein kinases [30]. However, the proliferation of VSMCs is due to increase in cell number associated with cell cycle proteins, DNA synthesis, and mitosis of cells, may involve some of the same enzymes as in the migration process [31]. There are some reports in the literature demonstrating that increase in Cx43 expression induces migratory and proliferative activity in H9c2 cells and proepicardial cells [32]. Cx43 might be one of the downstream target genes regulated by Wnt-3a [33]. Obviously, further studies are warranted to elucidate and dissect cellular and molecular mechanisms underlying Cx43-induced proliferation and migration of VSMCs.
JNK and MAP kinase have been reported to be important intracellular signaling pathways that regulate Cx43 expression [34, 35]. However, many of the signaling events relevant to cell proliferation and differentiation are mediated through activation of transcription factors via MAP kinases, which can be upregulated by Ang II [36]. Activation of the ERK, p38 and JNK signaling pathways may participate in the regulation of cardiac gap junctions [25, 37]. Here, our results confirm that Ang II can modulate the expression of Cx43 via the ERK1/2, p38 MAPK and JNK in hSV SMCs. However, the precise role of MAPKs in the regulation of Cx43 expression and activity in VSMCs is uncertain.
We hypothesized that Ang II mediates its effect through the ERK1/2, P38 and JNK pathways via the activation of transcription factor AP-1. AP -1 is part of a family of dimeric transcription factors comprised of either c-Jun homodimers or heterodimers of jun/fos family members [38]. Dominant-negative AP-1 (A-Fos) has been proven to inhibit AP-1 activity in epithelial tumor cell [39] and cardiac cells [40]. In cultured neonatal rat cardiomyocytes, amphetamine-activated Cx43 gene expression was through the JNK and AP-1 pathways [37]. The AP-1 and its promoter site are likely involved in the cytokine regulation of Cx43 [41]. Activation of ERK1/2 induces overexpression of c-fos mRNA and enhanced AP-1 DNA-binding activity [42]. Meanwhile, activation of p38 MAPK also results in phosphorylation of c-Jun and ATF-2 transcription factors, increasing their trans-activating AP-1 activity [43]. In our study, Ang II induced Cx43 expression via AP-1 transcriptional activity in hSV SMCs.
In summary, we have delineated cellular pathways to further understand the molecular mechanisms involved in the role of Cx43 in the proliferation and migration in hSV SMCs. These findings may explain, at least in part, the proliferative vasculopathy observed in vein graft disease. Further understanding of the expression of Cx43 in human venous bypass vessels has great clinical significance and could provide the basis for new approaches to better and effective treatment and to contain the morbidity and mortality associated with vein graft disease.
Acknowledgments
This study was supported by National Institutes of Health grants R01HL070885 (D.K.A) and R01HL073349 (D.K.A), and the State of Nebraska Tobacco Settlement Funds (D.K.A.). We thank Nebraska Heart Institute for providing us the venous bypass specimens.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deglise S, Martin D, Probst H, Saucy F, Hayoz D, Waeber G, et al. Increased connexin43 expression in human saphenous veins in culture is associated with intimal hyperplasia. J Vasc Surg. 2005;41:1043–52. doi: 10.1016/j.jvs.2005.02.036. [DOI] [PubMed] [Google Scholar]
- 2.Jia G, Mitra AK, Cheng G, Gangahar DM, Agrawal DK. Angiotensin II and IGF-1 regulate connexin43 expression via ERK and p38 signaling pathways in vascular smooth muscle cells of coronary artery bypass conduits. J Surg Res. 2007;142:137–42. doi: 10.1016/j.jss.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Jia G, Cheng G, Agrawal DK. Differential effects of insulin-like growth factor-1 and atheroma-associated cytokines on cell proliferation and apoptosis in plaque smooth muscle cells of symptomatic and asymptomatic patients with carotid stenosis. Immunol Cell Biol. 2006;84:422–9. doi: 10.1111/j.1440-1711.2006.01449.x. [DOI] [PubMed] [Google Scholar]
- 4.Jia G, Cheng G, Agrawal DK. Autophagy of vascular smooth muscle cells in atherosclerotic lesions. Autophagy. 2007;3:63–4. doi: 10.4161/auto.3427. [DOI] [PubMed] [Google Scholar]
- 5.Kwak BR, Mulhaupt F, Veillard N, Gros DB, Mach F. Altered pattern of vascular connexin expression in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2002;22:225–30. doi: 10.1161/hq0102.104125. [DOI] [PubMed] [Google Scholar]
- 6.Daemen MJ, Lombardi DM, Bosman FT, Schwartz SM. Angiotensin II induces smooth muscle cell proliferation in the normal and injured rat arterial wall. Circ Res. 1991;68:450–6. doi: 10.1161/01.res.68.2.450. [DOI] [PubMed] [Google Scholar]
- 7.Kwak BR, Mulhaupt F, Veillard N, Gros DB, Mach F. Altered pattern of vascular connexin expression in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2002;22:225–30. doi: 10.1161/hq0102.104125. [DOI] [PubMed] [Google Scholar]
- 8.Chanson M, Derouette JP, Roth I, Foglia B, Scerri I, Dudez T, et al. Gap junctional communication in tissue inflammation and repair. Biochim Biophys Acta. 2005;1711:197–207. doi: 10.1016/j.bbamem.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Plenz G, Ko YS, Yeh HI, Eschert H, Sindermann JR, Dorszewski A, et al. Upregulation of connexin43 gap junctions between neointimal smooth muscle cells. Eur J Cell Biol. 2004;83:521–30. doi: 10.1078/0171-9335-00417. [DOI] [PubMed] [Google Scholar]
- 10.Dodge SM, Beardslee MA, Darrow BJ, Green KG, Beyer EC, Saffitz JE. Effects of angiotensin II on expression of the gap junction channel protein connexin43 in neonatal rat ventricular myocytes. J Am Coll Cardiol. 1998;32:800–807. doi: 10.1016/s0735-1097(98)00317-9. [DOI] [PubMed] [Google Scholar]
- 11.Abebe W, Cavallari N, Agrawal DK, Rowley J, Thorpe PE, Hunter WJ, et al. Functional and morphological assessment of rat aorta stored in University of Wisconsin and Eurocollins solutions. Transplantation. 1993;56:808–16. doi: 10.1097/00007890-199310000-00006. [DOI] [PubMed] [Google Scholar]
- 12.Tanmahasamut P, Sidell N. Up-regulation of gap junctional intercellular communication and connexin43 expression by retinoic acid in human endometrial stromal cells. J Clin Endocrinol Metab. 2005;90:4151–6. doi: 10.1210/jc.2004-0663. [DOI] [PubMed] [Google Scholar]
- 13.Spinella F, Rosano L, Di Castro V, Nicotra MR, Natali PG, Bagnato A. Endothelin-1 decreases gap junctional intercellular communication by inducing phosphorylation of connexin 43 in human ovarian carcinoma cells. J Biol Chem. 2003;278:41294–301. doi: 10.1074/jbc.M304785200. [DOI] [PubMed] [Google Scholar]
- 14.Vigetti D, Moretto P, Viola M, Genasetti A, Rizzi M, Karousou E, et al. Matrix metalloproteinase 2 and tissue inhibitors of metalloproteinases regulate human aortic smooth muscle cell migration during in vitro aging. FASEB J. 2006;20:1118–30. doi: 10.1096/fj.05-4504com. [DOI] [PubMed] [Google Scholar]
- 15.Yan M, Liu DL, Chua YL, Chen C, Lim YL. Tyrosine kinase inhibitors suppress alpha1-adrenoceptor mediated contraction in human radial, internal mammary arteries and saphenous vein. Neurosci Lett. 2002;333:171–4. doi: 10.1016/s0304-3940(02)00877-7. [DOI] [PubMed] [Google Scholar]
- 16.Canham PB, Finlay HM, Boughner DR. Contrasting structure of the saphenous vein and internal mammary artery used as coronary bypass vessels. Cardiovasc Res. 1997;34:557–67. doi: 10.1016/s0008-6363(97)00056-4. [DOI] [PubMed] [Google Scholar]
- 17.Wong CW, Burger F, Pelli G, Mach F, Kwak BR. Dual benefit of reduced Cx43 on atherosclerosis in LDL receptor-deficient mice. Cell Commun Adhes. 2003;10:395–400. doi: 10.1080/cac.10.4-6.395.400. [DOI] [PubMed] [Google Scholar]
- 18.Mensink A, Brouwer A, van den Burg EH, Geurts S, Jongen WM, Lakemond CM, et al. Modulation of intercellular communication between smooth muscle cells by growth factors and cytokines. Eur J Pharmacol. 1996;310:73–81. doi: 10.1016/0014-2999(96)00368-8. [DOI] [PubMed] [Google Scholar]
- 19.Sandow SL, Tare M, Coleman HA, Hill CE, Parkington HC. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ Res. 2003;90:1108–1113. doi: 10.1161/01.res.0000019756.88731.83. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Simardm JM. Multiple connexins form gap junction channels in rat basilar artery smooth muscle cells. Circ Res. 1999;84:1277–1284. doi: 10.1161/01.res.84.11.1277. [DOI] [PubMed] [Google Scholar]
- 21.Cheng G, Kim MJ, Jia G, Agrawal DK. Involvement of chloride channels in IGF-I-induced proliferation of porcine arterial smooth muscle cells. Cardiovasc Res. 2007;73:198–207. doi: 10.1016/j.cardiores.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai W, Ruan LM, Wang YN, Chen JZ. Effects of angiotensin II on connexin 43 of VSMCs in arteriosclerosis. J Zhejiang Univ Sci B. 2006;7:648–53. doi: 10.1631/jzus.2006.B0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia G, Cheng G, Soundararajan K, Agrawal DK. Insulin-like Growth Factor-1 Receptors in Atherosclerotic Plaques of Symptomatic and Asymptomatic Patients with Carotid Stenosis: Effect of IL-12 and IFN-{gamma} Am J Physiol Heart Circ Physiol. 2007;292:H1051–7. doi: 10.1152/ajpheart.00801.2006. [DOI] [PubMed] [Google Scholar]
- 24.Bokkala S, Reis HM, Rubin E, Joseph SK. Effect of angiotensin II and ethanol on the expression of connexin43 in WB rat liver epithelial cells. Biochem J. 2001;357:769–77. doi: 10.1042/0264-6021:3570769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polontchouk L, Ebelt B, Jackels M, Dhein S. Chronic effects of endothelin 1 and angiotensin II on gap junctions and intercellular communication in cardiac cells. FASEB J. 2002;16:87–9. doi: 10.1096/fj.01-0381fje. [DOI] [PubMed] [Google Scholar]
- 26.Bokkala S, Reis HM, Rubin E, Joseph SK. Effect of angiotensin II and ethanol on the expression of connexin 43 in WB rat liver epithelial cells. Biochem J. 2001;357:769–77. doi: 10.1042/0264-6021:3570769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang LH, Chen JZ, Sun YL, Zhang FR, Zhu JH, Hu SJ, et al. Statins reduce connexin40 and connexin43 expression in atherosclerotic aorta of rabbits. Int J Cardiol. 2005;100:467–75. doi: 10.1016/j.ijcard.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Chadjichristos CE, Matter CM, Roth I, Sutter E, Pelli G, Luscher TF, et al. Reduced connexin43 expression limits neointima formation after balloon distension injury in hypercholesterolemic mice. Circulation. 2006;113:2835–43. doi: 10.1161/CIRCULATIONAHA.106.627703. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez-Alvarez R, Paino T, Herrero-Gonzalez S, Medina JM, Tabernero A. Tolbutamide reduces glioma cell proliferation by increasing Connexin43, which promotes the up-regulation of p21 and p27 and subsequent changes in retinoblastoma phosphorylation. Glia. 2006;54:125–34. doi: 10.1002/glia.20363. [DOI] [PubMed] [Google Scholar]
- 30.Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007;100:607–21. doi: 10.1161/01.RES.0000258492.96097.47. [DOI] [PubMed] [Google Scholar]
- 31.Berk BC. Vascular smooth muscle growth: autocrine growth mechanisms. Physiol Rev. 2001;81:999–1030. doi: 10.1152/physrev.2001.81.3.999. [DOI] [PubMed] [Google Scholar]
- 32.Liu X, Liu W, Yang L, Xia B, Li J, Zuo J, et al. Increased connexin 43 expression improves the migratory and proliferative ability of H9c2 cells by Wnt-3a overexpression. Acta Biochim Biophys Sin (Shanghai) 2007;39:391–8. doi: 10.1111/j.1745-7270.2007.00296.x. [DOI] [PubMed] [Google Scholar]
- 33.Li WE, Waldo K, Linask KL, Chen T, Wessels A, Parmacek MS, et al. An essential role for connexin43 gap junctions in mouse coronary artery development. Development. 2002;129:2031–42. doi: 10.1242/dev.129.8.2031. [DOI] [PubMed] [Google Scholar]
- 34.Cameron SJ, Malik S, Akaike M, Lerner-Marmarosh N, Yan C, Lee JD, et al. Regulation of epidermal growth factor-induced connexin43 gap junction communication by big mitogen-activated protein kinase1/ERK5 but not ERK1/2kinase activation. J Biol Chem. 2003;278:18682–18688. doi: 10.1074/jbc.M213283200. [DOI] [PubMed] [Google Scholar]
- 35.Petrich BG, Gong X, Lerner DL, Wang X, Brown JH, Saffitz JE, et al. c-Jun N-terminal kinase activation mediates downregulation of connexin43 in cardiomyocytes. Circ Res. 2002;91:640–647. doi: 10.1161/01.res.0000035854.11082.01. [DOI] [PubMed] [Google Scholar]
- 36.Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol. 2006;84:448–54. doi: 10.1111/j.1440-1711.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- 37.Shyu KG, Wang BW, Yang YH, Tsai SC, Lin S, Lee CC. Amphetamine activates connexin43 gene expression in cultured neonatal rat cardiomyocytes through JNK and AP-1 pathway. Cardiovasc Res. 2004;63:98–108. doi: 10.1016/j.cardiores.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 38.Hattori Y, Suzuki M, Hattori S, Kasai K. Vascular smooth muscle cell activation by glycated albumin (Amadori adducts) Hypertension. 2002;39:22–8. doi: 10.1161/hy1201.097300. [DOI] [PubMed] [Google Scholar]
- 39.Gerdes MJ, Myakishev M, Frost NA, Rishi V, Moitra J, Acharya A, et al. Activator protein-1 activity regulates epithelial tumor cell identity. Cancer Res. 2006;66:7578–88. doi: 10.1158/0008-5472.CAN-06-1247. [DOI] [PubMed] [Google Scholar]
- 40.Jeong MY, Kinugawa K, Vinson C, Long CS. A-Fos dissociates cardiac myocyte hypertrophy and expression of the pathological gene program. Circulation. 2005;111:1645–51. doi: 10.1161/01.CIR.0000160367.99928.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandez-Cobo M, Stewart D, Drujan D, De Maio A. Promoter activity of the rat connexin43 gene in NRK cells. J Cell Biochem. 2001;81:514–22. [PubMed] [Google Scholar]
- 42.Touyz RM, He G, Wu XH, Park JB, Mabrouk ME, Schiffrin EL. Src is an important mediator of extracellular signal-regulated kinase 1/2-dependent growth signaling by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients. Hypertension. 2001;38:6–64. doi: 10.1161/01.hyp.38.1.56. [DOI] [PubMed] [Google Scholar]
- 43.Viedt C, Soto U, Krieger-Brauer HI, Fei J, Elsing C, Kubler W, et al. Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II: involvement of p22phox and reactive oxygen species. Arterioscler Thromb Vasc Biol. 2000;20:940–8. doi: 10.1161/01.atv.20.4.940. [DOI] [PubMed] [Google Scholar]