

Abstract
Many neurological disorders are caused by expanded trinucleotide repeats1, including Machado-Joseph Disease (MJD)2 and Huntington Disease (HD)3. MJD and HD are caused by expanded CAG repeats within the ataxin-3 (ATXN3) and huntingtin (HTT) genes. Inhibiting expression of ATXN3 or HTT are promising therapeutic strategies, but indiscriminant inhibition of wild-type and mutant alleles may lead to toxicity. We hypothesized that expanded triplet repeat mRNA might be preferentially recognized by complementary oligomers. We observe selective inhibition of mutant ataxin-3 and HTT protein expression by peptide nucleic acid (PNA) and locked nucleic acid (LNA) oligomers targeting CAG repeats. Duplex RNAs were less selective, suggesting an advantage for single-stranded oligomers. Inhibiting mutant HTT expression protected cultured striatal neurons from an HD mouse model against glutamate-induced toxicity. Antisense oligomers that discriminate between wild-type and mutant genes on the basis of repeat length offer new options for treating MJD, HD, and other hereditary diseases.
Expanded trinucleotide repeats have been implicated in at least nineteen inherited diseases1, including MJD2, and HD3. These diseases are autosomal dominant disorders with most patients expressing both mutant and wild-type alleles. Production of the mutant protein can be toxic, possibly due to aggregation of the mutant protein or alteration of native protein-protein interactions.
MJD is one of the most common ataxias2. It is usually first diagnosed in adults, with patients eventually becoming wheelchair-bound or bedridden. There are no curative treatments. MJD is caused by expanded CAG repeats (12–39 repeats are normal, beyond 45 repeats indicates disease) within the ATXN3 gene.
HD has an incidence of 5–10 per 100,000 individuals in Europe and North America3,4. Unaffected individuals have up to 35 repeats, while HD patients can have from 36 to >100 repeats5. The disease is characterized by adult onset and progressive neurodegeneration. Like MJD, there are no curative treatments. HD is caused by the expansion of CAG trinucleotide repeats within the first exon of the HTT gene, leading to disruption of protein function and neurodegeneration.
Antisense oligonucleotides or double stranded RNAs have been proposed as a therapeutic strategy6–16. Mutant HTT and ataxin-3 proteins form interactions that are difficult to disrupt using traditional small molecule drugs17. Oligonucleotides and siRNAs, by contrast, affect phenotypes by reducing protein expression, providing a different therapeutic mechanism that avoids the problems faced by small molecules. siRNAs can inhibit HTT expression after infusion into the central nervous system10.
Most double-stranded or antisense oligonucleotides tested to date inhibit the mutant and wild-type protein expression indiscriminately6–10. HTT is known to play an essential role in embryogenesis, neurogenesis, and normal adult function18,19, raising concerns that agents inhibiting both mutant and wild-type HTT may induce significant side-effects in HD patients, especially if chronic administration is necessary. One strategy for distinguishing mutant from wild-type alleles for HD and other neurological diseases uses siRNAs that target single nucleotide or deletion polymorphisms11–16. These polymorphisms will often differ from patient to patient, necessitating development of a family of related compounds and complicating application of allele-specific RNAi in the clinic.
One challenge for therapeutic development for MJD or HD is to identify agents that will block the neurodegenerative effects of the mutant gene while preserving expression of the wild-type allele and normal biological function. To achieve this selectivity, we hypothesized that it might be possible to use single-stranded oligomers that discriminate between differences in the expanded mRNA sequence of wild-type and mutant alleles.
Triplet repeat sequences within RNA can form hairpin structures (Supplementary Fig. 1 online)20. The structures formed by wild-type and mutant mRNAs will possess different energies and stabilities, possibly enabling selective recognition of the mutant allele and subsequent selective inhibition of mutant protein expression. Alternatively, the expanded repeats create additional target sequence and more potential binding sites. For example, an allele with a wild-type repeat number of twenty would accommodate a maximum of three twenty-base oligomers, whereas a mutant allele with forty repeats would be large enough to accommodate six twenty-base oligomers.
To test our hypothesis, we synthesized peptide nucleic acid (PNA)-peptide conjugates targeting HTT mRNA (Supplementary Table 1 online, Fig. 1a,b). PNAs are a class of DNA/RNA mimic with an uncharged amide backbone that facilitates recognition of target sequences within RNA structure21. Unless otherwise noted, PNA conjugates were synthesized to contain a cationic peptide D-Lys8 at the C-terminus to promote the import of PNAs into cells22.
Figure 1. PNAs, LNAs, and inhibition of HTT expression by PNA-peptide conjugates.
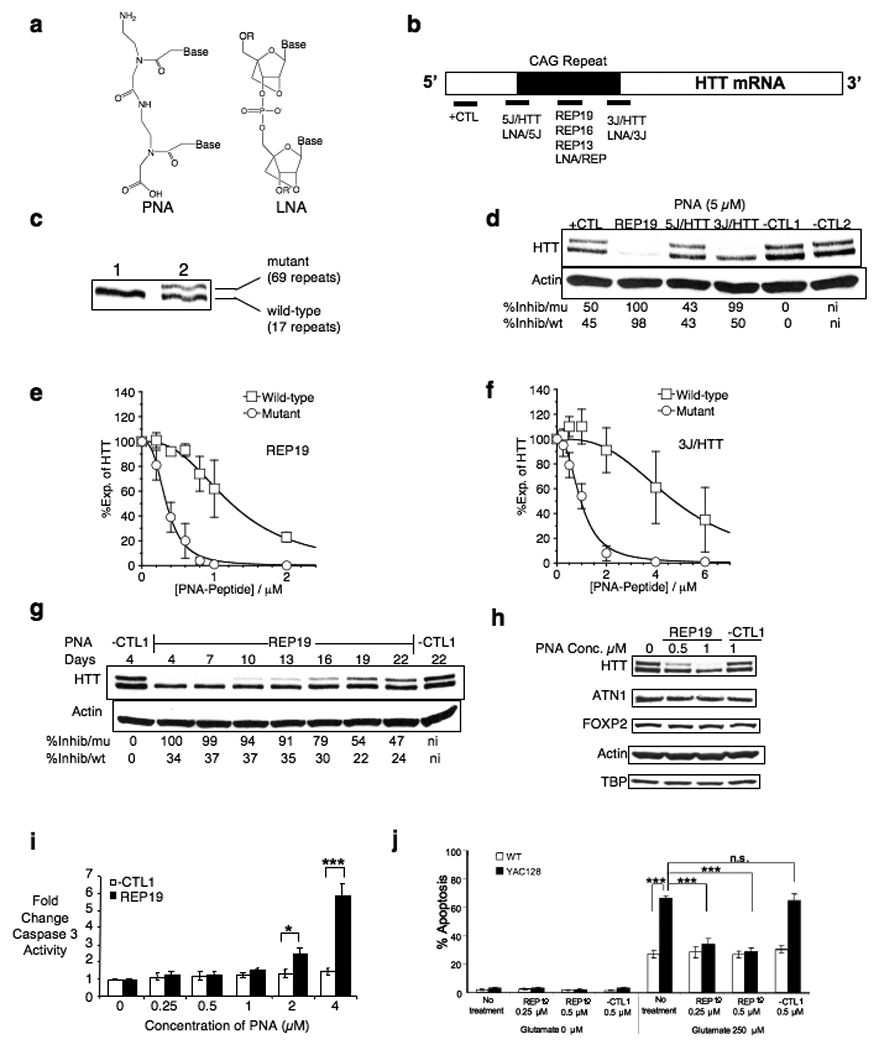
Unless otherwise noted, experiments use GM04281 fibroblast cells that are heterozygous for mutant HTT expression and graphs are quantifications of triplicate independent experiments. (a) Chemical structures of PNA and LNA. (b) Schematic of target sites for PNA and LNA oligomers within HTT mRNA. (c) Western analysis showing that wild-type and mutant HTT protein can be separated by gel electrophoresis. Lane 1 shows HTT from GM04795 cells, a fibroblast cell line that is homozygous for wild-type HTT. Lane 2 shows HTT from GM04281 cells, a fibroblast cell line that is heterozygous for mutant HTT. (d) Effect on HTT expression of adding 5 µM PNA-peptide conjugates. (e) Effect on HTT expression of adding (e) REP19 or (f) 3J/HTT. For (e) and (f), examples of western gels used for quantitation are shown in Supplementary Fig. 6 online. (g) Timecourse of inhibition of HTT expression by REP19 (1 µM). (h) Effect of adding REP19 on expression of other proteins with mRNAs that contain CAG repeats. (i) Effect of adding REP19 on cell toxicity measured by monitoring levels of caspase 3. *p < 0.05, ***p < 0.001 relative to negative control PNA/-CTL1. n = 3. (j) Glutamate-induced apoptosis of WT and YAC128 MSN. The fraction of TUNEL-positive is shown for WT (open bars) and YAC128 (filled bars) MSN. Experiments were repeated five times and data from a blinded cell count is shown. Data were evaluated using One-Way ANOVA. Statistical difference was considered to be significant if p ≤0.05. n.s. – not statistically significant.
We examined PNA-mediated inhibition of HTT expression in GM04281 patient-derived fibroblast cells (wild type allele/17 repeats, mutant allele/69 repeats) (Fig. 1c). We targeted 5J/HTT and 3J/HTT to the 5’ and 3’ junctions because complementarity to mRNA sequence outside the CAG repeat may further enhance the specificity for targeting mutant HTT mRNA relative to other cellular mRNAs. PNA conjugates REP19 and 3J/HTT inhibited expression of HTT protein (Fig. 1d) and were chosen for further analysis. Inhibition of mutant HTT expression by PNA REP19 and HTT/3J was characterized by IC50 values of 0.34 µM and 0.96 µM respectively (Fig. 1e,f, Supplementary Table 2 online) and 3.5 fold and 5-fold selectivities (wild-type IC50/mutant IC50) respectively. Inhibition decreased as the PNA target site was progressively adjusted downstream (thereby gradually losing complementarity to the CAG repeat) relative to HTT/3J (Supplementary Fig. 2 online). PNA conjugate REP19 selectively inhibited expression of mutant HTT relative to wild-type HTT for up to 22 days after a one-time addition of PNA to cells (Fig. 1g). Noncomplementary PNAs –CTL1 and –CTL2 did not inhibit HTT expression.
Many genes contain CAG repeats, including some that are essential for cellular function. At concentrations sufficient for selective inhibition of mutant HTT, addition of PNA conjugate REP19 did not affect expression of representative CAG repeat-containing genes including TATA box binding protein (TBP) (19 CAG repeats), ATN1 (15 CAG repeats), FOXP2 (40 consecutive glutamines encoded by mixed CAG and CAA codons), AAK1 (6 CAG repeats), and POU3F2 (6 CAG repeats) (Fig. 1h and Supplementary Fig. 3a online) and did not cause cellular toxicity or affect rates of cell proliferation (Supplementary Fig. 3b online). REP19 began to exhibit elevated toxic effects on cells when added at concentrations ≥ 2 µM, a concentration almost four-fold greater than that required for allele selective inhibition (Fig. 1i, Supplementary Fig. 3c online).
To test the consequences of selectively inhibiting expression of mutant HTT protein on phenotypes related to HD, we added PNA REP19 to primary neuronal cell (medium spiny neurons, MSN) cultures derived from YAC128 transgenic mice (Fig. 1j, Supplementary Fig. 4 online)23,24. In this model, full length human HTT mRNA containing 128 CAG repeats is expressed under control of its endogenous promoter in mice that also express wild-type murine HTT. MSN cells expressing mutant HTT protein are more susceptible to apoptosis upon addition of glutamate24. Addition of REP19 to striatal cultures was neuroprotective against glutamate-induced toxicity, reducing the percentage of apoptotic YAC128 cells to ~30 %, similar to levels seen in wild-type MSN. While not a perfect mimic of the situation in vivo because it is an engineered transgene model, this experiment offers a first indication that the strategy may exert a protective effect in neuronal cells.
The PNAs are complementary to both mRNA and chromosomal DNA and could, in theory, block transcription by binding to HTT DNA. It is known that the binding of PNAs to mRNA does not reduce mRNA levels25. By contrast, the binding of PNAs to DNA blocks transcription and reduces mRNA levels26. We observed that addition of PNA REP19 did not decrease levels of HTT mRNA (monitored by Q-PCR) or alter levels of RNA polymerase 2 at the HTT promoter (monitored by chromatin immunoprecipitation) (Supplementary Fig. 5 online). Indeed, efficient inhibition of both HTT alleles increased levels of HTT mRNA by a mechanism that does not involve enhancing the stability of HTT mRNA. These data are consistent with a mechanism that involves binding to mRNA and blocking translation rather than binding to DNA and inhibition of transcription.
An advantage of antisense oligomers is that there are many options for improving their activity through length modification or chemical modifications. To investigate the potential for optimizing allele-selective inhibition, we varied PNA length and peptide conjugation. PNA-peptide conjugates that were 16 and 13 bases in length (REP16 and REP13 respectively) were potent and selective inhibitors with IC50 values of 0.39 µM and 0.47 µM respectively (Fig. 2a,b). Conjugate REP13 did not affect expression of other proteins when used at 1 µM (Supplementary Fig. 7 online). These results suggest that shorter PNAs can achieve potent and selective inhibition and broaden the options for designing effective agents.
Figure 2. Effect of PNA modification, LNAs, and siRNAs on selectivity.
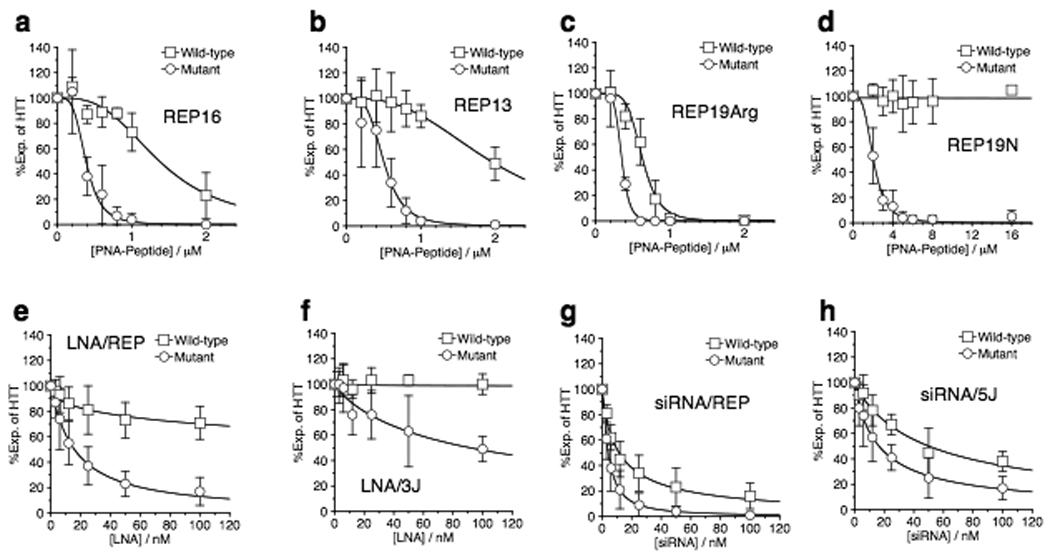
All quantitation of western analysis of protein levels in GM04281 fibroblast cells is derived from at least three independent experiments. Examples of western gels used for quantitation are shown in Supplementary Fig. 6 online. Effect on HTT expression of adding increasing concentrations of (a) REP16, (b) REP13, (c) REP19Arg, (d) REP19N, (e) LNA/REP, (f) LNA/3J, (g) siRNA/REP and, (h) siRNA/5J.
A simple chemical modification is replacement of D-arginine for D-lysine in the attached peptide. PNA REP19Arg was as potent an inhibitor (0.33 µM) as REP19 attached to D-Lys8 (0.34 µM) (Fig. 2c, Supplementary Table 2 online), but selectivity for mutant versus wild-type was reduced (1.9-fold for REP19Arg versus 3.5-fold for REP19). This result demonstrates that the composition of the peptide sequence affects selectivity for inhibition of mutant versus wild-type protein. The importance of cationic peptide sequence for recognition is not surprising, we have previously documented examples where replacement of an attached peptide that contains lysine with an analogous peptide that contains arginine significantly alters recognition complementary nucleic acids27.
Another straightforward modification is attachment of the peptide domain to the N rather than the C terminus of the PNA to afford PNA-peptide conjugate REP19N. In contrast to results showing lower selectivity with REP19Arg relative to REP19, REP19N showed greater selectivity than REP19. The IC50 value for inhibiting mutant HTT expression was 2.1 µM, with relatively no obvious inhibition of wild-type HTT at concentrations as high as 16 µM (Fig. 2d). This experiment demonstrates that improved selectivity can be achieved by varying conjugate design. We observed no inhibition of other proteins and only mild toxicity (Supplementary Fig. 7 online).
In contrast to PNAs, which have a neutral amide backbone, oligonucleotides have negatively charged phosphodiester backbones. Because of this basic difference in their chemical properties relative to PNAs, oligonucleotides will have a much different potential for developing antisense oligomers for therapy. Oligonucleotides are approved drugs and are being used in several clinical trials, and this clinical experience may offer practical advantages for their development for treating HD.
To explore whether oligonucleotides with phosphodiester backbones can achieve allele-selective inhibition, we tested single-stranded oligonucleotides that contain locked nucleic acid (LNA) bases28. LNA is an RNA analog that contains a methylene bridge between the 2'-oxygen and 4'-carbon of the ribose (Fig. 1a). LNA bases can be placed at any position and allow the thermal stability of oligonucleotides to be precisely tailored for any application. In contrast to the neutral amide backbone of PNAs, LNAs have a negatively charged phosphodiester backbone, allowing us to use cationic lipid to introduce LNAs into cells. LNA oligomers are being tested in clinical trials28.
We observed allele-selective inhibition of mutant HTT expression by LNA/REP or LNA/3J (Fig. 2e,f). Inhibition by LNA/REP and LNA/3J was characterized by IC50 values of 0.017 and 0.086 µM respectively (Supplementary Table 2 online). These IC50 values are lower than those achieved using PNA-peptide conjugates, but because the transfection protocols differ it is impossible to draw direct conclusions regarding relative potencies. Inhibition of wild-type HTT was ≤30 % at 100 nM, the maximum concentration tested. Concentrations of LNA that selectively blocked expression of mutant HTT did not affect other genes that contain CAG repeats (Supplementary Fig. 8 online). LNA REP caused a modest decrease in levels of HTT mRNA (Supplementary Fig. 8 online). In contrast to PNAs, LNAs that contain LNA bases spread throughout a DNA backbone can recruit RNase H29 and the resultant cleavage may explain the observed lower levels of mRNA.
The potency and widespread use of duplex RNAs (siRNAs)30 makes them a good benchmark for evaluating the effectiveness of PNAs and LNAs. To test whether siRNAs would also achieve selective inhibition of mutant HTT, we introduced duplex RNAs analogous in sequence to PNAs REP19, 5J/HTT, and 3J/HTT into GM04281 fibroblast cells. Like LNAs, and unlike PNA-peptide conjugates, siRNAs have a phosphodiester backbone and we used cationic lipid to assist their entry into cells. We observed inhibition of HTT expression by siRNA/REP and siRNA/5J (Fig. 2g,h) characterized by IC50 values of 0.005 and 0.018 respectively (Supplementary Table 2 online). siRNA/REP revealed a narrow window for selectivity with relatively low statistical significance (Fig. 2g) while siRNA/5J exhibited a selectivity of approximately 3-fold (Fig. 2h) (Supplementary Table 2 online). Our RNAs were not chemically modified and duplex RNAs with well-chosen modifications might achieve better selectivity.
Most HD patients have mutant mRNAs with 40–50 CAG repeats5. We chose to extend our studies to fibroblast patient-derived cell lines GM04869 (wild-type allele 15 repeats, mutant allele 47 repeats), GM04719 (wild-type allele 15 repeats, mutant allele 44 repeats) and GM04717 (wild-type allele 20 repeats, mutant allele 41 repeats) (Fig. 3a).
Figure 3. Selectivity is affected by the number of repeats.

All quantitation of western analysis of protein levels in fibroblast cells is derived from at least three independent experiments. Examples of Western gels used for quantitation are shown in Supplementary Fig. 9 online. (a) Separation by SDS-PAGE of wild-type and mutant HTT protein variants in four different patient-derived cell lines. For (b)–(g) effect on HTT protein expression of adding (b) REP19 to GM04869 cells, (c) REP19N to GM04869 cells, (d) REP19 to GM04719 cells, (e) REP19N to GM04719 cells, (f) REP19 to GM04717 cells, and (g) REP19N to GM04717 cells.
Upon addition of PNA REP19 to cells, we observed inhibition of mutant HTT in GM04869 (Fig. 3b), GM04719 (Fig. 3d), and GM04717 (Fig. 3f) cells with selectivities (wild-type IC50 value/mutant IC50 value) of 2.1, 1.8, and 1.2 respectively (Supplementary Table 2 online), values reduced relative to the 3.5 fold selectivity achieved in GM04281 cells (Fig. 1e). We observed slight decreases in the potency of inhibition of mutant HTT as the number of mutant repeats was decreased from 47 to 44 and 41.
Because we had previously observed that attachment of the D-Lys8 peptide to the PNA N terminus improves selectivity in GM04281 cells (Fig. 2d), we tested N-linked conjugate REP19N in the cell lines that possess mutant alleles with fewer CAG repeats. We observed inhibition of mutant HTT in GM04869 (Fig. 3c), GM04719 (Fig. 3e), and GM04717 (Fig. 3g) cells with selectivities (wild-type IC50 value/mutant IC50 value) of >3.5, >1.8, and >1.5 respectively (Supplementary Table 2 online). These data suggest that simple chemical modifications have the potential to extend allele-selective inhibition of HTT protein expression to a broader subset of HD patients.
A CAG repeat expansion within the gene encoding ataxin-3 is responsible for MJD2 and we chose ataxin-3 as a target to test whether allele-selective inhibition could be achieved with other repeat-containing genes. To examine the potential for allele-specific inhibition in MJD cells, we obtained patient-derived cell line GM06151 that is heterozygous for an expanded CAG repeat (wild type allele/24 repeats, mutant allele/74 repeats). The 74 repeats of the mutant allele is in the middle of the repeat range found in patient samples31.
We tested PNA conjugates that targeted the CAG repeat region (REP19), the 5’ junction (5J/ATX), and the 3’ junction (3J/ATX) (Fig. 4a, Supplementary Table 2 online). PNA conjugate REP19 selectively inhibited expression of mutant ataxin-3 with an IC50 value of 0.36 µM (Fig. 4b). Conjugates that target the 5’ and 3’ junctions were also selective inhibitors with IC50 values of 0.7 µM and 2.2 µM respectively (Fig. 4c,d). These data suggest that our strategy can be extended beyond HTT to other therapeutic targets. We also tested siRNA/REP. Similar to our observations for inhibition of HTT protein expression (Fig. 3), this siRNA was an inhibitor of ataxin-3 expression but yielded less selective reduction of the mutant protein (Fig. 4e). A failure of a repeat-targeted siRNA to selectively inhibit ataxin-3 expression had been reported previously11.
Figure 4. Potent and selective inhibition of mutant ataxin-3.

All data show analysis of ataxin-3 expression in GM06151 fibroblast cells. Examples of Western gels used for quantitation are shown in Supplementary Figure 10 online. (a) Schematic of target sites for PNAs within ataxin-3 mRNA, Inhibition of ataxin-3 expression by (b) PNA conjugate REP19, (c) PNA conjugate 3J/ATX, (d) PNA conjugate 5J/ATX, and (e) siRNA/REP.
One obstacle to therapy for HD or MJD will be delivery to the central nervous system because oligonucleotides are not distributed to the brain after intravenous or oral administration. Single stranded oligonucleotides can be delivered directly to the central nervous system and block gene expression32. While LNAs have not been used in animal models of HD or MJD, they have been administered into rodent brains by several methods (intrahecal, intracerebroventricular, and intrastriatal)33,34. Toxicity is minimal and potent inhibitory effects were observed for targeting deltoporhin II33 and miRNA-2134.
Much attention has been focused on allele-selective inhibition using siRNAs complementary to sequences that have single nucleotide or deletion polymorphism11–16. An advantage of this approach is that it involves familiar RNA interference (RNAi) technology. Unfortunately, patient populations have diverse polymorphisms, necessitating development of multiple siRNAs. Our benchmark patient-derived cell line GM04281 has none of the most commonly identified polymorphisms14 and is representative of patients who would be unlikely to benefit from clinical application of the SNP approach. We were able to perform experiments in GM09197 cells that have a deletion polymorphism and have been used to observe siRNA-mediated allele-selective inhibition16. LNA/REP offered better selectivity than the best allele-selective RNA (Supplementary Fig. 11 online).
Patient populations are heterogeneous and approaches that target polymorphisms, triplet repeats, or make no attempt at allele-selectivity may benefit different groups of patients. One advantage is that our approach employs one oligomer strand rather than the two needed for siRNAs, simplifying issues associated with clinical development such as compound synthesis and the potential for off-target effects. Conversely, while we demonstrate that optimizing the structure of the oligomer can lead to better selectivity over a wide range of repeats, it may be difficult to achieve adequate selectivity for patients with shorter repeats using our strategy.
Another important issue is the minimum efficacy sufficient for successful application in vivo. Complete inhibition of mutant protein expression might not be necessary to achieve beneficial therapeutic effects, even partial reduction of mutant protein levels may be adequate. Conversely, partial inhibition of wild-type protein may not have adverse consequences because the remaining wild-type protein may be sufficient for function. The window for therapy may be relatively broad, requiring less than complete inhibition of mutant protein expression and tolerating partial reduction in wild-type protein levels. Such information will help set the therapeutic window for drug development.
Our findings offer two lessons. The first is that single-stranded oligomers can discriminate among sequences inside cells on the basis of context – in this case the length of the repeat and the potential to form energetically different structures – rather than base differences. The second is that the potential for developing single-stranded oligomers as allele-selective treatment for genetic disease is greater than had been appreciated previously. There is broad potential for optimizing selectivity and potency. Antisense oligomers for other diseases are making good progress in clinical trials35 and appear to offer near-term potential for wider therapeutic application. Exploiting the ability of antisense oligomers to selectively recognize mutant nucleic acid sequences offers a promising strategy for developing therapies for HD, MJD, and other triplet repeat disorders.
METHODS
Cell Culture and Transfection
PNA-peptide conjugates were synthesized and purified as desccribed22,26. LNA oligonucleotides were provided by Sigma-Aldrich (Paris, France). siRNAs were purchased from Integrated DNA Technologies (IDT, Coralville, IA). Patient-derived fibroblast cell lines GM04281, GM06151, GM04719, GM04869, GM04717, GM04795, and GM09197 were obtained from the Coriell Institute (Camden, NJ). Cells were maintained at 37 °C and 5% CO2 in Minimal Essential Media Eagle (MEM) (Sigma, M4655) supplemented with 10% heat inactivated fetal bovine serum (Sigma) and 0.5% MEM nonessential amino acids (Sigma). Cells were plated in 6-well plates at 60,000 cells/well in supplemented MEM two days prior to transfection. Stock solutions of PNA-peptide conjugates were heated at 65 °C for 5 min before use to dissolve any aggregates that may have formed. PNA-peptide conjugates were diluted to the appropriate concentration using OptiMEM (Invitrogen, Carlsbad, CA) and then added to cells. After 24 h, the media containing PNA-peptides were removed and replaced by fresh supplemented MEM. Cells were typically harvested 4 days after transfection for protein assay. siRNAs or LNAs were transfected to cells using RNAiMAX (Invitrogen) according to the manufacturer's instructions. The appropriate amount of the lipid (3 µL for 100 nM oligonucleotides) were added to OptiMEM containing oligonucleotides and the oligonucleotide-lipid mixture (250 µL) were incubated for 20 min. OptiMEM was added to the mixture to a final volume of 1.25 mL and then added to cells. The media were exchanged 24 h later with fresh supplemented MEM.
Analysis of HTT and ataxin-3 expression
Cells were harvested with trypsin-EDTA solution (Invitrogen) and lysed. The protein concentration in each sample was quantified with BCA assay (Thermo Scientific, Waltham, MA). SDS-PAGE (separating gel: 5 % acrylamide-bisacrylamide/34.7:1, 450 mM Tris-acetate pH 8.8; stacking gel: 4% acrylamide-bisacrylamide/34.7:1, 150 mM Tris-acetate pH 6.8) (XT Tricine Running Buffer, Bio-Rad, Hercules, CA) was used to separate wild-type and mutant HTT proteins. Gels were run at 70V for 15 min followed by 100V for 4 h. For separation of HTT variants containing shorter CAG repeats, gels were run at 70 V for 15 min, then 110 V for 6 h. The electrophoresis apparatus was placed in ice-water bath to prevent overheating of the running buffer. We monitored expression of actin protein to ensure even loading on protein in each lane. In parallel with analysis for HTT expression, portions of each protein lysate sample were analyzed for β-actin expression by SDS-PAGE (7.5% acrylamide pre-cast gels; Bio-Rad). These gels were run at 70V for 15 min followed by 100V for 1 h. After gel electrophoresis, proteins were transferred to membrane (Hybond-C Extra; GE Healthcare Bio-Sciences, Piscataway, NJ). Ataxin-3 and β-actin were analyzed by SDS-PAGE (7.5 % acrylamide pre-cast gels; Bio-Rad). Primary antibodies specific for each protein were obtained and used at the indicated dilution ratio: anti-HTT antibody (MAB2166. 1:10000; Chemicon), anti-ataxin-3 antibody (MAB5360; 1:10000; Chemicon), and anti-β-actin antibody (1:10,000; Sigma).
HRP conjugate anti-mouse or anti-rabbit secondary antibody (1:10000 and 1:5000, respectively; Jackson ImmunoResearch Laboratories, West Grove PA) was used for visualizing proteins using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Protein bands were quantified using ImageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/, 1997–2007 ). The percentage of inhibition was calculated as a relative value to a control sample.
Neuronal cell glutamate susceptibility assay23,24
YAC128 mice (FVBN/NJ background strain) were obtained from Jackson Labs (stock number 004938). The male YAC128 mice were crossed to wild-type (WT) female FVBN/NJ mice and P1-P2 pups were collected and genotyped by PCR. The primary cultures of striatal medium spiny neurons (MSN) were established from YAC128 and control wild type pups. Striata were dissected, diced and digested with trypsin. After dissociation, neurons were plated on poly-L-lysine (Sigma) coated 12 mm round coverslips (Assistent) in Neurobasal-A medium supplemented with 2% B27, 1 mM glutamine and penicillin-streptomycin (all from Invitrogen) and kept at 37°C in a 5% CO2 environment. PNA was added to the 9-DIV (days in vitro) MSN. The 13-DIV MSN were exposed for 7 h to 250 µM glutamate in Neurobasal-A added to the culture medium. Immediately after the treatment with glutamate, neurons were fixed for 30 min in 4% paraformaldehyde plus 4% sucrose in PBS (pH 7.4), permeabilized for 5 min in 0.25% Triton-X-100, and stained by using the DeadEnd fluorometric TUNEL System (Promega). Nuclei were counterstained with 5 µM propidium iodine (PI) (Molecular Probes). Coverslips were extensively washed with PBS and mounted in Mowiol 4–88 (Polysciences). For quantification six to eight randomly chosen microscopic fields containing 100–300 MSN each were cell-counted for YAC128 and wild type cultures. The number of TUNEL-positive neuronal nuclei was calculated as a fraction of PI-positive neuronal nuclei in each microscopic field. The fractions of TUNEL-positive nuclei determined for each microscopic field were averaged and the results are presented as means ± SE (n= number of fields counted). MSN cells were supported in culture by surrounding glial cells, but only MSN cells were counted during the neuroprotection assay.
PNA conjugate REP19 was added at the concentration of 0.25 µM and 0.5 µM, 4 days before the glutamate application. Non-complementary PNA conjugate –CTL1 was added at 0.5 µM. MSN were exposed to 250 µM glutamate at 13-DIV for 7h, fixed, permeabilized and analyzed by TUNEL staining and PI counterstaining. The data are presented as mean ± SE (n = 6–8 microscopic fields, 100–300 MSN per field).
For protein assays, after the addition of PNA REP19 at 9-DIV, the 13-DIV MSN were washed with ice-cold PBS and solubilized for 30 min at 4°C in extraction buffer A (1% CHAPS, 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4 [pH 7.2], 5 mM EDTA, 5 mM EGTA, and protease inhibitors). Extracts were clarified by centrifugation for 20 min at 10000 × g at 4°C and the upper solutions were collected for SDS-PAGE. For selectively harvesting MSN, cells were incubated with cell dissociation solution (Sigma) for 4 min at 37°C and neuron basal-A medium with 10% FBS were added. MSN were detached from plate and most of the glial cells were not harvested. The combined solution was collected and lysed as previously described23,24.
Caspase-3 activity assay
Caspase-3 activity was measured by hydrolysis of acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) according to the manufacture instruction in a colorimetric assay kit (Sigma). Fibroblast cells in 6-well plate were harvested as previously described. Cell pellet was re-suspended in 15 µL of 1x lysis buffer without protease inhibitor and incubated for 30 min on ice. Protein concentration was determined by BCA assay (Thermo Scientific). 50 µg of cell extract was mixed with 2 mM Ac-DEVD-pNA substrate to a total volume of 100 µL of 1x assay buffer in a 96-well plate and incubated at 37 °C overnight. The absorbance was read at 405 nm.
Analysis of TBP, AAK1, ATN1, FOX2P, and POU3F2
The number of CAG repeats was estimated according to the published mRNA sequence in GeneBank. TATA box binding protein (TBP) (~19 CAG repeats, NM_003194), AAK1 (6 CAG repeats, NM_014911), ATN1 (15 CAG repeats, NM_001940), FOXP2 (~40 CAA⋯CAG repeats, NM_148899), and POU3F2 (~6 CAG repeats, NM_005604). Protein lysates were analyzed by SDS-PAGE (7.5% acrylamide pre-cast gels; Bio-Rad) followed by western blotting with anti-TBP antibody (1:2000; Sigma), anti-AAK1 antibody (1:1000; Abcam, Cambridge MA), anti-ATN1 antibody (1:300, Affinity Bioreagents, Golden CO), anti-FOXP2 antibody (1:1000, Abcam), or anti-POU3F2 antibody (1:1000; Abnova, Taipei, Taiwan).
Statistical analysis and curve fitting
Student t-test was performed for evaluating statistical significance between two study groups. Each data plot from dose response experiments for inhibition of HTT or ataxin-3 was fit to the following model equation, y=100(1−xm/(nm+xm)) where y is % inhibition of protein and x is concentration of synthetic oligomers. m and n are fitting parameters, where n is taken as the IC50 value.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the High-Q foundation, the National Institutes of Health (NIGMS 60642 and 73042 to DRC) (NINDS RO1NS056224 to IB), and the Robert A. Welch Foundation (I-1244 and I-1336) and Ataxia MJD research project. We thank Dr. Bethany Janowski for helpful comments and Yumei Li for help maintaining the YAC128 mouse colony.
Footnotes
COMPETING INTERESTS STATEMENT
D.R.C and J. H. have filed a patent application related to this research.
Bibliography
- 1.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 2.Paulson HL. Dominantly inherited ataxias: Lessons learned from Machado-Joseph disease/spinocerbeller ataxia type 3. Semin. Neurol. 2007;27:133–142. doi: 10.1055/s-2007-971172. [DOI] [PubMed] [Google Scholar]
- 3.Walker FO. Huntington’s disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 4.Gusella JF, MacDonald ME. Huntington’s disease: seeing the pathogenic process through a genetic lens. Trends Biochem. Sci. 2006;31:533–540. doi: 10.1016/j.tibs.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 5.Kremer B, et al. A worldwide study of the Huntington’s disease mutation: The sensitivity and specificity of measuring CAG repeats. New Engl. J. Med. 1994;330:1401–1406. doi: 10.1056/NEJM199405193302001. [DOI] [PubMed] [Google Scholar]
- 6.Boado RJ, Kazantsev A, Apostol BL, Thompson LM, Pardridge WM. Antisense-mediated down-regulation of the mutant human huntingtin gene. J. Pharmacol. Exp. Ther. 2000;295:239–243. [PubMed] [Google Scholar]
- 7.Harper SQ, et al. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA. 2005;102:5820–5825. doi: 10.1073/pnas.0501507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denovan-Wright EM, Davidson BL. RNAi: a potential therapy for the dominantly inherited nucleotide repeat diseases. Gene Ther. 2006;13:525–531. doi: 10.1038/sj.gt.3302664. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y-L, et al. Clinico-pathological rescue of a model mouse of Huntington’s disease by siRNA. Neurosci. Res. 2005;53:241–249. doi: 10.1016/j.neures.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 10.DiFiglia M, et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc. Natl. Acad. Sci. USA. 2007;104:17204–17209. doi: 10.1073/pnas.0708285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller VM, et al. Allele-specific silencing of dominant disease genes. Proc. Natl. Acad. Sci. USA. 2003;100:7195–7200. doi: 10.1073/pnas.1231012100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwarz DS, et al. Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet. 2006;2:1307–1318. doi: 10.1371/journal.pgen.0020140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez-Lebron E, Paulson HL. Allele-specific RNA interference for neurological disease. Gene Ther. 2006;13:576–581. doi: 10.1038/sj.gt.3302702. [DOI] [PubMed] [Google Scholar]
- 14.van Bilsen PHJ, et al. Identification and allele-specific silencing of the mutant huntingtin allele in Huntington’s disease patient-derived fibroblasts. Hum. Gene Ther. 2008;19:710–718. doi: 10.1089/hum.2007.116. [DOI] [PubMed] [Google Scholar]
- 15.Alves S, et al. Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease. PLoS One. 2008;3:e3341. doi: 10.1371/journal.pone.0003341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Engelman J, Friedlander RMJ. Allele-specific silencing of mutant Huntington’s disease gene. J. Neurochem. 2009;108:82–90. doi: 10.1111/j.1471-4159.2008.05734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Souza EB, Cload ST, Pendergrast PS, Sah DW. Novel therapeutic modalities to address nondrugable protein interaction targets. Neuropsychopharmacology Reviews. 2008;34:142–158. doi: 10.1038/npp.2008.115. [DOI] [PubMed] [Google Scholar]
- 18.Nasir J, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 19.White JK, et al. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat. Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- 20.Sobczak K, de Mezer M, Michlewski G, Krol J, Krzyzosiak WJ. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucl. Acids Res. 2003;31:5469–5482. doi: 10.1093/nar/gkg766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marin VL, Armitage BA. RNA guanine quadruplex invasion by complementary and homologous PNA probes. J. Am. Chem. Soc. 2005;127:8032–8033. doi: 10.1021/ja051102y. [DOI] [PubMed] [Google Scholar]
- 22.Hu J, Corey DR. Inhibiting gene expression with peptide nucleic acid (PNA)-peptide conjugates that target chromosomal DNA. Biochemistry. 2007;46:7581–7589. doi: 10.1021/bi700230a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slow EJ, et al. Selective striatal neuronal loss in a YAC128 mouse model for Huntington disease. Hum. Mol. Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 24.Tang T-S, et al. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. USA. 2005;102:2602–2607. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knudsen H, Nielsen PE. Antisense properties of duplex- and triplex-forming PNAs. Nucl. Acid. Res. 1996;24:494–500. doi: 10.1093/nar/24.3.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janowski BA, et al. Inhibiting transcription of chromosomal DNA with antigene peptide nucleic acids. Nat. Chem. Biol. 2005;1:210–215. doi: 10.1038/nchembio724. [DOI] [PubMed] [Google Scholar]
- 27.Corey DR. 48 000-fold acceleration of hybridization of chemically modified oligonucleotides to duplex DNA. J. Am. Chem. Soc. 1995;117:9373–9374. [Google Scholar]
- 28.Koch T, et al. Locked nucleic acid: Properties and therapeutic aspects. In: Kurreck J, editor. Published in Therapeutic Oligonucleotides. RSC Biomolecular Sciences, Royal Society of Chemistry; 2008. pp. 103–141. [Google Scholar]
- 29.Kurreck J, Wyszko E, Gillen C, Erdmann VA. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucl. Acids Res. 2002;30:1911–1918. doi: 10.1093/nar/30.9.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006;2:711–719. doi: 10.1038/nchembio839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maruyama H, et al. Molecular features of the CAG repeats and clinical manifestation of Machado-Joseph disease. Hum. Mol. Genet. 1995;4:807–812. doi: 10.1093/hmg/4.5.807. [DOI] [PubMed] [Google Scholar]
- 32.Smith RA, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 2006;116:2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wahlestedt C, et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. USA. 2000;97:5633–5638. doi: 10.1073/pnas.97.10.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corsten MF, et al. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 2007;67:8894–9000. doi: 10.1158/0008-5472.CAN-07-1045. [DOI] [PubMed] [Google Scholar]
- 35.Corey DR. RNAi learns from antisense. Nat. Chem. Biol. 2007;3:8–11. doi: 10.1038/nchembio0107-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.