

Abstract
Oligomeric interactions between Ca-ATPase polypeptide chains and their modulation by phospholamban (PLB) were measured in native cardiac sarcoplasmic reticulum (SR) microsomes. Progressive modification of Lys514 with fluorescein-5-isothiocyanate (FITC), which physically blocks access to the nucleotide binding site by ATP, demonstrates that Ca-ATPase active sites function independently of one another prior to the phosphorylation of PLB. However, upon PKA-dependent phosphorylation of PLB, a second-order dependence between residual enzyme activity and the fraction of active sites is observed, consistent with a dimeric functional complex. Complementary distance measurements were made using FITC or 5-iodoacetamido-fluorescein (IAF) bound to Cys674 within the N- or P-domains respectively, to detect structural coupling within oligomeric complexes. Accompanying the phosphorylation of PLB, neighboring Ca-ATPase polypeptide chains exhibit a 4 ± 2 Å decrease in the proximity between FITC sites within the N-domain and a 9 ± 3 Å increase in the proximity between IAF sites within P-domains. Thus, the phosphorylation of PLB induces spatial rearrangements between the N- and P-domain elements of proximal Ca-ATPase polypeptide chains which restore functional interactions between neighboring polypeptide chains and, in turn, result in increased rates of catalytic turnover. These results are interpreted in terms of a structural model, calculated through optimization of shape complementarity, desolvation, and electrostatic energies, which suggests a dimeric arrangement of Ca-ATPase polypeptide chains through the proximal association of N-domains that accommodates interaction with PLB. We suggest that the phosphorylation of PLB acts to release constraints involving interdomain subunit interactions that enhance catalytically important N-domain motions.
Ca-ATPases function to maintain low cytoplasmic calcium levels, acting to resequester calcium into the sarcoplasmic reticulum (SR) lumen after each contractile event, thus controlling relaxation rates in the heart and other muscle cells. Co-expression of the regulatory protein phospholamban (PLB) in slow-twitch skeletal muscle and the heart permits the modulation of Ca-ATPase transport function and thus the force and rate of muscle contraction. Specifically, in response to β-adrenergic stimulation, the activation of cAMP-dependent protein kinase (PKA) results in the phosphorylation of Ser16 in PLB, which releases inhibitory interactions with the Ca-ATPase that enhance transport activity at submicromolar (nonsaturating) calcium level. This release of inhibition is not accompanied by dissociation of PLB from the Ca-ATPase; rather, protein-protein interactions within the intact PLB-ATPase complex are altered (1-3). PLB imposes restrictions on large scale domain rearrangements of the Ca-ATPase that are critical to the formation of the second high-affinity calcium binding site associated with cooperative calcium binding and ATP utilization (1, 4). PLB phosphorylation acts to stabilize the hinge region within PLB between the transmembrane and cytosolic helices. At the same time the overall dimensions of the cytosolic domain of PLB are reduced, resulting in release of the PLB-ATPase interactions that restrict catalytically important motions of the nucleotide (N) domain in the Ca-ATPase (1, 5-8).
Specific binding interfaces between PLB and transmembrane helices TM2, TM4, and the nucleotide (N) domain of the Ca-ATPase have been identified using both cross-linking (9-11) and molecular modeling approaches (9, 12-15) (Figure 1). Cross-linking efficiencies are diminished upon calcium binding to the Ca-ATPase; in combination with kinetic measurements, these results indicate that PLB stabilizes the apo-form of the Ca-ATPase (i.e., the E2 state) through specific binding interactions that restrict large scale movements associated with calcium binding (15-17). Cryoelectron microscopy structural measurements indicate that one PLB is able to interact with two Ca-ATPase polypeptide chains (18). These structural measurements are consistent with functional measurements in both reconstituted and native SR membranes that have demonstrated that one PLB complexed with two Ca-ATPase is sufficient for complete inhibition (19, 20). These results suggest that PLB modulates allosteric interactions between Ca-ATPase polypeptide chains within an oligomeric complex (16, 21). Inhibition of enzyme activity is suggested to involve the modulation of specific contact points between the N-domains of neighboring Ca-ATPase polypeptide chains that act to uncouple the conformational coupling between oligomeric complexes that accelerate enzyme turnover through enhanced domain movements. To investigate this latter hypothesis, we have used fluorescein-5-isothiocyanate (FITC) to block ATP access to the active site of the Ca-ATPase to assess the functional role of oligomeric interactions between Ca-ATPase polypeptide chains in native SR microsomes isolated from porcine hearts. Complementary measurements used fluorescence resonance energy transfer (FRET) to assess changes in the distances between fluorescein moieties, site-specifically bound to either the nucleotide (N) or phosphorylation (P) domain elements of the Ca-ATPase, in response to the phosphorylation of PLB by cAMP-dependent protein kinase. Our results indicate that the Ca-ATPase functions as an oligomeric complex, whose structural coupling is disrupted by PLB. Phosphorylation of PLB by PKA results in large-scale domain movements that act to restore the functional coupling between Ca-ATPase polypeptide chains.
Figure 1. Structures of the Ca-ATPase and PLB.
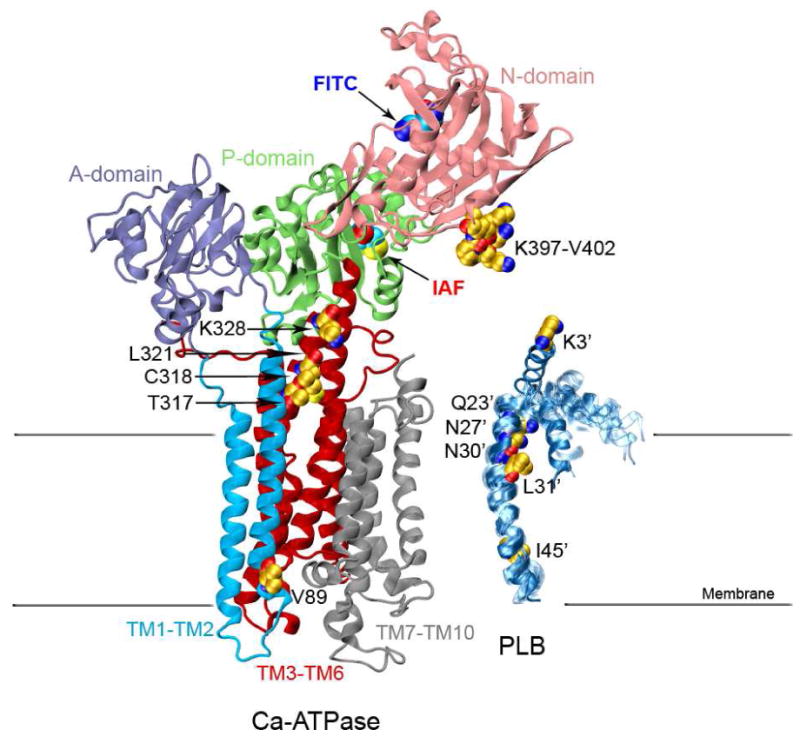
Structure of the Ca-ATPase (1su4.pdb) depicts headpiece, corresponding to discrete structural elements associated with the A-domain (residues 1-43 and 124-235; purple), N-domain (residues 360-600; pink), and P-domain (residues 330-359 and 601-739; green) relative to the transmembrane helices TM1-TM2 (residues 44-123; cyan), TM3-TM6 (residues 239-329 and 740-821, red), and TM7-TM10 (residues 831-994; gray), where domain boundaries are as previously described (72). The ten NMR structures of a PLB monomer are shown on the right (1fjk.pdb). Highlighted in the structures of Ca-ATPase and PLB are (yellow space-filling) side chains that have been identified through covalent cross-linking experiments which involve both endogenous amino acids (C or K) or engineered cysteines at sites in TM2 (i.e., V89), TM4 (T317, C318, L321, and K328), and the N-domain (i.e., K397DDKPV402) of the Ca-ATPase with sites on PLB (i.e., I45-L52, L31, N30, N27, Q23, or K3) (9-11). Labeling sites are indicated for FITC modification at K514 /K515 in the N-domain (blue oval) and IAF modification at C674 in the P-domain (red oval) of the Ca-ATPase.
Experimental Procedures
Materials
ATP (disodium salt), cAMP, cAMP-dependent protein kinase (PKA), calcium ionophore A23187, nonionic detergent C12E8, EGTA, and fluorescein-5-isothiocyanate (FITC) were purchased from Sigma (St. Louis, MO). 5-iodoacetamidofluorescein (IAF) was obtained from Invitrogen Molecular Probes (Eugene, OR). Calyculin A was purchased from Calbiochem (La Jolla, CA). MOPS was purchased from Thermal Fischer Scientific (Waltham, MA). Alkaline phosphatase was purchased from Worthington Biochemical Corporation (Lakewood, NJ). CaCl2 standard solutions were purchased from VWR International (West Chester, PA).
Preparation of Cardiac SR
SR membranes were isolated from pig hearts by several steps of differential centrifugation for removal of cell debris, mitochondria, and myosin prior to enrichment of the SR membrane vesicles on a sucrose density gradient containing 5-7% dextran T-10 as detailed previously (22). The protein concentration was determined by the Amido Schwartz assay.
PLB Phosphorylation and Dephosphorylation
For fluorescence measurements, activation by cAMP-dependent protein kinase (PKA) involved addition of 1 μM cAMP, 80 μg/ml bovine heart PKA, and 0.1 mM ATP added to SR microsomes in a buffered solution containing 0.5 μM free calcium for at least 10 min prior to making measurements. Maximal levels of phosphorylated PLB (4-5 nmol per mg SR) are retained for at least 2 hr allowing for stable fluorescence measurements which require only 10 min (22). For Ca-ATPase activity assays, cAMP and PKA were added to the assay medium for 10 minutes to ensure temperature stabilization prior to addition of 5 mM ATP to start Ca-ATPase activity reaction. For dephosphorylation of PLB, samples were similarly pre-treated for 10 min with 7 μg/ml PKA inhibitor (calyculin A) and 5 units/ml alkaline phosphatase Type VII-NL (Sigma).
ATPase Activity Assay
Calcium-dependent ATPase activity was measured at 25° C as previously described, using SR microsomes (0.1 mg / mL) in 25 mM MOPS (pH 7.0), 0.1 M KCl, 5 mM MgCl2, 6 μM A23187, 0.1 mM EGTA, and sufficient calcium to yield the desired free calcium concentration (23). Basal activity assayed in the presence of 1 mM EGTA with no added calcium. Steady state levels of phosphorylated Ca-ATPase were measured from γ-[32P]-ATP as previously described (22).
Gel Electrophoresis
Sodium dodecyl sulfate polyacrylamide gel electrophoresis was performed by the method of Laemmli (24), using 7.5% acrylamide gels with a 3% stacking gel. Fluorescence was detected prior to staining with Coomassie Blue under ultraviolet light (λex=366 nm), where fluorescence was detected subsequent to a Corning 3-70 long-pass filter.
Derivatization of the SR Ca-ATPase
The Ca-ATPase was specifically derivatized using either fluorescein-5-isothiocyanate (FITC) or 5-iodoacetamidofluorescein (IAF), essentially as previously described (25, 26). Specific labeling of Lys514 in SERCA2a involved incubation with the indicated amounts of FITC for ten minutes with cardiac microsomes (2.0 mg/mL) in 10 mM Tris (pH 9.2), 0.1 M KCl, and 0.3 M sucrose at 22 °C. The reaction was quenched by a 10-fold dilution into an ice cold buffer A comprised of 20 mM MOPS (pH 7.0), 0.3 M sucrose, and 1 mg/mL bovine serum albumin followed by centrifugation (100,000 × g); the resulting pellet was resuspended in 20 mM MOPS (pH 7.0, 0.3 M sucrose. Specific labeling of Cys674 in SERCA2a with IAF required the modification of other reactive cysteines through the preincubation of cardiac microsomes (2 mg/mL) in buffer A with N-ethyl maleimide (NEM) (80 μM) for 30 minutes at 22 °C. Following centrifugation at 100,000 × g to remove unbound NEM, pellets were resuspended in buffer A and diluted to 2 mg/mL prior to the addition of IAF. Following 10 minutes of incubation, samples were diluted ten-fold with buffer A. The stoichiometries of bound FITC or IAF was measured in the presence of 1% sodium dodecyl sulfate and 0.1 M NaOH where ε491 = 80,000 M-1 cm-1.
Fluorescence Measurements
FITC- or IAF-labeled Ca-ATPase were excited at 488 nm, and emitted fluorescence was detected after a Schott OG-530 long-pass filter using a Glan-Thompson polarizer set parallel or perpendicular to the vertically polarized excitation light. Fluorescence lifetime measurements utilized a Pockels cell to modulate the light, measuring both phase lag and demodulation of the modulated light relative to a glycogen standard, as previous described (27), where data was fit using the method of nonlinear least squares to a sum of two exponential decays (28). Steady-state polarization (P) was calculated from the ratio of the fluorescence intensities (I) measured with the polarizers on the excitation and emission sides of the sample in the vertical (v) or horizontal (h) position. P is calculated from the equation:
| (1) |
where g = Ihv/Ihh and corrects for the differing sensitivities of the detection system for vertically and horizontally polarized light (29).
Analysis of Spatial Separation Between Fluorescein-Labeled Ca-ATPase Polypeptide Chains
Decreases in the emission anisotropy [A = (1/P - 1/3)-1] of fluorescein bound to the Ca-ATPase upon increasing the labeling stoichiometry result from fluorescence resonance energy transfer (FRET), and provide information regarding subunit interactions between neighboring SERCA molecules (20, 30, 31), where for a Ca-ATPase dimer:
| (2) |
In this equation, A(A0,As,At) is the measured emission anisotropy, f is the labeling stoichiometry of fluorescein bound per mole of the Ca-ATPase, N is the apparent oligomeric size of the Ca-ATPase, A0 is the initial emission anisotropy in the absence of FRET, As is the emission anisotropy when all fluorescein labeling sites within the Ca-ATPase oligomeric complex are saturated, and At is the emission anisotropy after one FRET event. Fitting the data involves incrementally adjusting N by integral values and solving for A0, As, and At, which provide information regarding both the spatial separation and orientation of chromophores on adjacent Ca-ATPase polypeptide chains. Knowledge of A0, As, Af, and the Förster distance (R0) permits calculation of the average separation (rav) between fluorescein chromophores and the effective angle between donor and acceptor dipoles (θav) (30), where:
| (3) |
and
| (4) |
In fitting the emission anisotropy as a function of the fractional saturation of fluorophore sites, various integral values of N were specified, and for each value, a three-parameter fit was performed with optimal values of A0, At, and As. Minimal chi-squared values of the fits indicated which value of N best described the data. Solvent accessibilities of FITC or IAF were measured as a function of the quencher concentration [Q], using potassium iodide or acylamide, according to the Stern-Volmer equation:
| (5) |
where F0 and F are fluorescence in the absence and presence of quencher, respectively. KSV is the slope of a plot of F0/F versus [Q], and related to the solvent accessibility of the fluorophore.
Molecular Docking of the Ca-ATPase dimer
The Ca-ATPase dimer was modeled from the three-dimensional structure of the SERCA1 isoform, 1kju.pdb (32), the same structure previously used to propose a low-resolution (8-10 Å), ribbon-like model of the dimer from cryoelectron microscopy image experiments (33). The two monomers were docked with the ZDOCK software which uses a fast Fourier transform to exhaustively search the entire rotational and translational space of one monomer with respect to the other that remains fixed at the origin (34). The rotational search is performed by explicitly rotating the ligand around each of its three Cartesian angles by a certain increment. For every rotation, the algorithm rapidly scans the translational space using fast Fourier transform. The best structures are evaluated on the basis of shape complementarity, desolvation energy, and electrostatics (35). Based on the cryoelectron microscopy of the Ca-ATPase (33) and FRET measurements generated structures were required to meet some basic criteria regarding reasonable structures. For example: i) the two Ca-ATPase monomers should be aligned along an axis perpendicular to the plane of the membrane bilayers and ii) the two monomers should be related by a two-fold rotational center of symmetry; additionally, iii) the dimer interactions should not interfere with PLB bound to the Ca-ATPase as established by separate docking (see below). These three criteria were used to choose the best dimer structural model out of the 2000 solutions generated from the ZDOCK molecular docking algorithm.
Molecular Docking of PLB to Ca-ATPase monomer
Molecular docking calculations, using the program AutoDock, were carried out for the X-ray structures of the SERCA1 isoform of the Ca-ATPase (1su4.pdb and 2agv.pdb) solved at 2.6 Å and the NMR structure of PLB (1fjk.pdb) (36-39) using the program AutoDock (40). The transmembrane helix (domain II) and mobile N-terminal region (domain Ia) of PLB were separately docked to the Ca-ATPase structure to eliminate any conformational bias imposed by the initial configuration of the inter-helical loop (domain Ib) of PLB. This region is known to be very flexible, allowing multiple orientations of domain Ia with respect to domain II. The helical domains of PLB were treated as rigid bodies. Polar hydrogens and protonation states were assigned to the experimental coordinates, i.e., pH 7.0. The AMBER united atom charges and solvation parameters were added to the final protein file (41). The grid maps were calculated using AutoGrid version 4.1 (40). The grids were chosen to be sufficiently large to include not only the active site but also significant portions of the surrounding surface. Grid dimensions were 126 × 126 × 126 Å, with a spacing of 0.5 Å between the grid points and the center close to the Cα of residues Tyr317 and Lys397. The Lamarckian genetic search algorithm was chosen for the molecular docking calculations with the following run parameters (see (40, 42, 43) for details): the initial population of random individuals with a size of 50 individuals; a maximum number of 1.5 × 106 energy evaluations; a maximum number of generations of 27,000; mutation and crossover rates of 0.02 and 0.08, respectively. Elitism, which ensures that the top individual always survives into the next generations, was applied. A maximum of 300 iterations per local search was used. The probability of performing a local search on an individual was 0.06 where the maximum number of consecutive successes or failures before doubling or halving the search step was 4. A set of 100 docking simulations were performed for each domain of phospholamban, the results were clustered with a root-mean- square positional deviation of 2.0 Å.
Results
Selective Labeling of the Ca-ATPase by FITC
To investigate the functional coupling between Ca-ATPase polypeptide chains, we have measured the loss of catalytic activity with selective inactivation of Ca-ATPase polypeptide chains in the presence of PLB expressed in their native environment of cardiac SR membranes (44). This approach builds upon prior measurements demonstrating that FITC specifically and covalently modifies Lys514 in SERCA2a or the analogous site in SERCA1 (Lys515) to sterically block nucleotide binding (45, 46). Following FITC labeling, the Ca-ATPase remains conformationally competent as evidenced by the observation that small phosphorylester substrates (e.g., acetyl phosphate) are able to couple phosphoenzyme formation to calcium transport, albeit at much reduced catalytic rates (47, 48). Thus FITC modification provides a means to selectively titrate the number of available active sites without inducing tertiary structural changes associated with physical methods of inactivation (e.g., radiation inactivation) (49). In agreement with earlier results, FITC is observed to selectively modify the Ca-ATPase in cardiac SR microsomes at a site that is protected by the inclusion of nonhydrolyzable ATP analogs (Figures 2 and 3A). Maximal FITC incorporation corresponds to 3.5 ± 0.1 nmol/mg SR protein (Figure 3A), which is consistent with measurements of both Ca-ATPase abundance in cardiac SR microsomes based on its relative density on Coomassie stained gels, about 35% of the total protein and levels of steady state phosphoenzyme formation (2.0 ± 0.3 nmol per mg SR protein). Similar stoichiometries were obtained by specific derivatization of Cys674 by iodoacetamido fluorescein (IAF), where maximal levels of modification correspond to 3.7 ± 0.2 nmol per mg of microsomal protein (Figures 2 and 3B). IAF, in contrast to FITC, binds at a non-functional site as evidenced by the lack of Ca-ATPase inhibition upon IAF modification (25, 45, 46).
Figure 2. Specific Fluorescent Labeling of the Ca-ATPase.
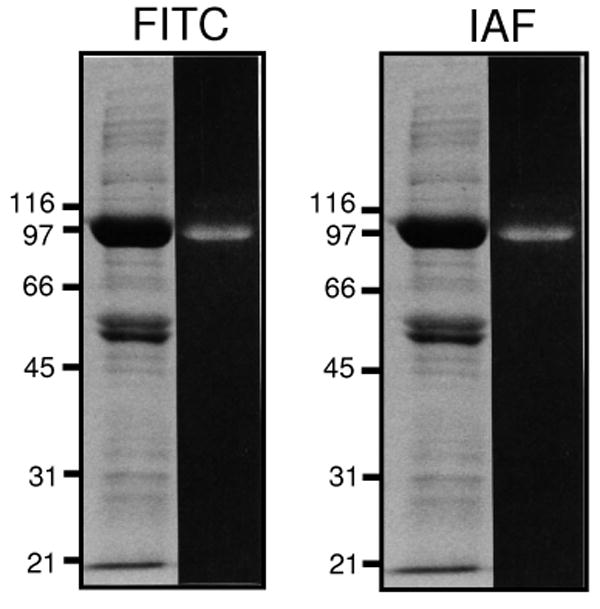
Fluorescent signals (right lanes) and Coomassie blue protein stain (left lanes) for FITC-labeled (A) or IAF-labeled (B) proteins following SDS-PAGE (7.5% acrylamide) separation of microsomal proteins (50 μg) isolated from porcine ventricles. Positions of protein standards are indicated, which correspond to galactosidease (116 kDa), phosphorylase a (97 kDa), albumin (66 kDa), ovalbumin (45 kDa), carbonic anhydrase (31 kDa), and trypsin inhibitor (21 kDa).
Figure 3. Functional Effects of Covalent Modification of Porcine SERCA2a in Cardiac Sarcoplasmic Reticulum.

Calcium-dependent ATPase activities (open symbols) or stoichiometries of fluorophore (FITC, panel A; IAF, panel B) labeling (closed symbols) were measured for cardiac SR microsomes in the absence (squares, circles) or presence (triangles) of 6 mM AMPPCP to block access to the nucleotide binding cleft. Specific labeling of Lys514, by FITC, or Cys674, by IAF in SERCA2a of cardiac SR membranes was performed and calcium-dependent ATPase activities were measured (25°C) as described in Materials and Methods.
PLB Modulates Functional Interactions Between Ca-ATPase Chains
Functional interactions between individual Ca-ATPase polypeptide chains under different conditions were examined by varying the ratio of modified-to-unmodified active sites in relation to the resulting ATPase activities. For example, cardiac SR exhibits a linear one-to-one relationship between the fraction of unmodified active sites and ATPase activity (Figure 4). In comparison, after PKA-mediated phosphorylation of PLB, a nonlinear and second order relationship is observed (i.e., Activity = F2), indicating that for every nucleotide site modified by FITC two Ca-ATPases are inactivated. Further, a virtually identical functional dependency is apparent in cardiac SR following addition of heparin, which provides an alternate means of enzyme activation by electrostatic charge disruption of interactions between PLB and the Ca-ATPase (50).
Figure 4. Phospholamban Modulates Functional Coupling Between Ca-ATPase Polypeptide Chains.

Rates of ATP hydrolysis were measured for the Ca-ATPase in cardiac SR vesicles in the absence (open circles) or presence of cAMP (1 μM) and PKA (80 μg/mL) (closed circles) or heparin (100 μM) (open triangle) in comparison with skeletal SR prior to (closed squares) or following (open squares) solubilization with C12E8 (6 μM). Access to the ATP binding cleft was blocked through the modification of either Lys515 (skeletal) or Lys514 (cardiac) with FITC to yield variable amounts of unmodified Ca-ATPase (F). Maximal stoichiometries of FITC were 3.4 ± 0.1 nmol/mg and 6.3 ± 0.1 nmol/mg, respectively, for cardiac and skeletal microsomes (see Figures S1 and S2 in Supporting Information). Inset: Normalized activity for cardiac SR in the presence of cAMP and PKA as a function of the squared concentration of unmodified (active) Ca-ATPase.
The functional inactivation of cardiac SR was compared with that of skeletal SR in which the homologous SERCA1 isoform is expressed in the absence of PLB. FITC modified (at Lys515) SERCA1 of skeletal SR exhibits a second-order functional inactivation identical to that of the Ca-ATPase in PKA-activated cardiac SR (Figures 4 and S1) (51). To test if these second-order inactivation profiles depend on interactions between Ca-ATPase polypeptide chains, parallel experiments were performed using skeletal SR solubilized with the nonionic detergent C12E8 under conditions previously demonstrated to result in SERCA1 monomers (52). Of note, under these conditions that disrupt protein interactions, full activity of unmodified SERCA1 was retained. In the case of C12E8-solubilized skeletal SR, the FITC inactivation profile reverts to first-order (Figure 4). Thus, the one-to-one relationship between enzyme function and active unmodified Ca-ATPase polypeptide chains in cardiac SR vesicles suggests that the SERCA2a active sites function independently until PLB is phosphorylated, which restores interactions between the two active sites. Together these results suggest that the phosphorylation state of PLB modulates the functional coupling between nucleotide sites on two Ca-ATPase polypeptide chains within an oligomeric complex (20).
Modulation of Ca-ATPase Structure by PLB
To directly address if modulation of functional coupling between active sites is dependent on changes in physical interactions between individual Ca-ATPases, we have utilized FRET measurements between the same fluorophore (homotransfer) on individual Ca-ATPase polypeptide chains in cardiac SR. This approach has previously been demonstrated to be sensitive to the spatial proximity of nearest neighbor Ca-ATPase proteins in skeletal SR (30). In the present study, measurements were made from the perspective of two distinct sites on SERCA2a, i.e., Lys514 within the N-domain, modified by FITC, and Cys674 within the P-domain, modified by IAF. For homotransfer measurements, fluorescein serves as an optimal fluorophore due to both its large quantum yield and extinction coefficient, but small Stokes shift. Thus a substantial spectral overlap between emission and absorption spectra results which permits fluorescein to serve as both an energy transfer donor and acceptor (30). Consequently, two proximal fluorescein molecules will undergo nonradiative energy transfer (homotransfer) much like that commonly measured using different donor and acceptor probes (heterotransfer). In the case of homotransfer the accompanying loss in the fluorescence polarization of fluorescein with increasing probe stoichiometry (increased FRET) provides a convenient means to assess intermolecular distances (30, 31, 53).
To detect any changes in spatial separation between nucleotide sites within Ca-ATPase polypeptide chains, FRET-induced depolarization of FITC was measured and plotted as changes in molecular anisotropy in the form of a Perrin equation, i.e., (1/P - 1/3)-1 (Figure 5). A progressive decrease in the anisotropy is observed as the fraction of FITC site occupation is increased, consistent with FRET-induced depolarization. Detergent solubilization of vesicles with C12E8 under conditions that produce monomeric Ca-ATPase results in substantially larger values for molecular anisotropy, which are independent of label stoichiometry, demonstrating that the measured depolarization is entirely due to FRET between sites on neighboring polypeptide chains. Upon PKA phosphorylation of PLB in native cardiac microsomes, the observed depolarization exhibits anisotropy values that are further decreased at each label stoichiometry relative to those measured prior to PLB phosphorylation, suggesting phosphorylation-induced increases in FRET, i.e., closer FITC proximity.
Figure 5. PLB Modulates Spatial Arrangement of Ca-ATPase Polypeptide Chains.

Anisotropy (1/P – 1/3)-1 of FITC- (top panel) or IAF- (bottom panel) labeled Ca-ATPase in cardiac SR microsomes (0.1 mg/mL) as a function of fractional labeling (f) of the probe sites (i.e., Lys514 or Cys674) (25). The maximal labeling stoichiometry is 3.4 ± 0.1 nmol FITC or 3.7 ± 0.2 nmol IAF per mg of microsomal protein (see Figure S1 in Supplemental Materials). Anisotropy was measured for 100 μg microsomes /ml in 20 mM MOPS (pH 7.0), 0.1 M KCl, 5 mM MgCl2, 0.1 mM EGTA, and 0.11 mM CaCl2 (Cafree is 0.5 μM) in the absence (open symbols) or presence (closed symbols) of PKA (80 μg/mL), cAMP (1 μM), and ATP (0.1 mM) before (circles) and after (squares) solubilization with C12E8 (6 mM). Excitation was at 485 nm and fluorescence emission was measured using a Schott OG530 cutoff filter. Lines represent nonlinear least squares fits to the data according to the models for interactions between neighboring fluorophores (see Eq. 2 in Experimental Procedures).
Similarly, with IAF, increasing probe modification also results in FRET-induced depolarization which is also entirely due to FRET as evidenced by the absence of any depolarization at each fractional label stoichiometry when SR vesicles are solubilized with detergent (Figure 5, lower panel). However, after PKA-induced phosphorylation of PLB there is an increase in anisotropy values are observed at each probe stoichiometry as compared with non-phosphorylated PLB, indicative of phosphorylation-induced decreases in FRET, i.e., farther distances between IAF molecules on neighboring Ca-ATPase polypeptide chains. Thus, phosphorylation of PLB induces opposite effects on the average distance between neighboring FITC molecules versus that of neighboring IAF molecules, suggesting a reorientation of nearby Ca-ATPase polypeptide chains with respect to one another.
For both FITC and IAF, phosphorylation-dependent changes in anisotropy are fully reversible with subsequent dephosphorylation of PLB mediated by alkaline phosphatase, which hydrolyzes the phosphoester linkage at Ser16 in PLB (Figure 6). Of note, the anisotropy value at the lowest labeling stoichiometry for FITC-labeled Ca-ATPase, where FRET-induced depolarization is minimal, is near the theoretical maximum (rigid limit) of 0.6 (Figure 5A), consistent with a highly ordered environment of fluorescein in the nucleotide binding cleft (45, 46). In comparison, the anisotropy of IAF is substantially smaller than that of FITC (Figure 5B), indicating some extent of rotational mobility of IAF that is consistent with its surface exposure as it is bound to Cys674 within a loop structure (Figure 1). PLB phosphorylation does not alter the average lifetime of either fluorophore or their respective solvent accessibilities, indicating that there are no substantial changes in the polarity or steric restrictions within the immediate environment of either fluorescein probe (Table 1). Thus, the PKA-dependent increases in homotransfer between FITC chromophores, with corresponding decreases between IAF chromophores, suggest spatial reorientations of domain elements in neighboring Ca-ATPase polypeptide chains rather than changes in local conformation around the probe site.
Figure 6. Reversible PLB Phosphorylation State.

Fluorescence polarization of FITC (top) or IAF (bottom) labeled Ca-ATPase (2.0 nmol/mg or F = 0.6) measured i) prior to activation (control), ii) following activation by addition of cAMP-dependent protein kinase (80 μg/mL), 5 mM ATP, and 1 μM cAMP (+PKA), and iii) subsequent treatment of activated microsomes with the PKA inhibitor calyculin (50 nM) and alkaline phosphatase (5 units/mL) (+Phosphatase). Asterisks represent statistically significant differences (p<0.005) in polarization values relative to controls.
Table 1.
Spatial Separation Between Fluorescein Binding Sites in Ca-ATPase Oligomeric Complex1
| Sample | <τ2> (ns) |
Ksv (M-1) | θav (deg) |
rav (Å) |
Oligomeric Size |
|
|---|---|---|---|---|---|---|
| Iodide | Acrylamide | |||||
| FITC (N-domain) | ||||||
| nonactivated | 3.8 ± 0.1 | 3.07 ± 0.07 | 0.37 ± 0.01 | 40 ± 2 | 24 ± 2 | 2 |
| PKA-activated | 3.4 ± 0.1 | 3.04 ± 0.02 | 0.37 ± 0.01 | 39 ± 2 | 20 ± 1 | 2 |
| IAF (P-domain) | ||||||
| nonactivated | 3.5 ± 0.1 | 9.9 ± 0.3 | 1.22 ± 0.06 | 38 ± 2 | 31 ± 2 | 2 |
| PKA-activated | 3.4 ± 0.1 | 9.6 ± 0.3 | 1.25 ± 0.07 | 35 ± 3 | 40 ± 2 | 2 |
Solution conditions are as described in the legend to Figure 4. Average lifetimes <τ> were measured as previously described (56), where the frequency response data was fit to a sum of two exponentials, where τ1= 1.7 ns and τ2 = 3.8 ns. The Stern-Volmer solvent accessibility (KSV) measured in the presence of charged (iodide) and neutral (acrylamide) quenchers was fit using Eq. 5. The average donor-acceptor distance (rav), the effective angle between the donor and acceptor absorption dipoles (θav), and the oligomeric size (n) were obtained from fits to the data in Figure 5), as described in Experimental Procedures.
Quantitative information regarding intermolecular distances and effective dipolar orientations between fluorescein moieties of FITC and IAF bound to Ca-ATPase polypeptide chains can be obtained by fitting the Perrin plots to a series of physical models in which the oligomeric state is set at various integer values (see Eq. 2 in Experimental Procedures) (Figure 5) (20, 30, 31). Optimal nonlinear least-squares fits to the data, assessed by the reduced chi-squared values (χR2), indicate that the average oligomeric state of the Ca-ATPase, regardless of the phosphorylation state of PLB, is a dimer. Upon PLB phosphorylation, there are no significant changes in the effective dipolar orientation between the fluorescein chromophores on proximal Ca-ATPase polypeptide chains (Table 1). These latter results are consistent with the presence of multiple dipole moments within individual fluorescein molecules and large amplitude domain motions of the Ca-ATPase, which are expected to minimize the sensitivity of energy transfer to changes in probe orientation (54-57). In comparison, there are significant changes in the intermolecular distance between fluorescein chromophores following the phosphorylation of PLB, which are apparent from the error surfaces associated with fitting the experimental data to a model (Figure 7). For example, upon PLB phosphorylation the mean distances recovered between FITC fluorophores bound to the N-domains on neighboring Ca-ATPase molecules decrease by 4 ± 2 Å, from 24 ± 2 Å to 20 ± 1 Å (Table 1). In contrast, in skeletal SR the spatial separation between FITC labeling sites on neighboring Ca-ATPase polypeptide chains was determined to be 42 ± 4 Å, in good agreement with previous measurements of 44 Å in skeletal SR (see Figure S2 in Supplemental Materials) (30). Thus these differences in FITC-FITC distances suggest conformational differences in Ca-ATPase dimers that bring neighboring N-domains into closer proximity in cardiac SR as compared with those in skeletal SR.
Figure 7. Phosphorylation of PLB Differentially Modifies Spatial Separation Between N- and P-Domains of Ca-ATPase Polypeptide Chains in Oligomeric Complex.

Depiction of error surfaces prior to (solid lines) or following (dashed lines) the phosphorylation of PLB by PKA for the distance (r) between FITC (left) or IAF (right) chromophores bound to proximal SERCA2a isoforms of the Ca-ATPase polypeptide chains labeled at Lys514 or Cys670, respectively. Normalized χ2N values were determined from nonlinear least-squares fits to the data depicted in Figure 5 following incremental adjustment of the spatial separation (distance) between either FITC (left) or IAF (right) chromophores. The horizontal dashed line at χ2N = 1.287 corresponds to the F-statistic, which represents one standard deviation relative to the best fit to the data. Experimental conditions are as described in the legend to Figure 5.
The intermolecular distances between IAF fluorophores bound to the P-domains on neighboring Ca-ATPase molecules are increased by 9 ± 3 Å upon PLB phosphorylation, from 31 ± 2 Å to 40 ± 2 Å (Figure 7). Comparative measurements of IAF-IAF distances in skeletal SR were less informative, since SERCA1 contains two IAF-reactive sites (Cys670 and Cys674) resulting in a substantial contribution of intramolecular FRET to the measured depolarization that precludes recovery of a single intersubunit distance (25) (Figure S2 in Supporting Information).
Discussion
We have examined the role of oligomeric interactions between individual Ca-ATPase polypeptide chains in native cardiac SR membranes and their modulation by the phosphorylation state of PLB. Assessing both functional and spatial interactions between Ca-ATPases, we find that prior to phosphorylation of PLB the active sites of neighboring Ca-ATPase monomers are uncoupled and function independently. However, the release of PLB inhibition of the Ca-ATPase, either by PKA-mediated PLB phosphorylation or heparin treatment, restores conformational coupling between individual Ca-ATPase polypeptide chains, which is accompanied by a reorientation of Ca-ATPase proteins within a dimeric unit. This reorientation is manifested by large-scale protein rearrangements that result in a 9 Å increase in the distance between labeled sites (Cys674) on the P-domain concomitant with a 4 Å decrease between labeled sites (Lys514) on the N-domain of SERCA2a in cardiac SR (Figure 7). These measurements indicate an important contribution to calcium transport of coupled domain movements between oligomeric complexes of the Ca-ATPase, in agreement with earlier kinetic measurements, suggesting that unphosphorylated PLB interferes with oligomeric interactions within Ca-ATPases important for calcium transport (16, 21). Further, PLB binding to the Ca-ATPase increases conformational barriers associated with the large-scale domain movements of the Ca-ATPase necessary for calcium activation.
Whereas prior measurements have recognized the presence of oligomeric interactions between Ca-ATPase proteins in both skeletal and cardiac membranes, their functional importance has remained uncertain based on observations that individual Ca-ATPase monomers retain independent active sites and functional competence following disruption of protein-protein contact interactions with detergent solubilization (49). Furthermore, much work aimed at understanding the regulation of the Ca-ATPase by PLB has been limited to the study of the each of these proteins in isolation, from e.g., their individual high-resolution structures and mutational analysis to identify key amino acids associated with the transport mechanism (58). More recently, the development of robust expression systems for the Ca-ATPase has permitted detailed functional measurements of the effect of PLB on SERCA2a function, identifying an important role for PLB in modulating the allosteric coupling between Ca-ATPase polypeptide chains within an oligomeric complex to affect rates of phosphoenzyme decomposition and hydrolysis (16, 21, 59-63). For example, Mahaney and Froehlich report very similar kinetic behavior between the monomeric form of the SERCA1 isoform of the Ca-ATPase in skeletal SR following detergent solubilization and the SERCA2a isoform of the Ca-ATPase co-expressed with PLB in insect microsomes or in cardiac SR (21, 62). In contrast, upon either the expression of SERCA2a in the absence of PLB or following the phosphorylation of PLB by PKA, the transient kinetics of the Ca-ATPase are similar to that observed for oligomeric complexes of SERCA1 (21). These latter results are consistent with the findings of the present study, which demonstrate that PLB modulates functional interactions between Ca-ATPase polypeptide chains within a dimeric protein complex (Figures 4-7; Table 1). Following the phosphorylation of PLB by PKA, we likewise observe a functional coupling between Ca-ATPase polypeptide chains similar to that observed in skeletal SR (Figure 4) (51). Combined, these results indicate the importance of the oligomeric complex involving two Ca-ATPase polypeptide chains in association with PLB, where the functional coupling between nucleotide binding sites is essential for maximal ATPase activity.
The ability of unphosphorylated PLB to uncouple the allosteric cooperativity between Ca-ATPase polypeptide chains acts to slow the rate-limiting step involving phosphoenzyme decomposition and is consistent with the importance of large-scale protein domain motions in the mechanism of calcium transport (32). Indeed, it has been noted that large-scale protein motions commonly underlie both allosteric cooperativity and catalysis and are, in many cases, rate limiting. In this respect, prior measurements have demonstrated i.) that large-scale domain motions of the Ca-ATPase are rate-limiting (64), and ii.) that PLB binding to the N-domain of the Ca-ATPase restricts domain mobility (10, 15, 56, 65-67). Previous work has demonstrated that upon phosphorylation of PLB there is an associated stabilization of the hinge domain of PLB that results in a shortening of the overall dimensions of the cytosolic domain, thus resulting in release of inhibitory interactions between PLB and the N-domain of the Ca-ATPase (1, 5-7, 67-70). Findings that a single PLB molecule interacting with two Ca-ATPase polypeptide chains are sufficient to fully inhibit transport activity (18, 19) lends further support to the hypothesis that phosphorylation of PLB acts to release structural constraints associated with oligomeric interactions between Ca-ATPase polypeptide chains that are necessary for optimal activity.
Understanding molecular mechanisms relating how PLB-dependent conformational changes in complex with the Ca-ATPase modulate function is currently limited by the availability of only a single low-resolution structure of a dimeric complex; this structure depicts a single PLB bound to both Ca-ATPase polypeptide chains in the homodimeric protein complex (18, 71). Building upon these earlier results, we have modeled the interactions between Ca-ATPase polypeptide chains through a series of molecular docking calculations using three-dimensional structures of the SERCA1 isoform of the Ca-ATPase (i.e., 1kju.pdb) and additionally, taking into consideration potential interaction sites with PLB with separate docking calculations. An optimal configuration involving the docking of two monomers of the Ca-ATPase was evaluated based on electrostatics, desolvation energies, and shape complementarity. Among 2000 structures generated, only two models met the criteria described in Experimental Procedures. One of these (Figure 8) better reproduced the residue-residue distance constraints from FRET experiments (Table 1) and allowed PLB-Ca-ATPase interactions (see Figure S4 in Supporting Information for a comparison of the two structure models).
Figure 8. Proposed Structural Model for the Ca-ATPase Homodimer.
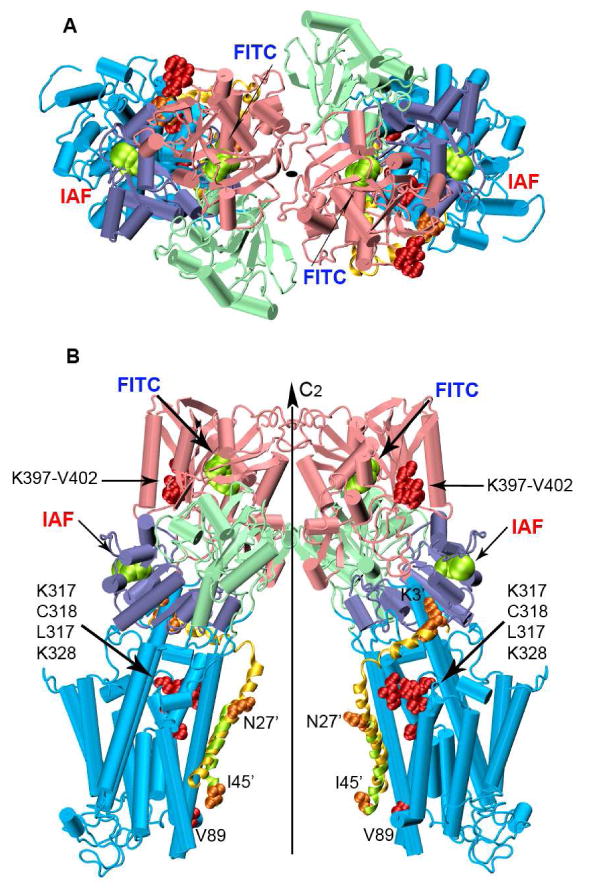
The two monomers of the Ca-ATPase are related by a two-fold rotational center of symmetry perpendicular to the cytoplasmic membrane. Top (A) and longitudinal (B) views of the dimer built from the crystallographic structure of SERCA1, i.e., 1kju.pdb. Domains are colored as in Figure 1. The lowest energy PLB conformer from the NMR ensemble (1fjk.pdb) was superimposed on the lowest energy docked PLB conformer (domain Ia and II; Figure S3), and does not consider known large-scale changes in the orientation of the N-domain of the Ca-ATPase upon PLB binding (10, 56, 67). The NMR-derived and the predicted PLB conformers are shown in yellow and green cartoon helices respectively. Amino acid residues involved in PLB-Ca-ATPase crosslinking are represented as spacefilling structures: orange for PLB and red for the Ca-ATPase. PLB residues have been marked with the prime symbol.
PLB interactions with the Ca-ATPase were modeled separately by docking known structures of PLB onto monomeric forms of the SERCA1 Ca-ATPase obtained for the apo-form and following calcium-activation (i.e., 2agv.pdb and 1su4.pdb). The transmembrane helix (i.e., domain II) and the cytoplasmic N-terminal portion (domain Ia) of PLB were docked separately to eliminate any bias imposed by the initial configuration of the flexible inter-helical loop (domain Ib), treating individual helical domains as rigid bodies (see Experimental Procedures). The two extremes of the most populated conformational cluster of structures show that there is substantial uniformity in the docking of transmembrane and cytosolic helices; in particular, the transmembrane helix of PLB binds within the groove formed by Ca-ATPase transmembrane helices M4, M6, and M9, as previously described (12, 13) (Figure S3 in Supporting Information).
Placement of these docked PLB complexes onto the structures of calculated homodimers of the Ca-ATPase suggests that the membrane spanning helix of PLB is located within the inner face of the complex between proximal Ca-ATPase polypeptide chains (Figure 8). Within the homodimeric complex (Figure 8), Ca-ATPase monomers are related by a two-fold rotational center of symmetry perpendicular to the plane of the membrane in agreement with EM studies of co-crystals of Ca-ATPase and PLB (Figure 8). Molecular interactions occur primarily through residues in the N-domain, with some involvement of P-domain side chains that are reflected by large reductions in calculated B-factors for the homodimeric complex (Figure S5 in Supporting Information). This model, based on individual high resolution structures of SERCA1 and PLB, can be taken as a starting point for defining the structure of Ca-ATPase-PLB oligomers. Use of this molecular docking algorithm, based on rigid body structures, does not take into account internal protein dynamics and induced fit of dimer units; nor does it consider large changes in domain positions of the Ca-ATPase upon association with PLB (10, 56). Nevertheless, this model (Figure 8), derived from docking of SERCA2 structures, exhibits a 37 Å spatial separation between alpha-carbons of Lys515, comparing favorably with the measured distances between FITC chromophores (42±2 Å) on neighboring Ca-ATPase polypeptide chains in skeletal SR (Figure S2), but fits less well to the smaller corresponding distance (Table 1) measured by FRET for cardiac SR (24±2 Å). We suggest that PLB, present in cardiac, but not skeletal SR, is primarily responsible for these inherent differences, and that PLB induces domain and concomitant dimeric rearrangements of the Ca-ATPase. The substantial homology (84% sequence identity) between SERCA2a and SERCA1 isoforms and the ability of PLB to fully regulate SERCA1 in both co-reconstituted or co-expression systems, suggests that isoform differences can be ruled out as an explanation for the structural differences in Ca-ATPase oligomers (1, 63). Previous reports have demonstrated that association of PLB with sites in the N-domain of the Ca-ATPase induce large domain reorientations that are sensitive to the phosphorylation of PLB which can bring nucleotide binding sites into closer proximity (10, 56, 67, 69). Indeed FRET measurements between sites within cytosolic and transmembrane domains of PLB have demonstrated that this distance remains the same whether PLB is reconstituted into proteoliposomes alone, or co-reconstituted with the Ca-ATPase providing further evidence that the Ca-ATPase undergoes structural changes that accommodate a compact and ordered PLB structure (7).
In conclusion, this model shows a potential mechanism for the functional coupling between Ca-ATPase active sites through the interaction of regions of the N- and P-domains of proximal Ca-ATPase polypeptide chains; furthermore, this model is compatible with modulation of this coupling by PLB through its modification of the Ca-ATPase dimeric interface to permit independent catalytic motions associated with transport function. This model is matched to experimental data taken under quiescent conditions and therefore reflects an oligomeric state that exists prior to the addition of ATP and enzymatic cycling; thus the model presented may constitute only one enzyme state with other possible arrangements of the chains occurring during cycling. Future work should combine experimental data taken under conditions that stabilize additional enzyme states compared with molecular modeling of the PLB-Ca-ATPase oligomers that considers molecular dynamics of Ca-ATPase domain reorientations.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (HL64031). Pacific Northwest National Laboratory is operated for the Department of Energy by Battelle Memorial Institute under contract DE-AC05-76RLO 1830.
Abbreviations used
- A-domain
actuator domain
- CPK
Corey, Pauling, and Koltun coloring conventions
- DTT
dithiothreitol
- EGTA
ethylene glycol bis(β-aminoethylether)-N,N,N′,N′-tetraacetic acid
- FITC
fluorescein-5-isothiocyanate
- FRET
fluorescence resonance energy transfer
- IAF
5-iodoacetamidofluorescein
- β-ME
2-mercaptoethanol
- MOPS
3-(N-morpholino) propanesulfonic acid
- N-domain
nucleotide binding domain
- P-domain
phosphorylation domain
- NMA
normal mode analysis
- PLB
phospholamban
- SERCA
sarco/endoplasmic reticulum Ca-ATPase
- TM
transmembrane
Footnotes
Supporting Information Available: Inactivation and FRET data for the SERCA1 isoform of the Ca-ATPase in skeletal SR is available as well as additional molecular models from docking calculations and normal mode analysis B-factors. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Li J, Bigelow DJ, Squier TC. Conformational changes within the cytosolic portion of phospholamban upon release of Ca-ATPase inhibition. Biochemistry. 2004;43:3870–3879. doi: 10.1021/bi036183u. [DOI] [PubMed] [Google Scholar]
- 2.Negash S, Yao Q, Sun H, Li J, Bigelow DJ, Squier TC. Phospholamban remains associated with the Ca2+- and Mg2+-dependent ATPase following phosphorylation by cAMP-dependent protein kinase. Biochem J. 2000;351:195–205. doi: 10.1042/0264-6021:3510195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asahi M, McKenna E, Kurzydlowski K, Tada M, MacLennan DH. Physical interactions between phospholamban and sarco(endo)plasmic reticulum Ca2+-ATPases are dissociated by elevated Ca2+, but not by phospholamban phosphorylation, vanadate, or thapsigargin, and are enhanced by ATP. J Biol Chem. 2000;275:15034–15038. doi: 10.1074/jbc.275.20.15034. [DOI] [PubMed] [Google Scholar]
- 4.Chen B, Mahaney JE, Mayer MU, Bigelow DJ, Squier TC. Concerted but noncooperative activation of nucleotide and actuator domains of the Ca-ATPase upon calcium binding. Biochemistry. 2008;47:12448–12456. doi: 10.1021/bi8014289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J, Boschek CB, Xiong Y, Sacksteder CA, Squier TC, Bigelow DJ. Essential role for Pro21 in phospholamban for optimal inhibition of the Ca-ATPase. Biochemistry. 2005;44:16181–16191. doi: 10.1021/bi051075o. [DOI] [PubMed] [Google Scholar]
- 6.Li J, Bigelow DJ, Squier TC. Phosphorylation by cAMP-dependent protein kinase modulates the structural coupling between the transmembrane and cytosolic domains of phospholamban. Biochemistry. 2003;42:10674–10682. doi: 10.1021/bi034708c. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Xiong Y, Bigelow DJ, Squier TC. Phospholamban binds in a compact and ordered conformation to the Ca-ATPase. Biochemistry. 2004;43:455–463. doi: 10.1021/bi035424v. [DOI] [PubMed] [Google Scholar]
- 8.Ha KN, Traaseth NJ, Verardi R, Zamoon J, Cembran A, Karim CB, Thomas DD, Veglia G. Controlling the inhibition of the sarcoplasmic Ca2+-ATPase by tuning phospholamban structural dynamics. J Biol Chem. 2007;282:37205–37214. doi: 10.1074/jbc.M704056200. [DOI] [PubMed] [Google Scholar]
- 9.Morita T, Hussain D, Asahi M, Tsuda T, Kurzydlowski K, Toyoshima C, Maclennan DH. Interaction sites among phospholamban, sarcolipin, and the sarco(endo)plasmic reticulum Ca(2+)-ATPase. Biochem Biophys Res Commun. 2008;369:188–194. doi: 10.1016/j.bbrc.2007.11.098. [DOI] [PubMed] [Google Scholar]
- 10.James P, Inui M, Tada M, Chiesi M, Carafoli E. Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature. 1989;342:90–92. doi: 10.1038/342090a0. [DOI] [PubMed] [Google Scholar]
- 11.Chen Z, Akin BL, Stokes DL, Jones LR. Cross-linking of C-terminal residues of phospholamban to the Ca2+ pump of cardiac sarcoplasmic reticulum to probe spatial and functional interactions within the transmembrane domain. J Biol Chem. 2006;281:14163–14172. doi: 10.1074/jbc.M601338200. [DOI] [PubMed] [Google Scholar]
- 12.Toyoshima C, Asahi M, Sugita Y, Khanna R, Tsuda T, MacLennan DH. Modeling of the inhibitory interaction of phospholamban with the Ca2+ ATPase. Proc Natl Acad Sci U S A. 2003;100:467–472. doi: 10.1073/pnas.0237326100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seidel K, Andronesi OC, Krebs J, Griesinger C, Young HS, Becker S, Baldus M. Structural characterization of Ca(2+)-ATPase-bound phospholamban in lipid bilayers by solid-state nuclear magnetic resonance (NMR) spectroscopy. Biochemistry. 2008;47:4369–4376. doi: 10.1021/bi7024194. [DOI] [PubMed] [Google Scholar]
- 14.Hutter MC, Krebs J, Meiler J, Griesinger C, Carafoli E, Helms V. A structural model of the complex formed by phospholamban and the calcium pump of sarcoplasmic reticulum obtained by molecular mechanics. Chembiochem. 2002;3:1200–1208. doi: 10.1002/1439-7633(20021202)3:12<1200::AID-CBIC1200>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 15.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 16.Waggoner JR, Huffman J, Froehlich JP, Mahaney JE. Phospholamban inhibits Ca-ATPase conformational changes involving the E2 intermediate. Biochemistry. 2007;46:1999–2009. doi: 10.1021/bi061365k. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z, Akin BL, Jones LR. Mechanism of reversal of phospholamban inhibition of the cardiac Ca2+-ATPase by protein kinase A and by anti-phospholamban monoclonal antibody 2D12. J Biol Chem. 2007;282:20968–20976. doi: 10.1074/jbc.M703516200. [DOI] [PubMed] [Google Scholar]
- 18.Young HS, Jones LR, Stokes DL. Locating phospholamban in co-crystals with Ca(2+)-ATPase by cryoelectron microscopy. Biophys J. 2001;81:884–894. doi: 10.1016/S0006-3495(01)75748-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrington DA, Yao Q, Squier TC, Bigelow DJ. Comparable levels of Ca-ATPase inhibition by phospholamban in slow-twitch skeletal and cardiac sarcoplasmic reticulum. Biochemistry. 2002;41:13289–13296. doi: 10.1021/bi026407t. [DOI] [PubMed] [Google Scholar]
- 20.Yao Q, Chen LT, Li J, Brungardt K, Squier TC, Bigelow DJ. Oligomeric interactions between phospholamban molecules regulate Ca-ATPase activity in functionally reconstituted membranes. Biochemistry. 2001;40:6406–6413. doi: 10.1021/bi002891t. [DOI] [PubMed] [Google Scholar]
- 21.Mahaney JE, Albers RW, Waggoner JR, Kutchai HC, Froehlich JP. Intermolecular conformational coupling and free energy exchange enhance the catalytic efficiency of cardiac muscle SERCA2a following the relief of phospholamban inhibition. Biochemistry. 2005;44:7713–7724. doi: 10.1021/bi048011i. [DOI] [PubMed] [Google Scholar]
- 22.Negash S, Chen LT, Bigelow DJ, Squier TC. Phosphorylation of phospholamban by cAMP-dependent protein kinase enhances interactions between Ca-ATPase polypeptide chains in cardiac sarcoplasmic reticulum membranes. Biochemistry. 1996;35:11247–11259. doi: 10.1021/bi960864q. [DOI] [PubMed] [Google Scholar]
- 23.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 24.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 25.Bishop JE, Squier TC, Bigelow DJ, Inesi G. (Iodoacetamido)fluorescein labels a pair of proximal cysteines on the Ca2+-ATPase of sarcoplasmic reticulum. Biochemistry. 1988;27:5233–5240. doi: 10.1021/bi00414a043. [DOI] [PubMed] [Google Scholar]
- 26.Pick U, Karlish SJ. Indications for an oligomeric structure and for conformational changes in sarcoplasmic reticulum Ca2+-ATPase labelled selectively with fluorescein. Biochim Biophys Acta. 1980;626:255–261. doi: 10.1016/0005-2795(80)90216-0. [DOI] [PubMed] [Google Scholar]
- 27.Gratton E, Limkeman M. A continuously variable frequency cross-correlation phase fluorometer with picosecond resolution. Biophys J. 1983;44:315–324. doi: 10.1016/S0006-3495(83)84305-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bevington P. Data Reduction and Error Analysis for the Physical Sciences. 56-64. McGraw-Hill; New York: 1969. pp. 56–64. [Google Scholar]
- 29.Lakowicz J. Principles of Fluorescence Specroscopy. 2nd. Kluwer Academic / Plenum Publishers; New York: 1999. [Google Scholar]
- 30.Highsmith S, Cohen JA. Spatial organization of CaATPase molecules in sarcoplasmic reticulum vesicles. Biochemistry. 1987;26:154–161. doi: 10.1021/bi00375a022. [DOI] [PubMed] [Google Scholar]
- 31.Runnels LW, Scarlata SF. Theory and application of fluorescence homotransfer to melittin oligomerization. Biophys J. 1995;69:1569–1583. doi: 10.1016/S0006-3495(95)80030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu C, Rice WJ, He W, Stokes DL. A structural model for the catalytic cycle of Ca(2+)-ATPase. J Mol Biol. 2002;316:201–211. doi: 10.1006/jmbi.2001.5330. [DOI] [PubMed] [Google Scholar]
- 33.Stokes DL, Pomfret AJ, Rice WJ, Glaves JP, Young HS. Interactions between Ca2+-ATPase and the pentameric form of phospholamban in two-dimensional co-crystals. Biophys J. 2006;90:4213–4223. doi: 10.1529/biophysj.105.079640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen R, Weng ZP. Docking unbound proteins using shape complementarity, desolvation, and electrostatics. Proteins. 2002;47:281–294. doi: 10.1002/prot.10092. [DOI] [PubMed] [Google Scholar]
- 35.Chen R, Weng Z. A novel shape complementarity scoring function for protein-protein docking. Proteins. 2003;51:397–408. doi: 10.1002/prot.10334. [DOI] [PubMed] [Google Scholar]
- 36.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature. 2000;405:647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 37.Obara K, Miyashita N, Xu C, Toyoshima I, Sugita Y, Inesi G, Toyoshima C. Structural role of countertransport revealed in Ca(2+) pump crystal structure in the absence of Ca(2+) Proc Natl Acad Sci U S A. 2005;102:14489–14496. doi: 10.1073/pnas.0506222102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lamberth S, Schmid H, Muenchbach M, Vorherr T, Krebs J, Carafoli E, Griesinger C. Structural role of countertransport revealed in Ca2+ pump crystal structure in the absence of Ca2+ P Natl Acad Sci USA. 2000;102:14489–14496. doi: 10.1073/pnas.0506222102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lamberth S, Schmid H, Muenchbach M, Vorherr T, Krebs J, Carafoli E, Griesinger C. NMR solution structure of phospholamban. Helv Chim Acta. 2000;83:2141–2152. [Google Scholar]
- 40.Morris G, Goodsell DS, Halliday RS, Huey R, Hart WE, Beleew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 41.Yang L, Tan CH, Hsieh MJ, Wang J, Duan Y, Cieplak P, Caldwell J, Kollman PA, Luo R. New-generation amber united-atom force field. J Phys Chem B. 2006;110:13166–13176. doi: 10.1021/jp060163v. [DOI] [PubMed] [Google Scholar]
- 42.Soares TA, Goodsell DS, Briggs JM, Ferreira R, Olson AJ. Docking of 4-oxalocrotonate tautomerase substrates: Implications for the catalytic mechanism. Biopolymers. 1999;50:319–328. doi: 10.1002/(SICI)1097-0282(199909)50:3<319::AID-BIP7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 43.Soares TA, Goodsell DS, Ferreira R, Olson AJ, Briggs JM. Ionization state and molecular docking studies for the macrophage migration inhibitory factor: the role of lysine 32 in the catalytic mechanism. J Mol Recognit. 2000;13:146–156. doi: 10.1002/1099-1352(200005/06)13:3<146::AID-JMR497>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 44.Mitchinson C, Wilderspin AF, Trinnaman BJ, Green NM. Identification of a labelled peptide after stoicheiometric reaction of fluorescein isothiocyanate with the Ca2+ - dependent adenosine triphosphatase of sarcoplasmic reticulum. FEBS Lett. 1982;146:87–92. doi: 10.1016/0014-5793(82)80710-2. [DOI] [PubMed] [Google Scholar]
- 45.Kirley TL, Wang T, Wallick ET, Lane LK. Homology of ATP binding sites from Ca2+ and (Na,K)-ATPases: comparison of the amino acid sequences of fluorescein isothiocyanate labeled peptides. Biochem Biophys Res Commun. 1985;130:732–738. doi: 10.1016/0006-291x(85)90477-2. [DOI] [PubMed] [Google Scholar]
- 46.Tate CA, Shin G, Walseth TF, Taffet GE, Bick RJ, Entman ML. Nucleotide specificity of cardiac sarcoplasmic reticulum.Inhibition of GTPase activity by ATP analogue in fluorescein isothiocyanate-modified calcium ATPase. J Biol Chem. 1991;266:16165–16170. [PubMed] [Google Scholar]
- 47.Bodley AL, Jencks WP. Acetyl phosphate as a substrate for the calcium ATPase of sarcoplasmic reticulum. J Biol Chem. 1987;262:13997–14004. [PubMed] [Google Scholar]
- 48.Teruel JA, Inesi G. Roles of phosphorylation and nucleotide binding domains in calcium transport by sarcoplasmic reticulum adenosinetriphosphatase. Biochemistry. 1988;27:5885–5890. doi: 10.1021/bi00416a010. [DOI] [PubMed] [Google Scholar]
- 49.Andersen JP. Monomer-oligomer equilibrium of sarcoplasmic reticulum Ca-ATPase and the role of subunit interaction in the Ca2+ pump mechanism. Biochim Biophys Acta. 1989;988:47–72. doi: 10.1016/0304-4157(89)90003-8. [DOI] [PubMed] [Google Scholar]
- 50.Xu ZC, Kirchberger MA. Modulation by polyelectrolytes of canine cardiac microsomal calcium uptake and the possible relationship to phospholamban. J Biol Chem. 1989;264:16644–16651. [PubMed] [Google Scholar]
- 51.Squier TC, Hughes SE, Thomas DD. Rotational dynamics and protein-protein interactions in the Ca-ATPase mechanism. J Biol Chem. 1988;263:9162–9170. [PubMed] [Google Scholar]
- 52.Yao Q, Chen LT, Bigelow DJ. Affinity purification of the Ca-ATPase from cardiac sarcoplasmic reticulum membranes. Protein Expr Purif. 1998;13:191–197. doi: 10.1006/prep.1998.0892. [DOI] [PubMed] [Google Scholar]
- 53.Erijman L, Weber G. Oligomeric protein associations: transition from stochastic to deterministic equilibrium. Biochemistry. 1991;30:1595–1599. doi: 10.1021/bi00220a022. [DOI] [PubMed] [Google Scholar]
- 54.Wu P, Brand L. Orientation factor in steady-state and time-resolved resonance energy transfer measurements. Biochemistry. 1992;31:7939–7947. doi: 10.1021/bi00149a027. [DOI] [PubMed] [Google Scholar]
- 55.Chen B, Squier TC, Bigelow DJ. Calcium activation of the Ca-ATPase enhances conformational heterogeneity between nucleotide binding and phosphorylation domains. Biochemistry. 2004;43:4366–4374. doi: 10.1021/bi0356350. [DOI] [PubMed] [Google Scholar]
- 56.Negash S, Huang S, Squier TC. Rearrangement of domain elements of the Ca-ATPase in cardiac sarcoplasmic reticulum membranes upon phospholamban phosphorylation. Biochemistry. 1999;38:8150–8158. doi: 10.1021/bi990599j. [DOI] [PubMed] [Google Scholar]
- 57.Huang S, Squier TC. Enhanced rotational dynamics of the phosphorylation domain of the Ca-ATPase upon calcium activation. Biochemistry. 1998;37:18064–18073. doi: 10.1021/bi981997+. [DOI] [PubMed] [Google Scholar]
- 58.Toyoshima C. Structural aspects of ion pumping by Ca(2+)-ATPase of sarcoplasmic reticulum. Arch Biochem Biophys. 2008;476:3–11. doi: 10.1016/j.abb.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 59.Froehlich JP, Heller PF. Transient-state kinetics of the ADP-insensitive phosphoenzyme in sarcoplasmic reticulum: implications for transient-state calcium translocation. Biochemistry. 1985;24:126–136. doi: 10.1021/bi00322a018. [DOI] [PubMed] [Google Scholar]
- 60.Froehlich JP, Taniguchi K, Fendler K, Mahaney JE, Thomas DD, Albers RW. Complex kinetic behavior in the Na,K- and Ca-ATPases. Evidence for subunit-subunit interactions and energy conservation during catalysis. Ann N Y Acad Sci. 1997;834:280–296. doi: 10.1111/j.1749-6632.1997.tb52259.x. [DOI] [PubMed] [Google Scholar]
- 61.Froehlich JP, Taylor EW. Transient state kinetic studies of sarcoplasmic reticulum adenosine triphosphatase. J Biol Chem. 1975;250:2013–2021. [PubMed] [Google Scholar]
- 62.Mahaney JE, Thomas DD, Froehlich JP. The time-dependent distribution of phosphorylated intermediates in native sarcoplasmic reticulum Ca2+-ATPase from skeletal muscle is not compatible with a linear kinetic model. Biochemistry. 2004;43:4400–4416. doi: 10.1021/bi035068g. [DOI] [PubMed] [Google Scholar]
- 63.Mahaney JE, Autry JM, Jones LR. Kinetics studies of the cardiac Ca-ATPase expressed in Sf21 cells: new insights on Ca-ATPase regulation by phospholamban. Biophys J. 2000;78:1306–1323. doi: 10.1016/S0006-3495(00)76686-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 65.Squier TC, Bigelow DJ, Thomas DD. Lipid fluidity directly modulates the overall protein rotational mobility of the Ca-ATPase in sarcoplasmic reticulum. J Biol Chem. 1988;263:9178–9186. [PubMed] [Google Scholar]
- 66.Bigelow DJ, Squier TC, Thomas DD. Temperature dependence of rotational dynamics of protein and lipid in sarcoplasmic reticulum membranes. Biochemistry. 1986;25:194–202. doi: 10.1021/bi00349a028. [DOI] [PubMed] [Google Scholar]
- 67.Pantano S, Carafoli E. The role of phosphorylation on the structure and dynamics of phospholamban: a model from molecular simulations. Proteins. 2007;66:930–940. doi: 10.1002/prot.21239. [DOI] [PubMed] [Google Scholar]
- 68.Bigelow DJ, Squier TC. Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim Biophys Acta. 2005;1703:121–134. doi: 10.1016/j.bbapap.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 69.Bigelow DJ, Squier TC. Coil-to-helix transition within phospholamban underlies release of Ca-ATPase inhibition in response to beta-adrenergic signaling. Current Enzyme Inhibition. 2006;2:19–27. [Google Scholar]
- 70.Sugita Y, Miyashita N, Yoda T, Ikeguchi M, Toyoshima C. Structural changes in the cytoplasmic domain of phospholamban by phosphorylation at Ser16: a molecular dynamics study. Biochemistry. 2006;45:11752–11761. doi: 10.1021/bi061071z. [DOI] [PubMed] [Google Scholar]
- 71.Stokes DL, Pomfret AJ, Rice WJ, Glaves JP, Young HS. Interactions between Ca2+-ATPase and the pentameric form of phospholamban in two-dimensional co-crystals. Biophysical journal. 2006;90:4213–4223. doi: 10.1529/biophysj.105.079640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sorensen TL, Moller JV, Nissen P. Phosphoryl transfer and calcium ion occlusion in the calcium pump. Science. 2004;304:1672–1675. doi: 10.1126/science.1099366. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.