

Abstract
Two quantitative trait loci (Fr-H1 and Fr-H2) for frost tolerance (FT) have been discovered on the long arm of chromosome 5H in barley. Two tightly linked groups of CBF genes, known to play a key role in the FT regulatory network in A. thaliana, have been found to co-segregate with Fr-H2. Here, we investigate the allelic variations of four barley CBF genes (HvCbf3, HvCbf6, HvCbf9 and HvCbf14) in a panel of European cultivars, landraces and H. spontaneum accessions. In the cultivars a reduction of nucleotide and haplotype diversities in CBFs compared with the landraces and the wild ancestor H. spontaneum, was evident. In particular, in cultivars the loss of HvCbf9 genetic variants was higher compared to other sequences. In order to verify if the pattern of CBF genetic variants correlated with the level of FT, an association procedure was adopted. The pairwise analysis of linkage disequilibrium (LD) among the genetic variants in four CBF genes was computed to evaluate the resolution of the association procedure. The pairwise plotting revealed a low level of LD in cultivated varieties, despite the tight physical linkage of CBF genes analysed. A structured association procedure based on a general liner model was implemented, including the variants in CBFs, of Vrn-H1, and of two reference genes not involved in FT (α-Amy1 and Gapdh) and considering the phenotypic data for FT. Association analysis recovered two nucleotide variants of HvCbf14 and one nucleotide variant of Vrn-H1 as statistically associated to FT.
Electronic supplementary material
The online version of this article (doi:10.1007/s00122-009-1138-7) contains supplementary material, which is available to authorized users.
Introduction
Plants can increase their freezing tolerance (FT) in response to low, non-freezing temperatures, a phenomenon known as cold acclimation or hardening. The molecular dissection of cold acclimation has revealed a complex process characterised by the coordinated up- or down-regulation of hundreds of Cold-Regulated (COR) genes which, in turn, are controlled by a complex regulatory network (Shinozaki and Yamaguchi-Shinozaki 2000; Thomashow 2001; Fowler and Thomashow 2002). In many plant species, including Triticeae, the C-repeat Binding Factor (CBF) genes are the key regulators of the signal cascade leading to the expression of COR genes. The CBF transcription factors recognise the cis-acting CRT/DRE (C-repeat/dehydration responsive element) element largely represented in the regulatory regions of COR genes (Stockinger et al. 1997; Skinner et al. 2005). Heterologous over-expression of CBF sequences in transgenic plants has been demonstrated to increase levels of FT (Takumi et al. 2008; Oh et al. 2007).
In barley (Hordeum vulgare), 20 CBF genes have been identified by Skinner et al. (2006), 11 of which were found to be arranged in two tight clusters in tandem on the long arm of chromosome 5H, within a genomic region previously identified as FT quantitative trait locus (Fr-H2) (Skinner et al. 2006; Francia et al. 2007). Several studies have reported that the expression of CBF and, in turn, of COR genes is reduced following induction of Vrn-H1 in response to vernalization, suggesting an interaction between vernalization and acclimation regulatory gene networks (Stockinger et al. 2007). In the Nure × Tremois mapping population, CBF gene clusters were reported to span 0.8 cM (Francia et al. 2007). In Triticum monococcum an orthologous genomic region containing similar CBF gene clusters was found (Miller et al. 2004). The role of CBFs in cold acclimation (Stockinger et al. 2007), the association of CBF expression level with FT (Vágújfalvi et al. 2005) and the tight co-segregation of the CBF gene clusters with the Fr-H2 QTL region make these genes the most appropriate candidates for one of the two major QTL controlling frost tolerance in Triticeae (Stockinger et al. 2007). The detailed analysis of the genomic structure of the CBF cluster in a panel of barley cultivars has revealed a non-conserved genomic organisation at the Fr-H2 locus: the cultivars Nure and Dicktoo (both winter type) share the same genomic organisation, while Morex and Tremois (both spring type) are characterised by a different genomic structure (Francia et al. 2007). For example, HvCbf10B gene is present in Dicktoo and Nure, though it is not harboured in Morex. As a consequence, the total number of the CBF genes at the Fr-H2 locus may vary among accessions (Francia et al. 2007). The role of each CBF gene and their regulatory network has been extensively studied in Arabidopsis thaliana where at least AtCbf1 and AtCbf3 have additive effect and redundant activities (Novillo et al. 2007).
The investigation of the genetic variants in cultivated and wild cereal crops allowed understanding important phenomena as domestication effects and selective sweep. Nucleotide and haplotype diversities are widely used estimators of the degree of loci polymorphism in populations. In barley a dramatic loss of genetic variants in cultivated varieties was reported in genes controlling pathogen resistances (Bundock and Henry 2004), germination (Russell et al. 2004) and in genes encoding enzymes (Kilian et al. 2006). Intensive selection and the resulting bottlenecks appear to be the major determinants of the narrowness of the modern gene pools (Badr et al. 2000; Salamini et al. 2002). In A. thaliana the natural purifying selection was pointed out to act on CBF genes to explain the clinal variation in freezing tolerance of the different geographical accessions (Hannah et al. 2006; Zhen and Ungerer 2007; Zhen and Ungerer 2008).
The level of linkage disequilibrium (LD) or the non-random association of alleles at distinct loci (Hill and Robertson 1968; Levontin and Kojima 1960), can change across genomic regions, depending on different local recombination rates and sequence compositions. As pointed out in previous studies in human and plants (Yu and Buckler 2006; Hayes and Szucs 2006; Rostoks et al. 2006), LD evaluation is a crucial step to carry out association analysis. In out-crossing plants as maize, LD decay extends from few bases up to 2,000 bp, in landraces and elite cultivars, respectively (Remington et al. 2001; Tenaillon et al. 2001; Palaisa et al. 2003; Yu and Buckler 2006), although, in some genomic regions, LD can extend till 500 kb (Jung et al. 2004). In A. thaliana, an inbreeding species, the extent of LD range is about 10 kb (Nordborg et al. 2005; Kim et al. 2007), nevertheless in highly isolated populations LD extends up to 50 cM (Garris et al. 2003). In cultivated barley, LD can be estimated as far as 60 cM (Rostoks et al. 2006), while in wild barley, despite the high rate of self-fertilisation, LD decays at a rate closer to that observed in out-crossing plants (Morrell et al. 2005).
Association mapping has been originally proposed in humans to dissect the genetics of complex diseases (Lander and Schork 2004; Templeton 1995). The aim of association mapping is to find in unrelated individuals significant association between genetic variants and phenotypes (Weiss and Clark 2002). In plants, association mapping offers the opportunity to dissect candidate genes that underlie important agronomic QTLs using large germoplasm collections instead of family based crosses (Laird and Lange 2006). Association mapping can be performed with a genome-wide molecular marker set or with markers designed to target candidate genes. The second approach requires low levels of LD and a high density of markers in order to detect association at high resolution (Rafalski 2002). This approach was used in wheat to refine the genomic region carrying candidate genes for resistance to Stagonospora nodarum blotch (Tommasini et al. 2007) and for finding molecular markers associate with kernel size (Breseghello and Sorrells 2006). In barley and maize, several studies were conducted to find statistical association between genetic variants and complex traits such as yield, flowering time and drought tolerance (Kraakman et al. 2004; Thornsberry et al. 2001; Pakniyat et al. 1999).
In this work, we explored the natural allelic variation in four CBF genes located at the Fr-H2 locus and in the Vrn-H1 gene using a representative collection of European cultivated barley, landraces and wild accessions. This study was designed (1) to assess existing allelic variants and the occurrence of domestication effects in key genes controlling a trait associated with plant adaptation to environment, and (2) to perform an association analysis between the genetic variants in the studied genes and the freezing tolerance phenotype.
Materials and methods
Plant material and DNA extraction
The genetic material used in this study (216 accessions in total) was selected from three different sources. A representative subset of 113 cultivars and locally adapted accessions extracted from the germplasm collection of the CRA-Genomic Research Centre of Fiorenzuola d’Arda were selected to represent widely cultivated germplasm across Europe, ensuring representation of likely current genetic variants in European barley. This set of genotypes was composed of modern cultivars (released between 1960 and 1980), of advanced cultivars (released after 1980) and of 12 European landraces. The growth habit of each cultivar considered in this study was obtained querying the European Barley Database (http://barley.ipk-gatersleben.de/ebdb.php3). For barley accessions missing in the database as well as for cultivars having contrasting passports, the vernalization requirement was assessed during experimental trials in Fiorenzuola d’Arda.
Sixty-seven barley landraces and old European cultivars previously selected out of 5,842 accessions for their different plant characters and described by Castiglioni et al. (1998) were obtained from the germplasm bank of Braunschweig (Germany). These accessions were used to investigate the genetic variants in barley populations locally adapted in a broad range of different environments.
Thirty-six Hordeum spontaneum originating from the Fertile Crescent were added as representative samples of the genetic variability present in wild populations (Badr et al. 2000). The genetic material considered is described in the supplementary Tables S1, S2 and S3.
Genomic DNA was isolated from about 60 mg of leaf tissue in 96-well microtube plates. Plant material was ground with MM300 Mixer Mill (Retsch®, Haan, Germany) and DNA was extracted with Promega Magnetic® 96 DNA Plant System following manufacturer’s instructions.
PCR amplification and SNP discovery
SNP analysis was carried out on five genes putatively involved in the adaptation of barley to low temperature: four amplicons from the CBF clusters on the long arm of the chromosome 5H (HvCbf3, HvCbf6, HvCbf9 and HvCbf14) and one corresponding to the Vrn-H1 gene located distally from the CBF clusters on the same chromosome. The CBF genes are named according to the classification of Miller et al. (2004), while following Badawi et al. (2007) the selected genes correspond to HvCbfIIIc-3, HvCbfIIIa-6, HvCbfIVd-9 and HvCbfIVc-14, respectively. PCR primers were designed using Primer3 software (http://frodo.wi.mit.edu) (Rozen and Skaletsky 2000) on the basis of the available sequences for HvCbf3, HvCbf9, HvCbf14 and Vrn-H1 (GenBank accession numbers AF239616 and EF409484, AY785877, DQ151545 and AY750995, respectively) in order to amplify most of the coding sequence and the 3′UTR. Two primer pairs with slightly different annealing positions were drawn for HvCbf3 and HvCbf9. One primer only was developed for HvCbf6 and used in association with the primer HvCBF6.006 (Skinner et al. 2005). Two reference sequences, α-Amy1 and Gapdh, described by Kilian et al. (2006) known to generate significant Tajima’s D values were also considered as references. They were amplified using two previously published primer pairs, B306–B307 and B604–B605 (Kilian et al. 2006). The list of primers used in this study and the length of expected sizes of amplicons are reported in Table S4.
All PCRs and sequencing reactions were carried out in 96-well plates (MicroAmp® 96-well plates, Applied Biosystems, Foster City, USA) in Eppendorf Mastercycler EPgradient thermalcyclers (Eppendorf, Hamburg, Germany) and each reaction was performed in 10 μl with the following mixture composition: 20 ng of DNA, 1.5 mM MgCl2, 0.4 μM of each primer and 0.25 U Taq HotStart DNA polymerase QIAGEN (Valencia, USA). The reaction mixtures for Vrn-H1 and HvCbf6 amplifications were supplemented with 3 mM of MgCl2 and 1× Q Solution QIAGEN (Valencia, USA), respectively. The following thermal protocol was used to amplify HvCbf3, HvCbf9 and HvCbf14: after an initial denaturation step at 95°C for 15 min, the reactions were subjected to 11 cycles of 95°C for 30 s, 60°C for 45 s and 72°C for 90 s and the annealing temperature was decreased by 0.7°C in each cycle. Then the reactions were subjected to 29 cycles of 95°C for 30 s, 53°C for 45 s and 72°C for 90 s. For the amplification of Vrn-H1 and HvCbf6, after an initial denaturation (95°C for 15 min), the thermal protocol was 35 cycles of 95°C for 30 s, 56°C for 45 s and 72°C for 90 s. A final elongation step of 10 min was applied for all amplification. Gapdh and α-Amy were amplified following the published protocol (Kilian et al. 2006).
PCR products were sequenced on both strands using ABI PRISM BigDye Primer Cycle sequencing kit v.3.1 (Applied Biosystems, Foster City, USA), following manufacturer’s instruction. Purified sequencing reactions were injected in ABI3730 DNA Analyzer (Applied Biosystems Foster City, USA). Raw electropherograms were analysed using Sequencing Analysis® software (Applied Biosystems, Foster City, USA) to obtain FASTA sequences. Subsequently, for each gene a consensus sequence for each accession was created, assembling forward and reverse sequences using Contig Assembly Program (CAP) (Huang 1992). The consensus sequence of each gene was aligned with web-based MultAlin Software (http://bioinfo.genopole toulouse.prd.fr/multalin/multalin.html) (Corpet 1988) and each FASTA alignment was used as input file for DnaSP 4.10 (Rozas et al. 2003) for SNPs detection and for all other analyses. Indel polymorphisms were included for inferring Vrn-H1 haplotypes, while for other genes, indels were not included.
Fluorescent AFLP reactions
AFLP reactions were conducted in fluorescence following the published protocol (Vos et al. 1995) with some modifications. Digestion, ligation and pre-amplification reactions were conducted as in the original protocol, while for selective fluorescent amplification, custom forward primers, 5′-labelled with 6-FAM (Applied Biosystems, Foster City, USA), were used instead of radioactive labelled primers. Selective amplification products, were diluted tenfold and 2 μl of each dilution was mixed with 10 μl of deionised formamide and 0.15 μl of GeneScan 1200-LIZ internal size standard (Applied Biosystems, Foster City, USA). Raw data were analysed using GeneMapper® 4.0 software (Applied Biosystems, Foster City, USA) to obtain genotyping tables containing only polymorphic peaks. Genotyping tables were then exported as tab-delimited files and appropriately formatted in Microsoft Excel® (Redmond, USA) to conduct phylogenetic and statistical analyses.
Data analyses
The degree of genetic polymorphism of each gene was investigated using two estimators, nucleotide diversity (π) and haplotype diversity (Hd). The estimators were calculated using the DnaSP software with Nei’s equation 10.5 and Nei’s unbiased equation, respectively (Nei 1987). Analysis of π and Hd was initially conducted for each group of accessions and, subsequently, considering the population structure.
To investigate the phylogenetic relationships among accessions, binary data obtained after scoring peaks in AFLP analysis were transformed in NEXUS format and analysed by the SplitsTree4 software (Huson and Bryant 2006). Trees based on Hamming’s genetic distance and neighbour-joining method, and Uncorrected-P genetic distance and neighbour-joining method, were constructed.
In order to test if the selected genes deviate from the neutrality (Kimura 1983), the D test of Tajima’s (1989) was applied accounting for population structure. Tajima’s D values were calculated with DnaSP, and when significant, the Tajima’s test was repeated within domesticated (D) and wild (W) accessions.
Linkage disequilibrium (LD) was estimated by squared allele–frequency correlations (r²) using the software package TASSEL 2.01 and 1,000 permutations (Bradbury et al. 2007). The significance of pairwise LD (P value) among all possible pairs was also evaluated by TASSEL with the rapid permutation test. Since r² has a large variance with rare alleles, only sites with an allele frequency (MAF) >0.1 were formatted appropriately and used to perform pairwise analysis of LD. To calculate the threshold beyond which LD values can be significant due to genetic linkage, square root of each pairwise r² among nucleotide variants detected in CBFs and in three physically unlinked genes (α-Amy; Gapdh and Vrn-H1) was calculated. The 95 percentile of this approximate normal distribution was assumed as r² threshold to declare the presence of LD among molecular markers (Breseghello and Sorrells 2006). Undetected amplicons of HvCbf3 and HvCbf9 were modelled as deletions of these genes in their respective genomes in LD analysis in order to find significant association with other molecular markers in the CBF genes.
Phenotypic analysis of frost tolerance
One-hundred-thirteen genotypes of European cultivars and locally adapted accessions were selected considering their different level of frost tolerance as assessed in repeated experiments conducted at Fiorenzuola during the past decade (Rizza et al. 2003). Frost tolerance was evaluated under controlled conditions (growth chamber),on plants at first leaf stage, cold acclimated for 4 weeks (3°C, 8 h light and 1°C, 16 h dark) and subjected to a freezing treatment according to Rizza et al. (2001). The freezing temperatures ranged between –10 and –14°C on the bases of previous experiences. A freezing stress at –10°C was able to discriminate within susceptible genotypes; –12°C between susceptible and resistant cultivars; while −14°C underline the behaviour of the very resistant genotypes. Three barley cultivars previously described in the literature and representative of different frost tolerance levels were included as controls in all experiments: Tremois (spring, frost susceptible—Francia et al. 2004), Nure (winter, frost resistant—Francia et al. 2004) and Pamina (facultative, very frost resistant—Rizza et al. 2003). The frost-induced damage was measured in leaves as a decrease in the maximum quantum efficiency of photosystem II (PSII) photochemistry, using the chlorophyll fluorescence parameter F v/F m, which is the ratio of variable (F v) to maximal (F m) fluorescence in dark adapted state. On the basis of the results of at least three independent experiments the genotypes were classified into five classes based on their level of frost tolerance: I: very good (not significantly different from Pamina), II: good (not significantly different from Nure); III: middle (less resistant than Nure, more resistant than Tremois); IV: low (not significantly different from Tremois); V: very low (more susceptible than Tremois).
Association analysis
The five FT groups served as the dependent variable for an association analysis to determine the effects of polymorphisms in CBFs and Vrn-H1. A structured association based on least squared general linear model (GLM) procedure was implemented in SAS® (Cary, NC, USA) modelling each polymorphism in the five candidate genes (CBFs and Vrn-H1) as a single factors consisting of two classes, one for each of the two SNP variants observed. Only those SNPs with a minor allele frequency >0.10 were considered. Likewise, a similar approach was used to include the genetic variants detected in the reference genes (α-Amy1 and Gapdh) in order to verify if spurious statistical association with genes not involved in FT occurred.
To account for population structure, principal component analysis was computed on AFLP matrix containing polymorphic loci scored in the 113 European genotypes of cultivars and of locally adapted accessions (Table S1) data set with NTSYSpc® (Exetersoftware, New York, USA). The first three principal components were included as regression variables in the association analysis. In addition, growth habit and ear type were also considered in the analysis.
The approach taken was to start with a saturated model, including all factors, and then progressively eliminating the least significant factor to finally arrive at a model with only plausibly important effects.
The initial, “saturated” model including selected nucleotide variants, plus additional explanatory factors, was
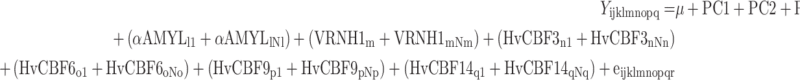 |
where Y is the FT score (I–V); μ a population mean, PC1 to PC3 are the linear regressions on the respective principal components accounting for population stratification; HABITi is the fixed effect associated with growth habit (winter, spring or alternative); EAR TYPEj is the fixed effect corresponding to whether the barley was classified a two- or six-row variety; (GAPDHk1 + GAPDHkNk) is the sum of the fixed effects of the Nk polymorphisms at the Gapdh reference gene; (αAMYLl1 + αAMYLlNl) is the sum of the fixed effects of the Nl polymorphisms at the α-Amyl l1 reference gene; (VRNH1ml + VRNH1mNm) is the sum of the fixed effects of the Nm polymorphisms in the Vrn-H1 gene; (HvCBF3n1 + HvCBF3nNn),… (HvCBF14q1 + HvCBF14qNq) are the sums of fixed effects of the polymorphisms at each of the four CBF genes and eijklmnopq is the random residual.
Backwards stepwise regression was used to eliminate non-significant effects and after, models with progressively fewer effects were implemented after elimination of the factors with the lowest level of significance (i.e., greatest P value). All factors with a P value below 0.40 were maintained in the final model. Inclusion of all significant marker loci in the models precludes the need for further multiple testing correction.
Results
CBF genes have a narrow gene pool in widely cultivated European barley
A representative subset of European cultivars, landraces and wild accessions of barley was investigated to detect genetic variants in five genes involved in FT plus two reference genes. The sequences of the amplicons were analysed to detect SNPs as well as to calculate π, Hd and the number of haplotype per gene (Table 1).
Table 1.
Nucleotide and haplotype diversity recorded in four CBF genes, in Vrn-H1 and in reference genes Gapdh and α-Amy1
| Gene | La | Sb | Number of haplotypes | Hdc | σ(Hd)d | πe × 10−3 | |
|---|---|---|---|---|---|---|---|
| HvCbf3 | All samples | 427 | 9 | 7 | 0.648 | 0.022 | 3.70 |
| H. spontaneum | 427 | 8 | 6 | 0.761 | 0.048 | 6.11 | |
| Landraces and old cultivars | 427 | 2 | 3 | 0.580 | 0.043 | 2.87 | |
| Modern and advanced cultivars | 427 | 3 | 4 | 0.628 | 0.022 | 3.27 | |
| HvCbf6 | All samples | 520 | 16 | 10 | 0.481 | 0.035 | 8.01 |
| H. spontaneum | 520 | 16 | 8 | 0.731 | 0.067 | 9.47 | |
| Landraces and old cultivars | 520 | 10 | 4 | 0.504 | 0.048 | 8.82 | |
| Modern and advanced cultivars | 520 | 9 | 4 | 0.373 | 0.047 | 7.21 | |
| HvCbf9 | All samples | 601 | 7 | 5 | 0.180 | 0.030 | 3.70 |
| H. spontaneum | 601 | 3 | 4 | 0.477 | 0.010 | 2.12 | |
| Landraces and old cultivars | 601 | 6 | 4 | 0.177 | 0.005 | 1.18 | |
| Modern and advanced cultivars | 601 | 1 | 2 | 0.058 | 0.030 | 0.23 | |
| HvCbf14 | All samples | 398 | 10 | 11 | 0.769 | 0.016 | 6.21 |
| H. spontaneum | 398 | 6 | 7 | 0.742 | 0.062 | 5.98 | |
| Landraces and old cultivars | 398 | 8 | 8 | 0.761 | 0.033 | 8.10 | |
| Modern and advanced cultivars | 398 | 7 | 7 | 0.636 | 0.045 | 4.67 | |
| Vrn-H1 | All samples | 600 | 4 | 14 | 0.701 | 0.009 | 2.00 |
| H. spontaneum | 600 | 4 | 12 | 0.819 | 0.056 | 3.44 | |
| Landraces and old cultivars | 600 | 4 | 4 | 0.706 | 0.014 | 2.89 | |
| Modern and advanced cultivars | 600 | 4 | 4 | 0.671 | 0.012 | 2.61 | |
| α-Amy1 | All samples | 663 | 8 | 6 | 0.133 | 0.031 | 0.97 |
| H. spontaneum | 663 | 8 | 6 | 0.308 | 0.099 | 2.87 | |
| Landraces and old cultivars | 663 | 4 | 2 | 0.144 | 0.056 | 1.10 | |
| Modern and advanced cultivars | 663 | 4 | 2 | 0.035 | 0.020 | 0.24 | |
| Gapdh | All samples | 765 | 15 | 5 | 0.682 | 0.013 | 3.56 |
| H. spontaneum | 765 | 13 | 5 | 0.739 | 0.048 | 3.30 | |
| Landraces and old cultivars | 765 | 12 | 3 | 0.654 | 0.024 | 3.16 | |
| Modern and advanced cultivars | 765 | 12 | 3 | 0.642 | 0.033 | 3.41 |
| Total | La | Sb | Average No. of haplotypes per gene | Hdc | σ(Hd)d | πe × 10−3 |
|---|---|---|---|---|---|---|
| All samples | 3974 | 59 | 8,3 | 0.513 | 0.022 | 4.02 |
| H. spontaneum | 3974 | 50 | 6,86 | 0.654 | 0.056 | 4.76 |
| Landraces and old cultivars | 3974 | 37 | 4,00 | 0.504 | 0.032 | 4.02 |
| Modern and advanced cultivars | 3974 | 31 | 3,71 | 0.435 | 0.030 | 3.09 |
The same estimates are reported below considering the overall nucleotide variants
aAmplicon size
bNo. of polymorphic sites
cNei’s unbiased haplotype diversity
dStandard deviation associated with haplotype diversity
eNucleotide diversity
A total of 3,974 bp of genomic DNA were sequenced from each accession for candidate and reference genes and the average number of substitutions was calculated for each group of accessions (Table 1). In H. spontaneum one SNP every 79 bp was found, while in landraces and in cultivars the same value was one SNP every 107 bp and one SNP every 128 bp, respectively. Heterozygote mutations were not detected and all SNPs resulted bi-allelic. A summary of the overall genetic variants detected in CBF and reference genes is reported in Table S5. All singletons (polymorphic sites present only once in the germplasm analysed) occurred in landraces and in H. spontaneum. When nucleotide variants of each gene were translated in haplotypes, the number of haplotypes varied depending on the loci, from 14 in Vrn-H1 to six in α-Amy1 (Table 1). All haplotypes detected in advanced and modern varieties were represented in H. spontaneum accessions. An exception was a particular haplotype of HvCbf14 found only in landraces and old cultivars, but not in H. spontaneum.
Three CBF genes, HvCbf3, HvCbf6 and HvCbf9 showed a reduction of allele diversity and of π in advanced and modern varieties compared to other groups of accessions (Table 1). Compared with H. spontaneum these genes were undergone a loss of nucleotide polymorphisms (1 − πd/πw) (Tenaillon et al. 2004) of 46, 24 and 89%, respectively. Haplotype analysis in HvCbf9 revealed an almost monomorphic sequence in European cultivars (Hd = 0.058), while in wild accessions the allelic gene pool was wider (Table 1). When the primer pair HvCBF3fw-HVCBF3rw was used to amplify HvCbf3 the amplification failed in 38% of modern and advanced cultivars, in 4% of landraces and old cultivars and in 20% of H. spontaneum accessions (Tables S1, S2, S3). An additional primer pair (HvCBF3fw2-HVCBF3rw2) with slightly different annealing positions was therefore designed to amplify the gene in the accessions where amplicons were undetected; nevertheless, no amplification products were found. Similarly, the primer pair HvCBF9fw-HVCBF9rw failed the amplification in 12% of modern and advanced cultivars, 15% of landraces and old cultivars and in 11% of H. spontaneum accessions (Tables S1, S2, S3). A further attempt at amplifying with a second primer pair (HvCBF9fw2-HvCBF9rw2) did not give any amplification products in the same accessions.
HvCbf14 showed a wider allelic diversity in landraces and old cultivars than in H. spontaneum; nevertheless, in advanced and modern varieties the nucleotide and haplotype diversity were lower (Table 1). Compared to the other CBFs analysed, the loss of nucleotide diversity of HvCbf14 in modern and advanced cultivars was lower (21%). On average, the loss of nucleotide diversity of CBF genes in modern and advanced European cultivars compared to H. spontaneum was of 45%. Vrn-H1 represents the best candidate gene that underlie the Fr-H1 QTL for FT in barley (Hayes et al. 1993; Laurie et al. 1995; Stockinger et al. 2007). Fourteen Vrn-H1 haplotypes were found in selected accessions, and a subset of 12 were detected in H. spontaneum. Unlike other genes studied, the most recurrent genetic variants detected in Vrn-H1 were indel polymorphisms (Table S5). In advanced and modern varieties, Vrn-H1 showed a reduction of Hd and π compared to other groups of accessions (Table 1).
Two-hundred-fifteen AFLP polymorphic peaks obtained with six primer combinations (E32M49, E32M55, E36M49, E36M55, E38M55 and E41M55) were used to infer the population structure of the genetic material studied. The tree obtained with AFLP data, based on Hamming’s genetic distance and neighbour-joining method showed 4 NJ-clades (Fig. 1). Other trees based on different genetic distances showed very similar topologies (data not shown). Accessions were split on the basis of their growth habit: in NJ-clades I and IV predominant accessions were spring, while in NJ-clades II and III predominant accessions were winter types (Fig. 1). Two subgroups of H. spontaneum clustered with winter and spring accessions in NJ-clade II and IV, respectively.
Fig. 1.

Clustering of selected accessions using neighbour-joining method and Hamming’ s genetic distance. The tree was constructed on the basis of 215 AFLP polymorphisms obtained with six primer combinations. Red circles indicate H. spontaneum, while spring and winter accessions are shown in violet and dark blue, respectively. The four nodes from which the NJ-clades considered depart are indicated with black squares
Significant Tajima’s D values in non-structured populations can be consistent with a molecular selection hypothesis (Tajima 1989). To evaluate if the narrowness of allelic gene pool of CBFs and reference genes in advanced and modern cultivars of barley was an effect of selection, population structure was introduced and Tajima’s test was applied within each NJ-clade. Among the four CBF genes considered, a significant and negative D value was found for HvCbf9 in NJ-clade IV. Other CBF genes and Vrn-H1 did not deviate significantly from the neutral hypothesis (Table 2). As previously reported, the two reference genes deviated from neutrality in some NJ-clades (Kilian et al. 2006). Gapdh showed significant and positive D values for NJ-clades I, III and IV and these cases of significance could be consistent with balancing selection hypothesis (Wright and Gaut 2004) while for α-Amy1 Tajima’s D was significant and negative in NJ-clade IV (Table 2).
Table 2.
Nucleotide diversity, haplotype diversity and Tajima’s D values recorded at seven barley genes considering genotypes assigned to four populations corresponding to the four major clades of the NJ-tree
| GENE | NJ-CLADE | Sa | Hdb | πc × 10−3 | Tajima’s D | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dd | LDSe | Wf | Dd | LDSe | Wf | Dd | LDSe | Wf | |||
| HvCbf3 | I | 3 | 1 | / | 0.72 | 0.40 | / | 4.01 | 1.73 | / | −1.16 |
| HvCbf3 | II | 2 | 2 | 8 | 0.52 | 0.63 | 0.84 | 3.30 | 3.17 | 8.50 | −1.21 |
| HvCbf3 | III | 2 | 1 | 4 | 0.63 | 0.46 | 0.80 | 3.18 | 2.00 | 6.64 | −0.31 |
| HvCbf3 | IV | 2 | 1 | 2 | 0.68 | 0.17 | 0.70 | 3.70 | 0.72 | 3.62 | 1.16 |
| HvCbf6 | I | 9 | 6 | / | 0.50 | 0.40 | / | 8.64 | 6.59 | / | 1.11 |
| HvCbf6 | II | 10 | 10 | 16 | 0.32 | 0.36 | 0.84 | 6.83 | 7.20 | 10.10 | −1.01 |
| HvCbf6 | III | 9 | 4 | 11 | 0.13 | 0.62 | 0.86 | 2.42 | 9.97 | 12.04 | −0.65 |
| HvCbf6 | IV | 9 | 9 | 12 | 0.30 | 0.24 | 0.59 | 5.19 | 4.88 | 8.33 | −0.49 |
| HvCbf9 | I | 0 | 0 | / | 0.00 | 0.00 | / | 0.00 | 0.00 | / | / |
| HvCbf9 | II | 1 | 1 | 3 | 0.36 | 0.44 | 0.70 | 1.45 | 1.76 | 3.47 | −0.50 |
| HvCbf9 | III | 0 | 0 | 3 | 0.00 | 0.00 | 0.64 | 0.00 | 0.00 | 3.00 | −1.71* |
| HvCbf9 | IV | 0 | 5 | 0 | 0.00 | 0.32 | 0.00 | 0.00 | 2.76 | 0.00 | −1.90** |
| HvCbf14 | I | 4 | 5 | / | 0.69 | 0.90 | / | 5.60 | 9.64 | / | 0.6 |
| HvCbf14 | II | 4 | 6 | 3 | 0.82 | 0.71 | 0.60 | 6.17 | 7.04 | 3.75 | 0.46 |
| HvCbf14 | III | 5 | 5 | 3 | 0.43 | 0.82 | 0.74 | 3.17 | 7.45 | 5.09 | −0.27 |
| HvCbf14 | IV | 4 | 5 | 4 | 0.74 | 0.82 | 0.77 | 5.94 | 7.73 | 6.90 | 0.49 |
| Vrn-h1 | I | 2 | 1 | / | 0.68 | 0.40 | / | 2.50 | 1.08 | / | 1.63 |
| Vrn-h1 | II | 3 | 4 | 2 | 0.52 | 0.69 | 0.71 | 2.31 | 2.99 | 2.87 | 0.38 |
| Vrn-h1 | III | 2 | 4 | 4 | 0.60 | 0.74 | 0.75 | 2.72 | 3.55 | 4.04 | 0.63 |
| Vrn-h1 | IV | 4 | 3 | 4 | 0.65 | 0.26 | 0.71 | 2.81 | 1.08 | 3.39 | 0.65 |
| α-Amy1 | I | 4 | 0 | / | 0.13 | 0.00 | / | 0.86 | 0.00 | / | −1.33 |
| α-Amy1 | II | 4 | 4 | 5 | 0.10 | 0.18 | 0.26 | 0.70 | 1.26 | 2.20 | −1.05 |
| α-Amy1 | III | 4 | 4 | 4 | 0.40 | 0.14 | 0.33 | 0.30 | 9.40 | 2.22 | −1.86** |
| α-Amy1 | IV | 4 | 0 | 5 | 0.70 | 0.00 | 0.20 | 0.49 | 0.00 | 1.67 | −1.86** |
| Gapdh | I | 10 | 10 | / | 0.61 | 0.70 | / | 9.63 | 9.70 | / | 1.73** |
| Gapdh | II | 10 | 10 | 12 | 0.73 | 0.64 | 0.80 | 11.76 | 5.77 | 9.64 | 1.33 |
| Gapdh | III | 12 | 10 | 11 | 0.66 | 0.69 | 0.86 | 10.91 | 11.34 | 7.31 | 1.91** |
| Gapdh | IV | 12 | 10 | 13 | 0.67 | 0.60 | 0.89 | 12.00 | 11.98 | 9.92 | 2.12** |
Note in some NJ-clades the deviation from neutrality of HvCbf9, α-Amy1 and Gapdh (marked with asterisks)
* 0.05 < P value < 0.1, ** P value < 0.05
aNo. of polymorphic sites
bNei’s unbiased haplotype diversity
cNucleotide diversity
dCultivars (advanced, modern and old varieties)
eLandraces
f H. spontaneum
To gain insight, for all cases of significance, Tajima’s D values were re-computed to assess if significant D values occurred when domesticated lines (D) and H. spontaneum (W) accessions were considered separately. The sequences of HvCbf9 within domesticated lines (D) in NJ-clade IV were monomorphic and Tajima’s D cannot be calculated, while within wild lines Tajima’s D was significant (Table 3). These wild lines were phenotyped for FT, revealing a heterogeneous group of accessions with different levels of tolerance to freezing. Differently in NJ-clades III and IV, D test was not significant for α-Amy1 in wild lines, and in domesticated lines only one haplotype is represented. Nevertheless, when domesticated and wild accessions were considered together D values were negative and significant suggesting that α-Amy1 may have undergone selection.
Table 3.
Significant Tajima’s D values of Table 2 calculated separately for domesticated (D) lines, H. spontaneum (W) and D + W groups
| Gene | Tajima’s D values | |||
|---|---|---|---|---|
| NJ-Clade | Within Wa | Within Db | Within D + W | |
| HvCbf9 | IV | −1.89** | Only one haplotype | −1.90** |
| α-Amy1 | III | −1.1 | Only one haplotype | −1.86** |
| α-Amy1 | IV | −1.70* | Only one haplotype | −1.86** |
| Gapdh | I | 1.59 | 2.36** | 1.73** |
| Gapdh | III | 2.38** | 0.55 | 1.91** |
| Gapdh | IV | 0.45 | 1.84** | 2.12** |
Significant Tajima’s D values for α-Amy1 is maintained only when domesticated and wild lines are considered together
* 0.05 < P value < 0.1
** P value < 0.05
a H. spontaneum
b Domesticated accessions
The pattern of polymorphisms of HvCbf14 and Vrn-H1 genes is statistically associated with freezing tolerance
Analysis of nucleotide variants in candidate and reference genes detected in European cultivars was conducted to find those with a MAF >0.1. Two SNPs of HvCbf3, nine SNPs of HvCbf6, and four SNPs of HvCbf14, were used to evaluate the extent of LD in cultivated accessions within the CBF cluster (Table S5). Undetected amplicons of HvCbf3 and HvCbf9 were modelled as deletions in their respective genomes and treated as polymorphisms in LD calculation and in association analysis.
The LD threshold beyond two sites to declare them in disequilibrium was fixed at r² = 0.32 on the basis of the statistical analysis of the 95 percentile of the square root distribution of the r 2 values (Breseghello and Sorrells 2006). The four CBF genes at Fr-H2 locus made a co-segregating group, reported to span in 0.8 cM in barley genetic maps: within this distance high levels of LD are expected in barley cultivars based on previous publications (Rostoks et al. 2006). Considering all modern and advanced European cultivars, pairwise plot of LD evidenced, as expected, blocks of significant associations at intra-genic level, showing that polymorphisms in HvCbf6 were almost all in complete disequilibrium (r² = 1) with each other (Fig. 2). On the contrary, considering inter-genic polymorphisms, pairwise analysis of r² showed low level of LD among physically linked CBF genes: almost all these pairwise comparison did not reach a value of r² > 0.1. The maximum levels of inter-genic pairwise LD were found among Cbf3_4 and nearly all SNPs of HvCbf6 and among Cbf3_4 and Cbf14_3 (Fig. 2). Since population structure may overestimate the real extent of LD (Rostoks et al. 2006), pairwise analysis of LD was recomputed within each NJ-clade; nevertheless, resulting plots maintained the same decay in each NJ-clade (data not shown). To provide further evidence of the low extent of LD among genetic variants in selected CBFs, an analysis of the number of haplotype blocks was conducted. As these genes are linked in a narrow region of chromosome 5H, haplotype blocks are expected to co-segregate in cultivated accessions. When each gene was considered by itself, a reduction of the number of haplotypes in cultivars was observed, while considering the combinations of haplotypes of the four CBFs, 27 combinations were identified in 113 cultivated accessions and 31 combinations were found in 36 wild accessions. Considering the overall polymorphisms making haplotypes blocks detected at the CBF locus, Hd values of 0.88 and 0.98 were observed in cultivars and in H. spontaneum, respectively. In other words, the absence of recurrent haplotypes blocks in cultivars at the CBF locus supports the rapid decay of LD at this locus, despite the global trend of chromosome 5H, where the extent of linkage disequilibrium spans wider genomic distances (Rostoks et al. 2006).
Fig. 2.

Linkage disequilibrium at CBF genes in modern and advanced cultivars. Each point in the LD matrix represents a comparison between a pair of polymorphic sites, with the r 2 values displayed above the diagonal and the P values for Fisher’s exact test below. The name of polymorphic sites and the colour codes for r 2 and P values are also reported. The LD threshold beyond two sites to declare them in disequilibrium was fixed at r² = 0.20 on the basis of the evaluation of LD among unlinked molecular markers (Breseghello and Sorrells 2006)
To gain insight into the FT variability in European barley cultivars, plants were phenotyped using non-disruptive fluorescent method based on chlorophyll fluorescence analysis (Rizza et al. 2001). Based on phenotypic data the genotypes were split in five discrete classes of frost tolerance indicated from I (highly tolerant) to V (low tolerant); the data are shown in Table S1. In order to verify the hypothesis of statistical association between genetic variants at CBF genes and FT classes, a structured association based on GLM procedure was conducted including in the model each one of the first three principal components explaining respectively 23, 16 and 6% of the variation. As expected, no statistical relationship among nucleotide variants of reference genes and FT was evidenced (Table 4). Using GLM test statistic, the SNPs Cbf14_10 and CBf14_7 of HvCbf14, located in the 3′UTR, and the nucleotide variant VRN_1 of Vrn-H1 were strongly associated with the trait of interest (Table 4). All three nucleotide variants were significant with P value < 0.005 and the two polymorphisms of HvCbf14 showed similar effects on the trait. One nucleotide variant of HvCbf6 showed a P value slightly inferior to 0.005. As well as reference genes, other CBF genes considered in this study do not contribute significantly to FT with the statistical test implemented.
Table 4.
List of the seven fixed effects and their level of significance retained in the final model after the backwards stepwise regression analysis
| Gene | Name of SNP | Allele | F value | P value |
|---|---|---|---|---|
| HvCbf14 | Cbf14_10 | C | 27.11 | <0.0001 |
| HvCbf14 | Cbf14_7 | C | 21.13 | <0.0001 |
| Vrn-H1 | VRN_1 | C | 6.89 | 0.0016 |
| HvCbf6 | Cbf6_13 | C | 5.64 | 0.0047 |
| HvCbf14 | Cbf14_10 | T | 1.59 | 0.2106 |
| HvCbf14 | Cbf14_7 | T | 1.41 | 0.2375 |
| Gapdh | GAPDH_4 | A | 1.15 | 0.3231 |
Fixed effects of nucleotide substitution at Cbf14_10 and Cbf14_7 as well as at VRN_1 represent significant cases
Discussion
The cluster of CBF genes located at the peak of the FT QTL Fr-H2 has relevance to the regulation of freezing tolerance in barley and in diploid wheat (Francia et al. 2004; Vágújfalvi et al. 2005; Båga et al. 2007). In recent years, the CBF gene clusters of few barley accessions have been the subjects of intense research. While several studies have considered the gene pool structures of these transcription factors in different barley populations (Francia et al. 2007; Stockinger et al. 2007), this study is the first systematic report on the sets of CBF alleles in a large collection of barley accessions.
Genetic variants of several CBF genes co-segregating with a FT QTL have been searched in a large panel of barley, including wild accessions and landraces, to assess if the signature of selection is molecularly and functionally evident. The CBF gene family has expanded itself in barley and in wheat genomes compared with other related species of temperate grasses (Badawi et al. 2007). CBF sequences of monocots cluster in several monophyletic clades, some of which contain only CBFs of temperate grasses, indicating the appearance of their common ancestors before the speciation and during the colonisation of temperate habitats (Badawi et al. 2007). As the freezing tolerance is a characteristic of temperate plants, the key CBF genes that in barley underlie this trait are expected to be peculiar and highly conserved in the Triticeae tribe and in the less related temperate species of the family. Following this principle, only four CBF genes were considered, three of which (HvCbf3, HvCbf9 and HvCbf14) having homologs currently found in temperate grasses only and one (HvCbf6) belonging to a CBF monophyletic clade deriving from a more ancient ancestral gene, HvCbf6 homologs have indeed been found in temperate and tropical grasses (Badawi et al. 2007).
It is generally agreed that crop plants contain less genetic variation than their wild ancestors (Doebley et al. 2006). Domestication in barley, as well as in other crops, acted upon some key loci, selecting favourable alleles at genes that control agronomic traits relevant for domestication (Salamini et al. 2002). In addition, domestication processes may involve genetic bottlenecks caused by the repeated reproduction of superior individuals (Doebley et al. 2006). As an outcome, neutral loci of domesticated populations can show a reduction of genetic variation compared with wild ancestor populations. When comparing the levels of nucleotide and haplotype diversities between cultivated varieties and wild accessions (Table 1), a loss of genetic variants is evident. The analysis of the CBF gene pools in the cultivated European germplasm reveals that sequences of HvCbf9 are almost monomorphic in cultivated varieties. As several oligonucleotides present in the coding sequence failed to detect HvCbf9 in some lines, we hypothesise the absence of this gene in some barley accessions. A similar situation holds for HvCbf3 which was not detected in one-third of European cultivars. Our results beg the question of CBF cluster structure in barley. Large deletions and a different organisation of orthologue genomic regions in unrelated accessions of barley have been already demonstrated, and BAC comparison of two barley accessions shows that some CBFs are absents in specific varieties (Francia et al. 2007; Skinner et al. 2005) showing that a pan-cluster of CBFs with some dispensable genes evolves and segregates in barley.
A structured population as well as variations in population size and selection might explain deviation from the neutral hypothesis. HvCbf9 and α-Amy1 show a strong reduction of nucleotide diversity in cultivated accessions and deviate significantly from the neutral hypothesis. To bypass possible effects of population structure, Tajima’s D values have been re-calculated within NJ-clades to avoid the effect of population structure and to find evidence of selection for HvCbf9; a significant D value within the NJ-clade IV was evident. When the Tajima’s test was applied only in wild accession the D value was again significant (Table 3). Most probably the excess of rare variants of HvCbf9 in the wild gene pool affects the test, supporting a rapid population expansion rather than natural selection targeted to the gene. However, the second hypothesis was also considered and NJ-clade IV lines were phenotyped (Table S3). Unexpectedly, they were characterised by a large variation in FT, but genotypes with rare HvCbf9 variants did not show a high level of tolerance; thus most probably this gene does not affect the trait. As selection targeted at individual loci reduce genetic diversity within and around selected loci (a phenomenon known as genetic hitchhiking; Smith and Haigh 1974; Palaisa et al. 2004), the strong reduction of alleles of HvCbf9 in cultivated varieties could have been generated by selection targeted to tightly linked CBF genes.
It is generally assumed that breeding programmes act simultaneously upon more loci that control a variety of traits, and, similarly to domestication bottlenecks, they contribute to reduce drastically genetic diversity in modern crops. Being one of the most important crops in the world, barley has been considered by several breeding programmes. The data presented here demonstrate that the genetic erosion of the barley gene pools caused by the intense breeding is continuing and increases progressively with time (Fig. 3).
Fig. 3.

Haplotype diversity (Hd) of five genes involved in FT (HvCbf3, HvCbf6, HvCbf9, HvCbf14 and Vrn-H1) plus two reference genes (α-Amy1 and Gapdh) recorded in two subgroups of European cultivars released before 1980 a and after 1980 b, respectively. More recent cultivars showed a loss of Hd, for almost all genes
Changing the approaches to CBF analyses and QTL genetic dissections in plants (Francia et al. 2007; Knox et al. 2008), association mapping is applied for the first time to a panel of barley varieties to reveal significant associations of genetic variants detected within the candidate CBF genes. Barley, as well as other temperate grasses, shows a broad range of phenotypic variation for freezing tolerance and the phenotyping of selected germplasm for this complex trait provides a useful tool to study candidate genes involved in the freezing response. On the basis of phenotypic data, selected European cultivars have been sorted in five discrete classes, each of which identifies groups of accessions with comparable tolerance to freezing stress.
In the map generated from the cross between the varieties Morex and Dicktoo, HvCbf14, HvCbf6 and HvCbf3 co-segregate together, while HvCbf9 is located in a separate but genetically linked cluster of CBF genes (Skinner et al. 2005). At the physical level, HvCbf6 and HvCbf3, but not HvCbf14, have been identified in a single BAC. The physical association between HvCbf6 and HvCbf3 is reflected in the levels of LD higher than a significant threshold (Fig. 2). On the basis of these data, the four CBFs considered in this study should span the length of the CBF gene cluster. Subtle genetic differences in unrelated lines allow using the association mapping approach to search for genes that underlie one or more traits. The resolution of an association mapping procedure is affected by the level of linkage disequilibrium present in the region that contains the candidate genes (Rafalski 2002). Based on the analysis of r 2 values, we reported for the fist time the levels of pairwise associations among CBF variants at the Fr-H2 locus. The data demonstrate the low levels of r 2 among CBF genes of European cultivars and also that LD decay allows sufficient mapping resolution to locate the genetic determinant of freezing tolerance within the CBF cluster. In rice and in other plant species, homogenization events internal to the CBF gene cluster seem to be a key driving force guiding CBF evolution via gene conversion and crossing over (Wang et al. 2005; Pennycooke et al. 2008). The same mechanisms may contribute to the erosion of linkage disequilibrium as well as to gene deletions; similar events may have played a role in the evolution of barley CBF genes.
In a collection of barley cultivars, different levels of genetic similarity correlate with specific alleles of loci involved in the control of growth habit (Fig. 1). This introduces a bias due to population stratification and phenotypic homogenization which ultimately affects negatively the association procedure. To circumvent this effect, principal component data were inserted in a general linear model as covariates to account for population stratification in association analysis. The association analysis between genetic variants of CBFs and freezing tolerance gave positive results for two genes and revealed that, more than other CBF variants, two nucleotide substitutions of HvCbf14 are statistically associated with freezing tolerance. Both polymorphisms fall in the 3′UTR of HvCbf14 and bioinformatics analysis indicates that this region is devoid of cis-elements affecting CBF gene expression. Interestingly, a number of freezing-tolerant wild lines from the NJ-clade IV have the same variants of HvCbf14, supporting the effectiveness of the statistical procedure. It is not, however, established that these polymorphisms are the causative variant(s) of FT; the coding sequence and cis-regulatory elements at the 5′of HvCbf14 are also good candidates, as well as other physically linked genes (i.e. another gene also affecting FT in the CBF cluster in high LD with HvCbf14 due to a common history of selection for FT) and/or regulatory elements located in this region of the cluster. A deletion in the first intron of Vrn-H1 gene was pointed out as the functional genetic variant associated with spring growth habit in barley and diploid wheat and, consequently, able to affect frost tolerance in these species (Yan et al. 2004; Fu et al. 2005; von Zitzewitz et al. 2005; Szucs et al. 2007). Even though considering the functional genetic variant of Vrn-H1 gene as a fixed effect the power of the statistical model would have increased, other genetic variants in the 3′UTR of Vrn-H1 were instead searched and inserted as fixed effects in the equation model to verify their capability to evidence the well-known role of Vrn-H1 in frost tolerance. One of them, named VRN_1, was revealed to be associated with frost tolerance (Table 4) providing a further control of the association procedure effectiveness. On the other side, this statistical association raises the question of the LD of this gene as in the barley population examined VRN_1 must be in high LD with the functional genetic variant previously discovered. Several studies have investigated on the extent of the LD in barley and their conclusions support this speculation as the LD decay extends over the genes and well far away (Rostoks et al. 2006); a remarkable exception is the relatively low level of LD within the CBF locus described in this report. The comparison of resistant and susceptible varieties of bread wheat (Cheyenne and Chinese spring, respectively, Vágújfalvi et al. 2005) as well as of barley (Nure and Tremois, respectively Stockinger et al. 2007) indicated the CBF14 gene as one of the CBF sequences whose expression is associated with frost tolerance. Dissection of the QTL for FT in T. monococcum points to the CBF sub-cluster region carrying TmCBF12, TmCBF14 and TmCBF15 genes as the only one explaining the variation in frost tolerance in the two accessions compared (Knox et al. 2008). The comparison between our results and those from diploid wheat suggests that the conservation of this sub-cluster region is the most relevant for FT, and highlights that, despite the high degree of homology among the CBF members, they have different physiological roles and/or relevance in trait control. In A. thaliana evidence indicates that genes AtCbf1, AtCbf2 and AtCbf3 have different physiological roles (Novillo et al. 2004, 2007). The results of our work, indicating that only HvCBF14 is associated with FT in a large germplasm collection, suggest that there are some degrees of specificity among the different CBFs, and that HvCbf14 more than HvCbf3, HvCbf6 and HvCbf9 is relevant for FT. This finding makes unlikely that FT is merely due to a high amount of any CBF mRNAs, rather that the presence of specific CBF mRNAs is required to promote a high level of cold hardening.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by grants from MIUR (Ministero dell’Università e della Ricerca Scientifica) “Progetto Industriale” and “Progetto EXBARDIV” (RBER06MR84). The authors are grateful to Francesco Salamini for his critical reading of this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Badawi M, Danyluk J, Boucho B, Houde M, Sarhan F. The CBF gene family in hexaploid wheat and its relationship to the phylogenetic complexity of cereal CBFs. Mol Genet Genomics. 2007;277:533–554. doi: 10.1007/s00438-006-0206-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badr A, Müller K, Schäfer-Pregl R, El Rabey H, Effgen S, Ibrahim HH, Pozzi C, Rohde W, Salamini F. On the origin and domestication history of Barley (Hordeum vulgare) Mol Biol Evol. 2000;17:499–510. doi: 10.1093/oxfordjournals.molbev.a026330. [DOI] [PubMed] [Google Scholar]
- Båga M, Chodaparambil SV, Limin AE, Pecar M, Fowler DB, Chibbar RN. Identification of quantitative trait loci and associated candidate genes for low-temperature tolerance in cold-hardy winter wheat. Funct Integr Genomics. 2007;7:53–68. doi: 10.1007/s10142-006-0030-7. [DOI] [PubMed] [Google Scholar]
- Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- Breseghello F, Sorrells ME. Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics. 2006;172:1165–1177. doi: 10.1534/genetics.105.044586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundock PC, Henry RJ. Single nucleotide polymorphism, haplotype diversity and recombination in the Isa gene of barley. Theor Appl Genet. 2004;109:543–551. doi: 10.1007/s00122-004-1675-z. [DOI] [PubMed] [Google Scholar]
- Castiglioni P, Pozzi C, Heun M, Terzi V, Müller KJ, Rohde W, Salamini F. An AFLP-based procedure for the efficient mapping of mutations and DNA probes in barley. Genetics. 1998;149:2039–2056. doi: 10.1093/genetics/149.4.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet F. Multiple sequence alignment with hierarchical clustering. Nucl Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebley JF, Gaut BS, Smith BD. The molecular genetics of crop domestication. Cell. 2006;127:1309–1321. doi: 10.1016/j.cell.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Fowler S, Thomashow MF. Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell. 2002;14:1675–1690. doi: 10.1105/tpc.003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia E, Rizza F, Cattivelli L, Stanca AM, Galiba G, Tóth B, Hayes PM, Skinner JS, Pecchioni N. Two loci on chromosome 5H determine low-temperature tolerance in a ‘Nure’ (winter) × ‘Tremois’ (spring) barley map. Theor Appl Genet. 2004;108:670–680. doi: 10.1007/s00122-003-1468-9. [DOI] [PubMed] [Google Scholar]
- Francia E, Barabaschi D, Tondelli A, Laidò G, Rizza F, Stanca AM, Busconi M, Fogher C, Stockinger EJ, Pecchioni N. Fine mapping of a HvCBF gene cluster at the frost resistance locus Fr-H2 in barley. Theor Appl Genet. 2007;115:1083–1091. doi: 10.1007/s00122-007-0634-x. [DOI] [PubMed] [Google Scholar]
- Fu D, Szűcs P, Yan L, Helguera M, Skinner JS, von Zitzewitz J, Hayes PM, Dubcovsky J. Large deletions within the first intron in VRN-1 are associated with spring growth habit in barley and wheat. Mol Genet Genomics. 2005;273:54–65. doi: 10.1007/s00438-004-1095-4. [DOI] [PubMed] [Google Scholar]
- Garris AJ, McCouch SR, Kresovich S. Population structure and its effect on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryzasativa L.) Genetics. 2003;165:759–769. doi: 10.1093/genetics/165.2.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah MA, Wiese D, Freund S, Fiehn O, Heyer AG, Hincha DK. Natural genetic variation of freezing tolerance in Arabidopsis. Plant Physiol. 2006;142:98–112. doi: 10.1104/pp.106.081141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes P, Szucs P. Disequilibrium and association in barley: thinking outside the glass. Proc Natl Acad Sci USA. 2006;103:18385–18386. doi: 10.1073/pnas.0609405103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes PM, Blake T, Chen TH, Tragoonrung S, Chen F, Pan A, Liu B. Quantitative trait loci on barley (Hordeum vulgare L.) chromosome 7 associated with components of winterhardiness. Genome. 1993;36:66–71. doi: 10.1139/g93-009. [DOI] [PubMed] [Google Scholar]
- Hill WG, Robertson A. Linkage disequilibrium in finite populations. Theor Appl Genet. 1968;38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- Huang X. A contig assembly program based on sensitive detection of fragment overlaps. Genomics. 1992;14:18–25. doi: 10.1016/S0888-7543(05)80277-0. [DOI] [PubMed] [Google Scholar]
- Huson DH, Bryant H. Application of phylogenetic network in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- Jung M, Ching A, Bhattramakki D, Dolan M, Tingey S, Morgante M, Rafalski A. Linkage disequilibrium and sequence diversity in a 500-kbp region around the adh1 locus in elite maize germplasm. Theor Appl Genet. 2004;109:681–689. doi: 10.1007/s00122-004-1695-8. [DOI] [PubMed] [Google Scholar]
- Kilian B, Ozkan H, Kohl J, von Haeseler A, Barale F, Deusch O, Brandolini A, Yucel C, Martin W, Salamini F. Haplotype structure at seven barley genes: relevance to gene pool bottlenecks, phylogeny of ear type and site of barley domestication. Mol Genet Genomics. 2006;276:230–241. doi: 10.1007/s00438-006-0136-6. [DOI] [PubMed] [Google Scholar]
- Kim S, Plagnol V, Hu TT, Toomajian C, Clark RM, Ossowski S, Ecker JR, Weigel D, Nordborg M. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat Genet. 2007;39:1151–1155. doi: 10.1038/ng2115. [DOI] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge, Massachusetts: Cambridge University Press; 1983. [Google Scholar]
- Knox AK, Li C, Vágújfalvi A, Galiba G, Stockinger EJ, Dubcovsky J. Identification of candidate CBF genes for the frost tolerance locus Fr-Am2 in Triticummonococcum. Plant Mol Biol. 2008;67:257–270. doi: 10.1007/s11103-008-9316-6. [DOI] [PubMed] [Google Scholar]
- Kraakman AT, Niks RE, Van den Berg PM, Stam P, Van Eeuwijk FA. Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics. 2004;168:435–446. doi: 10.1534/genetics.104.026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird NM, Lange C. Family-based designs in the age of large-scale gene-association studies. Nat Rev Genet. 2006;7:385–394. doi: 10.1038/nrg1839. [DOI] [PubMed] [Google Scholar]
- Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 2004;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- Laurie DA, Pratchett N, Snape JW, Bezant JH. RFLP mapping of five major genes and eight quantitative trait loci controlling flowering time in a winter × spring barley (Hordeum vulgare L.) cross. Genome. 1995;38:575–585. doi: 10.1139/g95-074. [DOI] [PubMed] [Google Scholar]
- Levontin RC, Kojima K. The evolutionary dynamics of complex polymorphisms. Evol Int J org Evol. 1960;14:458–472. [Google Scholar]
- Miller AK, Galiba G, Dubcovsky J. A cluster of 11 CBF transcription factors is located at the frost tolerance locus Fr-Am2 in Triticum monococcum. Mol Genet Genomics. 2004;275:193–203. doi: 10.1007/s00438-005-0076-6. [DOI] [PubMed] [Google Scholar]
- Morrell PL, Toleno DM, Lundy KE, Clegg MT. Low levels of linkage disequilibrium in wild barley (Hordeum vulgare ssp. spontaneum) despite high rates of self-fertilization. Proc Natl Acad Sci USA. 2005;102:2442–2447. doi: 10.1073/pnas.0409804102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987. [Google Scholar]
- Nordborg M, Hu TT, Ishino Y, Jhaveri J, Toomajian C, Zheng H, Bakker E, Calabrese P, Gladstone J, Goyal R, Jakobsson M, Kim S, Morozov Y, Padhukasahasram B, Plagnol V, Rosenberg NA, Shah C, Wall JD, Wang J, Zhao K, Kalbfleisch T, Schulz V, Kreitman M, Bergelson J. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 2005;3:196–203. doi: 10.1371/journal.pbio.0030196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novillo F, Alonso JM, Ecker JR, Salinas J. CBF2/DREB1C is a negative regulator of CBF1/DREB1B and CBF3/DREB1A expression and plays a central role in stress tolerance in Arabidopsis. Proc Natl Acad Sci USA. 2004;101:3985–3990. doi: 10.1073/pnas.0303029101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novillo F, Medina J, Salinas J. ArabidopsisCBF1 and CBF3 have a different function than CBF2 in cold acclimation and define different gene classes in the CBF regulon. Proc Natl Acad Sci USA. 2007;104:21002–21007. doi: 10.1073/pnas.0705639105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SJ, Kwon CW, Choi DW, Song SI, Kim JK. Expression of barley HvCBF4 enhances tolerance to abiotic stress in transgenic rice. Plant Biotechnol J. 2007;5:646–656. doi: 10.1111/j.1467-7652.2007.00272.x. [DOI] [PubMed] [Google Scholar]
- Palaisa KA, Morgante M, Williams M, Rafalski A. Contrasting effects of selection on sequence diversity and linkage disequilibrium at two phytoene synthase loci. Plant Cell. 2003;15:1795–1806. doi: 10.1105/tpc.012526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaisa K, Morgante M, Tingey S, Rafalski A. Longrange patterns of diversity and linkage disequilibrium surrounding the maize Y1 gene are indicative of an asymmetric selective sweep. Proc Natl Acad Sci USA. 2004;101:9885–9890. doi: 10.1073/pnas.0307839101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennycooke JC, Cheng H, Roberts SM, Yang Q, Rhee SY, Stockinger EJ. The low temperature-responsive, SolanumCBF1 genes maintain high identity in their upstream regions in a genomic environment undergoing gene duplications, deletions, and rearrangements. Plant Mol Biol. 2008;67:483–497. doi: 10.1007/s11103-008-9333-5. [DOI] [PubMed] [Google Scholar]
- Rafalski A. Applications of single nucleotide polymorphisms in crop genetics. Curr Opin Plant Biol. 2002;5:94–100. doi: 10.1016/S1369-5266(02)00240-6. [DOI] [PubMed] [Google Scholar]
- Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA. 2001;98:11479–11484. doi: 10.1073/pnas.201394398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizza F, Pagani D, Stanca AM, Cattivelli L. Use of chlorophyll fluorescence to evaluate the cold acclimation and freezing tolerance of winter and spring oats. Plant Breeding. 2001;120:389–396. doi: 10.1046/j.1439-0523.2001.00635.x. [DOI] [Google Scholar]
- Rizza F, Pagani D, Enneking D, Jestin L, Jaiser H, Einfeldt CHP, Oeynhausen F, Stanca AM, Cattivelli L (2003) Evaluation of winter genotypes from the Barley Core Collection (BCC) for frost tolerance in field trials and laboratory experiments. In: Marè C, Faccioli P, Stanca AM (eds) Proceedings of the cereal section meeting. From biodiversity to genomics: breeding strategies for small grain cereal in the third millennium, pp 223–225
- Rostoks N, Ramsay L, MacKenzie K, Cardle L, Bhat PR, Roose ML, Svensson JT, Stein N, Varshney RK, Marshall DF, Graner A, Close TJ, Waugh R. Recent history of artificial outcrossing facilitates whole-genome association mapping in elite inbred crop varieties. Proc Natl Acad Sci USA. 2006;103:18656–18661. doi: 10.1073/pnas.0606133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics methods and protocols: methods in molecular biology. Totowa: Humana Press; 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- Russell J, Booth A, Fuller J, Harrower B, Hedley P, Machray G, Powell W. A comparison of sequence-based polymorphism and haplotype content in transcribed and anonymous regions of the barley genome. Genome. 2004;47:389–398. doi: 10.1139/g03-125. [DOI] [PubMed] [Google Scholar]
- Salamini F, Ozkan H, Brandolini A, Schäfer-Pregl R, Martin W. Genetics and geography of wild cereal domestication in the near east. Nat Rev Genet. 2002;3:429–441. doi: 10.1038/nrg817. [DOI] [PubMed] [Google Scholar]
- Shinozaki K, Yamaguchi-Shinozaki K. Molecular responses to dehydration and low temperature: differences and cross-talk between two stress signalling pathways. Curr Opin Plant Biol. 2000;3:217–223. [PubMed] [Google Scholar]
- Skinner JS, von Zitzewitz J, Szucs P, Marquez-Cedillo L, Filichkin T, Amundsen K, Stockinger EJ, Thomashow MF, Chen TH, Hayes PM. Structural, functional, and phylogenetic characterization of a large CBF gene family in barley. Plant Mol Biol. 2005;59:533–551. doi: 10.1007/s11103-005-2498-2. [DOI] [PubMed] [Google Scholar]
- Skinner JS, Szucs P, von Zitzewitz J, Marquez-Cedillo L, Filichkin T, Stockinger EJ, Thomashow MF, Chen TH, Hayes PM. Mapping of barley homologs to genes that regulate low temperature tolerance in Arabidopsis. Theor Appl Genet. 2006;112:832–842. doi: 10.1007/s00122-005-0185-y. [DOI] [PubMed] [Google Scholar]
- Smith MJ, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–25. doi: 10.1017/S0016672300014634. [DOI] [PubMed] [Google Scholar]
- Stockinger EJ, Gilmour SJ, Thomashow MF. Arabidopsis thalianaCBF1 encodes an AP2 domain-containing transcriptional activator that binds to the C-repeat/DRE, a cis-acting DNA regulatory element that stimulates transcription in response to low temperature and water deficit. Proc Natl Acad Sci USA. 1997;94:1035–1040. doi: 10.1073/pnas.94.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger EJ, Skinner JS, Gardner KG, Francia E, Pecchioni N. Expression levels of barley Cbf genes at the Frost resistance-H2 locus are dependent upon alleles at Fr-H1 and Fr-H2. Plant J. 2007;51:308–321. doi: 10.1111/j.1365-313X.2007.0141.x. [DOI] [PubMed] [Google Scholar]
- Szucs P, Skinner JS, Karsai I, Cuesta-Marcos A, Haggard KG, Corey AE, Chen THH, Hayes PM. Validation of the VRN-H2/VRN-H1 epistatic model in barley reveals that intron length variation in VRN-H1 may account for a continuum of vernalization sensitivity. Mol Genet Genomics. 2007;277:249–261. doi: 10.1007/s00438-006-0195-8. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takumi S, Shimamura C, Kobayashi F. Increased freezing tolerance through up-regulation of downstream genes via the wheat CBF gene in transgenic tobacco. Plant Physiol Biochem. 2008;46:205–211. doi: 10.1016/j.plaphy.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Templeton AR. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping or DNA sequencing. V. Analysis of case/control sampling designs: Alzheimer’s disease and the apoprotein E locus. Genetics. 1995;140(1):403–409. doi: 10.1093/genetics/140.1.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon MI, Sawkins MC, Long AD, Gaut RL, Doebley JF, Gaut BS. Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp. mays L.) Proc Natl Acad Sci USA. 2001;98:9161–9166. doi: 10.1073/pnas.151244298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon MI, U’Ren J, Tenaillon O, Gaut BS. Selection versus demography: a multilocus investigation of the domestication process in maize. Mol Biol Evol. 2004;21:1214–1225. doi: 10.1093/molbev/msh102. [DOI] [PubMed] [Google Scholar]
- Thomashow MF. So what’s new in the field of plant cold acclimation? Lots! Plant Physiol. 2001;125:89–93. doi: 10.1104/pp.125.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES. Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet. 2001;28:286–289. doi: 10.1038/90135. [DOI] [PubMed] [Google Scholar]
- Tommasini L, Schnurbusch T, Fossati D, Mascher F, Keller B. Association mapping of Stagonospora nodorum blotch resistance in modern European winter wheat varieties. Theor Appl Genet. 2007;115:697–708. doi: 10.1007/s00122-007-0601-6. [DOI] [PubMed] [Google Scholar]
- Vágújfalvi A, Aprile A, Miller A, Dubcovsky J, Delugu G, Galiba G, Cattivelli L. The expression of several Cbf genes at the Fr-A2 locus is linked to frost resistance in wheat. Mol Genet Genomics. 2005;274:506–514. doi: 10.1007/s00438-005-0047-y. [DOI] [PubMed] [Google Scholar]
- von Zitzewitz J, Szűcs P, Dubcovsky J, Yan L, Francia E, Pecchioni N, Casas A, Chen THH, Hayes PM, Skinner JS. Molecular and structural characterization of barley vernalization genes. Plant Mol Biol. 2005;59:449–467. doi: 10.1007/s11103-005-0351-2. [DOI] [PubMed] [Google Scholar]
- Vos P, Hogers R, Bleeker M, Reijans M, Van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 1995;23:4407–4414. doi: 10.1093/nar/23.21.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Triezenberg SJ, Thomashow MF, Stockinger EJ. Multiple hydrophobic motifs in Arabidopsis CBF1 COOH terminus provide functional redundancy in trans-activation. Plant Mol Biol. 2005;58:543–559. doi: 10.1007/s11103-005-6760-4. [DOI] [PubMed] [Google Scholar]
- Weiss KM, Clark AG. Linkage disequilibrium and the mapping of complex human traits. Trends Genet. 2002;18:19–24. doi: 10.1016/S0168-9525(01)02550-1. [DOI] [PubMed] [Google Scholar]
- Wright SI, Gaut BS. Molecular population genetics and the search for adaptive evolution in plants. Mol Biol Evol. 2004;22:506–519. doi: 10.1093/molbev/msi035. [DOI] [PubMed] [Google Scholar]
- Yan L, Loukoianov A, Blechl A, Tranquilli G, Ramakrishna W, SanMiguel PJ, Bennetzen JL, Echenique V, Dubcovsky J. The wheat Vrn2 gene is a flowering repressor down-regulated by vernalization. Science. 2004;303:1640–1644. doi: 10.1126/science.1094305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Buckler ES. Genetic association mapping and genome organization of maize. Curr Opin Biotechnol. 2006;17:155–160. doi: 10.1016/j.copbio.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Zhen Y, Ungerer MC. Clinal variation in freezing tolerance among natural accessions of A. thaliana. New Phytol. 2007;177:419–427. doi: 10.1111/j.1469-8137.2007.02262.x. [DOI] [PubMed] [Google Scholar]
- Zhen Y, Ungerer MC. Relaxed selection on the CBF/DREB1 regulatory genes and reduced freezing tolerance in the southern range of Arabidopsis thaliana. Mol Biol Evol. 2008;25:2547–2555. doi: 10.1093/molbev/msn196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.