

Summary
Background
Interleukin (IL-6) and transforming growth factor (TGF)-β have been shown to play a role in skin development and maintenance.
Objectives
A link between these two cytokines has yet to be identified and therefore in this study we investigated the modulation of TGF-β1 and TGF-β type 2 receptor (TGF-βR2) by IL-6 in skin.
Methods
An IL-6 knockout (IL-6KO) fibroblast-populated lattice model and intradermal injections of IL-6 into unwounded IL-6KO mice were used to investigate the direct effects of IL-6 treatment on TGF-β and TGF-βR2 expression and to determine the signalling mechanism. In addition, IL-6KO and C57BL/6 control mice were wounded by a 4-mm punch biopsy to monitor expression of TGF-β1 and TGF-βR2 within a wound over time. The expression of TGF-β1 and TGF-βR2 was assessed by real-time quantitative polymerase chain reaction, enzyme-linked immunosorbent assay and immunohistology.
Results
Recombinant IL-6 treatment of IL-6KO lattices and intradermal injections of IL-6 showed a significant induction of TGF-β1 mRNA and protein, with TGF-β1 expression localized in the dermis, while TGF-βR2 expression was primarily in the epidermis in IL-6KO mice. During healing, the expression of TGF-β1 and TGF-βR2 mRNA was significantly greater in unwounded and 7-day-old wounds from wild-type mice; however, protein expression did not differ. Treatment with signal transduction inhibitors indicated that IL-6 modulates TGF-β through a mitogen-activated protein kinase/extracellular signal-regulated kinase (Mapk/Erk)-dependent mechanism.
Conclusion
These studies indicate that IL-6 has the ability to modulate the expression of TGF-β and TGF-βR2 to varying degrees in the skin, which may provide a possible mechanism for defining the role of IL-6 in skin maintenance and a new association of IL-6 with TGF-β in pathologies associated with fibrosis.
Keywords: collagen, fibroblast, IL-6, inflammation, skin, TGF-β
More than 6 million people in the U.S.A. develop nonhealing wounds arising from various injuries including diabetic ulcers, venous stasis and pressure ulcers, and burns.1 These chronic, delayed-healing wounds can be a result of diabetes, circulatory problems and obesity to name a few. The care of chronic wounds can become a very labour-intensive and expensive process with the estimated cost being $3 billion each year.2 While the restoration or augmentation of cutaneous wound healing has long been an elusive goal for healthcare professionals, advancements have led to a better understanding of the mechanisms involved in repair. Throughout the repair process, there is interaction and cooperation among many diverse cell types, providing coordination of events so that they proceed in a well-organized fashion.
These events, when simplified, include coagulation, inflammation, formation of a provisional collagen matrix (granulation tissue), re-epithelialization and remodelling (review3). The transforming growth factor (TGF)-β family comprises more than 30 different members, including three TGF-β isoforms, four activins and more than 20 bone morphogenetic proteins. TGF-β has been shown to be involved in many diverse cellular processes such as proliferation, differentiation, migration, adhesion and apoptosis.4 The three TGF-β isoforms, β1, β2 and β3, have a high degree of sequence homology. TGF-β is secreted as a latent complex that must be proteolytically processed and released from the latency associated peptide (LAP) located at the N-terminus of the molecule. Proteolytic cleavage is carried out by various proteases, such as plasmin and thrombin, or through physical interaction of the LAP region with thrombospondin-1.5 Signalling by mature TGF-β is initiated through type I and type II transmembrane receptors. Each isoform can bind with different affinity directly to the constitutively active type II receptor, which then recruits and phosphorylates the type I receptor, thereby activating signal transduction through type I receptor kinases. Activated type I receptor kinases propagate the signal by phosphorylation of intracellular effector proteins called Smads.4,6,7 Of the three subclasses of Smads, receptor-regulated Smads (R-Smad) and common-partner Smads (Co-Smads) translocate to the nucleus and regulate the transcription of genes, while inhibitory Smads (I-Smads) antagonize the regulatory function of R-Smads by blocking their interactions with the type I receptor.8
The TGF-β family of proteins consists of multifunctional regulators that have been linked to nearly every aspect of wound repair, affecting all cell types involved. For example, TGF-β1 has been shown to act as a mitogen and chemoattractant for cells such as fibroblasts, to promote collagen deposition and to induce myofibroblast differentiation.9-11 Of the three TGF isoforms, TGF-β1 and β2 play a more prominent role in adult wound healing, whereas TGF-β3 is elevated in fetal wounds resulting in scarless repair.12 Treatment with exogenous TGF-β1 and TGF-β2 can improve the healing process by improving tensile strength and the rate of repair, which are related to an increase in collagen synthesis and other matrix proteins.13,14 TGF-β has also been shown to regulate re-epithelialization, angiogenesis and the deposition of the extracellular matrix.11 Despite its positive role in wound healing, TGF-β has been linked to fibrotic diseases such as hypertrophic scarring,11 chronic liver disease,15 cancer,16 autoimmune disease17 and diabetic retinopathy.18
Interleukin (IL)-6 is a pleiotropic cytokine that is involved in the growth and differentiation of numerous cell types, including those of dermal and epidermal origin.19 Epidermal keratinocytes are a primary producer of IL-6 within the skin, while macrophages, Langerhans cells and fibroblasts in the dermis represent other sources of the cytokine.20 Increased levels of IL-6 have been associated with a number of skin pathologies, such as psoriasis,21 scleroderma22 and systemic lupus erythematosus.23 Overexpression of IL-6 in the skin of normal rats induces epidermal proliferation and inflammation,24 while transgenic mice overexpressing IL-6 display little more than a thickened stratum corneum.25
While the involvement of IL-6 in inflammation and certain disease states has been well established, relatively little is known about its role in the skin. An inflammatory response following cutaneous wounding has been shown to be a pre-requisite for healing, and inflammatory cytokines such as IL-6 might be intimately involved in this process. Indeed, IL-6-deficient (knockout, IL-6KO) transgenic mice display significantly delayed cutaneous wound healing compared with control animals.26,27 Wounds from IL-6KO mice also displayed multiple defects, including delayed re-epithelialization, greatly decreased granulation tissue, inhibited neovascularization and delayed wound closure.
Although it is clear that TGF-β and IL-6 play important roles in the proper healing of cutaneous wounds, a link between these two cytokines in the skin remains to be identified. Studies have shown that the downregulation of IL-6 results in abrogation of the proliferative response of lung fibroblasts to mitogenic stimuli such as TGF-β1.28 However, other studies have reported that TGF-β1 upregulates the expression of IL-6 in human lung fibroblasts28 and pancreatic stellate cells.29 In this study, the direct relationship between IL-6 and TGF-β was explored using IL-6-deficient fibroblasts cultured in a collagen lattice and IL-6KO skin treated intradermally with IL-6. Whether or not TGF-β and its receptor are significantly modulated during wound healing in wild-type C57 and IL-6KO mice was also investigated. It was found that IL-6 plays a role in modulating the expression of this growth factor and its receptor in IL-6KO dermal fibroblasts in the skin. Furthermore, the IL-6-specific signal transduction pathways that might lead to TGF-β induction in IL-6KO fibroblasts were investigated, and it was found that IL-6-induced expression of this growth factor appears to be dependent on the extracellular signal-regulated kinase (Erk)1/2 pathway and displays crosstalk with the janus kinase/signal transducers and activator of transcription (Jak/Stat) pathway.
Materials and methods
In vivo wounding and intradermal injections
Experimental animals were treated in accordance with the criteria outlined in the Public Health Service Policy on Humane Care and Use of Laboratory Animals and the ‘Guide for the Care and Use of Laboratory Animals’30 in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. IL-6-deficient (C57BL/6J-IL6tmKopf) or control (C57BL/6J) mice were wounded as previously described.26 Briefly, control or IL-6KO mice (Jackson Laboratories, Bar Harbor, ME, U.S.A.) weighing 22–28 g and approximately 8–12 weeks old were housed in polycarbonate cages containing hardwood chip bedding at room temperature (21 ± 2 °C) on a 12-h light/dark cycle. Mice were anaesthetized by intraperitoneal injection with 80 mg kg−1 ketamine/10 mg kg−1 xylazine (Vedco, St Joseph, MO, U.S.A.), and the dorsal skin was shaved and swabbed with povidone–iodine (Betadine®; VWR, West Chester, PA, U.S.A.) and 70% ethanol three times prior to the procedure. For wounding, 4-mm punch biopsies were performed on the shaved area. At various time points post-wounding, a 2–3 mm border of tissue around the wound site was excised and placed in Tri-Reagent (Molecular Resource Center Inc., Cincinnati, OH, U.S.A.) for RNA, RIPA buffer (50 nmol L−1 Tris–HCl, 150 nmol L−1 NaCl, 1 mmol L−1 ethylenediaminetetraacetic acid, 1% Triton, 1% sodium deoxycholate, 0·1% sodium dodecyl sulphate) with phenylmethylsulphonyl fluoride (PMSF; Sigma, St Louis, MO, U.S.A.) and protease inhibitors (cat. no. P8340; Sigma) for total protein isolation or 10% buffered formalin for histology. Unwounded skin from IL-6KO and C57BL/6 mice was taken and processed as above to serve as the day 0 control. For intradermal injections, mice were anaesthetized and shaved as described. Various concentrations of IL-6 (10 μL) and a saline control were injected just under the epidermal layer of skin of unwounded IL-6KO mice using a Hamilton microsyringe and a 30-gauge needle. Each injection site was excised with a 4-mm punch biopsy after 2 h for RNA or 18 h for protein and placed in Tri-Reagent or 10% buffered formalin, respectively.
Dermal fibroblast isolation and culture
IL-6KO dermis was collected and separated from the epidermis as described previously.31 Briefly, newborn mouse pups (0–48 h old) were euthanized by decapitation, rinsed in 70% ethanol, and the skin was removed. Skins were incubated dermis down in culture dishes containing Hank’s balanced saline solution + 3·6% dispase (Sigma) overnight at 4 °C. The dermis was separated from the epidermis and placed in a collagenase solution [1 mg mL−1 in phosphate-buffered saline (PBS)], incubated at 37 °C, vortexed and sterile filtered. Cells were adjusted to a concentration of 5 × 106 mL−1 and frozen in CryoStore media (VWR). Fibroblasts were resuspended in Dulbecco’s modified Eagle’s medium (DMEM) + 5% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, U.S.A.), plated at 8 × 103 cells cm−2 in 100-mm culture dishes and incubated at 37 °C, and 5% CO2 and 95% room air. The primary IL-6KO fibroblasts were frozen in 90% culture medium + 10% dimethyl sulphoxide (DMSO) or CryoStor CS-10 (VWR) until further culture.
Floating collagen lattice preparation
Free-floating type I collagen (Upstate Biotechnology Inc., Lake Placid, NY, U.S.A.) lattices were prepared essentially as previously described.32 IL-6KO fibroblasts were cultured in type I collagen with a final collagen concentration of 1·25 mg mL−1 and a cell density of 4·50 × 105 cells mL−1. Two millilitres of the collagen cell suspension was placed in the wells of a six-well tissue culture plate (BD, Bedford, MA, U.S.A.). After a 1-h polymerization at 37 °C, the lattices were freed from the walls of the well with a pipette tip, and 4 mL of 5 mmol L−1 glucose DMEM + 5% FBS was placed over the free-floating collagen lattice. The IL-6KO fibroblast-populated lattices were cultured at 37 °C and 5% CO2 and 95% room air for various numbers of days prior to treatment with recombinant mouse (rm) IL-6 (PeproTech, Rocky Hill, NJ, U.S.A.). Prior to treatment, the lattices were washed once with PBS, and the culture medium was replaced with serum-free DMEM (Invitrogen) containing 1 mg mL−1 bovine serum albumin, 60 IU mL−1 penicillin and 100 IU mL−1 streptomycin. Following a 1-h incubation in serum-free medium, treatments were applied to the cultures and incubated for 2 h for RNA. For protein, lattices were washed once with PBS, and the culture medium was replaced with serum-free DMEM and incubated for 1 h. Following a 1-h incubation, the lattices were treated with varying concentrations of IL-6 and incubated overnight. Treated IL-6KO lattices were placed in Tri-Reagent for RNA, RIPA buffer with PMSF and protease inhibitors for total protein isolation or in 10% buffered formalin for histology. Lattice culture supernatants were also collected after the overnight IL-6 treatment incubation and stored at −20 °C. Collagen lattice and supernatant total protein concentrations were measured using bicinchonic (Pierce, Rockford, IL, U.S.A.) or Bradford (BioRad, Hercules, CA, U.S.A.) assays, respectively.
Inhibitors
IL-6KO fibroblast free-floating collagen lattices were prepared as described. The lattices were cultured as described previously. Prior to treatment with inhibitors, the lattices were washed once in PBS, and the culture medium was replaced with 4 mL of serum-free medium for 1 h. The inhibitors used were LLL-3 (a gift from Dr Pui-Kai Li, Ohio State University, Columbus, OH, U.S.A.) and PD98059 (EMD Biosciences, San Diego, CA, U.S.A.). Inhibitors were used at a concentration of 20 and 30 μmol L−1 for LLL-333 and PD98059, respectively. One microlitre of each inhibitor or DMSO (vehicle) was added to the respective culture wells. The lattices were incubated for 30 min, and various concentrations of IL-6 were added to the lattices for a 2-h incubation. Following treatment the lattices were placed in Tri-Reagent.
Histology
Formalin-fixed paraffin-embedded sections of intradermal injection samples were dewaxed, rehydrated and incubated with anti-TGF-β1 (R&D Systems, Minneapolis, MN, U.S.A.) and TGF-βR2 (Santa Cruz, Santa Cruz, CA, U.S.A.) essentially as previously described.34 Blocking was accomplished with the Mouse on Mouse kit (Vector Labs, Burlingame, CA, U.S.A.) and nuclei were stained with 4,6-diamino-2-phenylindole (Sigma). Secondary antibodies conjugated to Alexa 546 were obtained from Rockland Immunochemical (Gilbertsville, PA, U.S.A.). Slides were washed and coverslipped, and staining was visualized by fluorescent microscopy using a Leica DM400B microscope.
Real-time quantitative polymerase chain reaction
Total RNA from mouse skin or dermal IL-6KO fibroblasts was prepared and cDNA synthesized as described previously.31 Primers for mouse genes were synthesized by Invitrogen. Quantitative polymerase chain reaction (PCR) analysis was performed on an ABI PRISM 7000 SDS, utilizing SYBR Green Mastermix (VWR) according to the manufacturer’s instructions. Quantitative values of genes of interest were normalized based on 28S rRNA content using the ΔΔCt method.35
Enzyme-linked immunosorbent assay analysis
Total TGF-β1 protein from tissue, lattice and lattice culture supernatant was measured using a sandwich ELISA (eBioscience, San Diego, CA, U.S.A.). The ELISA was performed according to the manufacturer’s instructions and analysed using a 96-well plate reader (Molecular Devices, Sunnyvale, CA, U.S.A.).
Statistical analysis
All experiments were replicated, and representative findings are shown. Statistical significance was determined by one-way ANOVA. When the F value was significant, the means were compared using Fisher’s post-hoc analysis. In all statistical comparisons, a P-value of < 0·05 was used to indicate a significant difference.
Results
Transforming growth factor-β expression in fibroblast-populated lattices
To investigate whether or not IL-6 modulates TGF-β expression, IL-6KO dermal fibroblasts were cultured in a type I collagen lattice, which serves as a useful model of unwounded dermis.36 Lattices were cultured for 5 days and treated with various concentrations of rmIL-6 (0–20 ng mL−1) for 2 h. Total RNA was isolated, and gene expression was assessed by quantitative PCR. As seen in Figure 1, TGF-β1 mRNA expression increased significantly up to fivefold in a dose–response manner with IL-6 treatment. However, IL-6 did not significantly induce TGF-βR2 mRNA expression in the lattices (Fig. 1). To confirm whether or not the observed induction of mRNA correlates with induction of TGF-β1 protein, IL-6KO fibroblast-populated lattices were cultured for 5 days, then treated with various concentrations of rmIL-6 (0–50 ng mL−1) overnight, and total protein was extracted from the lattice and culture supernatant. TGF-β1 protein in fibroblast-populated lattices and supernatant was determined by ELISA. Within the lattice, a significant dose-dependent increase in TGF-β1 protein levels up to twofold was shown in response to IL-6 treatment (Fig. 2). However, while IL-6 upregulated TGF-β1 protein in lattices, the culture supernatant did not show a similar increase (Fig. 2).
Fig 1.
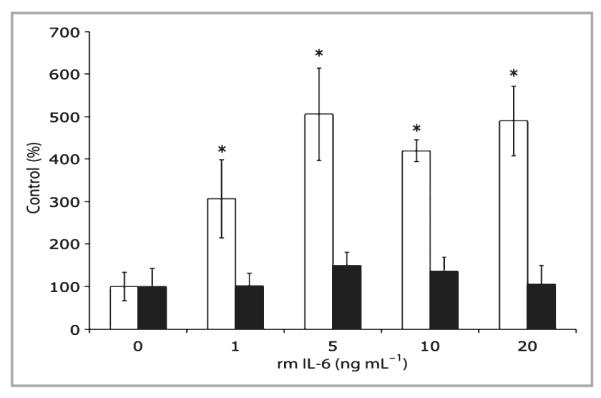
Interleukin (IL)-6 modulates transforming growth factor (TGF)-β in IL-6 knockout (IL-6KO) fibroblast-populated lattices. Floating collagen matrices were prepared using primary dermal fibroblasts from IL-6KO mice as described. The growth medium was replaced and cultures were treated with various concentrations of recombinant mouse (rm) IL-6 for 2 h. Total cellular RNA was prepared as described. Expression of mRNA was determined by real-time polymerase chain reaction utilizing an ABI 7000 SDS system, and accompanying software. Expression differences were determined relative to 28S rRNA utilizing the ΔΔCt method. Data are expressed as mean ± SD (n = 3 experiments per data point). *Significantly different from 0 ng mL−1 rmIL-6, P ≤ 0·0001; white bar, TGF-β1; black bar, TGF-β type 2 receptor.
Fig 2.
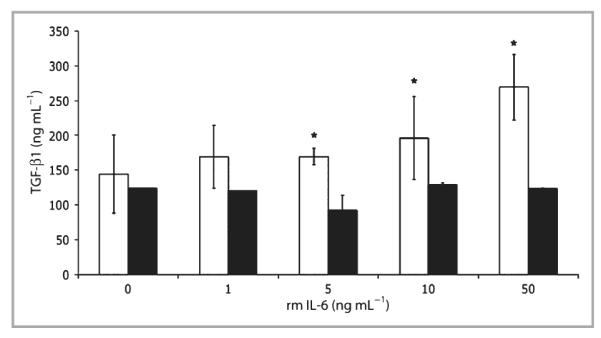
Transforming growth factor (TGF)-β1 protein expression in interleukin (IL)-6 knockout (KO) fibroblast-populated lattices. Floating collagen matrices were prepared using primary dermal fibroblasts from IL-6KO mice as described. The growth medium was replaced and cultures were treated with various concentrations of recombinant mouse (rm) IL-6 overnight. Total protein was prepared, and protein expression was determined with an enzyme-linked immunosorbent assay as per the manufacturer’s instructions (eBioscience). Data are expressed as mean ± SD (n = 9 lattices). *Significantly different from 0 ng mL−1 rmIL-6, P ≤ 0·05; white bar, lattice; black bar, supernatant.
Induction of transforming growth factor-β expression by intradermal injection of recombinant mouse interleukin-6
To investigate if local administration of IL-6 could directly influence TGF-β mRNA expression levels in vivo, various concentrations of IL-6 and a saline control (10 μL) were injected intradermally into unwounded IL-6KO mouse skin. Whole skin samples of injection sites were collected, total skin RNA was isolated and mRNA expression was assessed by quantitative PCR. TGF-β1 mRNA expression levels increased in a dose–response manner, with significantly increased expression (nearly twofold) at 100 ng mL−1 rmIL-6 (Fig. 3). TGF-βR2 mRNA expression was significantly increased (P < 0·03) in response to 10 ng mL−1 rmIL-6 but was not different from the control with 100 ng mL−1 of cytokine (Fig. 3). To determine the distribution of IL-6-induced TGF-β protein, intradermally injected IL-6-treated skin from IL-KO mice was collected 24 h post-treatment and placed in formalin for histological sectioning, and protein expression was assessed by immunohistology. TGF-β1 expression appeared to increase in response to high doses (100 ng mL−1) of rmIL-6 24 h after intradermal injection. TGF-β1 expression was localized essentially in the extracellular matrix, the hair follicles and in the epidermal region (Fig. 4a,b). However, TGF-βR2 did not show an appreciable induction in the dermis, but did appear to be upregulated in the epidermis (Fig. 4c,d).
Fig 3.
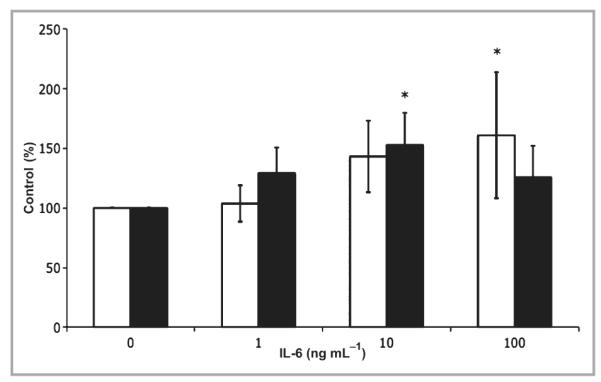
Intradermal interleukin (IL)-6 treatment modulates transforming growth factor TGF-β in IL-6 knockout (KO) mice. IL-6KO or C57BL/6 mice were injected with 10 μL of recombinant mouse (rm) IL-6 at various concentrations, and mRNA was extracted from the tissue as described. Expression of TGF-β1 and TGF-β type 2 receptor (TGF-βR2) mRNA was determined by real-time polymerase chain reaction utilizing an ABI 7000 SDS system and accompanying software. Expression differences were determined relative to 28S rRNA utilizing the ΔΔCt method. Data are expressed as mean ± SE (n = 6 animals per data point). *Different from 0 ng mL−1 rmIL-6, P < 0·05; white bar, TGF-β1; black bar, TGF-βR2.
Fig 4.

Immunohistology of transforming growth factor (TGF)-β1 and TGF-β type 2 receptor (TGF-βR2) in intradermal injection skin samples. Interleukin (IL)-6 knockout (KO) mice were injected with 10 μL of 0 ng mL−1 (a, c) or 100 ng mL−1 (b, d) recombinant mouse (rm) IL-6, and mRNA was extracted from the tissue as described. Formalin-fixed, paraffin-embedded tissue sections of treated skin samples were incubated with anti-TGF-β1 (a and b), anti-TGF-βR2 (c and d) antibodies, and detected using Alexflor 546 conjugated secondary antibodies. Stained slides were visualized under fluorescence using a Leica DM400B microscope and 20 × objective.
Interleukin (IL)-6 modulation of transforming growth factor-β in IL-6 knockout and C57BL/6 wounds
It was previously shown that IL-6KO mice display decreased granulation tissue and collagen deposition,37 processes which could be associated with TGF-β expression. Therefore, to investigate further if TGF-β expression is modulated by IL-6, IL-6KO and C57BL/6 control mice were wounded by full-thickness punch biopsy as described.38 Wounds were excised after 1, 3, 5, 7 and 9 days, and total skin RNA was isolated from the wounds. Unwounded skin was collected from IL-6KO and C57BL/6 mice to serve as the day 0 control. Figure 5(a,b) shows that TGF-β1 and TGF-βR2 mRNA expression was significantly higher at day 0 in C57BL/6 mice than in IL-6KO mice, but expression decreased following wounding in both C57BL/6 and IL-6KO mice. Both TGF-β1 and TGF-βR2 mRNA expression increased in wounded skin as healing progressed in both types of mice (Fig. 5a,b). While nonsignificant (P = 0·11), a trend can be observed at 1, 3 and 5 days post-wounding when both TGF-β1 and TGF-βR2 were expressed at higher levels in C57BL/6 mice compared with IL-6KO mice. As healing progressed, at 7 days post-wounding, TGF-β1 mRNA expression levels were significantly higher in C57BL/6 mice compared with IL-6KO mice but not significantly different at day 9 (P = 0·08), while TGF-βR2 mRNA expression levels were significantly higher in the KO mice. TGF-β1 protein levels were also assessed and, as seen in Figure 5(c), unwounded skin did not express TGF-β1 at detectable levels. However, by 1, 3 and 5 days post-wounding TGF-β1 protein expression levels increased significantly with peak expression by day 3, and were not significantly different between wild-type and IL-6KO mice. Interestingly, by days 7 and 9 post-wounding, TGF-β protein expression decreased to below the detection limit in both wild-type and IL-6KO mice (Fig. 5c).
Fig 5.

Wound transforming growth factor (TGF)-β mRNA and protein expression levels in interleukin (IL)-6 knockout (KO) vs. C57BL/6 mice. IL-6KO or C57BL/6 mice were wounded, and mRNA was extracted from the wound tissue as described. Expression of TGF-β mRNA was determined by real-time polymerase chain reaction utilizing an ABI 7000 SDS system and accompanying software. (a) TGF-β1. (b) TGF-β type 2 receptor. Expression differences were determined relative to 28S rRNA utilizing the ΔΔCt method. (c) Protein expression was determined with an enzyme-linked immunosorbent assay as per the manufacturer’s instructions. Data are expressed as mean ± SD (n = 6 animals per data point). *Significantly different from unwounded C57BL/6 mice, P ≤ 0·013; white bar, C57BL/6; black bar, IL-6KO.
Interleukin-6 receptor signal transduction inhibitors alter transforming growth factor-β1 expression
IL-6 receptor (IL-6R) signals primarily through two specific pathways, that of Jak/Stat and mitogen-activated protein kinase (Mapk)/Erk. To investigate a possible mechanism of how IL-6 modulates TGF-β1 expression, IL-6KO fibroblast-populated lattices were treated with IL-6R pathway-specific signal transduction inhibitors 30 min prior to being treated with various concentrations of rmIL-6. Fibroblast-populated lattices (5 days in culture) were treated with specific inhibitors for Jak/Stat (LLL-3)33 or Mapk/Erk (PD98059). Treatment with LLL-3 resulted in a robust dose-dependent increase in TGF-β1 mRNA expression in KO fibroblasts (Fig. 6) similar to that shown in Figure 1. Interestingly, Erk inhibition in the absence of IL-6 treatment (0 ng mL−1) appeared to increase TGF-β1 (approximately threefold), while subsequent rmIL-6 treatment resulted in a profound dose-dependent decrease in TGF-β1 mRNA expression (Fig. 6).
Fig 6.

Inhibition of the Erk1/2 signal transduction pathway affects transforming growth factor (TGF)-β1 expression. Floating collagen matrices were prepared using primary dermal fibroblasts from interleukin (IL)-6 knockout (KO) mice as described. The growth medium was replaced and cultures were treated with various concentration of LLL-3 (Stat3 inhibitor), PD98059 (Erk inhibitor) or vehicle [0·1% dimethyl sulphoxide (DMSO) in culture media] 30 min prior to recombinant mouse (rm) IL-6 treatment as described. Expression of mRNA was determined by real-time polymerase chain reaction utilizing an ABI 7000 SDS system and accompanying software. Expression differences were determined relative to 28S rRNA utilizing the ΔΔCt method. Data are expressed as mean ± SE (n = 6 lattices per data point). White bar, DMSO; grey bar, PD98059; black bar, LLL-3.
Discussion
The regeneration of dermal and epidermal tissue is a complex process that involves coordination of the activity of many cell types and follows a sequence of events that occurs in a fairly predictable fashion. These events can be categorized into inflammation, re-epithelialization, formation of granulation tissue (provisional matrix), remodelling of the newly formed tissue and wound contraction. Each phase of wound healing is regulated by numerous growth factors and cytokines, including tumour necrosis factor-α, IL-1β, IL-6 and TGF-β.39,40 IL-6 in particular plays an important role in healing and is essential for repair to proceed properly.26,27 The IL-6KO mouse displays numerous defects in skin repair, including delayed re-epithelialization, decreased granulation tissue formation and decreased wound contraction. As the goal of these experiments was to investigate the ability of IL-6 to modulate TGF-β expression in the skin, the IL-6KO mouse model was utilized. While exon 3 (receptor-binding portion) of the gene has been deleted in the KO mouse, it still expresses functional receptor. This model is very useful as endogenous IL-6 produced from normal mice could confound data by possibly activating feedback inhibitory mechanisms.41
TGF-β is also well known as an important component of the wound healing process. Of the three TGF-β isoforms, TGF-β1 seems to have the broadest spectrum of action. For instance, following injury TGF-β1 is released from platelets and can induce chemotaxis of the inflammatory cells into the wound bed.42 During and after inflammation, TGF-β1 has also been shown to induce both angiogenesis and extracellular matrix formation and is essential for initiating granulation tissue formation.42,43 The formation of the granulation tissue in part results from the effects of TGF-β1 on fibroblasts, which primarily make up the granulation tissue and are responsible for a myriad of functions, including matrix deposition and wound contraction (review44). The primary structural component of granulation tissue is collagen, which can be induced directly by TGF-β1.45-47 TGF-β1 can stimulate wound contraction by inducing fibroblasts to differentiate to myofibroblasts.10,47 These contractile cells express proteins such as α-smooth muscle actin (SMA), stress fibres, vinculin-containing adhesion complexes and fibronectin fibrils.10 These proteins promote the contractile force needed for wound contraction.10 Interestingly, α-SMA, which is known to be controlled by TGF-β148 also appears to be modulated by IL-6 as well.49
Previous studies concerning the relationship between IL-6 and TGF-β have not provided a definitive link between the mediators. Zhang et al.50 found that TGF-β1 signalling activity was significantly greater in renal proximal tubular epithelial cells in the presence of IL-6 than that induced by TGF-β1 alone. Conversely, other studies showed that TGF-β1 has the ability to decrease IL-6 and the expression of other cytokines.51-53 The data presented herein indicate that IL-6 specifically modulates the expression of TGF-β in the skin and IL-6KO fibroblasts.
To begin these investigations, IL-6KO fibroblast-populated collagen lattices were used to model granulation tissue and give insight into what might occur within the unwounded dermis. As mentioned, the dermis consists predominantly of fibroblasts and collagen. With increasing levels of IL-6, a profound increase in TGF-β1 mRNA expression levels was seen within the lattice (Fig. 1), while TGF-βR2 mRNA expression was not found at detectable levels with any concentration of IL-6. Intradermal injections of IL-6 into IL-6KO skin also resulted in a significant increase in TGF-β1 as well as TGF-βR2 mRNA (Fig. 3). The disparity of TGF-βR2 expression may be explained by the presence of epidermis in these whole skin intradermal samples. Indeed, each TGF-β isoform is well known to have specific spatial and temporal patterns of expression.13,42
Following injury, distinct expression patterns of TGF-β and its receptors form, corresponding with each phase of wound repair.13 Immunohistology of unwounded IL-6KO mouse skin treated with intradermal IL-6 injections indicated a marked increase in TGF-β1 mRNA expression with high concentrations of IL-6 (~100 ng mL−1) compared with the control samples (Fig. 4a,b). TGF-β1 protein staining was found in the epidermal, dermal and extracellular matrix regions. TGF-β is secreted as inactive ‘latent’ TGF-β that can be sequestered by the extracellular matrix, which probably accounts for the matrix staining seen in Figure 454 and protein expression in the collagen lattice (Fig. 2). These results are similar to earlier findings by Frank et al.,55 who found that in unwounded skin, TGF-β1 was expressed mainly in the dermis (and, to a lesser extent, the epidermis). Unlike TGF-β1, TGF-βR2 protein expression showed relatively little dermal staining with any concentration of IL-6 injected intradermally; however, staining was apparent in the epidermal regions of the skin (Fig. 4c,d). Indeed, TGF-βR2 expression has not been shown in human fibroblasts or endothelial cells in human unwounded skin, while it is upregulated 7–10 days postinjury in fibroblast and connective tissue cells.42 The present intradermal experiments were only acute exposures to IL-6, thus no significant increase in TGF-βR2 expression might be expected following such short-term treatment with IL-6.
To investigate IL-6 modulation of TGF-β expression in a wound, IL-6KO and C57BL/6 mice were wounded by punch biopsy, and mRNA expression levels of TGF-β1 and TGF-βR2 and TGF-β1 protein expression were analysed. Unwounded control mice (day 0) displayed significantly higher expression of TGF-β1 and TGF-βR2 mRNA compared with the IL-6KO mice. Yet 1 day post-wounding, this robust expression decreased similarly between mouse types (Fig. 5). As previously mentioned, TGF-β induces many processes such as proliferation, migration, differentiation and collagen production to facilitate the wound healing process.39 The decrease in expression observed herein may be indicative of a protective effect in which the premature differentiation of fibroblasts or deposition collagen induced by TGF-β is initially blocked to inhibit premature fibroblast differentiation and possibly collagen synthesis. TGF-β1 protein expression was also analysed in unwounded and wounded skin. While TGF-β1 mRNA expression was slightly higher in the control mice, the opposite was shown at the protein level with expression being higher in the IL-6KO mice post-wounding. While paradoxical, these observations do not appear to be novel for this gene. Kehrl et al.56 demonstrated that upon B- and T-lymphocyte activation, mRNA was significantly increased for 2 h following stimulation, whereas the rate of secreted TGF-β1 protein was only gradually increased over a period of 4 days. It has also been reported that human prostrate cancer cells secrete TGF-β2 predominantly with little TGF-β1 being secreted, whereas the TGF-β1 mRNA is higher than TGF-β2 expression.57 Treatment of keratinocytes with retinoic acid showed increased TGF-β1 transcription, but there was no accompanying increase in TGF-β1 protein expression.58 These studies seem to point toward post-transcriptional modulation of TGF-β1. Indeed, TGF-β1 along with β2 and β3 all share the properties of a 5′-untranslated region (UTR) and high GC, both of which allow TGF-β to undergo modifications.59 Long 5′-UTRs and mRNA with a high GC content have been shown to undergo post-transcriptional modification; however, the exact mechanism for how these sequences play a role in the further modification of the mRNA has yet to be elucidated.59 Also of note, as shown in Figure 5(a), mRNA expression of TGF-β1 at 7 and 9 days post-wounding was significantly higher in C57 mice compared with KO mice. However, there was little if any TGF-β1 protein expression detected in the wound at these time points post-wounding. Aside from post-transcriptional modification, there is the possibility that TGF-β1 could have been cleaved from the matrix by a protease such as matrix metalloproteinase, which is abundant in the wound, rendering it soluble. As the method employed examined primarily TGF bound to collagen in the dermis, diffusion may have affected the apparent concentration of the cytokine.
To understand further the mechanism of IL-6 induction of TGF-β1 expression, IL-6R signal transduction pathways were manipulated utilizing specific inhibitors in an IL-6-deficient fibroblast-populated lattice model (Fig. 6). The IL-6R signals through three pathways: Jak/Stat, Mapk/Erk and Akt/PkB (protein kinase B). Previous studies by our laboratory have determined that dermal fibroblasts appear to signal primarily through the Erk pathway with no apparent Stat3 or Akt activation.49 Therefore, the primary focus of these experiments was on Jak/Stat and Mapk/Erk due to extensive studies that show these two pathways to be commonly induced by IL-6.41 In order to block Stat3 DNA binding, a novel Stat3 inhibitor LLL-333 was utilized which inhibits the dimerization of the Stat proteins. Following treatment with LLL-3, IL-6-induced TGF-β1 mRNA expression showed a dose-dependent increase similar to that seen in Figure 2.
Conversely, when lattices were treated with the Mapk/Erk-specific inhibitor PD98059, IL-6 induced a dose-dependent decrease in TGF-β1 mRNA expression with increasing concentrations of IL-6. While no enzyme inhibitor possesses completely specific activity, PD98059 is known to have little if any cross-reactivity with Akt or Jak.60 Therefore, this seems to indicate that the IL-6R signals through the Mapk/Erk pathway to induce TGF-β1 mRNA expression in dermal fibroblasts. These results are similar to a recent study in which rat glioma cells were stimulated with basic fibroblast growth factor, TGF-β1 was induced, and this induction was sensitive to Mapk/Erk-specific inhibitors.61 Together, these findings suggest the importance of the Mapk/Erk signalling pathway in IL-6-induced TGF-β1 expression. A complex interaction exists between the Erk and Stat pathways associated with IL-6R signalling which has been termed the ‘signal orchestration model’.62 This model may explain the very pleiotropic nature of IL-6 and suggests that the overall balance of distinct signals could determine the final biological outputs by a ligand where the ablation of one signalling cascade could influence the other, such as with Jak/Stat and Mapk/Erk.62 Numerous studies have detailed the interactions between these two cytokine-induced signalling pathways such as those by David et al.63 and Chung et al.64 that suggest a functional or physical interaction between the two pathways that probably results in cross-phosphorylation of these molecules. While the essential role for tyrosine phosphorylation of Stat3 for dimerization, translocation and DNA binding is well known, past investigations have also shown the importance of serine phosphorylation, such as by Erk, in regulating the activities of Stat1 and Stat3.65-67 The data presented herein appear to support the ‘signal orchestration model’, as well as studies that indicate that Erk activity may be inhibitory toward Stat activation.64,67,68 For instance, at an rmIL-6 dose of 0 ng mL−1, Erk blockade induces TGF-β1 expression, while higher concentrations of the cytokine decrease expression in a dose-dependent manner (Fig. 6). This seems to indicate that Stat activation as a result of IL-6 stimulation, in the absence of Erk activity, inhibits IL-6-induced TGF-β1 expression. Interestingly, inhibition of Erk in the absence of IL-6 treatment, causes TGF-β expression to increase compared with that of the DMSO control (Fig. 6, grey bars). While the specific mechanism of this induction is unknown, it could be that an IL-6-independent TGF-β1-inducing pathway in this cell type is inhibited by basal Erk activity. Erk/Mapk activity has been shown to have varying affects on gene expression depending on the cell type and conditions.69-73 Therefore, low-level basal Erk activity may inhibit TGF-β expression to ensure that detrimental effects, such as persistent collagen expression or hyperkeratosis, do not occur in normal skin. However, once IL-6 expression is increased, as perhaps a result of barrier disruption, high levels of Erk/Mapk activation can paradoxically induce TGF-β expression. Additionally, DMSO alone (Fig. 6, white bars) seemed to blunt IL-6-induced TGF-β expression somewhat compared with that shown in Figure 2. Again the mechanism of this phenomenon is not known, but could be associated with the antioxidant properties of DMSO, as Erk is known to be inhibited by antioxidants.74-76
It is tempting to speculate that decreased wound healing in IL-6KO mice is associated with altered TGF-β1 expression; however, this is most probably not the case. Indeed, IL-6KO mice present with decreased re-epithelialization, decreased keratinocyte proliferation and differentiation, deceased collagen deposition and granulation tissue formation and delayed wound contraction,26,27,38 all of which can be linked with TGF-β expression (review39), yet Figure 5(c) indicates that the differences in TGF-β protein concentration were not found in the wound. In the absence of the presented in vitro and intradermal injection data, these wound data may lead the reader to believe that IL-6 does not modulate TGF-β in the skin. However, this result is not surprising as it is well known that the major source for TGF-β early in healing appears to be degranulating platelets,77 which produce cytokine levels that probably greatly exceed those produced by the dermal cells. Thus, it is likely that the very high levels of TGF-β protein that were detected in wounds (Fig. 5c) were produced by sources other than dermal or epidermal cells. Indeed, IL-6KO mice have not been found to have deficiencies in platelet-produced TGF-β. Thus, what this work has shown is that the wound-healing deficit in IL-6KO mice is most probably not related to the ability of this mouse strain to produce TGF-β, which seems to be modulated in the wound by numerous redundant processes.
Instead of healing, IL-6 may modulate skin TGF-β in the absence of the influence of platelets, in situations such as regulation of epidermal barrier homeostasis as indicated by Wang et al.78 or perhaps skin development. It is important to note that the dermis, keratinocyte layer and stratum corneum are considerably thinner in the IL-6KO mouse compared with the wild-type,38 all of which can also be linked with TGF-β function (review39). While Figure 5(c) does not show differences in TGF-β in undisturbed skin, it should be noted that IL-6 levels,79 as well as those of TGF-β (Fig. 5c) in unwounded skin are extremely low to nearly undetectable. Thus, differences in skin thickness may reflect involvement of IL-6 in TGF-β production during skin development. Like TGF-β, the TGF-β receptor was differentially modulated within unwounded skin but not so following wounding. Staining of intradermal injection samples showed TGF-βR2 to be predominantly localized within the epidermis, which consists primarily of keratinocytes. It may be speculated that perhaps during development IL-6 induces epidermal differentiation, then this paradoxically increases basal cell proliferation, with an end result being thicker epidermis and stratum corneum. This may also indicate that IL-6 has the ability to modulate the receptor in epidermal keratinocytes from unwounded skin, which may cause increased proliferation of these cells in the wound periphery. Indeed, the thickness of the epidermal sheet increases proximal to the wound edge in wild-type and less so in IL-6KO mice.38 However, further research will be required to elucidate definitively the role of IL-6 and its relationship to TGF-β in skin development.
Aside from a possible role in skin development, these data may indicate a new association of IL-6 with TGF-β in pathologies associated with fibrosis. Both IL-6 and TGF-β are well-known fibrotic cytokines, and numerous fibrotic skin conditions are characterized by the coexpression of these cytokines. For instance, skin surrounding regressing melanoma is highly fibrotic and displays increased expression of IL-6 and TGF-β levels, whereas the skin around halo naevi is not fibrotic and does not show elevated expression of these cytokines.80 Damage by ionizing radiation also causes fibrotic changes that appear to be linked to IL-6 and TGF-β expression (review81). Skin fibrosis, as a well-known result of systemic sclerosis and increased levels of Th2 cytokines such as IL-6, appears to be associated with elevated TGF-β production.82-84 Indeed, if levels of these cytokines decrease, the prognosis improves.85 When viewed in the context of the current study, the modulation of IL-6 may be a viable pathway for the development of a therapeutic intervention in fibrotic pathways. However, further investigation will be required as the exact relationship between IL-6 and TGF-β in collagen deposition and fibrosis is not yet known.
Overall, this study demonstrates an association between the IL-6 and TGF-β1 expression in the skin and IL-6KO fibroblasts. These results show that IL-6 has the ability to modulate significantly TGF-β1 mRNA expression in dermal fibroblasts and unwounded skin. However, an additional layer of regulation for this gene appears to be at play during wound healing, as protein levels do not necessarily correspond with mRNA. We provide further evidence that demonstrates that IL-6 modulates TGF-β1 through the Mapk/Erk pathway and may display crosstalk with Jak/Stat by inhibiting its activation through serine phosphorylation. Further studies are needed to determine the molecular mechanism of IL-6 signalling in repair and to determine if other repair-specific genes are modulated.
Acknowledgments
The authors wish to thank Bethany Mickle for her scientific and technical assistance. This work was funded by NIH/NIGMS grant R01 GM067745.
Footnotes
Conflicts of interest None declared.
References
- 1.Bandyopadhyay P. Novel therapies in diabetes mellitus. Drug News Perspect. 2006;19:499–507. [PubMed] [Google Scholar]
- 2.Menke A, Muntner P, Wildman RP, et al. Measures of adiposity and cardiovascular disease risk factors. Obesity (Silver Spring) 2007;15:785–95. doi: 10.1038/oby.2007.593. [DOI] [PubMed] [Google Scholar]
- 3.Bello YM, Phillips TJ. Recent advances in wound healing. J Am Med Assoc. 2000;283:716–18. doi: 10.1001/jama.283.6.716. [DOI] [PubMed] [Google Scholar]
- 4.Dennler S, Goumans MJ, ten Dijke P. Transforming growth factor beta signal transduction. J Leukoc Biol. 2002;71:731–40. [PubMed] [Google Scholar]
- 5.Verrecchia F, Mauviel A. Control of connective tissue gene expression by TGF beta: role of Smad proteins in fibrosis. Curr Rheumatol Rep. 2002;4:143–9. doi: 10.1007/s11926-002-0010-4. [DOI] [PubMed] [Google Scholar]
- 6.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 7.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–73. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Miyazawa K, Shinozaki M, Hara T, et al. Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells. 2002;7:1191–204. doi: 10.1046/j.1365-2443.2002.00599.x. [DOI] [PubMed] [Google Scholar]
- 9.Crowe MJ, Doetschman T, Greenhalgh DG. Delayed wound healing in immunodeficient TGF-beta 1 knockout mice. J Invest Dermatol. 2000;115:3–11. doi: 10.1046/j.1523-1747.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- 10.Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. 2000;257:180–9. doi: 10.1006/excr.2000.4869. [DOI] [PubMed] [Google Scholar]
- 11.Tredget EB, Demare J, Chandran G, et al. Transforming growth factor-beta and its effect on reepithelialization of partial-thickness ear wounds in transgenic mice. Wound Repair Regen. 2005;13:61–7. doi: 10.1111/j.1067-1927.2005.130108.x. [DOI] [PubMed] [Google Scholar]
- 12.Laplante AF, Germain L, Auger FA, et al. Mechanisms of wound reepithelialization: hints from a tissue-engineered reconstructed skin to long-standing questions. FASEB J. 2001;15:2377–89. doi: 10.1096/fj.01-0250com. [DOI] [PubMed] [Google Scholar]
- 13.Levine JH, Moses HL, Gold LI, et al. Spatial and temporal patterns of immunoreactive transforming growth factor beta 1, beta 2, and beta 3 during excisional wound repair. Am J Pathol. 1993;143:368–80. [PMC free article] [PubMed] [Google Scholar]
- 14.Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002;118:211–15. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 15.Annoni G, Weiner FR, Zern MA. Increased transforming growth factor-beta 1 gene expression in human liver disease. J Hepatol. 1992;14:259–64. doi: 10.1016/0168-8278(92)90168-o. [DOI] [PubMed] [Google Scholar]
- 16.Saunier EF, Akhurst RJ. TGF beta inhibition for cancer therapy. Curr Cancer Drug Targets. 2006;6:565–78. doi: 10.2174/156800906778742460. [DOI] [PubMed] [Google Scholar]
- 17.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–8. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 18.Gacka M, Adamiec J. The role of transforming growth factor-beta in the pathogenesis of diabetic retinopathy. Przegl Lek. 2006;63:296–8. [In Polish] [PubMed] [Google Scholar]
- 19.Sehgal PB. Interleukin-6: molecular pathophysiology. J Invest Dermatol. 1990;94:S2–6. doi: 10.1111/1523-1747.ep12874963. [DOI] [PubMed] [Google Scholar]
- 20.Paquet P, Pierard GE. Interleukin-6 and the skin. Int Arch Allergy Immunol. 1996;109:308–17. doi: 10.1159/000237257. [DOI] [PubMed] [Google Scholar]
- 21.Grossman RM, Krueger J, Yourish D, et al. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc Natl Acad Sci USA. 1989;86:6367–71. doi: 10.1073/pnas.86.16.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koch AE, Kronfeld-Harrington LB, Szekanecz Z, et al. In situ expression of cytokines and cellular adhesion molecules in the skin of patients with systemic sclerosis. Their role in early and late disease. Pathobiology. 1993;61:239–46. doi: 10.1159/000163802. [DOI] [PubMed] [Google Scholar]
- 23.Fugger L, Morling N, Bendtzen K, et al. IL-6 gene polymorphism in rheumatoid arthritis, pauciarticular juvenile rheumatoid arthritis, systemic lupus erythematosus, and in healthy Danes. J Immunogenet. 1989;16:461–5. doi: 10.1111/j.1744-313x.1989.tb00495.x. [DOI] [PubMed] [Google Scholar]
- 24.Sawamura D, Meng X, Ina S, et al. Induction of keratinocyte proliferation and lymphocytic infiltration by in vivo introduction of the IL-6 gene into keratinocytes and possibility of keratinocyte gene therapy for inflammatory skin diseases using IL-6 mutant genes. J Immunol. 1998;161:5633–9. [PubMed] [Google Scholar]
- 25.Turksen K, Kupper T, Degenstein L, et al. Interleukin 6: insights to its function in skin by overexpression in transgenic mice. Proc Natl Acad Sci USA. 1992;89:5068–72. doi: 10.1073/pnas.89.11.5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallucci RM, Sugawara T, Yucesoy B, et al. Interleukin-6 treatment augments cutaneous wound healing in immunosuppressed mice. J Interferon Cytokine Res. 2001;21:603–9. doi: 10.1089/10799900152547867. [DOI] [PubMed] [Google Scholar]
- 27.Lin ZQ, Kondo T, Ishida Y, et al. Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J Leukoc Biol. 2003;73:713–21. doi: 10.1189/jlb.0802397. [DOI] [PubMed] [Google Scholar]
- 28.Eickelberg O, Pansky A, Mussmann R, et al. Transforming growth factor-beta1 induces interleukin-6 expression via activating protein-1 consisting of JunD homodimers in primary human lung fibroblasts. J Biol Chem. 1999;274:12933–8. doi: 10.1074/jbc.274.18.12933. [DOI] [PubMed] [Google Scholar]
- 29.Aoki H, Ohnishi H, Hama K, et al. Existence of autocrine loop between interleukin-6 and transforming growth factor-beta1 in activated rat pancreatic stellate cells. J Cell Biochem. 2006;99:221–8. doi: 10.1002/jcb.20906. [DOI] [PubMed] [Google Scholar]
- 30.National Institutes for Health . Guide for the Care and Use of Laboratory Animals. NIH; Bethesda, MD: 1985. NIH publication no. 86-23. [Google Scholar]
- 31.Gallucci RM, Sloan DK, Heck JM, et al. Interleukin 6 indirectly induces keratinocyte migration. J Invest Dermatol. 2004;122:764–72. doi: 10.1111/j.0022-202X.2004.22323.x. [DOI] [PubMed] [Google Scholar]
- 32.Tomasek JJ, Halliday NL, Updike DL, et al. Gelatinase A activation is regulated by the organization of the polymerized actin cytoskeleton. J Biol Chem. 1997;272:7482–7. doi: 10.1074/jbc.272.11.7482. [DOI] [PubMed] [Google Scholar]
- 33.Song H, Wang R, Wang S, et al. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci USA. 2005;102:4700–5. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomasek JJ, McRae J, Owens GK, et al. Regulation of alpha-smooth muscle actin expression in granulation tissue myofibroblasts is dependent on the intronic CArG element and the transforming growth factor-beta1 control element. Am J Pathol. 2005;166:1343–51. doi: 10.1016/s0002-9440(10)62353-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Tomasek JJ, Gabbiani G, Hinz B, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 37.Luckett LR, Gallucci RM. Interleukin-6 (IL-6) modulates migration and matrix metalloproteinase function in dermal fibroblasts from IL-6KO mice. Br J Dermatol. 2007;156:1163–71. doi: 10.1111/j.1365-2133.2007.07867.x. [DOI] [PubMed] [Google Scholar]
- 38.Gallucci RM, Simeonova PP, Matheson JM, et al. Impaired cutaneous wound healing in interleukin-6-deficient and immunosuppressed mice. FASEB J. 2000;14:2525–31. doi: 10.1096/fj.00-0073com. [DOI] [PubMed] [Google Scholar]
- 39.Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83:835–70. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 40.Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci. 2004;9:283–9. doi: 10.2741/1184. [DOI] [PubMed] [Google Scholar]
- 41.Heinrich PC, Behrmann I, Haan S, et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gold LI, Sung JJ, Siebert JW, et al. Type I (RI) and type II (RII) receptors for transforming growth factor-beta isoforms are expressed subsequent to transforming growth factor-beta ligands during excisional wound repair. Am J Pathol. 1997;150:209–22. [PMC free article] [PubMed] [Google Scholar]
- 43.McCartney-Francis NL, Wahl SM. Transforming growth factor beta: a matter of life and death. J Leukoc Biol. 1994;55:401–9. doi: 10.1002/jlb.55.3.401. [DOI] [PubMed] [Google Scholar]
- 44.Martin P. Wound healing—aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- 45.Ghosh AK, Mori Y, Dowling E, et al. Trichostatin A blocks TGF-beta-induced collagen gene expression in skin fibroblasts: involvement of Sp1. Biochem Biophys Res Commun. 2007;354:420–6. doi: 10.1016/j.bbrc.2006.12.204. [DOI] [PubMed] [Google Scholar]
- 46.Lijnen HR, Maquoi E, Demeulemeester D, et al. Modulation of fibrinolytic and gelatinolytic activity during adipose tissue development in a mouse model of nutritionally induced obesity. Thromb Haemost. 2002;88:345–53. [PubMed] [Google Scholar]
- 47.Wang XJ, Han G, Owens P, et al. Role of TGF beta-mediated inflammation in cutaneous wound healing. J Investig Dermatol Symp Proc. 2006;11:112–17. doi: 10.1038/sj.jidsymp.5650004. [DOI] [PubMed] [Google Scholar]
- 48.Montesano R, Orci L. Transforming growth factor beta stimulates collagen-matrix contraction by fibroblasts: implications for wound healing. Proc Natl Acad Sci USA. 1988;85:4894–7. doi: 10.1073/pnas.85.13.4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gallucci RM, Lee EG, Tomasek JJ. IL-6 modulates alpha-smooth muscle actin expression in dermal fibroblasts from IL-6-deficient mice. J Invest Dermatol. 2006;126:561–8. doi: 10.1038/sj.jid.5700109. [DOI] [PubMed] [Google Scholar]
- 50.Zhang XL, Topley N, Ito T, et al. Interleukin-6 regulation of transforming growth factor (TGF)-beta receptor compartmentalization and turnover enhances TGF-beta1 signaling. J Biol Chem. 2005;280:12239–45. doi: 10.1074/jbc.M413284200. [DOI] [PubMed] [Google Scholar]
- 51.Becker C, Fantini MC, Schramm C, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 52.Karres I, Kremer JP, Steckholzer U, et al. Transforming growth factor-beta 1 inhibits synthesis of cytokines in endotoxin-stimulated human whole blood. Arch Surg. 1996;131:1310–16. doi: 10.1001/archsurg.1996.01430240064008. discussion 6–7. [DOI] [PubMed] [Google Scholar]
- 53.Chen Y, Lu L, Wang L. Study on gene expression of TGF beta 1 and its receptor in leukemia cells and the serum TGF beta 1 level in the patients with acute leukemia. Zhonghua Xue Ye Xue Za Zhi. 1998;19:576–80. [In Chinese] [PubMed] [Google Scholar]
- 54.Clark RA, Ashcroft GS, Spencer MJ, et al. Re-epithelialization of normal human excisional wounds is associated with a switch from alpha v beta 5 to alpha v beta 6 integrins. Br J Dermatol. 1996;135:46–51. [PubMed] [Google Scholar]
- 55.Frank S, Madlener M, Werner S. Transforming growth factors beta1, beta2, and beta3 and their receptors are differentially regulated during normal and impaired wound healing. J Biol Chem. 1996;271:10188–93. doi: 10.1074/jbc.271.17.10188. [DOI] [PubMed] [Google Scholar]
- 56.Kehrl JH, Wakefield LM, Roberts AB, et al. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. 1986;163:1037–50. doi: 10.1084/jem.163.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ikeda T, Lioubin MN, Marquardt H. Human transforming growth factor type beta 2: production by a prostatic adenocarcinoma cell line, purification, and initial characterization. Biochemistry. 1987;26:2406–10. doi: 10.1021/bi00383a002. [DOI] [PubMed] [Google Scholar]
- 58.Glick AB, Flanders KC, Danielpour D, et al. Retinoic acid induces transforming growth factor-beta 2 in cultured keratinocytes and mouse epidermis. Cell Regul. 1989;1:87–97. doi: 10.1091/mbc.1.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim SJ, Park K, Koeller D, et al. Post-transcriptional regulation of the human transforming growth factor-beta 1 gene. J Biol Chem. 1992;267:13702–7. [PubMed] [Google Scholar]
- 60.Dudley DT, Pang L, Decker SJ, et al. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–9. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dhandapani KM, Khan MM, Wade FM, et al. Induction of transforming growth factorbeta1 by basic fibroblast growth factor in rat C6 glioma cells and astrocytes is mediated by MEK/ERK signaling and AP-1 activation. J Neurosci Res. 2007;85:1033–45. doi: 10.1002/jnr.21182. [DOI] [PubMed] [Google Scholar]
- 62.Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;149:1–38. doi: 10.1007/s10254-003-0012-2. [DOI] [PubMed] [Google Scholar]
- 63.David M, Petricoin E, III, Benjamin C, et al. Requirement for MAP kinase (ERK2) activity in interferon alpha- and interferon beta-stimulated gene expression through STAT proteins. Science. 1995;269:1721–3. doi: 10.1126/science.7569900. [DOI] [PubMed] [Google Scholar]
- 64.Chung J, Uchida E, Grammer TC, et al. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17:6508–16. doi: 10.1128/mcb.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wen Z, Zhong Z, Darnell JE., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–50. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 66.Zhang X, Blenis J, Li HC, et al. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 1995;267:1990–4. doi: 10.1126/science.7701321. [DOI] [PubMed] [Google Scholar]
- 67.Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–37. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- 68.Sengupta TK, Talbot ES, Scherle PA, et al. Rapid inhibition of interleukin-6 signaling and Stat3 activation mediated by mitogen-activated protein kinases. Proc Natl Acad Sci USA. 1998;95:11107–12. doi: 10.1073/pnas.95.19.11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinez-Salgado C, Fuentes-Calvo I, Garcia-Cenador B, et al. Involvement of H- and N-Ras isoforms in transforming growth factor-beta1-induced proliferation and in collagen and fibronectin synthesis. Exp Cell Res. 2006;312:2093–106. doi: 10.1016/j.yexcr.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 70.Grimm OH, Gurdon JB. Nuclear exclusion of Smad2 is a mechanism leading to loss of competence. Nat Cell Biol. 2002;4:519–22. doi: 10.1038/ncb812. [DOI] [PubMed] [Google Scholar]
- 71.Kretzschmar M, Doody J, Timokhina I, et al. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–16. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neugarten J, Medve I, Lei J, et al. Estradiol suppresses mesangial cell type I collagen synthesis via activation of the MAP kinase cascade. Am J Physiol. 1999;277:F875–81. doi: 10.1152/ajprenal.1999.277.6.F875. [DOI] [PubMed] [Google Scholar]
- 73.Reunanen N, Foschi M, Han J, et al. Activation of extracellular signal-regulated kinase 1/2 inhibits type I collagen expression by human skin fibroblasts. J Biol Chem. 2000;275:34634–9. doi: 10.1074/jbc.C000175200. [DOI] [PubMed] [Google Scholar]
- 74.Lee SY, Lee JW, Lee H, et al. Inhibitory effect of green tea extract on beta-amyloid-induced PC12 cell death by inhibition of the activation of NF-kappaB and ERK/p38 MAP kinase pathway through antioxidant mechanisms. Brain Res Mol Brain Res. 2005;140:45–54. doi: 10.1016/j.molbrainres.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 75.Yang LY, Ko WC, Lin CM, et al. Antioxidant N-acetylcysteine blocks nerve growth factor-induced H2O2/ERK signaling in PC12 cells. Ann N Y Acad Sci. 2005;1042:325–37. doi: 10.1196/annals.1338.056. [DOI] [PubMed] [Google Scholar]
- 76.Arany I, Megyesi JK, Nelkin BD, et al. STAT3 attenuates EGFR-mediated ERK activation and cell survival during oxidant stress in mouse proximal tubular cells. Kidney Int. 2006;70:669–74. doi: 10.1038/sj.ki.5001604. [DOI] [PubMed] [Google Scholar]
- 77.Assoian RK, Komoriya A, Meyers CA, et al. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. 1983;258:7155–60. [PubMed] [Google Scholar]
- 78.Wang XP, Schunck M, Kallen KJ, et al. The interleukin-6 cytokine system regulates epidermal permeability barrier homeostasis. J Invest Dermatol. 2004;123:124–31. doi: 10.1111/j.0022-202X.2004.22736.x. [DOI] [PubMed] [Google Scholar]
- 79.Gallucci RM, O’Dell SK, Rabe D, et al. JP-8 jet fuel exposure induces inflammatory cytokines in rat skin. Int Immunopharmacol. 2004;4:1159–69. doi: 10.1016/j.intimp.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 80.Moretti S, Spallanzani A, Pinzi C, et al. Fibrosis in regressing melanoma versus nonfibrosis in halo nevus upon melanocyte disappearance: could it be related to a different cytokine microenvironment? J Cutan Pathol. 2007;34:301–8. doi: 10.1111/j.1600-0560.2006.00616.x. [DOI] [PubMed] [Google Scholar]
- 81.Muller K, Meineke V. Radiation-induced alterations in cytokine production by skin cells. Exp Hematol. 2007;35:96–104. doi: 10.1016/j.exphem.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 82.Smith EA. Connective tissue metabolism including cytokines in scleroderma. Curr Opin Rheumatol. 1992;4:869–77. [PubMed] [Google Scholar]
- 83.Christner PJ, Jimenez SA. Animal models of systemic sclerosis: insights into systemic sclerosis pathogenesis and potential therapeutic approaches. Curr Opin Rheumatol. 2004;16:746–52. doi: 10.1097/01.bor.0000137893.68929.86. [DOI] [PubMed] [Google Scholar]
- 84.Hasegawa M, Fujimoto M, Takehara K, et al. Pathogenesis of systemic sclerosis: altered B cell function is the key linking systemic autoimmunity and tissue fibrosis. J Dermatol Sci. 2005;39:1–7. doi: 10.1016/j.jdermsci.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 85.Matsushita T, Hasegawa M, Hamaguchi Y, et al. Longitudinal analysis of serum cytokine concentrations in systemic sclerosis: association of interleukin 12 elevation with spontaneous regression of skin sclerosis. J Rheumatol. 2006;33:275–84. [PubMed] [Google Scholar]